

Abstract
Chimeric antigen receptor (CAR)-T cells show great promise in treating cancers and viral infections. However, most protocols developed to expand T cells require relatively long periods of time in culture, potentially leading to progression toward populations of terminally differentiated effector memory cells. Here, we describe in detail a 9-day protocol for CAR gene transduction and expansion of primary rhesus macaque peripheral blood mononuclear cells (PBMCs). Cells produced and expanded with this method show high levels of viability, high levels of co-expression of two transduced genes, retention of the central memory phenotype, and sufficient quantity for immunotherapeutic infusion of 1–2 × 108 cells/kg in a 10 kg rhesus macaque. This 9-day protocol may be broadly used for CAR-T cell and other T cell immunotherapy approaches to decrease culture time and increase maintenance of central memory populations.
Keywords: CAR-T cells, transduction, expansion, retrovirus, PBMC, central memory, rhesus macaque
Graphical Abstract
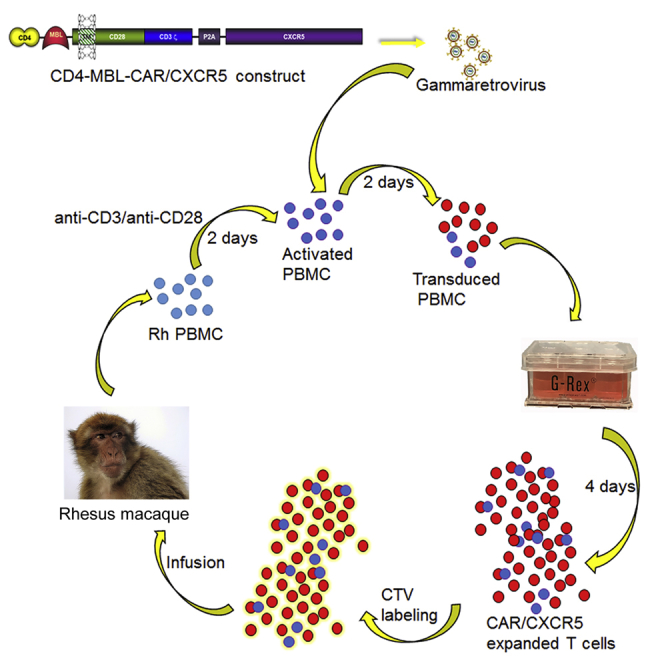
Introduction
Chimeric antigen receptor (CAR)-T cells have been used successfully in cancer immunotherapy, especially B cell leukemias.1, 2, 3 Engineered CAR-T cells also have the potential to treat viral infections such as HIV by specifically targeting viral envelope proteins on the surface of infected cells.4 Our studies have utilized a bispecific CAR encoding domains 1 and 2 of CD4, targeting the CD4 binding site of HIV and the carbohydrate recognition domain of mannose-binding lectin (MBL), which targets carbohydrates on HIV envelope glycoproteins, leading to enhanced potency against HIV.5 For studies in rhesus macaques, we modified the CAR to encode rhesus macaque CD4 and MBL motifs for the CAR to specifically bind simian immunodeficiency virus (SIV) envelope glycoproteins.6
HIV and SIV producing cells are concentrated within B cell follicles in secondary lymphoid tissues. However, cytotoxic T cells are typically in relatively low concentrations in follicles,7, 8 likely contributing to the concentration of virus within the follicle. Production of HIV- or SIV-specific CAR-T cells that include the follicular homing receptor CXCR5 could better target the follicular reservoir of infected cells.6, 9 For this reason, we set out to develop, and present here, a method to use gammaretroviral vectors encoding CD4-MBL-CAR/CXCR5 to transduce primary rhesus macaque peripheral blood mononuclear cells (PBMCs) and expand these cells to levels sufficient for therapeutic infusion.
In clinical trials, a failure of infused T cells to persist has been correlated with the absence of CD4+ T cells in the CD8+ T cell product.10 The use of PBMCs in the production of the CAR-T cells in this protocol allows the final cultures to contain both antigen-specific CD8+ T cells and CD4+ T cells. PBMCs are activated with anti-CD3 and anti-CD28 along with interleukin 2 (IL-2) at a relatively high density, allowing for cell to cell contact.11 After activation, the cells are transduced with a gammaretroviral vector containing the CD4-MBL CAR and CXCR5. The gammaretroviral approach is appealing because there are self-inactivating gammaretroviral vectors that are replication incompetent and induce long-term expression of the genes of interest following transduction.12 They are also a safe and efficient method of gene transfer. Initial results have shown the promise of this approach in clinical trials utilizing HIV-specific CAR-T cells produced by gammaretroviral transduction. A study by Scholler et al.13 demonstrated stable engraftment of the CAR-T cells with a calculated decay half-life that exceeded 16 years without vector-induced cell immortalization. However, the number of cells that were stably engrafted was low, and these cells did not specifically target viral reservoirs within B cell follicles. Further studies are needed to develop immunotherapy methods that target viral reservoirs and persist at high enough levels to lead to sustained remission of HIV infection.
Two commercial products have been instrumental in optimization of this CAR-T cell expansion protocol. The first is X-Vivo 15 medium, a Lonza product, which is designed for the growth of hematopoietic cells and has been used in the production of CAR-T cells for clinical trials.14, 15 This media has also been used to produce CAR-T cells for infusion into rhesus macaques.16 The second product is G-REX vessels, which have a gas permeable membrane at the base of the well that allows optimal gas exchange in large volumes of media, leading to enhanced cell survival and optimized nutrient availability.17 Use of these vessels allows undisturbed growth during the expansion phase of cell production. G-Rex vessels have been used for expansion of human antigen-specific T cells,18 human CAR-T cells,19 and T cell receptor (TCR)-transduced human T cells.20
Ideally, adoptively transferred T cells should express a less differentiated phenotype because those cell subsets circulate to lymphoid organs and are capable of robust expansion.21 In animal studies and human clinical trials, it is apparent that less differentiated T cells, defined as cells expressing the lymphoid homing molecules CD62L and CCR7, better contribute to engraftment and long-term persistence.22 In rhesus macaques, the less differentiated central memory phenotype is defined by expression of the co-receptor CD28 and the cell death receptor CD95.23 CD62L fails to reliably distinguish memory subsets in macaques.23 Studies suggest that cells produced in shorter time frames may have improved phenotype and cell function. In a study with CAR-T cells derived from human tumor cells, shortened ex vivo culture times of 3 to 5 days led to CAR-T cells with reduced differentiation and superior in vitro effector functions as compared to cells grown for 9 days.24 Rhesus macaque CAR-T cells grown for 8–17 days had a primarily central memory phenotype.16 Conversely, adoptively transferred cells, grown for extended periods of time, have been shown to localize to the lungs of the rhesus macaque rather than lymphoid tissues.25, 26 With these studies in mind, we developed a protocol for producing rhesus macaque CAR-T cells that utilizes a single rapid transduction step and a 4-day expansion step, leading to a shortened overall ex vivo culture time of 8 days. This method results in a transduced cell population, with a majority of the cells exhibiting a central memory phenotype.
The target dose of cells for infusion into a non-human primate is not firmly established, but previous adoptive transfer studies have used doses including 0.6–1.2 × 107 cells/kg,16 1–5 × 108 cells/kg,27 and 1.4–8 × 108 cells/kg.9 Based on these studies, we have set a target of 1–2 × 108 cells/kg for CAR/CXCR5 T cell immunotherapy in rhesus macaques. Using that guideline and the protocol outlined here, sufficient functional cells can be produced in a 9-day time frame for in vivo infusion studies in macaques, which will allow assessment of the ability of the transduced cells to control SIV. This protocol meets the challenge of producing enough cells to infuse into a 10 kg animal while minimizing culture time to avoid terminal differentiation and potential failure to engraft. Although the protocol is optimized for the production of rhesus macaque CAR-T cells, it could be modified to be used with other species.
Materials
Reagents
-
•
X-Vivo-15 medium (Lonza, 04-418Q)
-
•
Heat-inactivated fetal bovine serum (FBS; Hyclone, Sh30088.03)
-
•
Penicillin/streptomycin/glutamine (GIBCO, 10378-016)
-
•
IL-2 (NCI Preclinical Repository)
-
•
β mercaptoethanol (GIBCO, 21985-023)
-
•
RetroNectin (1 μg/μL) (TaKaRa, T100A)
-
•
BSA (Fraction V) (HyClone, SH 30574.02)
-
•
Anti-macaque CD3 (NHP Reagent Resource, Clone: FN18)
-
•
Anti-CD28 (NHP Reagent Resource, Clone: CD28.2)
-
•
PBS (GIBCO, 14190-144)
-
•
G-Rex 6-well plate (Wilson Wolf, P/N 80240M)
-
•
6-well plates, untreated (CytoOne, CC7672-7506)
-
•
15 mL and 50 mL conical centrifuge tubes (Thermo Scientific, 339650 and 339652)
-
•
Sterile pipets
Equipment
-
•
Beckman Allegra Centrifuge (Beckman, X13-R)
-
•
Swinging bucket rotor (Beckman, SX4750A Aries)
-
•
Microplate carriers with biocertified covers (Beckman, SX4750μ)
-
•
Aerosolve canisters to contain aerosol leakage (Beckman, 359232)
-
•
Laminar flow hood (Baker, Sterilgard e3)
Reagent Setup
-
•
Basic medium: X-Vivo 15 medium + 10% heat-inactivated FBS + 1 x penicillin/streptomycin/L-glutamine
-
•
Activation medium: basic medium + 50 IU/mL IL-2 + 5 μg/mL anti-CD28
-
•
Growth medium: basic medium + 50 IU/mL IL-2
-
•
Expansion medium: growth medium + 50 μM β mercaptoethanol
Procedure
Stimulation of PBMCs (Day 1)
Coat Plates with Anti-CD3
-
1.
Prepare a 10 μg/mL solution of anti-macaque CD3 (FN18) in PBS.
-
2.
Dispense 2 ml/well of a 6 well plate.
-
3.
Incubate at 37°C for 2 h or overnight at 4°C.
-
4.
Aspirate PBS/antibody.
-
5.
Wash two times with 2 mL PBS.
Stimulate PBMCs
-
6.
Thaw rhesus PBMCs in a waterbath until a small amount of ice remains.
-
7.
Gently add cells to a 15 mL conical. Rinse vial with 1 mL basic medium and add dropwise to cells.
-
8.
Add an additional 9 mL warm basic medium dropwise to cells.
NOTE: scale up with multiple vials of cells but never thaw more than 4 vials at one time.
-
9.
Spin at 600 × g for 5 min to pellet cells.
-
10.
Aspirate and resuspend pellet in a small amount of activation medium.
NOTE: the concentration should be greater than 2 × 106 cells/mL at this point.
-
11.
Count cells to determine live cell number.
-
12.
Dilute to 2 × 106 cells/mL in activation medium.
NOTE: activation medium contains anti-CD28 antibodies, a necessary co-stimulatory signal for T cell activation.
-
13.
Plate cells in the anti-CD3-coated plate. Add 3–6 × 106 cells/well (we usually use 4 × 106 cells in 2 mL media per well) and incubate for 2 days at 37°C, 5% CO2.
Preparation of RetroNectin-Coated Plates (Day 2)
NOTE: RetroNectin-mediated transduction requires binding to VLA-4 and/or VLA-5 integrin receptors. T cells express VLA-4 and activated T cell express VLA-5, leading to effective retronectin-mediated transduction. If using another type of cells for transduction, it is important to verify that the cells express these integrin receptors.
-
14.
Prior to coating, prepare a RetroNectin solution (1:100) by diluting with sterile PBS.
-
15.
Dispense an appropriate volume of sterile RetroNectin solution into each plate (2 mL/well in a 6-well plate) and allow the plate to rock for 2 h at room temperature. NOTE: non-treated, cell culture-grade tissue culture plates or dishes should be used in this step.
-
16.
Remove the RetroNectin solution and then block with 1 mL sterile 2% bovine serum albumin (BSA, Fraction V) in PBS. Allow the plate to rock at room temperature for 30 min.
-
17.
Aspirate BSA solution and wash the plate once with 2 mL PBS. After removing the wash solution, the plate is ready for use.
-
18.
The RetroNectin coated plate can be sealed with Parafilm and stored at 4°C for up to 1 week.
RetroNectin-Mediated Transduction Protocol (Day 3)
Attachment of Retroviral Transducing Vector
-
19.
Warm centrifuge to 32°C by running at 2,000 × g for about 30 min.
-
20.
Determine the amount of virus needed to transduce cells with an MOI of 0.5.
NOTE: see troubleshooting section for notes on determination of virus titer and MOI.
-
21.
Thaw the virus on ice or by gently swirling the vial in the 37°C water bath until only a small amount of ice remains.
CAUTION: to avoid degradation of the viral preparation, do not allow the contents to warm.
-
22.
Dilute retrovirus to an MOI of 0.5 in serum-free X-vivo medium. NOTE: decontaminate and discard any unused, thawed virus.
-
23.
Add 2 mL diluted retrovirus to each well of the RetroNectin-coated plate.
-
24.
For mock transduced cells, add only media to the RetroNectin-coated wells.
-
25.
Place plates in the prewarmed centrifuge in microplate carriers with biocertified covers.
-
26.
Centrifuge at 2,000 × g for 2 h at 32°C.
-
27.
Virus-coated plates can be used immediately or stored, with virus prep in the well, at 4°C overnight.
-
28.
For immediate use, aspirate the virus preparation from the prepared wells and add 2 mL growth medium.
CAUTION: do not allow the virus-coated wells to dry.
Transduction
-
29.
Collect the target cells and count the number of living cells.
NOTE: stimulated cells will be clumped, and this clumping may lead to difficulties in accurately counting the cells.
-
30.
Centrifuge at 600 × g for 5 min at 25°C to pellet cells.
-
31.
Aspirate media from the cell pellet and resuspend the cells in growth medium at a concentration of 1.5 × 106 cells/mL.
-
32.
Add 1 mL cell suspension to each virus-coated well (step 28) for a total of 1.5 × 106 cells/3 mL. (Note that each well already contains 2 mL growth medium.)
-
33.
Mock transduced cells are plated onto RetroNectin-coated wells that received no virus.
-
34.
Centrifuge at 1,000 × g for 10 min at 32°C.
-
35.
Incubate for 48 h at 37°C, 5% CO2.
Expansion of Transduced Cells (Day 5)
-
36.
Collect the cells from the transduction plates.
-
37.
Rinse each well with 1 mL media to remove adherent cells.
-
38.
Count to check the cell number and viability.
-
39.
Remove 1 × 106 cells for flow cytometry to assess gene expression and phenotype.
-
40.
Centrifuge the remaining cells at 600 × g for 5 min at 25°C.
-
41.
Aspirate the media and resuspend the cells in expansion media to a concentration of 1 × 106 cells/mL.
-
42.
Seed 5 mL of the cells in each GREX well.
-
43.
Carefully layer an additional 25 mL expansion media per well.
NOTE: add the cells in a small volume and then layer remaining media so that the cells remain on the bottom of the well.
-
44.
Incubate undisturbed at 37°C, 5% CO2 for 4 days.
NOTE: if additional days of culture are desired, remove 20 mL media without disturbing the cells and replace with 20 mL fresh expansion media.
Evaluation of Transduced Cell Phenotype and Expansion (Day 9)
-
45.
Collect cells from GREX wells by removing and discarding 20 mL media.
NOTE: do not disturb the cells.
-
46.
Pipet the remaining media up and down to dislodge the cells.
NOTE: media should be very cloudy if cells have grown well.
-
47.
Rinse well with 3 mL media to collect any remaining cells.
-
48.
Count to check for viability and cell number.
-
49.
Analyze samples with flow cytometry to determine gene expression and phenotype.
-
a.
Rhesus macaque PBMC phenotype is determined by using CD4 (the clone is reactive with endogenous rhCD4 and the rhCD4-MBL CAR), CD3, CD8, CD95, CD28, CCR7, CXCR5, and MBL.
NOTE: Table 1 contains additional information about antibodies in this panel.
-
b.
Viability was assessed with the Live/Dead Fixable Near IR Cell Stain Kit.
-
c.
A minimum of 150,000 events were acquired for each sample.
-
d.
Data were analyzed with FlowJo v10 (FlowJo, LLC).
-
50.
Cells can be used for in vitro assays of function, such as a transwell migration assay.6
-
51.
Cells can be infused into test animals by suspending in PBS containing 10% autologous serum at a density of 2 × 107 cells/mL. Cells can be stained with Cell Trace Violet (Invitrogen C34571) prior to infusion to allow tracking and assessment of proliferation.
Table 1.
Antibodies Used in Flow Cytometry Panel
| Antigen | Fluorophore | Clone | Concentration | Product | Company |
|---|---|---|---|---|---|
| MBL | AF 647 | 3E7 | 5 μg/mL | MA1-40145-S6 | Invitrogen |
| CXCR5 | PE | MU5UBEE | 0.625 μg/mL | 12-9185-42 | eBioscience |
| CD3 | AF 700 | SP34-2 | 2 μg/mL | 557917 | BD |
| CD4 | FITC | M-T477 | 2.5 μg/mL | 556615 | BD |
| CD8 | BV788 | RPA-T8 | 0.3125 μg/mL | 563824 | BD |
| CD28 | BV605 | CD28.2 | 10 μg/mL | 562976 | BD |
| CD95 | PerCP Cy5.5 | DX2 | 1 μg/mL | 561655 | BD |
| CCR7 | PECF594 | 150503 | 1.25 μg/mL | 562381 | BD |
| Live/dead | Near IR | L10119 | Invitrogen |
NOTE: keep the cells at 4°C until infusion
-
52.
Alternatively, cells can be frozen in 90% FBS, 10% DMSO for later use or analysis.
NOTE: we are currently infusing only freshly produced cells.
Timing
NOTE: this estimate is based on production of cells for infusion. The actual time would be reduced in a smaller experiment.
Day 1
-
•
Preparation of antibody-coated plates: 2 h
-
•
Media preparation: 0.5 h
-
•
Thawing and plating cells: 1 h
-
•
Flow cytometry: 2 h
Day 2
-
•
RetroNectin coating of plates: 2.5 h
Day 3
-
•
Warming centrifuge and supplies: 0.5 h
-
•
Viral coating of plates: 2.5 h
-
•
Transduction: 1 h
Day 5
-
•
Media preparation: 0.5 h
-
•
Collection and evaluation of cells: 1–1.5 h
-
•
Seeding GREX: 1 h
-
•
Flow cytometry: 2 h
Day 9
-
•
Collection and evaluation of cells: 2 h
-
•
Flow cytometry: 2 h
-
•
Freezing or preparation for infusion: 2–4 h
Troubleshooting
Media should be prepared fresh before use. Add IL-2 and β-mercaptoethanol on the day of use.
Each lot of FBS should be tested for its ability to support growth and transfection of 293T cells as well as transduction and expansion of rhesus PBMCs or other target cell populations.
Cells must be mitotically active to be transduced by a retrovirus. Look for visual clumping of the cells following anti-CD3/anti-CD28 stimulation. Failure to activate properly will lead to low transduction efficiency.
Look for high viability in your stimulated cells. If viability is poor after stimulation, the transduction is usually less successful. In order to achieve successful stimulation and transduction, it is essential that proper care is taken in freezing and transport of the PBMCs. Cells are shipped in liquid nitrogen and are quickly placed in long-term liquid nitrogen storage.
Good transduction depends on high-quality viral preparations. Retroviral vectors can be produced in the lab or outsourced to a viral vector service. A functional titer should be determined prior to transduction of primary cells so that consistent amounts of virus can be used in each experiment. Functional viral titer can be determined by titration of the viral supernatant on 293T cells using an adaptation of the Addgene fluorescence titering protocol (https://www.addgene.org/protocols/fluorescence-titering-assay/) in which expression of the transduced genes is determined by flow cytometry. Briefly, cells are plated at a density of 6 × 105 cells/well in a 6-well plate and incubated for 24 h. Media is removed and 2-fold dilutions of the virus preparation, in DMEM + 10% FBS, are added to each well. After 48 h, cells are removed with trypsin, and expression is analyzed by flow cytometry. Calculations of titer (transduction units/mL) are based on transduction levels of 20% or less using the following calculation:
Viral preparations can be damaged by freezing and thawing. Store the virus in single-use aliquots and do not refreeze. Thaw the virus quickly and store on ice until needed.
MOI may need to be determined empirically for each viral vector and cell type combination. The choice of promoter, enhancer, and envelope proteins may all impact transduction efficiency and expression of transgene in target cells. To determine optimal MOI (ratio of infectious virions to cells in culture), transduce primary PBMCs with serial dilutions of the virus preparation. The ideal MOI will allow maximal transduction of target cells with minimal toxicity.
There is animal to animal variability in both transduction efficiency and the cell expansion rate. We recommend a trial study to determine the ability of a particular cell preparation to be transduced and expanded.
If desired, the mitotic activity of the cells can be experimentally determined both in vitro and in vivo by one of the following methods: Ki67 can be included as part of the flow cytometry panel. Ki67 is a nuclear protein present in cells that are undergoing division.28 Alternatively, the cells can be stained with carboxyfluorescein succinimidyl ester (CFSE) or CellTrace violet (CTV) (Invitrogen products C34570 and C34571 respectively). These fluorescent dyes intensely stain the interior of the cell, and the fluorescent signal is reduced with each cell division. The dye can be monitored by flow cytometry, and distinct generations of cells can be identified.
This protocol can be modified to produce CAR-T cells from other species with alterations in stimulating antibodies and cytokines. The protocol must be modified to meet good manufacturing practice (GMP) standards prior to applying it to production of an immunotherapy product for clinical trials.
Anticipated Results
Cell Production
In preliminary studies, several types of media were evaluated to produce CAR/CXCR5 T cells, including X-Vivo 15 (Lonza), Immunocult (StemCell), Prime XV T cell expansion medium and Prime XV T cell CDM (Irvine Scientific), Stemline (Sigma), GT-T551 (Takara), and RPMI (GIBCO). Compared to RPMI, the use of X-Vivo 15 led to superior growth of cells and was obtained at a reasonable price point and, thus, was chosen for use in our protocol. As shown in Figure 1, use of X-Vivo media, containing 10% FBS, supports the growth of the transduced cells better than the standard T cell medium, RPMI, 10% FBS media during the expansion phase of cell production. Using PBMCs from three different animals and starting with equal numbers of cells in either RPMI or X-Vivo media, a comparison was made of the fold increase in cell number during expansion. Compared to the day 5 cell population, the day 9 population grown in X-Vivo media increased from 10.3- to 19.9-fold, and cells grown in RPMI increased from 5.8- to 14.3-fold. Despite animal-to-animal variability in expansion and differences in the number of cells seeded in these experiments (1.5 × 106 cells for 13,081; 5 × 106 cells for 10,002 and 571), we found that the use of X-Vivo media led to superior expansion of the cells.
Figure 1.

X-Vivo Media Provides Better Support for CAR-T Cell Expansion Than RPMI
Comparison of fold increase in cell numbers from days 5 to 9 using PBMCs from three different animals grown in either X-Vivo or RPMI media. The red bars represent RPMI + 10% FBS, while the blue bars represent X-Vivo + 10% FBS. For each animal, equal numbers of cells were grown in either RPMI or X-Vivo for the entire 8 days of ex vivo culture. Cell numbers were determined by trypan blue exclusion on a countess cell counter. Fold increase was determined during expansion from days 5 through 9. Data were not statistically analyzed due to low sample size.
With the protocol presented in this paper, we routinely achieved at least an 8-fold expansion of cells (median 11.5 fold, range 8.4–17.4-fold) while undergoing an average of at least 3.5 cell doublings (range 3.1–4.1) in a 4-day period of time. With a starting density of 5 × 106 cells per GREX well, we achieved a median density of 47.1 × 106 cells per well (range 34.5−86.9 × 106) (Figure 2A) at day 9. The cells produced showed high viability (median 87.5%, range 71.4%–94.8%) (Figure 2B) by trypan blue exclusion at the end of the 9-day protocol. Viability is usually consistent or improves following expansion in the GREX well. While there is some animal-to-animal variability in the expansion of the cells, all have produced sufficient cell density so that, with 50–100 × 106 cells on day 1, we can produce sufficient cells for infusion of 1–2 × 108 cells/kg into a 10 kg rhesus macaque by day 9.
Figure 2.
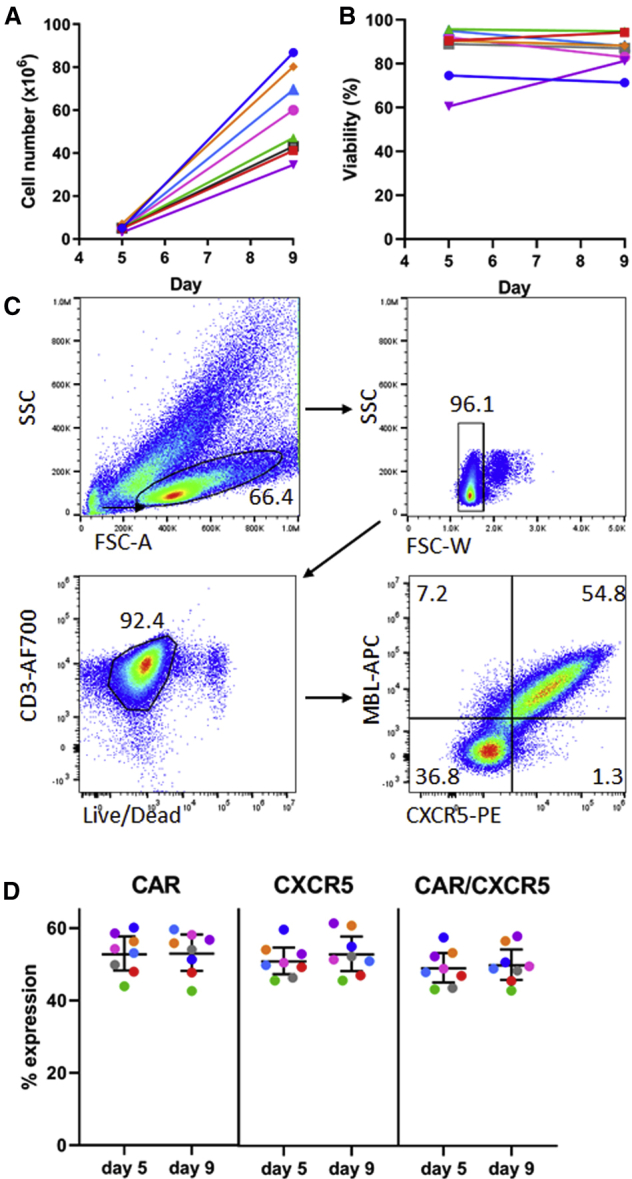
9-Day Transduction and Expansion Protocol Yields Sufficient Cells for Infusion, Which Are Viable and Maintain Co-expression of Two Transduced Genes
(A) Total number of cells in culture on days 5 and 9 of the expansion protocol and (B) viability of the transduced cells using PBMCs from seven different animals. Cell number and viability was monitored by trypan blue exclusion counting on the countess cell counter. (C) Representative flow plots showing gating strategy and expression of both the CD4-MBL CAR and CXCR5 in transduced cells on day 9. (D) Expression of CAR, CXCR5, and co-expression of CAR and CXCR5 on days 5 and 9 in cells from seven different animals. The bars represent the mean value and 95% confidence interval.
The transduced genes are efficiently co-expressed (Figure 2C) with a mean co-expression of the CD4-MBL CAR and CXCR5 of 45.3% (95% CI 41.5%–49%) on day 5 and 46.0% (95% CI 42.1%–49.9%) on day 9 (Figure 2D) with the single retroviral transduction described here. Total CAR expression (mean 53.0% on day 5 and 53.2% on day 9) and total CXCR5 expression (mean 47.0% on day 5 and 48.8% on day 9) are similar, demonstrating efficient co-expression of the two genes of interest. A second round of transduction has proven to be unnecessary in our hands (data not shown). These results compare favorably with the previously described 22%–75% efficiency of lentiviral transductions of rhesus macaque T cells.16
The cells that were used in the development of this protocol were collected prior to SIV infection or during chronic infection. When using PBMCs from SIV-infected animals treated with a combination of three anti-retroviral (ART) drugs—dolutegravir, (2.5 mg/kg/day) tenofovir disproxil fumarate (5.1 mg/kg/day), and emtricitabine (40 mg/kg/day)—we have found that the cells are somewhat resistant to transduction by gammaretroviruses. It is likely that this resistance is due to the prolonged intracellular half-life of the ART drugs interfering with reverse transcription and integration. This finding supports similar low transduction efficiencies seen in lentivirus-mediated gene transfer using cells from simian/human immunodeficiency virus (SHIV)-infected ART-treated pigtailed macaques.29 Figure 3 demonstrates the impact of prior ART treatment on gammaretroviral transduction of rhesus macaque PBMCs. There was a 3.6-fold decrease in the mean level of co-expression of CAR and CXCR5 using cells from animals on ART as compared to cells from the same animal prior to ART treatment (ART median 15.4%, range 10.2%–25.6%; pre-ART median 56.2%, range 50.5%–61.5%). Given this reduction in transduction efficiency, the protocol will need modification for use with ART-suppressed animals. Vectors that are not impacted by ART drugs could be used to produce the virus. Alternatively, ART could be stopped for several days prior to collection of the cells in order to reduce the intracellular level of the drugs prior to culture or alternative methods of gene transfer could be used.
Figure 3.
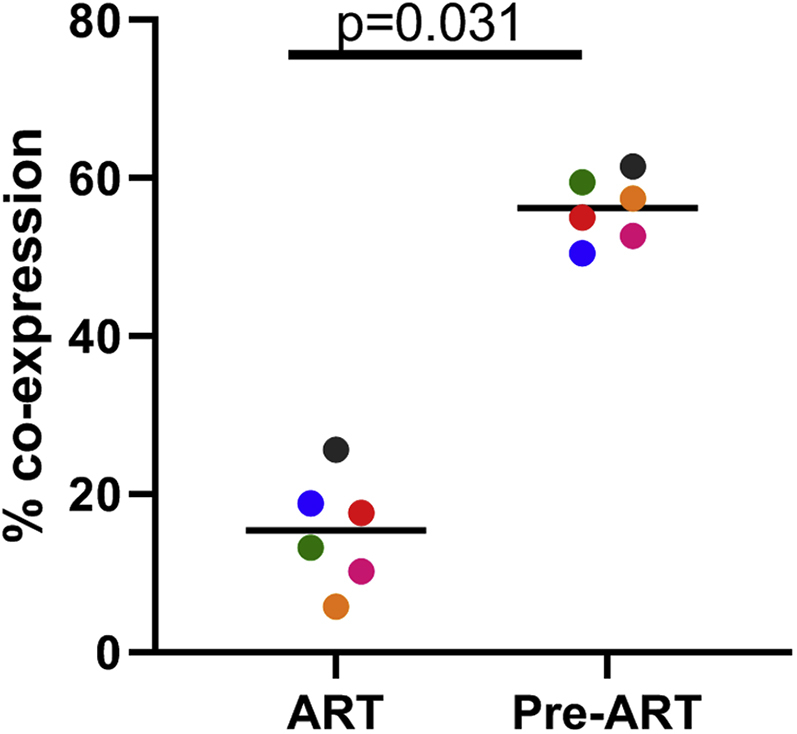
Treatment of Macaques with Antiretroviral Drugs Prior to Collection of PBMCs Leads to a Reduction of Transduction Efficiency with Gammaretroviral Vectors
PBMCs from six animals using cells collected prior to treatment with anti-retroviral drugs (Pre-ART) or during antiretroviral treatment (ART) were used in the 9-day transduction and expansion protocol. On day 9, cells were evaluated for expression of MBL, representing the CAR, and CXCR5 by flow cytometry. A Wilcoxon matched pairs signed rank test was used to determine significance. Colors denote cells from six individual animals. The bar represents the median value.
Cell Phenotype
The cells produced by the method were analyzed by flow cytometry to determine their memory phenotype. In uncultured cells, prior to transduction and expansion, naive, central memory, and effector memory populations were identified, but only central memory and effector memory were seen by day 5 (not shown) and day 9 in both the mock and CAR/CXCR5 transduced total cell population as well as the subset of CAR/CXCR5+ transduced cells (Figure 4A). In a study with cells from six individual animals, both the mock and transduced cells retained a primarily central memory phenotype, as identified by CD28+ CD95+ expression profile on day 9 (mock: mean 58.5%, 95% CI 49%–68%; total transduced: mean 66.4%, 95% CI 57%–76%; CAR/CXCR5 transduced subset: mean 67.5%, 95% CI 53%–82%) (Figure 4B). The central memory cells express CCR7 (Figure 4C), a molecule needed for migration into the lymph node. In an analysis of cells from six animals, the majority of the central memory cells express CCR7 (mock: mean 66%, 95% CI 48.8%–83.6%; total transduced population: mean 67.2%, 95% CI 52.9%–81.5%; CAR/CXCR5 transduced subset: mean 65.5, 95% CI 49%–82%) (Figure 4D). This outcome, with maintenance of a high central memory population, is an improvement over the primarily effector memory population produced using a longer culture time when rhesus macaque T cells were transduced with a gammaretrovirus to express CXCR5.9 The central memory population is comparable to that seen with a protocol that used lentiviral polybrene transduction with bead activation of rhesus macaque T cells and culture times of 8 to 17 days to produce CD20-targeting CAR-T cells.16 Overall, the protocol described here, with its rapid expansion and relatively low levels of IL-2, allows us to produce a population of cells that are not primarily terminally differentiated effector or effector memory cells, as identified by loss of CD28 expression, and which, through CCR7 expression,30 may be capable of migration into the lymph node in vivo.
Figure 4.

The Majority of CD4-MBL CAR/CXCR5 Transduced Cells are T central memory (TCM)
(A) Representative flow cytometry plots of the starting cell population, day 9 mock, day 9 total transduced, and CAR/CXCR5 transduced subset of rhesus PBMCs. For panels 1–3, gates were set on live, CD3+, and CD8+ cells. For the transduced subset, gates were set on live, CD3+, MBL+, and CD8+ cells. Central memory was defined as CD28+CD95+. Effector memory was defined as CD28−, CD95+. (B) The percentages of central memory cells on day 9 in mock and total transduced cells and the CAR/CXCR5 subset of transduced cells from six individual animals. Mean and 95% confidence intervals are shown with bars. (C) Representative flow plot of CCR7 expression in cells identified as CD28+/CD95+. (D) CCR7 expression levels in the central memory subset of mock, total transduced cells and the CAR/CXCR5 subset of transduced cells from six individual animals. Mean and 95% confidence intervals are shown with bars.
Cell Function
The function of CAR/CXCR5 transduced T cells was indicated in vitro by assessing the ability of the transduced cells to control the level of SIV in infected rhesus PBMCs. PBMCs, isolated from SIV-infected rhesus macaques, were transduced with CAR/CXCR5 and expanded with the 9-day protocol. SIV RNA levels were quantified by RT-PCR measurement of gag mRNA in the cell pellet of equal numbers of mock transduced and CAR/CXCR5 transduced cells. When SIV-infected PBMCs were transduced with the CAR/CXCR5 construct, they controlled the SIV infection significantly better than mock transduced cells as demonstrated by an SIV gag level 139 times lower in the CAR/CXCR5 cells (median 1.03, range 0.20–6.1) than in the mock transduced cells (median 143.0, range 6.6–2,129) (Figure 5). These results likely indicate an active suppression of the virally infected cells by the transduced cells during the expansion phase of the protocol. In addition, cells from three of the animals were transduced with only the CXCR5 construct and had virus levels comparable to the mock transduced cells (median 257.2, range 82.5–1,169.9). Thus, these data indicate that the suppressive effect was due to the CAR. These data are supported by similar findings showing that CD4-MBL-CAR/CXCR5 cells suppressed SIV replication using a traditional in vitro viral suppression assay.6 Together, these findings indicate that the CAR is functional in the CAR/CXCR5 T cells post-expansion.
Figure 5.
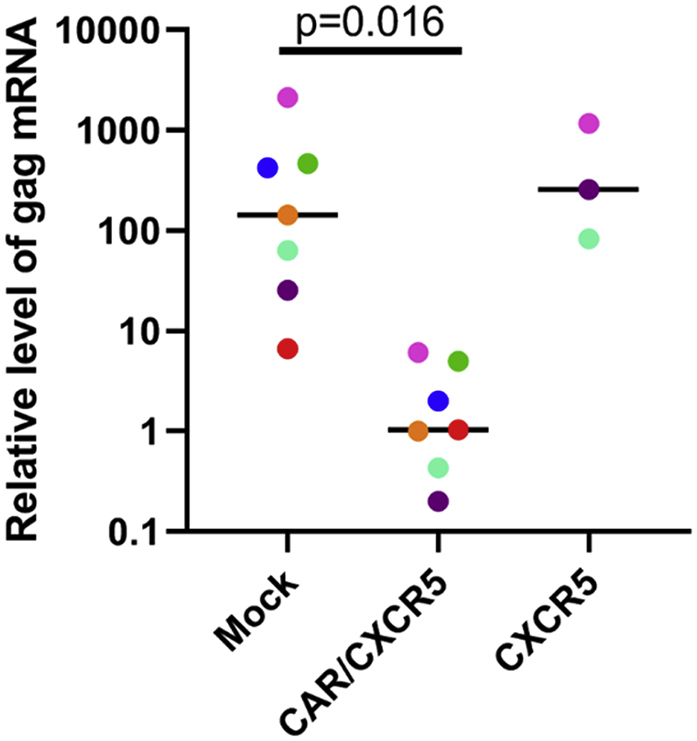
Virus Production in PBMCs Was Reduced in Cells Transduced with CAR/CXCR5 Compared to Mock Transduced Cells
PBMCs isolated from rhesus macaques infected with SIV mac 251 were transduced with CAR/CXCR5 or CXCR5 only using the 9-day protocol. Mock samples were treated identically without exposure to the transducing retrovirus. At the conclusion of the transduction and expansion procedure, equal numbers of mock and transduced cells were evaluated for the presence of viral RNA. Cell pellets were analyzed for the presence of gag mRNA relative to the housekeeping gene beta-actin by RT-PCR. Colors denote matched transduced or mock transduced cells from seven individual animals. A Wilcoxon matched pairs signed rank test was used to determine significance of the difference between mock and CAR/CXCR5 samples. The bar represents the median value.
Expression of the CXCR5 gene facilitates homing of transduced cells to the CXCR5 ligand, CXCL13. Function of the transduced CXCR5 molecule was demonstrated in an in vitro trans-well migration assay. The cells were placed in the upper chamber, while the lower chamber contained either no chemokine or the CXCR5-specific chemokine CXCL13. After 4 h, cells that had migrated to the lower chamber were counted by flow cytometry. Specific migration was determined by subtracting the number of cells that migrated to media alone from the number of cells that migrated to the CXCL13 and then dividing by the number of input cells. CAR/CXCR5 transduced cells migrated to CXCL13 to a significantly greater extent than did the mock transduced cells (p = 0.016), with a median of 12.6% of the transduced cells migrating to CXCL13 in the 4-h assay (range 7.9%–19.4%) and a median of only 2.4% of the mock transduced cells migrating (range 0%–5%) to CXCL13 (Figure 6B). Thus, these data indicate CXCR5 expressed on the CAR/CXCR5 T cells is functional post-expansion. Since migration into the B cell follicle is mediated through the chemokine receptor, CXCR5,31 and its ligand, CXCL13,32 demonstration of migration to CXCL13 suggests that the CAR/CXCR5 cells will migrate to the B cell follicle of secondary lymphoid tissues when infused into a macaque.
Figure 6.
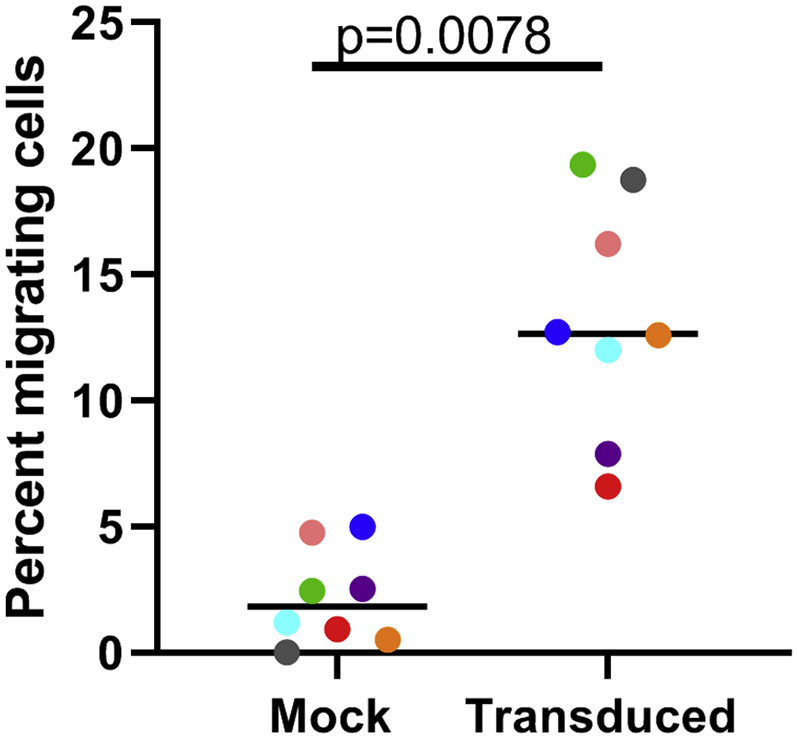
Transduced Cells Demonstrated Specific Migration to the CXCR5 Ligand, CXCL13, In Vitro
Transduced Rhesus macaque PBMCs (one million cells in 100 μL X-Vivo-15 containing 0.1% BSA) were placed in the upper chamber of a transwell plate with a 5.0 μm membrane. The lower chamber contained X-Vivo/1% BSA only or X-Vivo/1% BSA and the chemokine CXCL13 (2.5 μg/mL). After incubating for 4 h at 37°C, cells were collected and counted on a CytoFLEX flow cytometer (Beckman). All samples were normalized with the addition of AccuCheck counting beads. Specific migration was determined by subtracting the number of cells that migrated to X-Vivo/BSA alone from the number that migrated to the chemokine and then dividing by the number of input cells. Colors denote cells from seven individual animals. A Wilcoxon matched pairs signed rank test was used to determine significance. The bar represents the median value.
Conclusions
This protocol outlines a production strategy that results in sufficient numbers of viable and functional rhesus macaque CAR-T cells to allow infusion of 1–2 × 108 cells/kg for in vivo efficacy testing. The relatively rapid time frame allows maintenance of the desired central memory phenotype.
Author Contributions
M.S.P. optimized protocols, conducted the experiments, and drafted the manuscript; K.P.H. assisted with protocol development; G.T.H. assisted with development of flow cytometry panels and edited the manuscript; A.K.R. provided guidance with statistical analysis; E.G.R. provided study oversight, assisted with flow cytometry panels, and supplied rhesus macaque PBMCs used in the studies; E.A.B. designed the CAR vectors used to produce the CAR-T cells, obtained funding, and assisted with drafting of the manuscript; E.C. provided study oversight, obtained funding for the project, and assisted with drafting the manuscript; P.J.S. provided study oversight, obtained funding for the project, and assisted with drafting the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
This study was supported by NIH grants 5R01AI096966-06S1 (P.S., E.C., and E.B.), 1UM1AI26617 (P.S., E.C., and E.B.), and P51OD011106/P51RR000167 (E.R.), MN REACH grant 5U01HL127479-03 (P.S.), 1R01A143380-01 (P.S. and E.B.), and 1UM14126617 (P.S. and E.C.); as well as funds provided by the NIAID Division of Intramural Research and the NIH Intramural AIDS Targeted Antiviral Program. Anti-CD3 and anti-CD28 used in these studies was provided by the NIH Nonhuman Primate Reagent Resource (R24 OD010976 and U24 AI126683). IL-2 used in these studies was provided by The NCI Preclinical Repository.
We thank Chi Phan and Jhomary Alegria-Berrocal at the University of Minnesota for assistance with gammaretroviral production, Andrea Weiler at the University of Wisconsin-Madison for conducting viral load assays, and Kim Weisgrau at the University of Wisconsin-Madison for isolation of rhesus macaque PBMCs. We would also like to thank Dr. Scott McIvor at the University of Minnesota, Dr. Leslie Kean at Harvard Medical School, Dr. Catherine Bollard at the Children’s Research Institute, Dr. Christopher Peterson at the Fred Hutchinson Cancer Center, Dr. Matthew Trivett at NCI, Dr. Agne Taraseviciute at Seattle Children’s Hospital, and Dr. Conrad Russell Cruz at the Children’s Research Institute for their help in developing these methods.
Contributor Information
Mary S. Pampusch, Email: pampu002@umn.edu.
Pamela J. Skinner, Email: skinn002@umn.edu.
References
- 1.Klebanoff C.A., Rosenberg S.A., Restifo N.P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat. Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim W.A., June C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168:724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sadelain M., Rivière I., Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhlmann A., Peterson C.W., Kiem H.P. Chimeric antigen receptor T-cell approaches to HIV cure. Curr. Opin. HIV AIDS. 2018;13:446–453. doi: 10.1097/COH.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghanem M.H., Bolivar-Wagers S., Dey B., Hajduczki A., Vargas-Inchaustegui D.A., Danielson D.T., Bundoc V., Liu L., Berger E.A. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. 2018;20:407–419. doi: 10.1016/j.jcyt.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Haran K.P., Hajduczki A., Pampusch M.S., Mwakalundwa G., Vargas-Inchaustegui D.A., Rakasz E.G., Connick E., Berger E.A., Skinner P.J. Simian immunodeficiency virus (SIV)-specific chimeric antigen receptor-T cells engineered to target B cell follicles and suppress SIV replication. Front. Immunol. 2018;9:492. doi: 10.3389/fimmu.2018.00492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connick E., Mattila T., Folkvord J.M., Schlichtemeier R., Meditz A.L., Ray M.G., McCarter M.D., Mawhinney S., Hage A., White C., Skinner P.J. CTL fail to accumulate at sites of HIV-1 replication in lymphoid tissue. J. Immunol. 2007;178:6975–6983. doi: 10.4049/jimmunol.178.11.6975. [DOI] [PubMed] [Google Scholar]
- 8.Connick E., Folkvord J.M., Lind K.T., Rakasz E.G., Miles B., Wilson N.A., Santiago M.L., Schmitt K., Stephens E.B., Kim H.O. Compartmentalization of simian immunodeficiency virus replication within secondary lymphoid tissues of rhesus macaques is linked to disease stage and inversely related to localization of virus-specific CTL. J. Immunol. 2014;193:5613–5625. doi: 10.4049/jimmunol.1401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayala V.I., Deleage C., Trivett M.T., Jain S., Coren L.V., Breed M.W., Kramer J.A., Thomas J.A., Estes J.D., Lifson J.D., Ott D.E. CXCR5-Dependent Entry of CD8 T Cells into Rhesus Macaque B-Cell Follicles Achieved through T-Cell Engineering. J. Virol. 2017;91:e02507–e02516. doi: 10.1128/JVI.02507-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Patel S., Jones R.B., Nixon D.F., Bollard C.M. T-cell therapies for HIV: Preclinical successes and current clinical strategies. Cytotherapy. 2016;18:931–942. doi: 10.1016/j.jcyt.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Q., Wang Y., Lo A.S.-Y., Gomes E.M., Junghans R.P. Cell density plays a critical role in ex vivo expansion of T cells for adoptive immunotherapy. J. Biomed. Biotechnol. 2010;2010:386545. doi: 10.1155/2010/386545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vargas J.E., Chicaybam L., Stein R.T., Tanuri A., Delgado-Cañedo A., Bonamino M.H. Retroviral vectors and transposons for stable gene therapy: advances, current challenges and perspectives. J. Transl. Med. 2016;14:288–302. doi: 10.1186/s12967-016-1047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.-T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan J., Yang J.F., Deng B.P., Zhao X.J., Zhang X., Lin Y.H., Wu Y.N., Deng Z.L., Zhang Y.L., Liu S.H. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia. 2017;31:2587–2593. doi: 10.1038/leu.2017.145. [DOI] [PubMed] [Google Scholar]
- 15.Brentjens R.J., Rivie I., Park J.H., Davila M.L., Wang X., Stefanski J., Taylor C., Yeh R., Bartido S., Borquez-ojeda O. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;18:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taraseviciute A., Tkachev V., Ponce R., Turtle C.J., Snyder J.M., Liggitt H.D., Myerson D., Gonzalez-Cuyar L., Baldessari A., English C. Chimeric antigen receptor T cell–mediated neurotoxicity in nonhuman primates. Cancer Discov. 2018;8:750–763. doi: 10.1158/2159-8290.CD-17-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajgain P., Mucharla R., Wilson J., Welch D., Anurathapan U., Liang B., Lu X., Ripple K., Centanni J.M., Hall C. Optimizing the production of suspension cells using the G-Rex “M” series. Mol. Ther. Methods Clin. Dev. 2014;1:14015. doi: 10.1038/mtm.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vera J.F., Brenner L.J., Gerdemann U., Ngo M.C., Sili U., Liu H., Wilson J., Dotti G., Heslop H.E., Leen A.M., Rooney C.M. Accelerated production of antigen-specific T cells for preclinical and clinical applications using gas-permeable rapid expansion cultureware (G-Rex) J. Immunother. 2010;33:305–315. doi: 10.1097/CJI.0b013e3181c0c3cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakazawa Y., Huye L.E., Salsman V.S., Leen A.M., Ahmed N., Rollins L., Dotti G., Gottschalk S.M., Wilson M.H., Rooney C.M. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol. Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin J., Gkitsas N., Fellowes V.S., Ren J., Feldman S.A., Hinrichs C.S., Stroncek D.F., Highfill S.L. Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J. Transl. Med. 2018;16:13. doi: 10.1186/s12967-018-1384-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redeker A., Arens R. Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post-transfer vaccination. Front. Immunol. 2016;7:345. doi: 10.3389/fimmu.2016.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klebanoff C.A., Gattinoni L., Restifo N.P. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J. Immunother. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitcher C.J., Hagen S.I., Walker J.M., Lum R., Mitchell B.L., Maino V.C., Axthelm M.K., Picker L.J. Development and homeostasis of T cell memory in rhesus macaque. J. Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 24.Ghassemi S., Nunez-Cruz S., O’Connor R.S., Fraietta J.A., Patel P.R., Scholler J., Barrett D.M., Lundh S.M., Davis M.M., Bedoya F. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018;6:1100–1109. doi: 10.1158/2326-6066.CIR-17-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Minang J.T., Trivett M.T., Bolton D.L., Trubey C.M., Estes J.D., Li Y., Smedley J., Pung R., Rosati M., Jalah R. Distribution, persistence, and efficacy of adoptively transferred central and effector memory-derived autologous simian immunodeficiency virus-specific CD8+ T cell clones in rhesus macaques during acute infection. J. Immunol. 2010;184:315–326. doi: 10.4049/jimmunol.0902410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolton D.L., Minang J.T., Trivett M.T., Song K., Tuscher J.J., Li Y., Piatak M., Jr., O’Connor D., Lifson J.D., Roederer M., Ohlen C. Trafficking, persistence, and activation state of adoptively transferred allogeneic and autologous Simian Immunodeficiency Virus-specific CD8(+) T cell clones during acute and chronic infection of rhesus macaques. J. Immunol. 2010;184:303–314. doi: 10.4049/jimmunol.0902413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger C., Sommermeyer D., Hudecek M., Berger M., Balakrishnan A., Paszkiewicz P.J., Kosasih P.L., Rader C., Riddell S.R. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 2015;3:206–216. doi: 10.1158/2326-6066.CIR-14-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruno S., Darzynkiewicz Z. Cell cycle dependent expression and stability of the nuclear protein detected by Ki-67 antibody in HL-60 cells. Cell Prolif. 1992;25:31–40. doi: 10.1111/j.1365-2184.1992.tb01435.x. [DOI] [PubMed] [Google Scholar]
- 29.Younan P.M., Peterson C.W., Polacino P., Kowalski J.P., Obenza W., Miller H.W., Milless B.P., Gafken P., DeRosa S.C., Hu S.L., Kiem H.P. Lentivirus-mediated Gene Transfer in Hematopoietic Stem Cells Is Impaired in SHIV-infected, ART-treated Nonhuman Primates. Mol. Ther. 2015;23:943–951. doi: 10.1038/mt.2015.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bromley S.K., Thomas S.Y., Luster A.D. Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol. 2005;6:895–901. doi: 10.1038/ni1240. [DOI] [PubMed] [Google Scholar]
- 31.Förster R., Mattis A.E., Kremmer E., Wolf E., Brem G., Lipp M. A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996;87:1037–1047. doi: 10.1016/s0092-8674(00)81798-5. [DOI] [PubMed] [Google Scholar]
- 32.Legler D.F., Loetscher M., Roos R.S., Clark-Lewis I., Baggiolini M., Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]