

Abstract

One of the attractive properties of artemisinins is their extremely fast-killing capability, quickly relieving malaria symptoms. Nevertheless, the unique benefits of these medicines are now compromised by the prolonged parasite clearance times and the increasing frequency of treatment failures, attributed to the increased tolerance of Plasmodium falciparum to artemisinin. This emerging artemisinin resistance threatens to undermine the effectiveness of antimalarial combination therapies. Herein, we describe the medicinal chemistry efforts focused on a cGMP-dependent protein kinase (PKG) inhibitor scaffold, leading to the identification of novel chemical entities with very potent, similar to artemisinins, fast-killing potency against asexual blood stages that cause disease, and activity against gametocyte activation that is required for transmission. Furthermore, we confirm that selective PKG inhibitors have a slow speed of kill, while chemoproteomic analysis suggests for the first time serine/arginine protein kinase 2 (SRPK2) targeting as a novel strategy for developing antimalarial compounds with extremely fast-killing properties.
Introduction
Malaria, an infectious disease caused by parasites of the genus Plasmodium (Plasmodium falciparum and Plasmodium vivax are responsible for most of the clinical cases), is a major healthcare challenge, especially in developing countries. According to the 2018 World Health Organization (WHO) global malaria report, in 2017, there were an estimated 219 million cases of malaria, an increase of about 8 million cases over 2015, with deaths reaching 435 000, a number similar to the previous year. It is clear that the steep decline in mortality and disease burden observed between 2000 and 2015 has now been replaced by a plateau. Even more disturbing is the fact that of these people, more than two-thirds were children under 5 years of age and expectant mothers.1 Malaria control programs are currently focused on two pillars, namely, disease prevention by vector control and disease treatment with artemisinin-combination therapies (ACTs).2,3 Artemisinins (1, Chart 1) are extremely fast-killing agents, quickly relieving malaria symptoms. Nevertheless, the unique properties of these medicines are compromised by prolonged parasite clearance times and the increasing frequency of treatment failures, attributed to the increased tolerance of P. falciparum to artemisinin.4−7 These emerging problems have started to raise concerns about the effectiveness of this widely administered class of antimalarial drugs.8,9 As a consequence, the development of new bioactive molecules endowed with novel mechanisms of action has garnered the attention of both academia and industry. However, the biggest challenge in developing medicines to replace artemisinins is the identification of new chemical entities displaying parasite killing kinetics as fast as artemisinins. Such an achievement is a very tough and demanding task, given that no compound, to the best of our knowledge, with fast-killing properties similar to or better than those of artemisinin derivatives (1 and artesunate, Chart 1) has been reported in the literature thus far.
Chart 1. Structures of Artemisinin-Based Drugs and Compounds with Potent Inhibitory Activity against PKG (2–6).

The cyclic GMP-activated serine–threonine protein kinase, PKG, has been shown to play an essential role in all of the key stages of the complex parasite life cycle, including blood stage replication in the human host as well as gametogenesis and ookinete motility in the mosquito vector.10−12 In addition, it has been shown to be key for sporozoite motility, liver cell invasion, and late liver stage development.13−15 In the blood stages, PKG regulates the release of proteins from apical organelles and the mobilization of calcium required for merozoite egress and invasion.10 Using phosphoproteomics, PKG has also been shown to act as a crucial signaling hub in a number of the malaria parasite’s core processes required for egress and invasion.16 Thus, it can be inferred that targeting PKG is a tractable and multifaceted strategy for malaria intervention, and developing PKG inhibitors should be considered as a component of a promising alternative approach to combat malaria.
The in vivo proof of principle of using PKG inhibitors against malaria has been established recently, where an imidazopyridine PKG inhibitor (2, Chart 1) was able to clear infection in the GSK P. falciparum humanized mouse model and block transmission.17,18 The development of these compounds was based on structure–activity relationship (SAR) studies using the imidazopyridine compound 3 (Chart 1) as a lead. Compound 3 was originally developed by Merck for the treatment of coccidiosis caused by Eimeria infection,19 with compound 4 serving as the starting point.20 Thiazoles (e.g., compounds 5 and 6, Chart 1) constitute another class of PKG inhibitors,21 identified in the context of scaffold-hopping approaches conducted on the pyrrole analogue 3 (Chart 1).20
Despite the very promising antimalarial potential of PKG inhibitors, parasite reduction ratio (PRR) studies using the most potent and selective imidazopyridine and thiazole derivatives 2 (in a previous study)17 and 5 (in the context of this study, Chart 1), respectively, clearly showed that both analogues suffer from slow parasite killing kinetics. Mindful of the aforementioned, the aim of this study was to refine those structural determinants to provide the thiazole pharmacophore with fast-killing activity through the application of molecular diversity-oriented SAR and (bio)isosterism approaches. Toward this end, state-of-the-art medicinal chemistry strategies accompanied by cell-based assays and chemoproteomic approaches were applied. The present research delineates the optimization and the mode of action of a novel series of thiazole derivatives endowed with fast-killing properties which are similar to or slightly better than artesunate (Chart 1), the best fast-killing drug available so far. It aspires at the same time to create novel chemotype leads for further development, with the ultimate goal of identifying novel fast-killing agents with “druglike” properties against malaria.
Results and Discussion
Chemistry
The reference compounds 2–5 were synthesized according to previously published procedures.17−21 The new thiazole derivatives designed were synthesized following modified procedures described in the literature (scheme 1–3). Special emphasis was placed on the development of routes that are amenable to parallel synthesis for the exploration of extended SAR studies. First, a library of 52 compounds (6–57) with a molecular diversity at the 2-position of the pyrimidine ring and 2-position of the core-thiazole ring were made (Schemes 1 and 2). Starting from 4-methyl-2-(methylthio/chloro)pyrimidines a1 and a2, deprotonation followed by reaction with 4-fluoro-N-methoxy-N-methylbenzamide b gave the respective ketones c1 and c2, which upon α-chlorination with sulfuryl chloride in chloroform afforded the chlorides d1 and d2, respectively.22,23 Condensation of d1 and d2 with either numerous different substituted thioamides or thiourea in refluxing ethanol produced the corresponding 2-substituted thiazoles of structure e, g, and j (Schemes 1 and 2).22,23 The thioether group of e1 was subsequently oxidized to furnish derivatives of formula f (Scheme 1).22,23 An SNAr reaction between f and different (hetero)aromatic or aliphatic amines afforded the majority of the final compounds. Where the SNAr reaction was found to be inefficient, a Buchwald-type C-N palladium-catalyzed cross-coupling was performed using the respective chlorides e2 (Scheme 1).24 A few derivatives (9–18) were synthesized by slightly modifying the aforementioned synthetic procedure (Scheme 2).
Scheme 1. Synthetic Procedure Followed for the Preparation of the Thiazole Derivatives 6–8 and 19–57.

Reagents and conditions: (i) LDA, THF, −78 °C to rt, (ii) SO2Cl2, CHCl3, 0 °C to rt, (iii) EtOH, reflux, (iv) Oxone, MeOH:H2O, rt, (v) NaH, THF, rt or iPrOH, catalytic HCl/dioxane, 120 °C or DMSO, 100 °C (depending on amine’s basicity), (vi) Pd2(dba)3, XantPhos, t-BuOK, toluene, reflux, (vii) LiOH·H2O, THF:EtOH:H2O, reflux.
Scheme 3. Synthetic Route Used for the Preparation of the Thiazole Derivatives 58–60.

Reagents and conditions: (i) chloroacetaldehyde, acetone, reflux, (ii) NBS, DMF, rt, (iii) LDA, −78 °C to rt, (iv) Pd(OAc)2, XPhos or cataCXiumA, CsCO3, THF:H2O or toluene:H2O, reflux, (v) NIS, catalytic CF3COOH, CH3CN, rt or I2, n-BuLi, THF, −78 °C to rt, (vi) Pd(PPh3)4, CuI, DMF, 70 °C, (vii) Pd2(dba)3, XantPhos, t-BuOK, toluene, reflux, (viii) 4 M HCl/dioxane, dioxane, rt.
Scheme 2. Synthetic Procedure Followed for the Preparation of the Thiazole Derivatives 9–18.

Reagents and conditions: (i) EtOH, reflux, (ii) CuCl2, t-BuONO, CH3CN, rt, (iii) morpholine or pyrrolidine, THF, rt, (iv) m-CPBA, CH2Cl2, rt, (v) 1-Boc-4-(4-aminophenyl)piperazine, TFA, iPrOH, reflux, (vi) TFA or 4 N HCl/dioxane, iPrOH, reflux, (vii) RCOCl or RSO2Cl, Et3N, CH2Cl2, or HATU, DIPEA, DMF.
Regarding the synthesis of analogues 58–60 (Scheme 3), the 4-bromo-2-(4-piperidinyl)thiazole analogue l was initially built from N-Boc-piperidinethioamide. Thiazole ring formation followed by bromination at 5-position using NBS gave k,25 which subsequently afforded l under halogen-dance conditions.26 A Suzuki cross-coupling reaction between l and the substituted trifluoroborates produced analogues of formula m, which upon iodination and Stille cross-coupling reactions furnished n and o, respectively. A Buchwald cross-coupling24 between o and 4-amino-2-methylpyridine or 4-aminopyrimidine followed by Boc-deprotection under acidic conditions finally led to the final products 58–60. The isostere of the thiazole derivative 28A, oxazole analogue 28B, was made according to the synthetic procedure depicted in Scheme S1.
SAR Studies and Identification of Compounds with Fast-Killing Properties
Our efforts toward developing novel antimalarial compounds with potent fast-killing properties were first focused on performing SAR exploration using compound 6(21) as the starting point (Chart 1). The in vitro inhibition of recombinant PKG activity as well as the antiparasitic activity of each compound synthesized were evaluated using a kinase inhibition assay and a cell-based P. falciparum asexual blood stage growth inhibition assay, respectively. In parallel, several compounds were subjected to a preliminary characterization of the main physicochemical parameters potentially affecting the “druglike” properties of a bioactive molecule (lipophilicity, aqueous solubility, cytotoxicity, cardiotoxicity, plasma protein binding, permeability, and metabolic clearance). Our ultimate goal was to determine those structural and molecular features conferring fast-acting potency as well as favorable biopharmaceutical properties at the same time, and to come up with a promising lead compound that would be amenable to further optimization/development.
The first structural part of compound 6 investigated was the substitution at position 2 of the thiazole ring (Table 1). It seems that the substitution at this position does not significantly affect the inhibition of PKG activity since both the unsubstituted amino derivatives 11–13 and other structural motifs (7, 9, 14, 16–18) are well tolerated, with the exception of the amide derivative 15. On the other hand, 2-substitution plays a significant role in the antiplasmodial activity exhibited in cells (Pf EC50) since the unsubstituted analogues 11 and 12 are less potent compared to 7 and 6, respectively. The pyrrolidine (10) and acetyl (14) derivatives displayed similar cell potency, while other acetyl (15, 16) and sulfonamide (17, 18) derivatives either did not improve or abolished potency. It was also concluded from this SAR study that the basic nitrogen of piperidine of reference compounds 6 and 7 is crucial for cell potency as decrease of its basicity significantly reduced potency (8). Of note, the most potent analogues of this library (7, 10, and 14) showed a very promising metabolic stability (clearance in mouse, rat, and human liver microsomes), permeability (artificial membrane permeability, AMP), and cardiotoxicity (hERG inhibition) profile, whereas their cytotoxicity liability was subject to further optimization. In general, in the AMP permeability assay, compounds with values >0.05 × 10–5 cm/s are considered high permeable, between 0.01 and 0.05 × 10–5 moderate permeable, whereas <0.01 × 10–5 cm/s low permeable.27
Table 1. SAR Studies Conducted at Position 2 of the Thiazole Ring (R) and Piperidine Ring (X).
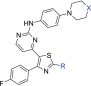

With the objective of improving both antiparasitic activity and cytotoxicity, our interest then focused on investigating the impact of the amino substitution of the pyrimidine ring of 6 and 7 on their biopharmaceutical profile. Toward this end, the bulky and lipophilic 4-piperazinyl-phenyl ring was replaced with heteroaromatic substituents endowed with molecular diversity (Table 2). From the experimental data shown in Table 2, it can be deduced that although all compounds tested are very potent PKG inhibitors in vitro, the activity in cells is strictly dependent on their electrostatic and shape/steric complementarities. The most active compounds that emerged were the pyrimidine and 2-methylpyridine derivatives 28A and 31, respectively. Notably, in the case of 28A, the number and the position of nitrogen atoms in the heteroaromatic ring are of paramount importance since the respective pyridine (25), pyrazine (26), and pyridazine (27) analogues exhibited much lower cell activity (Table 2). In addition, small substituents at the pyrimidine ring (29, 30) did not favor an increase in potency, while isosterism (different five-membered aromatic rings, 19–23) was not applicable in this series, with the result that 19–23 offered at least 10-fold decreased potency. Moreover, in a scaffold-hopping attempt, the thiazole core ring was replaced by its respective isostere oxazole (28B, Table 2). Interestingly, this modification in spite of the better solubility achieved (226 μM), resulted in decreased PKG inhibitory activity (IC50 = 89.1 nM), abolishing at the same time the antiparasitic activity (EC50 > 5 μM). Regarding 31, small substituents around the 2-methylpyridine ring (32, 34) were not detrimental, without however offering any significant improvement in potency. Finally, other (poly)substituted pyridine derivatives (35–40) did not give the desired result. After further evaluating the most potent analogues of this library (28A and 31), it was found that the incorporation of pyrimidine and 2-methylpyridine, besides increasing potency (compared to reference compound 6), also delivered molecules with very good solubility, permeability, and metabolic stability, although at the expense of potent inhibition against the cardiac ion channel hERG (IC50 values for 28A and 31 are 0.6 and 1.0 μM, respectively, Table 2). Moreover, the cytotoxicity/cell activity safety windows of both analogues were significantly improved (36 and 25 for compounds 28A and 31, respectively, compared to 6.8 and 5.4 for the reference compounds 6 and 7, respectively).
Table 2. SAR Studies Conducted at Position 2 of the Pyrimidine Ring of the Thiazole Scaffold (Aromatic Substitution, Ar).


Another desirable pharmacological feature of this series concerns their potential to act as transmission-blocking agents (Tables 1 and 2). The capability of compounds to prevent mature male and female gametocytes from activation and progressing to extracellular gametes (both of which are considered indicators of gametocyte functionality) was estimated using specialized bioassays.28 The activation of male gametocytes to differentiate into mature microgametes was evaluated by measuring levels of exflagellation (extrusion of rapidly waving flagellated microgametes from the infected erythrocyte), whereas the activation of female gametocytes was based on the specific expression of the Pfs25 protein at the surface of the female gamete following activation. The potent transmission-blocking effect exerted by the majority of the compounds can likely be ascribed to the powerful specific PKG inhibitory activity exhibited by the analogues tested. It has been previously demonstrated that PKG is essential for gametocyte activation and their transformation to gametes.1231, being one of the most promising compounds of this series, endowed with a balanced physicochemical and cell activity profile, also potently inhibited the activation of both male and female gametocytes with EC50 values of 0.30 and 0.40 μM, respectively.
Notwithstanding the liability of inhibiting hERG, 28A and 31 were subsequently tested in terms of their fast-acting properties in a parasite reduction ratio (PRR) study, using artesunate (fast rate of killing), pyrimethamine (moderate rate of killing), and atovaquone (slow rate of killing) for comparison. Surprisingly, both compounds exhibited an extremely fast-killing effect, displaying 87 and 93% clearance of parasites in the first 24 h, respectively. As depicted in Figure 1, this effect is similar to or slightly better than artesunate (Figure 1), the best fast-killing drug available so far.
Figure 1.

PRR study with compounds 28A and 31. Artesunate (fast rate of killing), pyrimethamine (moderate rate of killing), and atovaquone (slow rate of killing) have also been included.
Encouraged by the unprecedented fast-killing potency of 31, which is accompanied by a favorable aqueous solubility, permeability, and metabolic stability profile (Table 2), this compound was initially tested for other (besides hERG) secondary pharmacology-related liabilities (Table 3). It was found that no agonistic or antagonistic activity was exerted against several receptors and ion channels tested, while 31, except for inhibiting acetylcholinesterase (IC50 = 0.40 μM), did not exhibit any inhibitory activity against human monoamine oxidase A (MAOA), phosphodiesterase, and organic anion transport polypeptide OATP1B1 (Table 3).
Table 3. In Vitro Evaluation of Compound 31 Effect against Other Enzymes, Ion Channels, and Receptors.
| protein | activity (μM) |
|---|---|
| human monoamine oxidase A (MAOA) inhibitor | IC50 > 100 |
| β2 adrenoreceptor human agonist | EC50 > 100 |
| β2 adrenoreceptor human antagonist | IC50 > 100 |
| human PXR (NR1I2) agonist | EC50 > 50 |
| human KCNQ1/KCNE1 (Kv7.1/MinK) blocker | IC50 > 25 |
| human aryl hydrocarbon receptor (AhR) agonist | EC50 > 50 |
| organic anion transport polypeptide OATP1B1 inhibitor | IC50 > 30 |
| acetylcholinesterase (AChE) inhibitor | IC50 = 0.40 |
| phosphodiesterase 3A (PDE3A) inhibitor | IC50 > 100 |
| human CaV1.2 (L-type) calcium channel blocker | IC50 > 30 |
Our approach then focused on the identification of a more developable fast-killing agent, with similar or better antiparasitic activity in cells, and even further reduced cytotoxicity and/or potency against hERG. Our strategy to this end was based on the reduction of the numbers of aromatic rings of 31, on the grounds that the more aromatic rings a compound has, the more chance there is for this agent to exert toxicity. For synthetic reasons, our efforts were first focused on replacing the 2-methylpyridine heteroaromatic ring of 31 with a large number of nonaromatic/aliphatic groups (Table 4). Compound 5 (Chart 1) was included for comparison. According to the data depicted in Table 4, these structural modifications resulted in a great improvement of cytotoxicity (41–57), which was however counterbalanced by the significant drop (10- to 50-fold) in the antiplasmodial potency observed. Although the permeability of all derivatives was maintained at acceptable levels (Table 4, AMP values), none of the newly synthesized compounds 41–57 displayed better activity than 5, 28A, and 31.
Table 4. SAR Studies Conducted at Position 2 of the Pyrimidine Ring of the Thiazole Scaffold (Nonaromatic Substitution, R).
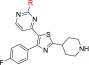

Additionally, PRR studies conducted with 5 clearly showed that it is a slow-acting compound (Figure 2), rendering the heteroaromatic substitution at this specific position of the thiazole-pyrimidine scaffold a key player toward the identification of fast-killing antimalarial compounds.
Figure 2.

PRR study with compounds 5 and 28A. Artesunate (fast rate of killing), pyrimethamine (moderate rate of killing), and atovaquone (slow rate of killing) have also been included.
After having experimentally confirmed that position 2 of the pyrimidine ring is not amenable to aliphatic/nonaromatic substitution, furnishing slow-acting analogues, a preliminary exploration of position 4 of the thiazole ring was performed. By use of similar fundamental design principles, the lipophilic para-fluorophenyl group of 28A and 31 was replaced by the smaller cyclopropyl group endowed with an sp2 character (Table 5). Somewhat surprisingly, despite the outstanding improvement in cytotoxicity noted by 58 (EC50 = 43.7 μM), the antiplasmodial potency was decreased by 23-fold (EC50 = 3.45 μM) compared to 28A (EC50 = 0.15 μM). Intriguingly, the same group preserved (compound 59, IC50 = 0.27 μM) the antiparasitic activity of 31 (IC50 = 0.16 μM), attenuating at the same time by 5-fold the hERG inhibitory activity and slightly (2.5-fold) the cytotoxicity (Table 5). Compound 59 also showed very good solubility, permeability, and metabolic clearance properties (Table 5). Finally, the potency was maintained (EC50 = 0.19 μM) and the cytotoxicity (EC50 = 20 μM, cytotoxicity/cell activity safety window = 105) was yet further improved by substituting the 4-position of the thiazole ring with the piperidinylmethyl group (compound 60, Table 5).
Table 5. Preliminary SAR Studies Conducted at Position 4 of the Thiazole Ring (R).
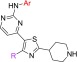

Furthermore, PRR studies using compounds 59 and 60 proved that both analogues are also endowed with potent fast-killing properties (with 59 displaying a better fast-killing profile than 60), exhibiting an effect similar to that of artesunate (Figure 3).
Figure 3.

PRR study with compounds 59 and 60. Artesunate (fast rate of killing), pyrimethamine (moderate rate of killing), and atovaquone (slow rate of killing) have also been included.
Additional Target Identification by Chemoproteomics
With the purpose of providing new insights into the mechanism of action of this series and specifically to investigate whether these potent fast-killing kinetics are attributed to a PKG-selective or an off-target (other than PKG) or a synergistic effect, assays utilizing the P. falciparum PKG gatekeeper mutant line (T618Q, Table 6)17 followed by chemoproteomic approaches were deployed. Initially, representative thiazole derivatives (26, 28A, 31, 32) were evaluated in the aforementioned transgenic cell line to determine whether PfPKG is their primary target in the asexual blood stages. In this transgenic cell line, the threonine residue at position 618 of the PfPKG catalytic site, which functions as a gatekeeper residue forming a small lipophilic pocket which is essential for the binding of small molecule inhibitors in the active site of the enzyme, has been replaced by the larger glutamine residue. As a consequence, entry to this small lipophilic pocket has been disrupted in the mutant line, preventing inhibitor binding to the catalytic site of PKG.17,29 Compound 5, which has been demonstrated by previous studies to exhibit high levels of potency against PKG in vitro and a remarkable selectivity over other human kinases,21 was also included for comparison.
Table 6. Inhibitory Activity of Representative Compounds against Plasmodium falciparum WT and Transgenic PKG T618Q Cell Lines at 48 and 72 h.


The results presented in Table 6 show that the potent inhibitory activity of 5 against parasite growth (EC50 = 110 nM) in the WT cell line at 72 h is significantly reduced (40-fold) in the T618Q mutant line (at 72 h). These experimental data strongly confirm that the potent antiparasitic activity in cells for this derivative is attributed to its selective inhibitory activity against PKG. Notably, the fact that 5 also exhibits a significantly decreased activity in the WT cell line at 48 h (EC50 = 1.02 μM) compared to 72 h (10-fold, Table 6) substantiates the PRR studies performed with 5 (Figure 2), classifying this analogue as a slow-acting agent. In particular, the loss of activity of 5 in the P. falciparum PKG gatekeeper mutant line (T618Q) at both 48 and 72 h (Table 6) further demonstrates that selective PKG inhibition is accompanied by the exhibition of slow parasite killing kinetics. In contrast, all of the new thiazole derivatives tested exerted a similar effect in both WT and mutant cell lines both at 48 and 72 h (Table 6), a signature of off-target (other than PKG) activity conferring the potent fast-killing properties of this series.
The above-mentioned results motivated us to investigate in more detail the molecular mechanism through which this powerful fast kill rate is exerted. Therefore, a chemoproteomic approach (Kinobeads profiling) was applied to identify the target(s) inhibited by one of the most potent derivatives of the series developed (31, Table 2). Compound 50 (Table 4), exhibiting an in vitro inhibitory potency against PKG (IC50 = 12.6 nM) comparable to 31, but a much weaker antiplasmodial activity in cells (EC50 = 4.03 μM), was also included to have a reliable monitoring framework in terms of the proteins whose inhibition is accompanied by potent antiparasitic/antimalarial activity and a fast-killing effect. In that way, any potential misinterpretation of the results could be avoided. Kinobeads represent a selection of immobilized promiscuous ATP-competitive kinase inhibitors,30,31 to affinity capture potential kinase target proteins from a P. falciparum protein extract. Using this approach, the activity of the compounds against 54 endogenous P. falciparum kinases was analyzed (Figure 4a). To explore additional potential kinase (Figure 4b) and nonkinase targets (Table S1), the compounds were attached to a bead matrix via their amine moiety and pull down assays were performed. All of the experiments were performed either without or with excess of 31 or 50 to identify target proteins for which capture is competitively inhibited. Both compounds were added in concentrations between 0.08 and 20 μM aiming at establishing a competition-binding curve and determining a half-maximal inhibitory concentration (IC50). The IC50 values obtained in this kind of experiment are representative of target affinity but are also affected by the target protein affinity for the bead-immobilized ligands. The latter effect can be deduced by determining the apparent dissociation constants (Kdapp), which are largely nondependent on the bead ligand, thus representing the depletion of the target proteins by the beads.32 The proteins captured by the beads were finally quantified by using isotope tagging of tryptic peptides and analyzed by LC-MS/MS.32
Figure 4.

Chemoproteomics profiling of compounds 31 and 50. (a) Both compounds were profiled on Kinobeads, which represent a combination of immobilized promiscuous ATP-competitive kinase inhibitors, in a P. falciparum protein extract. A total of 54 P. falciparum kinases were analyzed. The concentration of the “free” compounds used for competition, compound 50 and compound 31, was between 0.08 and 20 μM over six samples. (b) Compounds 50 and 31 were profiled with a bead matrix generated by immobilizing either compound 50 or compound 31 to beads, and competed with the respective “free” analogue over six concentrations between 0.08 and 20 μM. The Heatmaps show the protein kinases affected by any of the two compounds in two independent experiments, respectively. The values shown are apparent pKd values (blue: decreasing apparent pKd values; white: no competition; gray: protein not identified).
Based on our pharmacological results, both 31 (Table 2) and 50 (Table 4) are potent PKG inhibitors in vitro, but only 31 exhibits a strong effect in the P. falciparum growth inhibition assay at 48 h. Consequently, proteins preferentially binding to 31 and not to 50 are more likely to be potential targets that could lead to fast killing of the parasites. Capture experiments using Kinobeads showed that besides PKG, both 31 and 50 also inhibit calcium-dependent protein kinase 4 (CDPK4) with Kdapp values of 50 and 60 nM, respectively (Figure 4). Both compounds were also shown to bind to CDPK1 (Figure 4), with 31 exhibiting a much more potent inhibitory activity than 50 (Kdapp = 0.05 μM and Kdapp = 2.1 μM for 31 and 50, respectively). The importance of CDPK1 in the viability of P. falciparum during the erythrocytic and sporozoite stages, as well as its regulating role in parasite motility during egress and invasion in response to calcium transitions have been particularly underlined elsewhere.33 Three additional proteins which showed competition by both 31 and 50 were not considered as efficacy targets: (a) zinc finger (CCCH type) protein (Table S1), as the apparent Kdapp values were high (3.9 μM for 31 versus 12.6 μM for 50), (b) glycogen synthase kinase 3 (GSK3, Figure 4) (Kdapp values of 0.09 versus 5.3 μM for 31 and 50, respectively), and (c) casein kinase 1 (CK1, Figure 4b) (Kdapp values of 0.15 versus 1.5 μM for 31 and 50, respectively), as a second PfPKG inhibitor with activity against P. falciparum was tested and did not show any activity against GSK3 and CK1 (data not shown). As a follow-up to the above experiments, comparison of 31 and 50 revealed a difference in binding to only one protein, the serine/arginine protein kinase 2 (SRPK2 or CLK2, Figure 4a). The Kdapp values of 31 and 50 for SRPK2 are 0.19 μM and >20 μM, respectively. Notably, when tested on the immobilized compounds (Figure 4b), SRPK2 was found to not bind to 50 attached to beads, which suggests no affinity for the compound. In contrast, SRPK2 was found to bind to 31 attached to beads, with 31 strongly competing the protein with an apparent Kd value of 0.05 μM.
Altogether, the results of our target identification experiments suggest SRPK2 as a protein kinase target in which the potent fast-killing properties of the most active derivatives developed herein could be attributed to. Although we cannot rule out a synergistic SRPK2/CDPK1,4/PKG effect, such a scenario seems to be less likely due to the inhibitory effect also displayed by 50 against CDPK1, CDPK4, and PKG. Therefore, our study highlights for the first time SRPK2 targeting as a tractable approach for the development of potential fast-killing antimalarial drugs. Recent global kinomic and phospho-proteomic analyses of the human malaria parasite P. falciparum have emphasized the necessity of SRPK2 in parasite proliferation during the erythrocytic asexual cycle.34,35 A transcriptomic study by Hoeijmakers et al.36 indicates the expression profile of SRPK2 (27–45 h post invasion) is more extensive than that of PKG (32–45 h post invasion) in blood stages, which supports the idea that inhibition of SRPK2 might mediate the fast kill phenotype of compound 31 since it is likely active against a greater proportion of the 48 h blood stage cycle. We have previously assumed that the slow kill phenotype of specific PKG inhibitors is due to the narrow window of expression just prior to egress and invasion. Previous work has implicated Plasmodium SRPKs (CLKs) such as SRPK2 (CLK2) in the regulation of alternative splicing of mRNA.37 CLK1 and CLK2 (both located in the nucleus)34 are orthologues of a yeast SR protein kinase Sky1p that phosphorylates SR proteins which bind to RNA and play a key role in RNA splicing. SR kinases phosphorylate SR proteins in a serine/arginine-rich domain, thereby influencing their activity and localization. CLK1–CLK4 have been shown to phosphorylate SR protein orthologues34,38 and are expressed in blood stages and gametocytes.38 It is therefore possible that breakdown of the regulation of mRNA splicing by inhibition of SRPK2 contribute significantly to the fast kill phenotype of compound 31. There is a precedent for targeting SRPK/CLK to treat disorders such as Duchenne muscular dystrophy39 and Alzheimer’s disease40 either by disrupting alternative splicing or by correcting the aberrant splicing observed in some diseases.37 During the review process of the current article, an interesting research paper was published, highlighting CLK3 as a multistage cross-species malarial target, the inhibition of which could offer both a prophylactic and transmission-blocking effect.41
Collectively, starting from “hits” (4–6, Chart 1) acting through a well-validated target (PKG) playing a crucial role in all of the key stages of the complex parasite life cycle but offering a low parasite killing rate, we were able to develop novel powerful fast kill entities comparable to artemisinins by refocusing on a kinase polypharmacology strategy. Whole transcriptome and kinome screens suggest that Plasmodium infection dramatically alters signaling networks within both the circulation and hepatocytes.42−46 Recent evidence also suggests that signaling alterations in infected cells may affect the response of cells to extrinsic stimuli and provide new targets for therapeutic intervention which are unique to infected cells.47 Given that kinases are critical enzymes in cell signaling, protein regulation, cellular transport, secretory processes, and many other cellular pathways in malaria transmission, infection, and spread, the development of bioactive molecules targeting multiple kinases has the potential to offer a superior effect compared to a single agent. In addition, targeting simultaneously more than one parasite component may limit the development of resistance to a single therapeutic. The exploitation of the polypharmacology of kinase inhibitors has already become a major focus for the development of more efficient anticancer therapeutics and is currently a relatively untapped resource for the repurposing of drugs for use against malaria and other infectious diseases.48 In addition, it has long been recognized that Plasmodium protein kinases are attractive targets for antimalarial chemotherapy.49,50 The Plasmodium kinome is made up of between 6551 and 99 protein kinases,52 comprising a single PKG and a family of four SRPK-like kinases (CLKs).53
Conclusions and Future Plans
In this article, the development of powerful fast-acting agents with killing rates similar to or better than artemisinins is described, and the structural and molecular characteristics providing such unique properties are highlighted. Compounds 31, 59, and 60 could be considered promising lead compounds for further optimization in the search for identifying novel antimalarial agents with new mechanisms of action and a strong fast-killing profile which are missing from the therapeutic arsenal against malaria. In parallel, the present study confirms that selective PKG inhibition is accompanied by low parasite killing rates, while it brings to light for the first time the tractability of targeting SRPK2/PKG in developing powerful fast-kill chemotypes with curative and transmission-blocking properties against malaria. Our efforts are currently being focused on further refinement of the structural features of 31, 59, and 60, aiming at extracting a new generation of fast-kill antimalarial chemical entities with an optimal developability profile. Elucidation of the involvement of SRPK2 inhibition in mediating the fast-kill phenotype of these compounds is also underway and will utilize recombinant expression and immunoprecipitation of the native kinase from parasite extracts, as described elsewhere.38
Experimental Section
General Chemistry Information
All starting materials were purchased from commercial sources and used as received or synthesized via literature procedures. Solvents were dried using a commercial solvent purification system and stored under nitrogen. All final compounds were characterized by 1H NMR spectroscopy and LCMS. 1H NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer at 293 K. Purity was determined by HPLC (Acquity UPLC BEH C18 1.7 μ 2.1 mm × 50 mm) at 35 °C. All compounds tested present a purity >95%, except for a couple of derivatives that presented a purity of >90%. Method: acetate NH4 25 mM + 10% ACN at pH 6.6/ACN, 0–0.2 min 100:0; 0.2–1.0 min 10:90; 1.0–1.8 min 10:90; 1.8–2.0 min 100:0. Flow: 0.8 mL/min. The UV detection wavelength was 254 and 210 nm. Positive-ion mass spectra (high-resolution mass spectroscopy) was acquired using a QSTAR Elite (AB Sciex Instruments) mass spectrometer, equipped with a turbospray source, over a mass range of 250–700, with a scan time of 1 s. The elemental composition was calculated using Analyst QS 2.0 software.
Compounds 5–8 were synthesized using previously described procedures.21
Synthesis of Final Compounds 9 and 10
To a microwave vial containing a magnetic stirring bar were added compounds of formula i (0.38 mmol), 1-Boc-4-(4-aminophenyl)piperazine (116 mg, 0.42 mmol), iPrOH (4 mL), and trifluoroacetic acid (TFA, 44 μL, 0.57 mmol). The vial was capped and stirred in a Biotage microwave reactor at 105 °C for 2 h. Additional TFA (44 μL, 0.57 mmol) was added, and the reaction mixture further stirred in the microwave reactor at 105 °C for 2 h to afford the Boc-protected analogues of 9 and 10. To the resulting crude mixtures were then added 4 N HCl in dioxane (6 mL). The reaction mixtures were stirred at room temperature for 3 h and concentrated under reduced pressure. The crude materials were purified by semipreparative HPLC to furnish the desired final products.
4-[4-(4-Fluorophenyl)-2-(morpholin-4-yl)-1,3-thiazol-5-yl]-N-[4-(piperazin-1-yl)phenyl]pyrimidin-2-amine Trifluoroacetate (9)
Orange solid. Yield = 5%. 1H NMR (400 MHz, DMSO-d6) δ 9.40 (s, 1H), 8.69 (br s, 2H), 8.10 (d, J = 5.38 Hz, 1H), 7.61 (d, J = 9.10 Hz, 2H), 7.56 (dd, J = 5.57, 8.82 Hz, 2H), 7.30 (t, J = 8.92 Hz, 2H), 6.94 (d, J = 9.10 Hz, 2H), 6.23 (d, J = 5.38 Hz, 1H), 3.74 (t, J = 4.82 Hz, 4H), 3.51 (t, J = 4.80 Hz, 4H), 3.25 (s, 8H). MS: m/e 518 (MH+). Purity was determined as >95% by HPLC (266 nm). Rt: 0.99 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(pyrrolidin-1-yl)-1,3-thiazol-5-yl]-N-[4-(piperazin-1-yl)phenyl]pyrimidin-2-amine Trifluoroacetate (10)
Pale yellow solid. Yield = 3%. 1H NMR (400 MHz, DMSO-d6) δ 9.40 (s, 1H), 8.71 (br s, 2H), 8.05 (d, J = 5.48 Hz, 1H), 7.62 (d, J = 9.09 Hz, 2H), 7.55 (dd, J = 5.58, 8.80 Hz, 2H), 7.31 (t, J = 8.90 Hz, 2H), 6.94 (d, J = 9.13 Hz, 2H), 6.16 (d, J = 5.48 Hz, 1H), 3.47 (t, J = 6.20 Hz, 4H), 3.26 (s, 8H), 2.02 (t, J = 6.60 Hz, 4H). MS: m/e 502 (MH+). Purity was determined as >95% by HPLC (269 nm). Rt: 0.99 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[2-Amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]-N-[4-(4-methylpiperazin-1-yl)phenyl]pyrimidin-2-amine Trifluoroacetate (11)
To a microwave vial containing a magnetic stirring bar were added compound j (70 mg, 0.23 mmol), 4-(4-methylpiperazino)aniline (44 mg, 0.23 mmol), iPrOH (4 mL), and 4 N HCl in dioxane (57 μL, 0.23 mmol). The vial was capped and stirred in a Biotage microwave reactor at 170 °C for 30 min. The reaction mixture was concentrated under reduced pressure, and the crude material was purified by semipreparative HPLC. Yellow/orange solid. Yield = 18%. 1H NMR (400 MHz, DMSO-d6) δ 9.60 (br, 1H), 9.37 (s, 1H), 8.07 (d, J = 5.44 Hz, 1H), 7.70 (br s, 2H), 7.61 (d, J = 9.08 Hz, 2H), 7.55 (dd, J = 5.58, 8.80 Hz, 2H), 7.28 (t, J = 8.90 Hz, 2H), 6.93 (d, J = 9.14 Hz, 2H), 6.22 (d, J = 5.42 Hz, 1H), 3.73 (d, J = 13.34 Hz, 2H), 3.53 (d, J = 12.20 Hz, 2H), 3.24–3.12 (m, 2H), 2.94–2.85 (m, 5H). MS: m/e 462 (MH+). Purity was determined as >95% by HPLC (288 nm). Rt: 0.90 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[2-Amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]-N-[4-(piperazin-1-yl)phenyl]pyrimidin-2-amine (12)
To a round-bottom flask containing a magnetic stirring bar were added compound j (1.0 g, 3.26 mmol), 1-Boc-4-(4-aminophenyl)piperazine (1.09 g, 3.91 mmol), iPrOH (20 mL), and trifluoroacetic acid (TFA, 0.30 mL, 3.91 mmol). The reaction mixture was stirred at 100 °C for 2 h. Additional TFA (0.30 mL, 3.91 mmol) was added and the reaction mixture was further stirred at 100 °C for 2 h, concentrated under reduced pressure, diluted with EtOAc, and washed with saturated NaHCO3 solution. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified on silica gel cartridge (0–100% EtOAc in cyclohexane) to give the Boc-protected derivative of 12 (1.17 g, 66%) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 9.26 (s, 1H), 8.05 (d, J = 5.36 Hz, 1H), 7.61–7.52 (m, 6H), 7.27 (t, J = 8.90 Hz, 2H), 6.87 (d, J = 9.13 Hz, 2H), 6.20 (d, J = 5.36 Hz, 1H), 3.46 (t, J = 4.90 Hz, 4H), 3.00 (t, J = 5.09 Hz, 4H), 1.42 (s, 9H). MS: m/e 548 (MH+). To a stirred solution of the Boc-protected carbamate (100 mg, 0.18 mmol) in anhydrous 1,4-dioxane (8 mL) was added 4 N HCl in dioxane (8 mL). The reaction mixture was stirred at room temperature for 4 h, concentrated and passed through an SCX-2 cartridge. The impurities were first eluted using MeOH, and the desired product was subsequently eluted using 7 N NH3 in MeOH solution. The resulting solution was concentrated under reduced pressure, and the crude material was purified by semipreparative HPLC. Bright yellow solid. Yield = 40%. 1H NMR (400 MHz, DMSO-d6) δ 9.22 (s, 1H), 8.05 (d, J = 5.34 Hz, 1H), 7.60–7.53 (m, 6H), 7.28 (t, J = 8.90 Hz, 2H), 6.84 (d, J = 9.05 Hz, 2H), 6.20 (d, J = 5.34 Hz, 1H), 5.76 (s, 1H), 3.01–2.91 (m, 4H), 2.89–2.77 (m, 4H). MS: m/e 448 (MH+). Purity was determined as >95% by HPLC (292 nm). Rt: 0.96 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, acetate NH4 25 mM + 10% ACN at pH 6.6/ACN).
4-[2-Amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]-N-[4-(morpholin-4-yl)phenyl]pyrimidin-2-amine (13)
To a microwave vial containing a magnetic stirring bar were added compound j (70 mg, 0.23 mmol), 4-morpholinoaniline (41 mg, 0.23 mmol), iPrOH (4 mL), and 4 N HCl in dioxane (57 μL, 0.23 mmol). The vial was capped and stirred in a Biotage microwave reactor at 170 °C for 30 min. The reaction mixture was concentrated under reduced pressure and purified by semipreparative HPLC. Pale brown solid. Yield = 4%. 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.06 (d, J = 5.36 Hz, 1H), 7.61–7.53 (m, 6H), 7.28 (t, J = 8.92 Hz, 2H), 6.87 (d, J = 9.11 Hz, 2H), 6.21 (d, J = 5.35 Hz, 1H), 3.75 (t, J = 4.74 Hz, 4H), 3.04 (t, J = 4.78 Hz, 4H). MS: m/e 449 (MH+). Purity was determined as >95% by HPLC (296 nm). Rt: 1.13 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, acetate NH4 25 mM + 10% ACN at pH 6.6/ACN).
N-[4-(4-Fluorophenyl)-5-{2-[4-(piperazin-1-yl)anilino]pyrimidin-4-yl}-1,3-thiazol-2-yl]acetamide Trifluoroacetate (14)
To a stirred solution of tert-butyl 4-[4-({4-[2-amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)phenyl]piperazine-1-carboxylate (100 mg, 0.18 mmol) in anhydrous CH2Cl2 (10 mL) were added acetyl chloride (9 μL, 0.13 mmol) and dry triethylamine (18 μL, 0.13 mmol). The reaction mixture was stirred at room temperature for 4 h under N2 atmosphere. Additional acetyl chloride (3.9 μL, 0.055 mmol) and triethylamine (7.6 μL, 0.055 mmol) were added and the mixture was further stirred at room temperature overnight and concentrated under reduced pressure to give the crude BOC-protected 14 (MS: m/e 590 (MH+)), which was dissolved in anhydrous 1,4-dioxane (8 mL) and 4 N HCl in dioxane solution (8 mL). The mixture was stirred at room temperature for 4 h, concentrated under reduced pressure, and the residue was purified by semipreparative HPLC. Orange solid. Yield = 22%. 1H NMR (400 MHz, DMSO-d6) δ 12.44 (s, 1H), 9.50 (s, 1H), 8.67 (br s, 2H), 8.21 (d, J = 5.26 Hz, 1H), 7.64–7.58 (m, 4H), 7.31 (t, J = 8.90 Hz, 2H), 6.92 (d, J = 9.10 Hz, 2H), 6.40 (d, J = 5.26 Hz, 1H), 3.26 (br s, 8H), 2.20 (s, 3H). MS: m/e 490 (MH+). Purity was determined as >95% by HPLC (258 nm). Rt: 0.95 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-[4-(4-Fluorophenyl)-5-{2-[4-(piperazin-1-yl)anilino]pyrimidin-4-yl}-1,3-thiazol-2-yl]cyclopropanecarboxamide Trifluoroacetate (15)
To a stirred solution of tert-butyl 4-[4-({4-[2-amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)phenyl]piperazine-1-carboxylate (80 mg, 0.15 mmol) in anhydrous CH2Cl2 (10 mL) were added cyclopropanecarbonyl chloride (13.2 μL, 0.15 mmol) and dry triethylamine (20.3 μL, 0.15 mmol). The reaction mixture was stirred at room temperature for 18 h under N2 atmosphere. The reaction mixture was concentrated under reduced pressure to give the Boc-protected carbamate of 15 (MS: m/e 616 (MH+) as a crude residue, which was dissolved in anhydrous 1,4-dioxane (8 mL) and 4 N HCl in dioxane solution (8 mL). The resulting mixture was stirred at room temperature for 4 h, concentrated under reduced pressure, and the crude material was purified by semipreparative HPLC. Bright yellow solid. Yield = 11%. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (br s, 2H), 8.29 (d, J = 5.52 Hz, 1H), 7.77 (br s, 2H), 7.55 (dd, J = 5.60, 8.77 Hz, 2H), 7.29 (t, J = 8.89 Hz, 2H), 7.07 (d, J = 8.98 Hz, 2H), 6.99 (d, J = 9.02 Hz, 2H), 6.64 (d, J = 5.52 Hz, 1H), 3.36–3.34 (m, 4H), 3.27–3.21 (m, 4H), 1.97–1.91 (m, 1H), 0.94–0.90 (m, 2H), 0.88–0.82 (m, 2H). MS: m/e 516 (MH+). Purity was determined as >95% by HPLC (254 nm). Rt: 0.93 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-[4-(4-Fluorophenyl)-5-{2-[4-(piperazin-1-yl)anilino]pyrimidin-4-yl}-1,3-thiazol-2-yl]-1-methylpiperidine-4-carboxamide Trifluoroacetate (16)
To a stirred solution of 1-methylpiperidine-4-carboxylic acid (13 mg, 0.09 mmol) in anhydrous DMF (10 mL) under N2 atmosphere were added HATU (52 mg, 0.14 mmol), DIPEA (48 μL, 0.27 mmol), and tert-butyl 4-[4-({4-[2-amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)phenyl]piperazine-1-carboxylate (50 mg, 0.09 mmol). The resulting mixture was stirred at room temperature for 2 h and then heated at 60 °C for 72 h. In addition, 1-methylpiperidine-4-carboxylic acid (13 mg, 0.09 mmol), HATU (35 mg, 0.09 mmol), and DIPEA (32 μL, 0.18 mmol) were added and the mixture was further stirred at 60 °C for 48 h. LCMS showed product to starting material ratio as 1:1. Hence, NaH (60% dispersion in mineral oil, 7.3 mg, 0.18 mmol) was added with the mixture being stirred at room temperature for further 4 h. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give the Boc-protected 16 as a pale brown solid (MS: m/e 673 (MH+)). This pale brown solid was dissolved in 4 N HCl in dioxane (10 mL) and stirred at room temperature for 4 h, concentrated under reduced pressure, and purified by semipreparative HPLC. Orange solid. Yield = 17%. 1H NMR (400 MHz, DMSO-d6) δ 12.63 (s, 1H), 9.48 (s, 1H), 9.43 (br, 1H), 8.73 (br s, 2H), 8.23 (d, J = 5.26 Hz, 1H), 7.62–7.57 (m, 4H), 7.31 (t, J = 8.89 Hz, 2H), 6.92 (d, J = 9.07 Hz, 2H), 6.42 (d, J = 5.24 Hz, 1H), 3.26 (s, 8H), 3.04–2.95 (m, 2H), 2.83–2.76 (m, 3H), 2.54 (s, 3H), 2.14–2.08 (m, 2H), 1.91–1.78 (m, 2H). MS: m/e 573 (MH+). Purity was determined as >95% by HPLC (260 nm). Rt: 0.86 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-[4-(4-Fluorophenyl)-5-{2-[4-(piperazin-1-yl)anilino]pyrimidin-4-yl}-1,3-thiazol-2-yl]propane-1-sulfonamide Trifluoroacetate (17)
To a stirred solution of tert-butyl 4-[4-({4-[2-amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)phenyl]piperazine-1-carboxylate (150 mg, 0.27 mmol) and Et3N (46 μL, 0.33 mmol) in anhydrous CH2Cl2 (25 mL) at 0 °C under N2 atmosphere was added 1-propanesulfonyl chloride (32 μL, 0.28 mmol). The reaction mixture was stirred at 0 °C for 30 min and then allowed to warm to room temperature and stirred at room temperature for 5 h. In addition, Et3N (46 μL, 0.33 mmol) was added and the mixture was stirred under reflux for a further 2 h and concentrated under reduced pressure to give the Boc-protected carbamate of 17 (160 mg) as a yellow oil (MS: m/e 654 (MH+)). The oil was dissolved in anhydrous 1,4-dioxane (3 mL) and 4 N HCl in dioxane (7 mL), stirred at room temperature for 4 h, and concentrated under reduced pressure. The crude residue was purified by semipreparative HPLC. Orange solid. Yield = 47%. 1H NMR (400 MHz, DMSO-d6) δ 13.08 (br s, 1H), 9.52 (s, 1H), 8.80 (br s, 2H), 8.16 (d, J = 5.32 Hz, 1H), 7.66 (dd, J = 5.40, 8.82 Hz, 2H), 7.59 (d, J = 9.12 Hz, 2H), 7.41 (t, J = 8.86 Hz, 2H), 6.92 (d, J = 9.12 Hz, 2H), 6.10 (d, J = 5.30 Hz, 1H), 3.25 (s, 8H), 3.06 (t, J = 7.59 Hz, 2H), 1.73 (sextet, J = 7.50 Hz, 2H), 0.99 (t, J = 7.48 Hz, 3H). MS: m/e 554 (MH+). Purity was determined as >95% by HPLC (274 nm). Rt: 0.85 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-[4-(4-Fluorophenyl)-5-{2-[4-(piperazin-1-yl)anilino]pyrimidin-4-yl}-1,3-thiazol-2-yl]piperidine-4-sulfonamide Trifluoroacetate (18)
To a stirred solution of tert-butyl 4-[4-({4-[2-amino-4-(4-fluorophenyl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)phenyl]piperazine-1-carboxylate (80 mg, 0.15 mmol) in anhydrous CH2Cl2 (10 mL) were added 4-chlorosulfonyl-piperidine-1-carboxylic acid tert-butyl ester (41 mg, 0.15 mmol) and dry triethylamine (20.3 μL, 0.15 mmol). The reaction mixture was stirred at room temperature for 18 h under N2 atmosphere. In addition, 4-chlorosulfonyl-piperidine-1-carboxylic acid tert-butyl ester (41 mg, 0.15 mmol) and dry Et3N (20.3 μL, 0.15 mmol) were added. The mixture was stirred under reflux overnight, concentrated under reduced pressure, and purified on silica gel cartridge (0–100% EtOAc in cyclohexane, then 0–20% MeOH in EtOAc) to give the di-Boc-protected 18 (MS: m/e 795 (MH+) as a pale brown solid. This solid was subsequently dissolved in anhydrous 1,4-dioxane (8 mL) and 4 N HCl in dioxane solution (8 mL), stirred at room temperature for 4 h, and concentrated under reduced pressure. The crude residue was purified by semipreparative HPLC. Burgundy solid. Yield = 42%. 1H NMR (400 MHz, DMSO-d6) δ 13.26 (br s, 1H), 9.52 (s, 1H), 8.76 (br s, 3H), 8.46–8.31 (m, 1H), 8.18 (d, J = 5.28 Hz, 1H), 7.65 (dd, J = 5.40, 8.66 Hz, 2H), 7.59 (d, J = 9.02 Hz, 2H), 7.43 (t, J = 8.82 Hz, 2H), 6.92 (d, J = 9.06 Hz, 2H), 6.11 (d, J = 5.26 Hz, 1H), 3.41–3.38 (m, 2H), 3.33–3.29 (m, 1H), 3.26 (br s, 8H), 2.99–2.89 (m, 2H), 2.20–2.15 (m, 2H), 1.90–1.79 (m, 2H). MS: m/e 595 (MH+). Purity was determined as >90% by HPLC (274 nm). Rt: 0.83 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
General Procedure for the Removal of Ethyl Carbamate Group: Synthesis of the Final Compounds 19–40
In a microwave vial, the respective N-ethyl carbamate-protected derivatives 61−82 (0.067 mmol), LiOH·H2O (0.042 mg, 1.01 mmol), THF (1 mL), ethanol (1 mL), and water (1 mL) are added and the mixture is stirred at 105 °C overnight. EtOAc (15 mL) and an aqueous solution of NaOH (1 N, 5 mL) are added, the two phases are separated, the aqueous phase is washed with EtOAc (5 mL), and the combined organic phases are washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the desired compounds, which are purified by semipreparative HPLC to afford pure materials.
N-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)-1,3,4-thiadiazol-2-amine (19)
White solid. Yield = 60%. 1H NMR (400 MHz, DMSO-d6) δ 1.65 (br dd, J = 11.87, 3.54 Hz, 2H), 1.93–2.11 (m, 2H), 2.63 (td, J = 12.06, 2.15 Hz, 2H), 2.96–3.10 (m, 2H), 3.10–3.22 (m, 2H), 6.70 (d, J = 5.05 Hz, 1H), 7.15–7.42 (m, 2H), 7.57–7.76 (m, 2H), 8.49 (d, J = 5.05 Hz, 1H), 8.96–9.20 (m, 1H). MS: m/e 440 (MH+). Purity was determined as 94.4% by HPLC (280 nm). Rt: 0.90 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)-5-methyl-1,3,4-thiadiazol-2-amine (20)
White solid. Yield = 59%. 1H NMR (400 MHz, DMSO-d6) δ .65–1.79 (m, 2H), 2.07 (br d, J = 11.87 Hz, 2H), 2.60 (s, 3H), 2.71 (br t, J = 11.49 Hz, 2H), 3.12 (br d, J = 11.87 Hz, 2H), 3.16–3.40 (m, 2H), 6.73 (d, J = 5.31 Hz, 1H), 7.22–7.33 (m, 2H), 7.58–7.64 (m, 2H), 8.43 (s, 1H), 8.48 (d, J = 5.31 Hz, 1H). MS: m/e 454 (MH+). Purity was determined as 98.8% by HPLC (281 nm). Rt: 0.91 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)-4-methylthiazol-2-amine (21)
Yellow solid. Yield = 39%. 1H NMR (400 MHz, DMSO-d6) δ 1.20 (t, J = 7.07 Hz, 1H), 1.66 (br dd, J = 12.13, 3.54 Hz, 2H), 2.04 (br d, J = 10.36 Hz, 2H), 2.27 (d, J = 1.01 Hz, 3H), 2.65 (td, J = 11.87, 2.02 Hz, 2H), 2.94–3.11 (m, 3H), 4.05–4.07 (m, 1H), 6.63 (d, J = 5.05 Hz, 1H), 6.72 (d, J = 1.01 Hz, 1H), 7.27–7.34 (m, 2H), 7.59–7.65 (m, 2H), 8.44 (d, J = 5.31 Hz, 1H). MS: m/e 454 (MH+). Purity was determined as 95.3% by HPLC (230 nm). Rt: 1.02 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)-3-methylisoxazol-5-amine (22)
White solid. Yield = 57%. 1H NMR (400 MHz, DMSO-d6) δ 0.90–1.37 (m, 1H), 1.63 (qd, J = 11.96, 3.79 Hz, 2H), 1.94–2.08 (m, 2H), 2.11–2.21 (m, 3H), 2.55–2.68 (m, 2H), 3.03 (br d, J = 12.38 Hz, 2H), 3.09–3.18 (m, 1H), 5.90 (s, 1H), 6.75 (d, J = 5.05 Hz, 1H), 7.15–7.41 (m, 2H), 7.52–7.73 (m, 2H), 8.46 (d, J = 5.31 Hz, 1H). MS: m/e 437 (MH+). Purity was determined as 95.3% by HPLC (279 nm). Rt: 0.97 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(1,5-Dimethyl-1H-pyrazol-3-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (23)
Yellow solid. Yield = 33%. 1H NMR (400 MHz, DMSO-d6) δ 1.56–1.67 (m, 2H), 2.00 (br d, J = 10.86 Hz, 2H), 2.21 (s, 3H), 2.53–2.70 (m, 3H), 2.99–3.23 (m, 3H), 3.61 (s, 3H), 6.23 (s, 1H), 6.47 (d, J = 5.31 Hz, 1H), 7.25–7.31 (m, 2H), 7.57–7.62 (m, 2H), 8.27 (d, J = 5.05 Hz, 1H), 9.68 (s, 1 H). MS: m/e 450 (MH+). Purity was determined as 99.5% by HPLC (241 nm). Rt: 0.96 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(1-(piperidin-4-yl)-1H-pyrazol-3-yl)pyrimidin-2-amine (24)
Bright yellow solid. Yield = 81%. 1H NMR (400 MHz, DMSO-d6) δ 9.64 (br s, 1H), 8.29 (d, J = 5.15 Hz, 1H), 7.82 (br s, 1H), 7.62 (dd, J = 5.62, 8.62 Hz, 2H), 7.56 (s, 1H), 7.31 (t, J = 8.88 Hz, 2H), 6.44 (br s, 1H), 4.24 (br s, 2H), 3.35–3.20 (m, 6H), 2.87 (dtd, J = 2.60, 12.58, 34.36 Hz, 4H), 2.21–2.15 (m, 2H), 2.10–2.01 (m, 2H), 1.95–1.77 (m, 4H). MS: m/e 505 (MH+). Purity was determined as >95% by HPLC (279 nm). Rt: 0.66 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(pyridin-2-yl)pyrimidin-2-amine (25)
White solid. Yield = 25%. 1H NMR (400 MHz, DMSO-d6) δ 1.63 (qd, J = 12.04, 4.04 Hz, 2H), 1.95–2.07 (m, 2H), 2.56–2.68 (m, 2H), 2.99–3.07 (m, 2H), 3.13 (tt, J = 11.62, 3.79 Hz, 2H), 6.66 (d, J = 5.31 Hz, 1H), 7.00 (ddd, J = 7.20, 4.93, 1.01 Hz, 1H), 7.22–7.36 (m, 2H), 7.62 (dd, J = 8.59, 5.56 Hz, 2H), 7.70 (ddd, J = 8.59, 7.20, 1.89 Hz, 1H), 8.04 (d, J = 8.59 Hz, 1H), 8.29 (dt, J = 4.80, 1.01 Hz, 1H), 8.36–8.47 (m, 1H), 9.86 (s, 1 H). MS: m/e 433 (MH+). Purity was determined as 97.3% by HPLC (293 nm). Rt: 1.01 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(pyrazin-2-yl)pyrimidin-2-amine (26)
White solid. Yield = 28%. 1H NMR (400 MHz, DMSO-d6) δ 1.18–1.35 (m, 1H), 1.62 (qd, J = 12.00, 3.92 Hz, 2H), 1.94–2.06 (m, 2H), 2.61 (td, J = 12.06, 2.15 Hz, 2H), 2.98–3.08 (m, 2H), 3.09–3.24 (m, 1H), 6.69 (d, J = 5.05 Hz, 1H), 7.24–7.37 (m, 2H), 7.59–7.67 (m, 2H), 8.25 (d, J = 2.53 Hz, 1H), 8.35 (dd, J = 2.53, 1.77 Hz, 1H), 8.45 (d, J = 5.30 Hz, 1H), 9.38 (d, J = 1.52 Hz, 1H), 10.37 (br s, 1H). MS: m/e 434 (MH+). Purity was determined as 96.1% by HPLC (271 nm). Rt: 0.95 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)pyridazin-3-amine (27)
White solid. Yield = 34%. 1H NMR (400 MHz, DMSO-d6) δ 1.13–1.29 (m, 1H), 1.59–1.80 (m, 2H), 2.03 (br d, J = 12.13 Hz, 2H), 2.59–2.71 (m, 2H), 3.01–3.08 (m, 2H), 3.15 (tt, J = 11.62, 3.79 Hz, 1H), 6.73 (d, J = 5.31 Hz, 1H), 7.29 (t, J = 8.84 Hz, 2H), 7.55–7.64 (m, 3H), 8.21 (dd, J = 9.09, 1.52 Hz, 1H), 8.46 (d, J = 5.30 Hz, 1H), 8.86 (dd, J = 4.55, 1.52 Hz, 1H), 10.59 (s, 1H). MS: m/e 434 (MH+). Purity was determined as 99.5% by HPLC (271 nm). Rt: 0.87 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(pyrimidin-4-yl)pyrimidin-2-amine (28A)
White solid. Yield = 24%. 1H NMR (400 MHz, DMSO-d6) δ 1.18–1.36 (m, 1H), 1.64 (qd, J = 12.08, 3.92 Hz, 2H), 1.92–2.09 (m, 2H), 2.56–2.73 (m, 2H), 2.94–3.08 (m, 2H), 3.11–3.19 (m, 1H), 6.81 (d, J = 5.31 Hz, 1H), 7.25–7.34 (m, 2H), 7.58–7.67 (m, 2H), 7.99–8.05 (m, 1H), 8.51 (dd, J = 5.68, 1.89 Hz, 2H), 8.77–8.81 (m, 1H), 10.29–10.76 (m, 1H). MS: m/e 434 (MH+). Purity was determined as 97.1% by HPLC (285 nm). Rt: 0.92 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(2-methylpyrimidin-4-yl)pyrimidin-2-amine (29)
Off-white solid. Yield = 29%. 1H NMR (400 MHz, MeOH-d4) δ 8.43 (d, J = 5.27 Hz, 1H), 8.37 (d, J = 6.04 Hz, 1H), 8.08 (d, J = 6.04 Hz, 1H), 7.61 (dd, J = 5.38, 8.68 Hz, 2H), 7.24 (t, J = 8.70 Hz, 2H), 6.80 (d, J = 5.25 Hz, 1H), 3.43–3.36 (m, 3H), 3.04 (td, J = 2.80, 12.50 Hz, 2H), 2.58 (s, 3H), 2.33 (dd, J = 2.08, 13.43 Hz, 2H), 2.06–1.95 (m, 2H). MS: m/e 448 (MH+). Purity was determined as >95% by HPLC (293 nm). Rt: 0.68 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-(2-Ethoxypyrimidin-4-yl)-4-[4-(4-fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-amine Trifluoroacetate (30)
Pale yellow solid. Yield = 13%. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.65 (d, J = 7.60 Hz, 1H), 8.52 (d, J = 5.25 Hz, 1H), 8.38 (d, J = 9.60 Hz, 1H), 8.30 (d, J = 5.79 Hz, 1H), 7.68 (d, J = 5.80 Hz, 1H), 7.62 (dd, J = 5.52, 8.82 Hz, 2H), 7.32 (t, J = 8.86 Hz, 2H), 6.80 (d, J = 5.24 Hz, 1H), 4.33 (q, J = 7.06 Hz, 2H), 3.50–3.37 (m, 3H), 3.14–3.02 (m, 2H), 2.28 (dd, J = 2.78, 14.20 Hz, 2H), 2.01–1.90 (m, 2H), 1.32 (t, J = 7.06 Hz, 3H). MS: m/e 478 (MH+). Purity was determined as >95% by HPLC (292 nm). Rt: 0.86 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(2-methylpyridin-4-yl)pyrimidin-2-amine (31)
White solid. Yield = 37%. 1H NMR (400 MHz, DMSO-d6) δ 1.56–1.70 (m, 2H), 1.98–2.09 (m, 2H), 2.41 (s, 3H), 2.61 (td, J = 12.06, 2.40 Hz, 2H), 2.98–3.07 (m, 2H), 3.07–3.19 (m, 1H), 3.30 (s, 1H), 6.65 (d, J = 5.05 Hz, 1H), 7.31 (t, J = 8.84 Hz, 2H), 7.46 (dd, J = 5.81, 2.02 Hz, 1H), 7.56–7.73 (m, 3H), 8.20 (d, J = 5.81 Hz, 1H), 8.43 (d, J = 5.05 Hz, 1H), 10.10 (s, 1H). MS: m/e 447 (MH+). Purity was determined as 97.6% by HPLC (287 nm). Rt: 0.95 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(2,6-Dimethylpyridin-4-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (32)
White solid. Yield = 37%. 1H NMR (400 MHz, DMSO-d6) δ 1.62 (dd, J = 12.00, 3.41 Hz, 2H), 2.03 (br d, J = 10.36 Hz, 2H), 2.36 (s, 6H), 2.62 (td, J = 12.06, 2.40 Hz, 2H), 2.97–3.09 (m, 2H), 3.09–3.18 (m, 1H), 3.30 (s, 1H), 6.62 (d, J = 5.31 Hz, 1H), 7.27–7.35 (m, 2H), 7.46 (s, 2H), 7.60–7.66 (m, 2H), 8.42 (d, J = 5.05 Hz, 1H), 10.03 (s, 1H). MS: m/e 461 (MH+). Purity was determined as 98.5% by HPLC (289 nm). Rt: 0.95 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-({4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)pyridin-2-ol Trifluoroacetate (33)
Pale yellow solid. Yield = 8%. 1H NMR (400 MHz, DMSO-d6) δ 11.23 (br s, 1H), 10.08 (s, 1H), 8.66 (d, J = 9.50 Hz, 1H), 8.46–8.38 (m, 2H), 7.63 (dd, J = 5.52, 8.82 Hz, 2H), 7.35–7.27 (m, 3H), 7.09 (d, J = 2.08 Hz, 1H), 6.66 (d, J = 5.24 Hz, 1H), 6.50 (dd, J = 2.07, 7.28 Hz, 1H), 3.50–3.37 (m, 3H), 3.12–3.03 (m, 2H), 2.27 (dd, J = 2.39, 14.13 Hz, 2H), 2.01–1.89 (m, 2H). MS: m/e 449 (MH+). Purity was determined as >95% by HPLC (285 nm). Rt: 0.74 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(2-methoxy-6-methylpyridin-4-yl)pyrimidin-2-amine Trifluoroacetate (34)
Off-white solid. Yield = 30%. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.64 (br s, 1H), 8.43 (d, J = 5.24 Hz, 1H), 8.37 (br s, 1H), 7.63 (dd, J = 5.54, 8.80 Hz, 2H), 7.31 (t, J = 8.88 Hz, 2H), 7.18 (d, J = 1.13 Hz, 1H), 7.11 (s, 1H), 6.63 (d, J = 5.23 Hz, 1H), 3.80 (s, 3H), 3.49–3.37 (m, 3H), 3.12–3.09 (m, 2H), 2.33 (s, 3H), 2.27 (dd, J = 2.80, 14.54 Hz, 2H), 2.00–1.89 (m, 2H). MS: m/e 477 (MH+). Purity was determined as >95% by HPLC (297 nm). Rt: 0.69 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N-(5-Fluoro-6-methylpyridin-2-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (35)
White solid. Yield = 26%. 1H NMR (400 MHz, DMSO-d6) δ 1.24 (s, 1H), 1.63 (br dd, J = 11.87, 3.54 Hz, 2H), 2.01 (br d, J = 10.61 Hz, 2H), 2.38 (d, J = 2.78 Hz, 3H), 2.55–2.70 (m, 2H), 2.83–3.08 (m, 2H), 3.09–3.22 (m, 1H), 6.64 (d, J = 5.31 Hz, 1H), 7.25–7.33 (m, 2H), 7.53–7.64 (m, 3H), 7.87 (dd, J = 8.84, 3.28 Hz, 1H), 8.39 (d, J = 5.05 Hz, 1H), 9.87 (s, 1H). MS: m/e 465 (MH+). Purity was determined as 97.9% by HPLC (241 nm). Rt: 1.11 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-Ethyl-5-fluoro-6-methylpyridin-2-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (36)
White solid. Yield = 36%. 1H NMR (400 MHz, DMSO-d6) δ 1.24 (s, 1H), 1.28 (t, J = 7.58 Hz, 3H), 1.72 (qd, J = 12.13, 3.79 Hz, 2H), 2.10 (br s, 2H), 2.37 (d, J = 3.03 Hz, 3H), 2.61–2.71 (m, 2H), 2.72–2.84 (m, 2H), 3.12–3.24 (m, 3H), 6.58 (d, J = 5.31 Hz, 1H), 7.31 (t, J = 8.84 Hz, 2H), 7.56–7.69 (m, 2H), 8.01 (d, J = 5.05 Hz, 1H), 8.38 (d, J = 5.05 Hz, 1H), 9.81 (s, 1H). MS: m/e 493 (MH+). Purity was determined as 98.8% by HPLC (299 nm). Rt: 1.21 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-Cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (37)
White solid. Yield = 36%. 1H NMR (400 MHz, DMSO-d6) δ 0.95 (dd, J = 4.93, 2.15 Hz, 2H), 1.19 (dd, J = 8.34, 2.27 Hz, 2H), 1.56–1.71 (m, 2H), 1.99–2.09 (m, 2H), 2.10–2.21 (m, 1H), 2.37 (d, J = 3.03 Hz, 4H), 2.62 (br d, J = 2.02 Hz, 2H), 2.98–3.06 (m, 2H), 3.07–3.16 (m, 1H), 6.54 (d, J = 5.05 Hz, 1H), 7.31 (t, J = 8.84 Hz, 2H), 7.60–7.71 (m, 3H), 8.35 (d, J = 5.05 Hz, 1H), 9.74 (s, 1 H). MS: m/e 505 (MH+). Purity was determined as 98.3% by HPLC (240 nm). Rt: 1.22 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(4-Cyclobutyl-5-fluoro-6-methylpyridin-2-yl)-4-(4-(4-fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine (38)
White solid. Yield = 34%. 1H NMR (400 MHz, DMSO-d6) δ 1.51–1.68 (m, 2H), 1.83–1.94 (m, 1H), 1.96–2.16 (m, 3H), 2.23–2.44 (m, 8H), 2.59–2.69 (m, 2H), 3.03 (br d, J = 12.38 Hz, 2H), 3.07–3.18 (m, 1H), 3.68–3.83 (m, 1H), 6.56 (d, J = 5.05 Hz, 1H), 7.28–7.35 (m, 2H), 7.63 (br d, J = 3.03 Hz, 2H), 8.10 (d, J = 4.80 Hz, 1H), 8.37 (d, J = 5.31 Hz, 1H), 9.79 (s, 1H). MS: m/e 519 (MH+). Purity was determined as 89.9% by HPLC (243 nm). Rt: 1.32 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(pyridin-4-ylmethyl)pyrimidin-2-amine (39)
Pale yellow solid. Yield = 37%. 1H NMR (400 MHz, CDCl3) δ 9.82 (br, 1H), 8.62 (d, J = 5.82 Hz, 2H), 8.15 (d, J = 5.24 Hz, 1H), 7.57 (dd, J = 5.38, 8.74 Hz, 2H), 7.37 (d, J = 5.34 Hz, 2H), 7.15 (t, J = 8.68 Hz, 2H), 6.47 (d, J = 5.24 Hz, 1H), 5.71 (br, 1H), 4.71 (d, J = 5.95 Hz, 2H), 3.62–3.55 (m, 2H), 3.39–3.31 (m, 1H), 3.17–3.10 (m, 2H), 2.41 (dd, J = 3.34, 14.06 Hz, 2H), 2.29–2.18 (m, 2H). MS: m/e 447 (MH+). Purity was determined as >95% by HPLC (233 nm). Rt: 0.63 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
2-({4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)-1-(pyridin-4-yl)ethanol (40)
Pale yellow solid. Yield = 16%. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (d, J = 6.00 Hz, 2H), 8.15 (d, J = 5.12 Hz, 1H), 8.00 (br, 1H), 7.58 (dd, J = 5.56, 8.76 Hz, 2H), 7.35 (d, J = 6.00 Hz, 2H), 7.29 (t, J = 8.86 Hz, 2H), 6.29 (d, J = 4.96 Hz, 1H), 5.69 (br, 1H), 4.83–4.78 (m, 1H), 3.56–3.49 (m, 1H), 3.41–3.23 (m, 5H), 3.03–2.95 (m, 2H), 2.26–2.17 (m, 2H), 1.94–1.82 (m, 2H). MS: m/e 477 (MH+). Purity was determined as >95% by HPLC (236 nm). Rt: 0.87 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, acetate NH4 25 mM + 10% ACN at pH 6.6/ACN).
General Procedure for the Removal of Ethyl Carbamate Group: Synthesis of the Final Compounds 41–57
In a microwave vial, the respective N-ethyl carbamate-protected derivatives 83−99 (0.067 mmol), LiOH·H2O (0.042 mg, 1.01 mmol), THF (1 mL), ethanol (1 mL), and water (1 mL) are added and the mixture is stirred at 105 °C overnight. EtOAc (15 mL) and an aqueous solution of NaOH (1 N, 5 mL) are added, the two phases are separated, the aqueous phase is washed with EtOAc (5 mL), and the combined organic phases are washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the desired compounds, which are purified by preparative HPLC to afford pure materials.
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-amine (41)
Pale yellow solid. Yield = 16%. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 5.18 Hz, 1H), 7.57 (dd, J = 5.54, 8.84 Hz, 2H), 7.28 (t, J = 8.90 Hz, 2H), 6.76 (s, 2H), 6.23 (d, J = 5.18 Hz, 1H), 4.40 (br, 1H), 3.16–3.06 (m, 3H), 2.68 (td, J = 2.39, 12.17 Hz, 2H), 2.03 (dd, J = 2.80, 12.96 Hz, 2H), 1.65 (ddd, J = 3.84, 12.02, 24.50 Hz, 2H). MS: m/e 356 (MH+). Purity was determined as >95% by HPLC (228 nm). Rt: 0.68 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
2-({4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)ethanol (42)
Pale yellow solid. Yield = 58%. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 5.12 Hz, 1H), 7.58 (dd, J = 5.56, 8.80 Hz, 2H), 7.29 (t, J = 8.88 Hz, 2H), 7.16 (t, J = 5.52 Hz, 1H), 6.77 (br, 1H), 6.26 (d, J = 4.92 Hz, 1H), 4.66 (br, 1H), 3.54–3.48 (m, 2H), 3.40–3.20 (m, 5H), 2.86 (td, J = 2.30, 12.24 Hz, 2H), 2.13 (dd, J = 2.36, 13.60 Hz, 2H), 1.78 (ddd, J = 3.70, 12.76, 25.06 Hz, 2H). MS: m/e 400 (MH+). Purity was determined as >95% by HPLC (337 nm). Rt: 0.70 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(2-methoxyethyl)pyrimidin-2-amine (43)
Off-white solid. Yield = 73%. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 5.16 Hz, 1H), 7.57 (dd, J = 5.55, 8.84 Hz, 2H), 7.31–7.23 (m, 3H), 6.27 (br s, 1H), 3.45–3.39 (m, 3H), 3.34–3.27 (m, 2H), 3.26 (s, 3H), 3.12–3.00 (m, 3H), 2.61 (td, J = 2.34, 12.52 Hz, 2H), 2.00 (dd, J = 2.14, 13.14 Hz, 2H), 1.60 (ddd, J = 3.88, 12.16, 24.34 Hz, 2H). MS: m/e 414 (MH+). Purity was determined as >95% by HPLC (295 nm). Rt: 0.79 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(tetrahydrofuran-2-ylmethyl)pyrimidin-2-amine (44)
Off-white solid. Yield = 71%. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 5.15 Hz, 1H), 7.57 (dd, J = 5.56, 8.80 Hz, 2H), 7.30–7.24 (m, 3H), 6.27 (br s, 1H), 3.96 (t, J = 5.76 Hz, 1H), 3.79–3.73 (m, 1H), 3.64–3.58 (m, 1H), 3.27–3.16 (m, 2H), 3.12–2.98 (m, 3H), 2.59 (td, J = 2.26, 12.12 Hz, 2H), 1.99 (dd, J = 2.20, 12.56 Hz, 2H), 1.92–1.74 (m, 3H), 1.64–1.54 (m, 3H). MS: m/e 440 (MH+). Purity was determined as >95% by HPLC (295 nm). Rt: 0.83 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
N′-{4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}-N,N-dimethylethane-1,2-diamine (45)
Pale yellow solid. Yield = 95%. 1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 5.08 Hz, 1H), 7.95 (br, 1H), 7.58 (dd, J = 5.54, 8.82 Hz, 2H), 7.29 (t, J = 8.92 Hz, 2H), 7.09 (br, 1H), 6.26 (d, J = 5.08 Hz, 1H), 3.21–3.10 (m, 5H), 2.75 (td, J = 2.45, 12.24 Hz, 2H), 2.39 (t, J = 6.86 Hz, 2H), 2.17 (s, 6H), 2.09–2.05 (m, 2H), 1.70 (ddd, J = 4.16, 12.28, 24.66 Hz, 2H). MS: m/e 427 (MH+). Purity was determined as >95% by HPLC (293 nm). Rt: 0.61 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
1-Cyclopropyl-N′-{4-[4-(4-fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}-N,N-dimethylethane-1,2-diamine (46)
Off-white solid. Yield = 66%. 1H NMR (400 MHz, DMSO-d6) δ 8.12 (d, J = 5.00 Hz, 1H), 7.57 (dd, J = 5.56, 8.80 Hz, 2H), 7.28 (t, J = 8.90 Hz, 2H), 6.70 (br s, 1H), 6.26 (d, J = 3.70 Hz, 1H), 3.48–3.35 (m, 2H), 3.10–2.99 (m, 3H), 2.59 (td, J = 2.32, 12.16 Hz, 3H), 2.31 (s, 6H), 2.00–1.95 (m, 2H), 1.90–1.85 (m, 1H), 1.59 (ddd, J = 3.90, 12.12, 24.28 Hz, 2H), 0.78–0.68 (m, 1H), 0.58–0.50 (m, 1H), 0.41–0.33 (m, 1H), 0.31–0.23 (m, 1H), 0.09–0.01 (m, 1H). MS: m/e 467 (MH+). Purity was determined as >95% by HPLC (294 nm). Rt: 0.64 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-[2-(pyrrolidin-1-yl)ethyl]pyrimidin-2-amine (47)
Pale yellow solid. Yield = 71%. 1H NMR (400 MHz, DMSO-d6) δ 8.29 (br, 1H), 8.17 (d, J = 5.14 Hz, 1H), 7.58 (d, J = 5.54, 8.82 Hz, 2H), 7.30 (t, J = 8.88 Hz, 3H), 6.32 (br s, 1H), 3.46–3.35 (m, 6H), 3.05 (td, J = 2.86, 12.60 Hz, 2H), 2.82 (br, 5H), 2.24 (dd, J = 2.62, 14.08 Hz, 2H), 1.97–1.86 (m, 2H), 1.81–1.74 (m, 4H). MS: m/e 453 (MH+). Purity was determined as >95% by HPLC (293 nm). Rt: 0.62 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-[2-(4-methylpiperazin-1-yl)ethyl]pyrimidin-2-amine (48)
Sticky yellow solid. Yield = 75%. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (d, J = 5.08 Hz, 1H), 7.77 (br, 1H), 7.58 (dd, J = 5.54, 8.80 Hz, 2H), 7.29 (t, J = 8.90 Hz, 2H), 7.11 (br, 1H), 6.27 (br s, 1H), 3.42–3.32 (m, 4H), 3.03 (td, J = 2.76, 12.50 Hz, 2H), 2.48–2.32 (m, 8H), 2.23 (dd, J = 2.52, 13.96 Hz, 2H), 2.19 (s, 3H), 1.96–1.85 (m, 5H). MS: m/e 482 (MH+). Purity was determined as >95% by HPLC (285 nm). Rt: 0.60 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(2-morpholinoethyl)pyrimidin-2-amine (49)
White solid. Yield = 37%. 1H NMR (400 MHz, DMSO-d6) δ 1.51–1.69 (m, 2H), 1.95–2.05 (m, 2H), 2.37–2.42 (m, 4H), 2.45 (br t, J = 6.95 Hz, 3H), 2.62 (td, J = 12.06, 2.40 Hz, 2H), 2.99–3.21 (m, 4H), 3.36–3.40 (m, 1H), 3.53–3.60 (m, 4H), 6.27 (br s, 1H), 7.12 (br s, 1H), 7.25–7.32 (m, 2H), 7.55–7.61 (m, 2H), 8.13 (d, J = 5.05 Hz, 1H). MS: m/e 469 (MH+). Purity was determined as 99.6% by HPLC (295 nm). Rt: 0.92 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)-N-(2-thiomorpholinoethyl)pyrimidin-2-amine (50)
White solid. Yield = 31%. 1H NMR (400 MHz, DMSO-d6) δ 1.58 (dd, J = 12.00, 3.41 Hz, 2H), 1.99 (br d, J = 10.11 Hz, 2H), 2.48 (br s, 3H), 2.54–2.65 (m, 7H), 2.65–2.72 (m, 5H), 2.86–3.05 (m, 2H), 3.05–3.16 (m, 1H), 6.27 (br s, 1H), 7.10 (br s, 1H), 7.25–7.31 (m, 2H), 7.54–7.60 (m, 2H), 8.12 (d, J = 5.05 Hz, 1H). MS: m/e 485 (MH+). Purity was determined as 100% by HPLC (236 nm). Rt: 1.00 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
trans-4-({4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}amino)cyclohexanol (51)
Pale yellow solid. Yield = 37%. 1H NMR (400 MHz, DMSO-d6) δ 8.85 (br, 1H), 8.14 (d, J = 5.13 Hz, 1H), 7.58 (dd, J = 5.55, 8.78 Hz, 2H), 7.29 (t, J = 8.88 Hz, 2H), 7.16 (d, J = 6.72 Hz, 1H), 6.29 (br, 1H), 4.54 (d, J = 4.27 Hz, 1H), 3.43–3.36 (m, 4H), 3.08–3.00 (m, 2H), 2.24 (dd, J = 2.37, 14.02 Hz, 2H), 2.01–1.90 (m, 2H), 1.84–1.81 (m, 4H), 1.33–1.10 (m, 5H). MS: m/e 454 (MH+). Purity was determined as >95% by HPLC (236 nm). Rt: 0.90 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, acetate NH4 25 mM + 10% ACN at pH 6.6/ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-N-(1-methylpyrrolidin-3-yl)pyrimidin-2-amine (52)
Pale yellow sticky solid. Yield = 20%. 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 5.20 Hz, 1H), 7.58 (dd, J = 5.36, 8.72 Hz, 2H), 7.14 (t, J = 8.72 Hz, 2H), 6.37 (d, J = 5.24 Hz, 1H), 5.40 (d, J = 7.32 Hz, 1H), 4.56–4.49 (m, 1H), 3.25 (dt, J = 2.86, 12.34 Hz, 2H), 3.17 (tt, J = 3.80, 11.78 Hz, 1H), 2.88–2.76 (m, 4H), 2.60 (dd, J = 3.40, 9.66 Hz, 1H), 2.46–2.40 (m, 5H), 2.21–2.16 (m, 2H), 1.85–1.72 (m, 3H). MS: m/e 439 (MH+). Purity was determined as >95% by HPLC (234 nm). Rt: 0.62 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]-2-(pyrrolidin-1-yl)pyrimidine (53)
Off-white solid. Yield = 58%. 1H NMR (400 MHz, DMSO-d6) δ 8.19 (d, J = 5.16 Hz, 1H), 7.58 (dd, J = 5.56, 8.82 Hz, 2H), 7.28 (t, J = 8.90 Hz, 2H), 6.28 (d, J = 5.14 Hz, 1H), 3.49–3.40 (m, 4H), 3.12–2.98 (m, 3H), 2.59 (td, J = 2.30, 12.09 Hz, 2H), 1.99 (dd, J = 2.12, 12.34 Hz, 2H), 1.94–1.90 (m, 4H), 1.60 (ddd, J = 3.88, 12.12, 24.30 Hz, 2H). MS: m/e 410 (MH+). Purity was determined as >95% by HPLC (239 nm). Rt: 0.88 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
1-{4-[4-(4-Fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidin-2-yl}pyrrolidin-3-ol (54)
Off-white solid. Yield = 57%. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J = 9.02 Hz, 1H), 8.39 (d, J = 9.32 Hz, 1H), 8.22 (d, J = 5.54 Hz, 1H), 7.61 (dd, J = 5.52, 8.80 Hz, 2H), 7.31 (t, J = 8.90 Hz, 2H), 6.36 (d, J = 5.54 Hz, 1H), 4.40 (br s, 1H), 3.62–3.52 (m, 3H), 3.45–3.38 (m, 4H), 3.11–3.02 (m, 2H), 2.25 (dd, J = 2.50, 14.24 Hz, 2H), 2.06–1.88 (m, 4H). MS: m/e 426 (MH+). Purity was determined as >95% by HPLC (239 nm). Rt: 0.73 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
2-[4-(Azetidin-1-yl)piperidin-1-yl]-4-[4-(4-fluorophenyl)-2-(piperidin-4-yl)-1,3-thiazol-5-yl]pyrimidine (55)
Sticky yellow solid. Yield = 79%. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (d, J = 5.16 Hz, 1H), 7.57 (dd, J = 5.56, 8.84 Hz, 2H), 7.44 (br, 1H), 7.29 (t, J = 8.92 Hz, 2H), 6.34 (d, J = 5.12 Hz, 1H), 4.36–4.33 (m, 2H), 3.47–3.36 (m, 5H), 3.05 (td, J = 2.52, 12.45 Hz, 4H), 2.24 (dd, J = 3.08, 14.52 Hz, 2H), 2.10–2.04 (m, 2H), 1.98–1.87 (m, 4H), 1.76–1.71 (m, 2H), 1.24–1.08 (m, 3H). MS: m/e 479 (MH+). Purity was determined as >90% by HPLC (296 nm). Rt: 0.67 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, 0.1% formic acid in water/0.1% formic acid in ACN).
4-(1-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)piperidin-4-yl)morpholine (56)
White solid. Yield = 43%. 1H NMR (400 MHz, DMSO-d6) δ 1.16–1.36 (m, 2H), 1.60 (qd, J = 12.04, 4.04 Hz, 2H), 1.82 (br d, J = 10.61 Hz, 2H), 1.94–2.04 (m, 2H), 2.47 (br s, 4H), 2.55–2.63 (m, 2H), 2.86 (br t, J = 11.49 Hz, 2H), 2.97–3.22 (m, 3H), 3.32 (br s, 2H), 3.53–3.60 (m, 4H), 4.56 (br d, J = 12.88 Hz, 2H), 6.31 (d, J = 5.05 Hz, 1H), 7.24–7.32 (m, 2H), 7.53–7.61 (m, 2H), 8.21 (d, J = 5.31 Hz, 1H). MS: m/e 509 (MH+). Purity was determined as 93.8% by HPLC (239 nm). Rt: 1.00 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-((1-(4-(4-(4-Fluorophenyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-yl)piperidin-4-yl)methyl)morpholine (57)
White solid. Yield = 31%.1H NMR (400 MHz, DMSO-d6) δ 0.95–1.14 (m, 2H), 1.59 (dd, J = 12.13, 3.54 Hz, 2H), 1.75 (br d, J = 13.14 Hz, 3H), 1.99 (br d, J = 10.36 Hz, 2H), 2.13 (d, J = 7.07 Hz, 2H), 2.27–2.38 (m, 4H), 2.54–2.65 (m, 3H), 2.71–2.91 (m, 2H), 2.96–3.05 (m, 1H), 2.97–3.05 (m, 1H), 3.05–3.17 (m, 1H), 3.57 (t, J = 4.42 Hz, 4H), 4.55 (br d, J = 13.14 Hz, 2H), 6.29 (d, J = 5.31 Hz, 1H), 7.24–7.31 (m, 2H), 7.54–7.59 (m, 2H), 8.20 (d, J = 5.05 Hz, 1H). MS: m/e 523 (MH+). Purity was determined as 100% by HPLC (240 nm). Rt: 1.10 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
Synthesis of Final Compounds 58–60
In a microwave vial with a stirring bar, tert-butyl 4-(5-(2-chloropyrimidin-4-yl)-4-cyclopropylthiazol-2-yl)piperidine-1-carboxylate or tert-butyl 4-(5-(2-chloropyrimidin-4-yl)-4-(piperidin-1-ylmethyl)thiazol-2-yl)piperidine-1-carboxylate (o) (0.048 mmol), 4-aminopyrimidine or 2-methyl-4-aminopyridine (0.059 mmol), Pd2(dba)3 (0.003 mmol), Xantphos (0.005 mmol), and potassium tert-butoxide (0.095 mmol) are put and air is removed under reduced pressure. Then, anhydrous and degassed toluene (1.5 mL) is added and the reaction mixture is refluxed for 2 h under nitrogen. The reaction is monitored by TLC. Toluene is distilled off under reduced pressure, and the residue is purified on silica gel cartridge eluted with EtOAc:iPrOH 95:5, affording the desired compounds as yellow solids (72–88% yield), which were used directly in the next step. The Boc-protected thiazole derivatives of the previous step (0.042 mmol) dissolved in dry dioxane (0.6 mL) are treated with 4 N HCl/dioxane (0.84 mmol), and the mixture is stirred at room temperature for 1.5 h, after which LCMS showed the complete consumption of the starting material. Dioxane is distilled off, the residue is washed with diethyl ether, and is dried in vacuum to give the desired hydrochloride salt as a yellow solid.
4-(4-Cyclopropyl-2-(piperidin-4-yl)thiazol-5-yl)-N-(pyrimidin-4-yl)pyrimidin-2-amine Hydrochloride (58)
Yellow solid. Yield = 89%. 1H NMR (400 MHz, DMSO-d6) δ 0.99–1.15 (m, 4H), 1.91 (br d, J = 9.85 Hz, 2H), 2.18 (br d, J = 11.62 Hz, 2H), 2.85 (s, 1H), 3.02 (br d, J = 10.61 Hz, 2H), 3.25–3.41 (m, 3H), 7.51 (d, J = 5.31 Hz, 1H), 8.32–8.40 (m, 1H), 8.69 (d, J = 6.57 Hz, 1H), 8.72 (d, J = 5.56 Hz, 1H), 8.96 (s, 2H), 11.25 (br s, 1H). MS: m/e (free base) 380 (MH+). Purity was determined as 95.7% by HPLC (290 nm). Rt: 0.67 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
4-(4-Cyclopropyl-2-(piperidin-4-yl)thiazol-5-yl)-N-(2-methylpyridin-4-yl)pyrimidin-2-amine Dihydrochloride (59)
Yellow solid. Yield = 90%.1H NMR (400 MHz, DMSO-d6) δ 1.02–1.15 (m, 4H), 1.93 (br d, J = 10.11 Hz, 2H), 2.18 (br d, J = 11.87 Hz, 2H), 2.61–2.67 (m, 3H), 3.02 (br d, J = 9.85 Hz, 2H), 3.27–3.43 (m, 5H), 7.62 (d, J = 5.31 Hz, 1H), 8.02 (br d, J = 6.06 Hz, 1H), 8.17 (br s, 1H), 8.51 (d, J = 7.07 Hz, 1H), 8.78 (d, J = 5.31 Hz, 1H), 8.99 (br s, 2H), 11.33 (s, 1H). MS: m/e (free base) 393 (MH+). Purity was determined as 99.1% by HPLC (298 nm). Rt: 0.60 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
N-(2-Methylpyridin-4-yl)-4-(4-(piperidin-1-ylmethyl)-2-(piperidin-4-yl)thiazol-5-yl)pyrimidin-2-amine Trihydrochloride (60)
Yellow solid. Yield = 80%. 1H NMR (400 MHz, DMSO-d6) δ 1.34–1.49 (m, 1H), 1.62–1.73 (m, 1H), 1.73–1.89 (m, 4H), 1.97–2.15 (m, 2H), 2.23–2.35 (m, 2H), 2.66 (s, 3H), 3.03–3.15 (m, 2H), 3.16–3.27 (m, 2H), 3.43–3.53 (m, 4H), 3.57 (s, 2H), 4.91–5.03 (m, 2H), 7.52 (d, J = 5.31 Hz, 1H), 8.18–8.26 (m, 2H), 8.52 (d, J = 6.82 Hz, 1H), 8.84 (d, J = 5.31 Hz, 1H), 9.20–9.39 (m, 2H), 10.13–10.25 (m, 1H), 11.89 (br s, 1H). MS: m/e (free base) 450 (MH+). Purity was determined as 96.5% by HPLC (299 nm). Rt: 0.47 min (Acquity UPLC BEH C18 1.7 μm, 3 mm × 50 mm, CH3COO–NH4+ 25 mM + 10% acetonitrile at pH 6.6/acetonitrile).
In Vitro Whole Cell Drug Susceptibility Studies
Plasmodium falciparum 3D7A and NF54 strains (from the Malaria Research and Reference Reagent Resource Center MR4) were grown in complete medium [RPMI 1640 (Sigma), 25 mM HEPES and NaHCO3)] supplemented with 2 g/L d-sucrose, 0.3 g/L l-glutamine, and 0.150 mM hypoxanthine and with 5 g/L AlbuMAX II. Parasitized red blood cells (RBC) with 3D7A P. falciparum strain, (0.5% parasitemia, 2% hematocrit) in RPMI-1640, 5% AlbuMAX, and 5 μM hypoxanthine were exposed to threefold serial dilutions of the compounds (nine serial dilutions 5 μM as maximal concentration). After incubating the plates for 24 h at 37 °C and 5% CO2/5%O2/90% N2, [3H]-hypoxanthine (0.2 μCi to each well, from a stock solution of 3H-hypoxanthine of 0.025 μCi/μL in RPMI-1640) was added and the incubation of the plates was continued for another 24 h period. Thereafter, parasites were harvested on a glass fiber filter, filters were dried, and the incorporation of [3H]-hypoxanthine was determined using melt-on scintillator sheets. A microbeta counter was used for measuring radioactivity, while data normalization was performed by incorporation of the positive control (compound-free parasitized red blood cells). IC50 values were determined using Grafit 7 program.
In Vitro PKG Assay
The expression and purification of recombinant PKG as well as the PKG inhibitory activity assay were performed as previously described (see also the Supporting Information).54
Male/Female Gametocyte Functional Viability Assay
(A) Gametocyte production. Gametocyte cultures were produced as previously described (see also the Supporting Information).28 (B) Dual gamete formation assay. Gametocyte activation was triggered by reduced temperature and the addition of ookinete medium containing xanthurenic acid supplemented with the antibody anti-Pfs25-Cy3 at a final concentration of 1/2000 (from 1 mg/mL stock). Plates were analyzed to detect exflagellation centers. “Activated” cultures were then incubated (protected from light) at 26 °C for 24 h (in a thermo regulated incubator) to increase the fluorescent signal emitted by female gametes. The plates were then analyzed to record female activated gametes.28 (C) Measured parameters. The activation of male gametes is based on light change detections provoked by flagella movements causing movement of surrounding cells. A 10-frame video was taken and subsequently analyzed to determine the changes in cell position based on pixels change. The activation of female gametes was based on the detection of fluorescent Cy3-Anti Pfs25 antibody (as primary parameter), followed by selection of events according to their size, roundness, and the intensity of the fluorescence (see also the Supporting Information).55
In Vitro Parasite Reduction Ratio
In vitro PRR testing was conducted as previously described.56 The limiting dilution technique was used to quantify the number of parasites remaining viable after drug treatment. P. falciparum strain 3D7A (Malaria Research and Reference Reagent Resource Center, MR4, BEI Resources; Cat. No. MRA-102) was treated with a 10 × EC50 (antiparasitic activity in cells) drug concentration. Parasites were treated for 120 h. Parasite samples were collected from the treated culture every 24 h (24, 48, 72, 96, and 120 h time points). The number of viable parasites was determined by counting the number of wells with growth after 21 and 28 days, using [3H]-hypoxanthine incorporation (see also the Supporting Information).
Chemoproteomics-Target Identification Experiments
Kinobeads were prepared as described.31,32 Sepharose beads were derivatized with 31 or 50 at a concentration of 1 mM as described.30 The chemoproteomic affinity capturing experiments were performed as previously described.32 The experimental setup was such that 10 samples are measured in parallel (TMT 10-plex)57 to generate values for the affinity of the beads to the bound proteins and to generate IC50 values in a single experiment. Apparent dissociation constants were determined by taking into account the protein depletion by the beads.32 Proteins were digested according to a modified single-pot solid-phase sample preparation (SP3) protocol.58,59 Peptides were labeled with isobaric mass tags (TMT10, Thermo Fisher Scientific, Waltham, MA) using the 10-plex TMT reagents, enabling relative quantification of 10 conditions in a single experiment.57,60 LC-MS/MS measurements on Q Exactive Orbitrap or Orbitrap Fusion Lumos mass spectrometers (Thermo Fisher Scientific) was performed as described elsewhere.61 Analytical procedures and raw data tables for the chemoproteomics experiments can be found in the Supporting Information files 1 and 2, respectively.
Glossary
Abbreviations
- PKG
cGMP-dependent protein kinase
- SRPK2
serine/arginine protein kinase 2
- CDPK1,4
calcium-dependent protein kinase 1,4
- GSK3
glycogen synthase kinase 3
- CK1
casein kinase 1
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01099.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The Tres Cantos Open Lab Foundation and the Wellcome Trust.
The authors declare no competing financial interest.
Supplementary Material
References
- www.who.int/news-room/fact-sheets/detail/malaria (accessed February 25, 2019).
- Gogtay N.; Kannan S.; Thatte U. M.; Olliaro P. L.; Sinclair D. Artemisinin-based combination therapy for treating uncomplicated plasmodium vivax malaria. Cochrane Database Syst. Rev. 2013, 10, CD008492 10.1002/14651858.CD008492.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price R. N.; Douglas N. M. Artemisin combination therapy for malaria: Beyond good efficacy. Clin. Infect. Dis. 2009, 49, 1638–1640. 10.1086/647947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; et al. Artemisinin resistance: Current status and scenarios for containment. Nat. Rev. Macrobiol. 2010, 8, 272–280. 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- Straimer J.; Gnädig N. F.; Witkowski B.; Amaratunga C.; Duru V.; Ramadani A. P.; Dacheux M.; Khim N.; Zhang L.; Lam S.; Gregory P. D.; Urnov F. D.; Mercereau-Puijalon O.; Benoit-Vical F.; Fairhurst R. M.; Mènard D.; Fidock D. A. Drug resistance. K13-propeller mutations confer artemisin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga C.; Lim P.; Suon S.; Sreng S.; Mao S.; Sopha C.; Sam B.; Dek D.; Try V.; Amato R.; Blassborn D.; Song L.; Tullo G. S.; Fay M. P.; Anderson J. M.; Tarning J.; Fairhurst R. M. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in cambodia: a multisite prospective cohort study. Lancet Infect. Dis. 2016, 16, 357–365. 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley E. A.; Dhorda M.; Fairhurst R. M.; Amaratunga C.; Lim P.; Suon S.; Sreng S.; Anderson J. M.; Mao S.; Sam B.; Sopha C.; Chuor C. M.; Nguon C.; Sovannaroth S.; Pukrittayakamee S.; Jittamala P.; Chotivanich K.; Chutasmit K.; Suchatsoonthorn C.; Runcharoen R.; Hien T. T.; Thuy-Nhien N. T.; Thanh N. V.; Phu N. H.; Htut Y.; Han K. T.; Aye K. H.; Mokuolu O. A.; Olaosebikan R. R.; Folaranmi O. O.; Mayxay M.; Khanthavong M.; Hongvanthong B.; Newton P. N.; Onyamboko M. A.; Fanello C. I.; Tshefu A. K.; Mishra N.; Valecha N.; Phyo A. P.; Nosten F.; Yi P.; Tripura R.; Borrmann S.; Bashraheil M.; Peshu J.; Faiz M. A.; Ghose A.; Hossain M. A.; Samad R.; Rahman M. R.; Hasan M. M.; Islam A.; Miotto O.; Amato R.; MacInnis B.; Stalker J.; Kwiatkowski D. P.; Bozdech Z.; Jeeyapant A.; Cheah P. Y.; Sakulthaew T.; Chalk J.; Intharabut B.; Silamut K.; Lee S. J.; Vihokhern B.; Kunasol C.; Imwong M.; Tarning J.; Taylor W. J.; Yeung S.; Woodrow C. J.; Flegg J. A.; Das D.; Smith J.; Venkatesan M.; Plowe C. V.; Stepniewska K.; Guerin P. J.; Dondorp A. M.; Day N. P.; White N. J. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley C. H. Observing in real time the evolution of artemisin resistance in Plasmodium falciparum. BMC Med. 2015, 13, 67–70. 10.1186/s12916-015-0316-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Fairhurst R. M.; Slutsker L.; Macarthur J. R.; Breman J. G.; Guerin P. J.; Wellems T. E.; Ringwald P.; Newman R. D.; Plowe C. V. The thread of artemisinin-resistant malaria. N. Engl. J. Med. 2011, 365, 1073–1075. 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet M.; Collins M. O.; Smith T. K.; Thompson E.; Sebastian S.; Volkmann K.; Schwach F.; Chappell L.; Gomes A. R.; Berriman M.; Rayner J. C.; Baker D. A.; Choudhary J.; Billker O. Phosphoinositide metabolism links Cgmp-dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasite. PLoS Biol. 2014, 12, e1001806 10.1371/journal.pbio.1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz C. A.; Allocco J.; Powles M. A.; Yeung L.; Donald R. G.; Anderson J. W.; Liberator P. A. Characterization of Plasmodium falciparum cGMP-dependent protein kinase (PfPKG): antiparasitic activity of a PKG inhibitor. Mol. Biochem. Parasitol. 2006, 146, 78–88. 10.1016/j.molbiopara.2005.10.020. [DOI] [PubMed] [Google Scholar]
- McRobert L.; Taylor C. J.; Deng W.; Fivelman Q. L.; Cummings R. M.; Polley S. D.; Billker O.; Baker D. A. Gametogenesis in malaria parasites is mediated by the cGMP-dependent protein kinase. PLoS Biol. 2008, 6, e139 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon R. W.; Taylor C. J.; Bex C.; Schepers R.; Goulding D.; Janse C. J.; Waters A. P.; Baker D. A.; Billker O. A cyclic GMP signaling module that regulates gliding motility in a malaria parasite. PLoS Pathog. 2009, 5, e1000599 10.1371/journal.ppat.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor H. M.; McRobert L.; Grainger M.; Sicard A.; Dluzewski A. R.; Hopp C. S.; Holder A. A.; Baker D. A. The malaria parasite cyclic GMP-dependent protein kinase plays a central role in blood-stage schizogony. Eukaryotic Cell 2010, 9, 37–45. 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindasamy K.; Jebiwott S.; Jaijyan D. K.; Davidow A.; Ojo K. K.; Van Voorhis W. C.; Brochet M.; Billker O.; Bhanot P. Invasion of hepatocytes by plasmodium sporozoites requires cGMP-dependent protein kinase and calcium dependent protein kinase 4. Mol. Microbiol. 2016, 102, 349–363. 10.1111/mmi.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. M.; Solyakov L.; Bottrill A. R.; Flueck C.; Siddiqui F. A.; Singh S.; Mistry S.; Viskaduraki M.; Lee K.; Hopp C. S.; Chitnis C. E.; Doerig C.; Moon R. W.; Green J. L.; Holder A. A.; Baker D. A.; Tobin A. B. Phosphoproteomics reveals malaria parasite protein kinase G as a signaling hub regulating egress and invasion. Nat. Commun. 2015, 6, 7285 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D. A.; Stewart L. B.; Large J. M.; Bowyer P. W.; Ansell K. H.; Jiménez-Díaz M. B.; El Bakkouri M.; Birchall K.; Dechering K. J.; Bouloc N. S.; Coombs P. J.; Whalley D.; Harding D. J.; Smiljanic-Hurley E.; Wheldon M. C.; Walker E. M.; Dessens J. T.; Lafuente M. J.; Sanz L. M.; Gamo F. J.; Ferrer S. B.; Hui R.; Bousema T.; Angulo-Barturén I.; Merritt A. T.; Croft S. L.; Gutteridge W. E.; Kettleborough C. A.; Osborne S. A. A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission. Nat. Commun. 2017, 8, 430–438. 10.1038/s41467-017-00572-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Large J. M.; Birchalla K.; Bouloca N. S.; Merritta A. T.; Smiljanic-Hurleya E.; Tsagrisa D. J.; Wheldona M. C.; Ansella K. H.; Coombsa P. J.; Kettleborougha C. A.; Whalleya D.; Stewartb L. B.; Bowyerb P. W.; Baker D. A.; Osborne S. A. Potent inhibitors of malarial p. falciparum protein kinase G: improving the cell activity of a series of imidazopyridines. Bioorg. Med. Chem. Lett. 2019, 29, 509–514. 10.1016/j.bmcl.2018.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G.-B.; Qian X.; Feng D.; Fisher M.; Brown C. M.; Gurnett A.; Leavitt P. S.; Liberator P. A.; Misura A. S.; Tamas T.; Schmatz D. M.; Wyvratta M.; Biftua T. Synthesis and SAR studies of potent imidazopyridine anticoccidial agents. Bioorg. Med. Chem. Lett. 2007, 17, 3558–3561. 10.1016/j.bmcl.2007.04.041. [DOI] [PubMed] [Google Scholar]
- Qian X.; Liang G.-B.; Feng D.; Fisher M.; Crumley T.; Rattray S.; Dulski P. M.; Gurnett A.; Leavitt P. S.; Liberator P. A.; Misura A. S.; Samaras S.; Tamas T.; Schmatz D. M.; Wyvratta M.; Biftua T. Synthesis and SAR Studies of diarylpyrrole anticoccidial agents. Bioorg. Med. Chem. Lett. 2006, 16, 2817–2821. 10.1016/j.bmcl.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Tsagris D. J.; Birchalla K.; Bouloca N.; Largea J. M.; Merritta A.; Smiljanic-Hurleya E.; Wheldona M.; Ansella K. H.; Kettleborougha C.; Whalleya D.; Stewart L. B.; Bowyer P. W.; Baker D. A.; Osborne S. A. Trisubstituted thiazoles as potent and selective inhibitors of Plasmodium falciparum protein kinase G (PfPKG). Bioorg. Med. Chem. Lett. 2018, 28, 3168–3173. 10.1016/j.bmcl.2018.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W.-S.; Nam S.-W.; Ahn E.-K.; Park B.-S.; Lee S.-E.; Kim T.-J.; Choi I.-Y. Synthesis and fungicidal activity of N-[4-(4-fluoro)phenyl-2-piperidin-4-ylthiazol-5-yl]pyrimidin-2-yl-N-phenylamines on phytophthora capsici. J. Korean Soc. Appl. Biol. Chem. 2010, 53, 206–214. 10.3839/jksabc.2010.033. [DOI] [Google Scholar]
- Choi W. S.; et al. Synthesis and antifungal activity of 5-[2-(alkylamino)pyrimidin-4-yl]-4-phenylthiazol-2-cycloalkylamine derivatives on phytophthora capsici. J. Korean Soc. Appl. Biol. Chem. 2011, 54, 395–402. 10.3839/jksabc.2011.062. [DOI] [Google Scholar]
- Hameed S. P.; Solapure S.; Patil V.; Henrich P. P.; Magistrado P. A.; Bharath S.; Murugan K.; Viswanath P.; Puttur J.; Srivastava A.; Bellale E.; Panduga V.; Shanbag G.; Awasthy D.; Landge S.; Morayya S.; Koushik K.; Saralaya R.; Raichurkar A.; Rautela N.; Choudhury N. R.; Ambady A.; Nandishaiah R.; Reddy J.; Prabhakar K. R.; Menasinakai S.; Rudrapatna S.; Chatterji M.; Jiménez-Díaz M. B.; Martínez M. S.; Sanz L. M.; Coburn-Flynn O.; Fidock D. A.; Lukens A. K.; Wirth D. F.; Bandodkar B.; Mukherjee K.; McLaughlin R. E.; Waterson D.; Rosenbrier-Ribeiro L.; Hickling K.; Balasubramanian V.; Warner P.; Hosagrahara V.; Dudley A.; Iyer P. S.; Narayanan S.; Kavanagh S.; Sambandamurthy V. K. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat. Commun. 2015, 6, 6715 10.1038/ncomms7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauffer D.; Li P.; Waal N.; McGinty K.; Tang Q.; Ronkin S.; Farmer L.; Shannon D.; Jacobs D.. C-Met Protein Kinase Inhibitors. WO2009/045992A2. [Google Scholar]
- Schnürch M.; Khan A. F.; Mihovilovic M. D.; Stanetty P. Polyarylated thiazoles via a combined halogen dance – cross-coupling strategy. Eur. J. Org. Chem. 2009, 3228–3236. 10.1002/ejoc.200900092. [DOI] [Google Scholar]
- Masungi C.; Mensch J.; Van Dijck A.; Borremans C.; Willems B.; Mackie C.; Noppe M.; Brewster M. E. Parallel artificial membrane permeability assay (PAMPA) combined with a 10-day multiscreen Caco-2 cell culture as a tool for assessing new drug candidates. Pharmazie 2008, 3, 194–199. 10.1691/ph.2008.7327. [DOI] [PubMed] [Google Scholar]
- Ruecker A.; Mathias D. K.; Straschil U.; Churcher T. S.; Dinglasan R. R.; Leroy D.; Sinden R. E.; Delvesa M. J. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob. Agents Chemother. 2014, 58, 7292–7302. 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D.; Zhou T.; Lafleur K.; Nevado C.; Caflisch A. Kinase selectivity potential for inhibitors targeting the ATP binding site: a network analysis. Bioinformatics 2010, 26, 198–204. 10.1093/bioinformatics/btp650. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 2007, 25, 1035–1044. 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- Bergamini G.; Bell K.; Shimamura S.; Werner T.; Cansfield A.; Müller K.; Perrin J.; Rau C.; Ellard K.; Hopf C.; Doce C.; Leggate D.; Mangano R.; Mathieson T.; O’Mahony A.; Plavec I.; Rharbaoui F.; Reinhard F.; Savitski M. M.; Ramsden N.; Hirsch E.; Drewes G.; Rausch O.; Bantscheff M.; Neubauer G. A selective inhibitor reveals PI3K gamma dependence of T(H)17 cell differentiation. Nat. Chem. Biol. 2012, 8, 576–582. 10.1038/nchembio.957. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Hopf C.; Savitski M. M.; Dittmann A.; Grandi P.; Michon A. M.; Schlegl J.; Abraham Y.; Becher I.; Bergamini G.; Boesche M.; Delling M.; Dümpelfeld B.; Eberhard D.; Huthmacher C.; Mathieson T.; Poeckel D.; Reader V.; Strunk K.; Sweetman G.; Kruse U.; Neubauer G.; Ramsden N. G.; Drewes G. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- Kato N.; Sakata T.; Breton G.; Le Roch K. G.; Nagle A.; Andersen C.; Bursulaya B.; Henson K.; Johnson J.; Kumar K. A.; Marr F.; Mason D.; McNamara C.; Plouffe D.; Ramachandran V.; Spooner M.; Tuntland T.; Zhou Y.; Peters E. C.; Chatterjee A.; Schultz1 P. G.; Ward G. E.; Gray N.; Harper J.; Winzeler E. A. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat. Chem. Biol. 2008, 4, 347–356. 10.1038/nchembio.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S.; Kern S.; Halbert J.; Przyborski J. M.; Baumeister S.; Dandekar T.; Doerig C.; Pradel G. Two nucleus-localized CDK-like kinases with crucial roles for malaria parasite erythrocytic replication are involved in phosphorylation of splicing factor. J. Cell Biochem. 2011, 112, 1295–1310. 10.1002/jcb.23034. [DOI] [PubMed] [Google Scholar]
- Solyakov L.; Halbert J.; Alam M. M.; Semblat J.-P.; Dorin-Semblat D.; Reininger L.; Bottrill A. R.; Mistry S.; Abdi A.; Fennell C.; Holland Z.; Demarta C.; Bouza Y.; Sicard A.; Nivez M.-P.; Eschenlauer S.; Lama T.; Thomas D. C.; Sharma P.; Agarwal S.; Kern S.; Pradel G.; Graciotti M.; Tobin A. B.; Doerig C. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat. Commun. 2011, 2, 565 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- www.plasmodb.org (accessed August 26, 2019). [Google Scholar]
- Yeoh L. M.; Lee V. V.; McFadden G. I.; Ralph S. A. Alternative splicing in apicomplexan parasites. MBio 2019, 10, e02866 10.1128/mBio.02866-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern S.; Agarwal S.; Huber K.; Gehring A. P.; Strödke B.; Wirth C. C.; Brügl T.; Abodo L. O.; Dandekar T.; Doerig C.; Fischer R.; Tobin A. B.; Alam M. M.; Bracher F.; Pradel G. Inhibition of the SR protein-phosphorylating CLK kinases of Plasmodium falciparum impairs blood stage replication and malaria transmission. PLoS One 2014, 9, e105732 10.1371/journal.pone.0105732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y.; Ninomiya K.; Okuno Y.; Toyomoto M.; Nishida A.; Koike Y.; Ohe K.; Kii I.; Yoshida S.; Hashimoto N.; Hosoya T.; Matsuo M.; Hagiwara M. Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci. Rep. 2017, 7, 46126 10.1038/srep46126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P.; Karthikeyan C.; Moorthy N. S.; Waiker D. K.; Jain A. K.; Trivedi P. Human CDC2-like kinase 1 (CLK1): a novel target for Alzheimer’s disease. Curr. Drug Targets 2014, 15, 539–550. 10.2174/1389450115666140226112321. [DOI] [PubMed] [Google Scholar]
- Alam M. M.; Sanchez-Azqueta A.; Janha O.; Flannery E. L.; Mahindra A.; Mapesa K.; Char A. B.; Sriranganadane D.; Brancucci N. M. B.; Antonova-Koch Y.; Crouch K.; Simwela N. V.; Millar S. B.; Akinwale J.; Mitcheson D.; Solyakov L.; Dudek K.; Jones C.; Zapatero C.; Doerig C.; Nwakanma D. C.; Vázquez M. J.; Colmenarejo G.; Lafuente-Monasterio M. J.; Leon M. L.; Godoi P. H. C.; Elkins J. M.; Waters A. P.; Jamieson A. G.; Álvaro E. F.; Ranford-Cartwright L. C.; Marti M.; Winzeler E. A.; Gamo F. J.; Tobin A. B. Validation of the protein kinase PfCLK3 as a multistage cross-species malarial drug target. Science 2019, 365, eaau1682 10.1126/science.aau1682. [DOI] [PubMed] [Google Scholar]
- Albuquerque S. S.; Carret C.; Grosso A. R.; Tarun A. S.; Peng X.; Kappe S. H. I.; Prudêncio M.; Mota M. M. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics 2009, 10, 270. 10.1186/1471-2164-10-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arang N.; Kain H. S.; Glennon E. K.; Bello T.; Dudgeon D. R.; Walter E. N. F.; Gujral T. S.; Kaushansky A. Identifying host regulators and inhibitors of liver stage malaria infection using kinase activity profiles. Nat. Commun. 2017, 8, 1232 10.1038/s41467-017-01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudêncio M.; Rodrigues C. D.; Hannus M.; Martin C.; Real E.; Goncalves L. A.; Carret C.; Dorkin R.; Rohl I.; Jahn-Hoffmann K.; Luty A. J. F.; Sauerwein R.; Echeverri C. J.; Mota M. M. Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in plasmodium sporozoite infection. PLoS Pathog. 2008, 4, e1000201 10.1371/journal.ppat.1000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy S. C.; Harrison T.; Hamm H. E.; Lomasney J. W.; Mohandas N.; Haldar K. Erythrocyte G protein as a novel target for malarial chemotherapy. PLoS Med. 2006, 3, e528 10.1371/journal.pmed.0030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard A.; Semblat J. P.; Doerig C.; Hamelin R.; Moniatte M.; Dorin-Semblat D.; Spicer J. A.; Srivastava A.; Retzlaff S.; Heussler V.; Waters A. P.; Doerig C. Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cell. Microbiol. 2011, 13, 836–845. 10.1111/j.1462-5822.2011.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennon E. K. K.; Dankwa S.; Smith J. D.; Kaushansky A. Opportunities for host-targeted therapies for malaria. Trends Parasitol. 2018, 34, 843–860. 10.1016/j.pt.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souma T.; Thomsona B. R.; Heinenc S.; Carota I. A.; Yamaguchi S.; Onaya T.; Liua P.; Ghosha A. K.; Lid C.; Ereminad V.; Honge Y. K.; Economides A. N.; Vestweberg D.; Petersh K. G.; Jin J.; Quaggina S. E. Context-dependent functions of angiopoietin 2 are determined by the endothelial phosphatase VEPTP. Proc. Natl. Acad. Sci. USA 2018, 115, 1298–1303. 10.1073/pnas.1714446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerig C.; Abdi A.; Bland N.; Eschenlauer S.; Dorin-Semblat D.; Fennell C.; Halbert J.; Holland Z.; Nivez M. P.; Semblat J. P.; Sicard A.; Reininger L. Malaria: targeting parasite and host cell kinomes. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 604–612. 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Lucet I. S.; Tobin A.; Drewry D.; Wilks A. F.; Doerig C. Plasmodium kinases as targets for new-generation antimalarials. Future Med. Chem. 2012, 4, 2295–2310. 10.4155/fmc.12.183. [DOI] [PubMed] [Google Scholar]
- Ward P.; Equinet L.; Packer J.; Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics 2004, 5, 79 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan A. N.; Krupa A. A genomic perspective of protein kinases in Plasmodium falciparum. Proteins 2005, 58, 180–189. 10.1002/prot.20278. [DOI] [PubMed] [Google Scholar]
- Talevich E.; Tobin A. B.; Kannan N.; Doerig C. An evolutionary perspective on the kinome of malaria parasites. Philos. Trans. R. Soc., B 2012, 367, 2607–2618. 10.1098/rstb.2012.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W.; Parbhu-Patel A.; Meyer D. J.; Baker D. A. The role of two novel regulatory sites in the activation of the cGMP-dependent protein kinase from Plasmodium falciparum. Biochem. J. 2003, 374, 559–565. 10.1042/bj20030474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Blanco C.; Lelièvre J.; Delves M. J.; Bardera A. I.; Presa J. L.; López-Barragán M. J.; Ruecker A.; Marques S.; Sinden R. E.; Herrerosa E. Imaging-based high-throughput screening assay to identify new molecules with transmission-blocking potential against Plasmodium falciparum female gamete formation. Antimicrob. Agents Chemother. 2015, 59, 3298–3305. 10.1128/AAC.04684-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L. M.; Crespo B.; De-Cózar C.; Ding X. C.; Llergo J. L.; Burrows J. N.; García-Bustos J. F.; Gamo F.-J. P. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PLoS ONE 2012, 7, e30949 10.1371/journal.pone.0030949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner T.; Sweetman G.; Savitski M. F.; Mathieson T.; Bantscheff M.; Savitski M. M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014, 86, 3594–3601. 10.1021/ac500140s. [DOI] [PubMed] [Google Scholar]
- Hughes C. S.; Foehr S.; Garfield D. A.; Furlong E. E.; Steinmetz L. M.; Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol. Syst. Biol. 2014, 10, 757. 10.15252/msb.20145625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggridge S.; Sorensen P. H.; Morin G. B.; Hughes C. S. Extending the compatibility of the SP3 paramagnetic bead processing approach for proteomics. J. Proteome Res. 2018, 17, 1730–1740. 10.1021/acs.jproteome.7b00913. [DOI] [PubMed] [Google Scholar]
- Werner T.; Becher I.; Sweetman G.; Doce C.; Savitski M. M.; Bantscheff M. High-resolution enabled TMT 8-plexing. Anal. Chem. 2012, 84, 7188–7194. 10.1021/ac301553x. [DOI] [PubMed] [Google Scholar]
- Sridharan S.; Kurzawa N.; Werner T.; Günthner I.; Helm D.; Huber W.; Bantscheff M.; Savitski M. M. Proteome-wide solubility and thermal stability profiling reveals distinct regulatory roles for ATP. Nat. Commun. 2019, 10, 1155 10.1038/s41467-019-09107-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.