

Abstract
Aims
Fibroblast to myofibroblast trans‐differentiation with altered bioenergetics precedes cardiac fibrosis (CF). Either prevention of differentiation or promotion of de‐differentiation could mitigate CF‐related pathologies. We determined whether 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A (HMG‐CoA) reductase inhibitors—statins, commonly prescribed to patients at risk of heart failure (HF)—can de‐differentiate myofibroblasts, alter cellular bioenergetics, and impact the human ventricular fibroblasts (hVFs) in HF patients.
Methods and results
Either in vitro statin treatment of differentiated myofibroblasts (n = 3–6) or hVFs, isolated from human HF patients under statin therapy (HF + statin) vs. without statins (HF) were randomly used (n = 4–12). In vitro, hVFs were differentiated by transforming growth factor‐β1 (TGF‐β1) for 72 h (TGF‐72 h). Differentiation status and cellular oxygen consumption rate (OCR) were determined by α‐smooth muscle actin (α‐SMA) expression and Seahorse assay, respectively. Data are mean ± SEM except Seahorse (mean ± SD); P < 0.05, considered significant. In vitro, statins concentration‐dependently de‐differentiated the myofibroblasts. The respective half‐maximal effective concentrations were 729 ± 13 nmol/L (atorvastatin), 3.6 ± 1 μmol/L (rosuvastatin), and 185 ± 13 nmol/L (simvastatin). Mevalonic acid (300 μmol/L), the reduced product of HMG‐CoA, prevented the statin‐induced de‐differentiation (α‐SMA expression: 31.4 ± 10% vs. 58.6 ± 12%). Geranylgeranyl pyrophosphate (GGPP, 20 μmol/L), a cholesterol synthesis‐independent HMG‐CoA reductase pathway intermediate, completely prevented the statin‐induced de‐differentiation (α‐SMA/GAPDH ratios: 0.89 ± 0.05 [TGF‐72 h + 72 h], 0.63 ± 0.02 [TGF‐72 h + simvastatin], and 1.2 ± 0.08 [TGF‐72 h + simvastatin + GGPP]). Cellular metabolism involvement was observed when co‐incubation of simvastatin (200 nmol/L) with glibenclamide (10 μmol/L), a KATP channel inhibitor, attenuated the simvastatin‐induced de‐differentiation (0.84 ± 0.05). Direct inhibition of mitochondrial respiration by oligomycin (1 ng/mL) also produced a de‐differentiation effect (0.33 ± 0.02). OCR (pmol O2/min/μg protein) was significantly decreased in the simvastatin‐treated hVFs, including basal (P = 0.002), ATP‐linked (P = 0.01), proton leak‐linked (P = 0.01), and maximal (P < 0.001). The OCR inhibition was prevented by GGPP (basal OCR [P = 0.02], spare capacity OCR [P = 0.008], and maximal OCR [P = 0.003]). Congruently, hVFs from HF showed an increased population of myofibroblasts while HF + statin group showed significantly reduced cellular respiration (basal OCR [P = 0.021], ATP‐linked OCR [P = 0.047], maximal OCR [P = 0.02], and spare capacity OCR [P = 0.025]) and myofibroblast differentiation (α‐SMA/GAPDH: 1 ± 0.19 vs. 0.23 ± 0.06, P = 0.01).
Conclusions
This study demonstrates the de‐differentiating effect of statins, the underlying GGPP sensitivity, reduced OCR with potential activation of KATP channels, and their impact on the differentiation magnitude of hVFs in HF patients. This novel pleiotropic effect of statins may be exploited to reduce excessive CF in patients at risk of HF.
Keywords: Statins, Cardiac fibrosis, Mitochondria, Geranylgeranyl pyrophosphate, De‐differentiation
1. Introduction
Excessive cardiac fibrosis is a major predisposing factor leading to mechanical and electrical dysfunctions in heart failure (HF).1 Fibrosis requires activation of fibroblast trans‐differentiation into myofibroblasts,2 marked by increased α‐smooth muscle actin (α‐SMA) expression and excessive extracellular matrix secretion and deposition.1 Excessive/chronic fibrosis may result from the persistent presence of activated myofibroblasts.3, 4 Indeed, myofibroblast accumulation within pathologic lesions is a feature of various fibrotic disorders,5 including progressive HF and cardiac hypertrophy.6 Therefore, defining specific mechanisms that can de‐differentiate myofibroblasts back to fibroblasts could pave the way to mitigating fibrosis‐associated HF.
Statins are inhibitors of 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A (HMG‐CoA) reductase commonly prescribed for their lipid‐lowering effects in patients at risk of HF. Several prior in vitro studies have demonstrated statin‐induced prevention of differentiation to myofibroblasts,7, 8, 9 but their ability to de‐differentiate already‐differentiated myofibroblasts is unclear. Further, the impact of statins on human ventricular fibroblasts (hVFs) in HF patients is also not known. In hVFs/myofibroblasts, we determined whether de‐differentiation of myofibroblasts could be attained by HMG‐CoA reductase inhibition. Moreover, the effects of statins on mitochondrial energetics of hVFs are unknown especially in view of recent demonstration that mitochondrial bioenergetics increase with myofibroblast differentiation.10 Therefore, we tested the hypothesis that statins will reduce cellular respiration and induce de‐differentiation of human ventricular myofibroblasts and their population will be reduced by statin therapy in HF patients. Likewise, many pleiotropic effects of statins, independent of cholesterol‐synthesis pathways, have been reported11, 12 that are predominantly via geranylgeranyl pyrophosphate (GGPP) signalling.13, 14 Therefore, to elucidate the bioenergetics‐related mechanisms underlying the statin‐induced de‐differentiation of myofibroblasts, we tested in vitro the involvement of GGPP and the reported molecular sensors of cellular metabolism, ATP‐sensitive K+ channels.15
2. Methods
2.1. Materials
All materials information is provided in the Supporting Information, Table S1.
2.2. Study approval
The study was approved by the Institutional Review Board and adhered to the Health Insurance Portability and Accountability Act and the institution's patient privacy and security guidelines. The study conformed to the Declaration of Helsinki principles.
2.3. Study population
Human ventricular fibroblasts, isolated from trauma victims free of any structural heart disease, were used for in vitro control studies. New York Heart Association Class III and IV HF patients who underwent either cardiac transplantation or left ventricular assist device implantation were included to determine the in vivo statin effects on hVFs. Following written consent, left ventricular tissues were obtained at the time of surgery. Patients were grouped into HF without statin therapy (HF) and HF on statin therapy for at least 1 year (HF + statin). As summarized in the Supporting Information, Table S2, patients from the HF (n = 12) and HF + statin (n = 12) groups were well matched for major clinical characteristics. Assays were performed on randomly chosen hVF samples.
2.4. Isolation of ventricular fibroblasts
Left ventricular fibroblasts were isolated from human cardiac tissues as reported earlier.16 Cardiac tissues were transferred in an ice‐cold Dulbecco's phosphate‐buffered saline. The ventricular tissue block was cleaned off all non‐myocardial portions in a 60 mm culture dish in laminar flow hood and cut into 1 mm blocks, transferred to a 25 cm2 TPP tissue culture flask (MidSci, St. Louis, MO), washed thrice with Dulbeccos phosphate‐buffered saline, twice with FM‐b (ScienCell Inc., Carlsbad, CA) containing penicillin/streptomycin, spread evenly into 20–30 blocks per flask, and cultured in 5 mL FM‐2 media (ScienCell Inc.) with 5% foetal bovine serum and penicillin/streptomycin. The flask was inverted, with the bottom (surface with tissue) up, in the incubator (37°C; 21% O2; 5% CO2), and blocks were allowed to adhere for ~4 h. After 2–3 h, the flasks were turned to the normal orientation. After changing media every 2 days, in 2 weeks, fibroblasts migrated from the tissue explants and reached 70% confluency. The cells were trypsinized and transferred to 150 cm2 TPP tissue culture flasks (MidSci) with FM‐2 media (ScienCell Inc.) containing 5% foetal bovine serum and penicillin/streptomycin and grown to 70% confluency. These initial cultures were split, and Passages 2 and 3 were stored in liquid nitrogen until experiments were conducted.
2.5. Study design for statin‐induced myofibroblast de‐differentiation
Differentiation was characterized by expression of α‐SMA, COL III, or SPRY1. In vitro statin effects (de‐differentiation) on differentiated myofibroblasts were determined either from hVFs of HF patients or after TGF‐β1‐induced differentiation in normal hVFs. These hVFs were plated at a density of 4000 cells/cm2 and, after 24 h, replaced with fresh complete FM‐2 media containing TGF‐β1 (5 ng/mL) to stimulate differentiation to myofibroblasts. After 72 h, TGF‐β1 was removed, and fresh medium was added, with or without appropriate concentrations of respective statins, to induce de‐differentiation. Following another 72 h, the cells were subjected to immunoblotting. The signalling mechanisms underlying statin‐induced de‐differentiation were determined by repeating the experiments in the presence of mevalonic acid (MVA, 300 μmol/L), GGPP (20 μmol/L), or glibenclamide (10 μmol/L). To confirm the role of bioenergetics in myofibroblast de‐differentiation, differentiated myofibroblasts were cultured in the presence of oligomycin (1 ng/mL), an inhibitor of mitochondrial respiration.17 hVFs patients were randomly selected and plated at a density of 8000 cells/cm2 and subjected to immunoblotting after 24 to 48 h.
2.6. Immunological methods
Standard western blotting protocols were followed.16 The separated proteins were probed for α‐SMA, COL III, SPRY1, paxillin, or total OXPHOS complex subunits with respective antibodies (Supporting Information, Table S3 ). For immunohistochemistry, a previously reported protocol was used.16 hVF samples were fixed, permeabilized, blocked, and incubated with primary antibody‐polyclonal anti‐α‐SMA antibody, followed by secondary antibodies conjugated with Alexa 488 and acquired confocal images.
2.7. Seahorse assay
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of hVFs were measured in the non‐buffered Dulbeccos modified Eagles medium containing 10 mmol/L glucose, 4 mmol/L l‐glutamine, and 2 mmol/L sodium pyruvate under basal conditions and in response to 3.5 μmol/L oligomycin A, 1 μmol/L fluoro‐carbonyl cyanide phenylhydrazone (FCCP), and 14 μmol/L antimycin A (AA) with the XF96 Extracellular Flux Analyzer.10 Respiratory parameters were calculated as previously described.18 hVFs patients were plated at 20 000 cells per well and subjected to Seahorse assay after 24 to 48 h. In vitro statin effects on OCR and ECAR were determined following myofibroblast de‐differentiation study design.
2.8. Enzymatic activity of mitochondrial OXPHOS complexes
Fibroblast lysate was prepared from about 5 × 106 cells according to the previously published protocol.19 Briefly, cells were detached using 0.05% (w/v) trypsin–EDTA and washed two times in phosphate‐buffered saline by centrifuging at 1000 g for 5 min at 4°C. The fibroblast pellet was suspended in 20 mmol/L hypotonic potassium phosphate buffer (pH 7.5) by using 50 μL Hamilton syringe until it had an appearance of homogeneous solution. Cell lysates were snap‐frozen in liquid nitrogen and stored at −80°C until analysis. The functional activity of mitochondrial OXPHOS Complexes I–V was measured in cell lysates as previously described.
2.9. Measurement of ADP/ATP ratio
The ratio of ADP to ATP in hVFs was measured by ADP/ATP ratio luminescent kit and Tecan plate reader. hVFs, differentiated by TGF‐β1 (5 ng/mL for 72 h) and further cultured for 72 h TGF‐β1‐free with or without statins were harvested and plated at a density of 50 000 cells/cm2 in a 96‐well plate coated with Collagen I (15 μg/cm2). Following overnight incubation, the ratio of ADP/ATP was determined.
2.10. Statistics
Categorical variables were analysed by Fisher's exact test, and continuous variables were analysed by two‐sample t‐test or one‐way analysis of variance. Appropriateness of normality assumption was validated using Shapiro–Wilk and Kolmogorov–Smirnov. P < 0.05 was considered significant.
3. Results
3.1. Statin‐induced myofibroblast de‐differentiation in vitro
To determine whether statins can de‐differentiate myofibroblasts and find the effective concentration, we tested both lipophilic (atorvastatin and simvastatin) and hydrophilic (rosuvastatin) statins on already‐differentiated myofibroblasts by measuring the expression of α‐SMA, a marker of differentiated myofibroblasts. Figure 1 shows immunoblots depicting the effect of in vitro treatment for 72 h with (Figure 1A ) atorvastatin [10 nmol/L to 3 μmol/L], (Figure 1B ) rosuvastatin [30 nmol/L to 10 μmol/L], or (Figure 1C ) simvastatin [1 nmol/L to 500 nmol/L] on differentiated myofibroblasts, and their α‐SMA expression. The data were fit by four‐parameter logistic curves that showed the concentration‐dependent effects of atorvastatin (Figure 1D ), rosuvastatin (Figure 1E ), and simvastatin (Figure 1F ) on α‐SMA/α/β‐tubulin ratio, normalized to % maximal differentiation. For concentration for half‐maximal inhibition (IC50) calculation of atorvastatin and rosuvastatin, the highest concentration used was considered to have reached near maximum possible effect, and any further increase in concentration was assumed to have ±5% change from the measured maximum effect (asymptote). The respective IC50 values were 729 ± 13 nmol/L (atorvastatin), 3.6 ± 1 μmol/L (rosuvastatin), and 185 ± 13 nmol/L (simvastatin); n = 3 per concentration of each statin.
Figure 1.
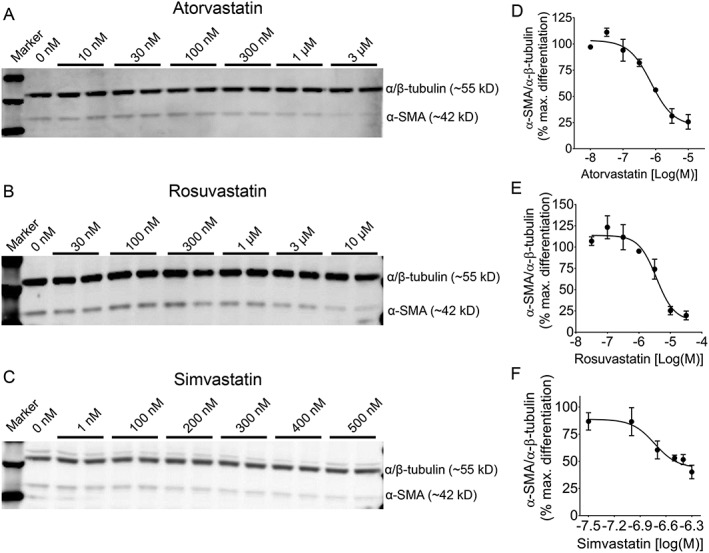
Concentration‐dependent de‐differentiation effect of statins in human ventricular myofibroblasts. Representative immunoblots showing the effect of in vitro treatment for 72 h with (A) atorvastatin (10 nmol/L to 3 μmol/L), (B) rosuvastatin (30 nmol/L to 10 μmol/L), or (C) simvastatin (1 to 500 nmol/L) on differentiated myofibroblasts, as determined by the expression of α‐SMA (α‐smooth muscle actin), a marker of differentiated myofibroblasts. In the right panel, respective non‐linear regression curves show the concentration‐dependent effects of corresponding statin [(D) atorvastatin, (E) rosuvastatin, and (F) simvastatin] on α‐SMA/α/β‐tubulin ratio, normalized to % maximal differentiation. For concentration for half‐maximal inhibition (IC50) analysis of atorvastatin and rosuvastatin, the highest concentration used was considered to have reached a near maximum possible effect, and any further increase in concentration was assumed to have ±5% change from the measured maximum effect (asymptote). The respective IC50 values were 729 ± 13 nmol/L (atorvastatin), 3.6 ± 1 μmol/L (rosuvastatin), and 185 ± 13 nmol/L (simvastatin); n = 3 per concentration; four‐parameter logistic fit. Data are mean ± SEM.
3.2. Signalling mechanisms in statin‐induced de‐differentiation
To determine the underlying mechanisms of statin‐induced de‐differentiation, we treated the already‐differentiated myofibroblasts with single concentration of statins in vitro in the presence or absence of signalling molecules or pharmacological inhibitors and assessed α‐SMA or COL III protein expressions. As depicted in Figure 2A , the de‐differentiating effect of single concentration of in vitro statin (simvastatin 200 nmol/L for 72 h) on already‐differentiated myofibroblasts (Diff) from HF patients, as α‐SMA expression decreased significantly by 64% (Diff + Simva). Immunocytochemistry confirmed simvastatin‐induced reduction in α‐SMA+ (red staining) fibroblasts compared with the differentiated (TGF) group (Figure 2B ). For further mechanistic studies, the differentiated myofibroblasts from patients were simulated in vitro by TGF‐β1‐induced differentiation in normal fibroblasts from trauma victims. Simvastatin (200 nmol/L) was applied for 72 h following TGF‐β1‐induced differentiation (TGF‐72 h + 72 h) in vitro, which also reproduced the same de‐differentiation effect (in Figures 2C , S2 ), as both α‐SMA and COL III expressions were significantly decreased by 87% and 142%, respectively (TGF‐72 h + Simva). MVA (300 μmol/L), the reduced product of HMG‐CoA, prevented the statin‐induced de‐differentiation (Figures 2C , S2 ), as expression of neither α‐SMA (31.4 ± 10% vs. 58.6 ± 12%) nor COL III (40 ± 3% vs. 37 ± 27%) decreased when simvastatin was co‐administered with MVA (TGF‐72 h + Simva + MVA), suggesting the role of HMG‐CoA reductase inhibition in de‐differentiation. To determine the signalling pathway downstream of MVA involved in statin‐induced de‐differentiation, we supplemented statins with GGPP (20 μmol/L), an intermediate in the cholesterol synthesis‐independent HMG‐CoA reductase pathway,13, 14 which completely prevented the statin‐induced de‐differentiation (Figure 2D ). The mean ratios of respective α‐SMA to GAPDH densities were 0.89 ± 0.05 (TGF‐72 h + 72 h), 0.63 ± 0.02 (TGF‐72 h + simvastatin), and 1.2 ± 0.08 (TGF‐72 h + simvastatin + GGPP); n = 3. To determine the association between altered bioenergetics10 and statin‐induced de‐differentiation, we evaluated the role of KATP channels, the molecular sensors of cellular metabolism.15 Interestingly, co‐incubation of simvastatin (200 nmol/L) with glibenclamide (10 μmol/L), a KATP channel inhibitor, attenuated the simvastatin‐induced de‐differentiation (Figure 2D ) (α‐SMA/GAPDH ratio: 0.84 ± 0.05 [TGF‐72 h + simvastatin + glibenclamide]; n = 3). Direct inhibition of mitochondrial respiration by oligomycin (1 ng/mL) also produced a de‐differentiation effect like statins (Figure 2D) with reduced α‐SMA expression (α‐SMA/GAPDH ratio: 0.33 ± 0.02 [TGF‐72 h + oligomycin]; n = 3). Similarly, as shown in Figure 2E , co‐administration of atorvastatin (300 nmol/L) with glibenclamide attenuated the atorvastatin‐induced de‐differentiation, as evident from the reversal of the atorvastatin‐induced reduced α‐SMA expression (α‐SMA/α/β‐tubulin ratios: 0.95 ± 0.03 [TGF‐72 h + 72 h], 0.57 ± 0.04 [TGF‐72 h + atorvastatin], and 0.93 ± 0.1 [TGF‐72 h + atorvastatin + glibenclamide]; n = 3).
Figure 2.
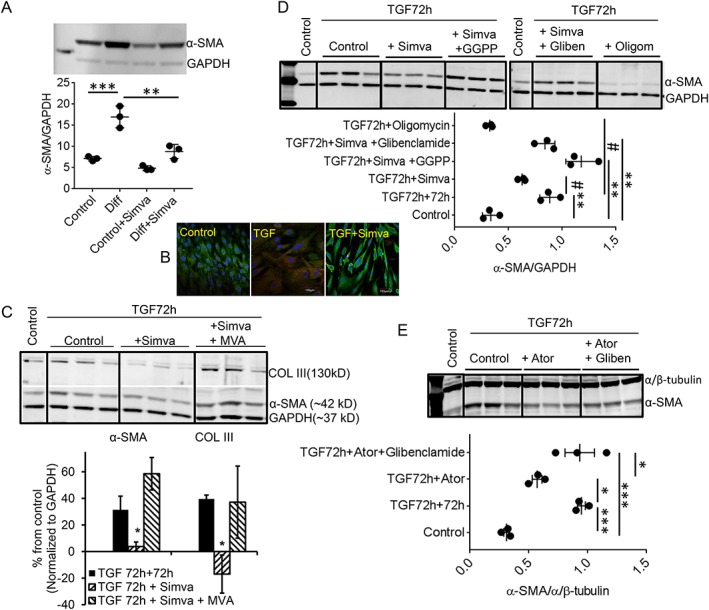
Mechanisms of statin‐induced de‐differentiation. (A) Representative immunoblot showing the effect of a single concentration of in vitro statin (simvastatin 200 nmol/L for 72 h) on already‐differentiated myofibroblasts (Diff) isolated from HF patients, with significant decrease (64%) in α‐SMA expression (Diff + Simva). (B) Representative immunocytochemistry showed a higher population of α‐SMA+ (red) cells in the differentiated (TGF) cells compared with control or in vitro treatment of simvastatin (TGF + Simva) (green: vimentin). (C) Representative immunoblots from in vitro differentiation. Simvastatin, applied for 72 h following TGF‐β1‐induced differentiation (5 ng/mL for initial 72 h) in vitro (simulating in vivo differentiated hVFs from HF patients), reproduced similar de‐differentiation, as expression of both α‐SMA and COL III were significantly decreased by 87% and 142%, respectively. Mevalonic acid (MVA, 300 μmol/L) prevented the statin‐induced de‐differentiation, as expression of both α‐SMA (31.4 ± 10% vs. 58.6 ± 12%) and COL III (40 ± 3% vs. 37 ± 27%) did not decrease when simvastatin was co‐administered with MVA. (D) Representative immunoblots depict complete prevention of simvastatin‐induced decreased α‐SMA expression by either GGPP (20 μmol/L) or glibenclamide (10 μmol/L). The effect of statin on the expression of a‐SMA is mimicked by oligomycin (1 ng/mL) in the same time frame. Individual data points column graph displays respective mean ratio of α‐SMA to GAPDH densities. (E) Representative immunoblot depicts significant reduction of atorvastatin‐induced decreased α‐SMA expression by glibenclamide (10 μmol/L). Individual data points column graph displays respective mean ratio of α‐SMA to α/β‐tubulin densities. * P < 0.05 vs. control; *** P < 0.001 vs. control; # P < 0.05 vs. TGF‐72 h + 72 h; * P < 0.05 vs. TGF‐72 h + Ator; considered as significant, n = 3 each; one‐way ANOVA. TGF‐72 h + 72 h = TGF‐β1 (5 ng/mL) treatment for 72 h to differentiate into myofibroblasts, and subsequent 72 h culture was without TGF‐β1. Data are mean ± SEM. Ator, atorvastatin; GGPP, geranylgeranyl pyrophosphate; Gliben, glibenclamide; Oligom, oligomycin; Simva, simvastatin.
3.3. In vitro, statins reduced cellular respiration
Myofibroblast differentiation is associated with increased mitochondrial OXPHOS capacity.10 Therefore, we determined the cellular respiration using Seahorse assay, and data are reported as pmol O2/min/μg protein. Similar to statin‐induced de‐differentiation of myofibroblasts in vitro, simvastatin significantly altered both OCR and ECAR. Basal OCR was significantly decreased in the simvastatin‐treated hVFs vs. the differentiated control hVFs (TGF‐72 h) (0.144 ± 0.026 vs. 0.278 ± 0.104, P = 0.002) (Figure 3A,B ). Simvastatin reduced the ATP‐linked OCR (0.083 ± 0.018 vs. 0.160 ± 0.059, P = 0.01), the proton leak‐linked OCR (0.017 ± 0.008 vs. 0.074 ± 0.041, P = 0.01), and the maximal OCR (0.250 ± 0.031 vs. 0.501 ± 0.142, P < 0.001) (Figure 3B ) but did not significantly affect spare capacity OCR (0.150 ± 0.027 vs. 0.278 ± 0.214, P = 0.348) or non‐mitochondrial OCR (0.044 ± 0.009 vs. 0.056 ± 0.020, P = 0.082) in the control differentiated (TGF) and in the simvastatin‐treated cells (TGF + Simva) Figure 3B ). The inhibitory effect of simvastatin on the mitochondrial OCR was reversed by GGPP (Figure 3A,B ). GGPP (20 μmol/L) significantly increased the basal OCR (0.193 ± 0.053, P = 0.020), the spare capacity OCR (0.233 ± 0.091, P = 0.008), and the maximal OCR (0.377 ± 0.131, P = 0.003), without any change in the non‐mitochondrial OCR (0.049 ± 0.01, P = 0.082) and in the simvastatin‐treated hVF cells (TGF + Simva+GGPP) (Figure 3B ). There was a trend towards increased ATP‐linked OCR (0.037 ± 0.010, P = 0.081) and proton leak‐linked OCR (0.036 ± 0.027, P = 0.058), but it did not reach statistical significance. The simvastatin treatment of the differentiated hVFs in vitro reduced ECAR after addition of FCCP (0.066 ± 0.011 vs. 0.093 ± 0.010, P < 0.001) and AA (0.056 ± 0.014 vs. 0.097 ± 0.008, P < 0.001) (Figure 3C,D ). GGPP reversed ECAR to a similar level as control differentiated hVFs (FCCP: 0.081 ± 0.015, P = 0.005; AA: 0.080 ± 0.014, P = 0.018). In accordance with decreased cellular respiration, both atorvastatin (100 and 300 nmol/L) and rosuvastatin (300 nmol/L and 1 μmol/L) also increased the ADP/ATP ratio (Figure 3E ) in these cells, confirming the pan‐statin effect of decreased bioenergetics during de‐differentiation. Also, simvastatin treatment significantly reduced functional activity of Complex V (34.76 ± 1.58 vs. 44.25 ± 1.58 nmol/min/μg protein, P = 0.01) while activities of Complex I (8.16 ± 7.15 vs. 10.04 ± 6.77 nmol/min/μg protein, P = 0.86), Complex II (20.11 ± 1.31 vs. 22.45 ± 3.37 nmol/min/μg protein, P = 0.55), Complex III (5.16 ± 0.45 vs. 5.73 ± 0.47 nmol/min/μg protein, P = 0.43), or Complex IV (13.99 ± 2.95 vs. 14.72 ± 3.49 nmol/min/μg protein, P = 0.88) were not affected (Figure 3F ).
Figure 3.
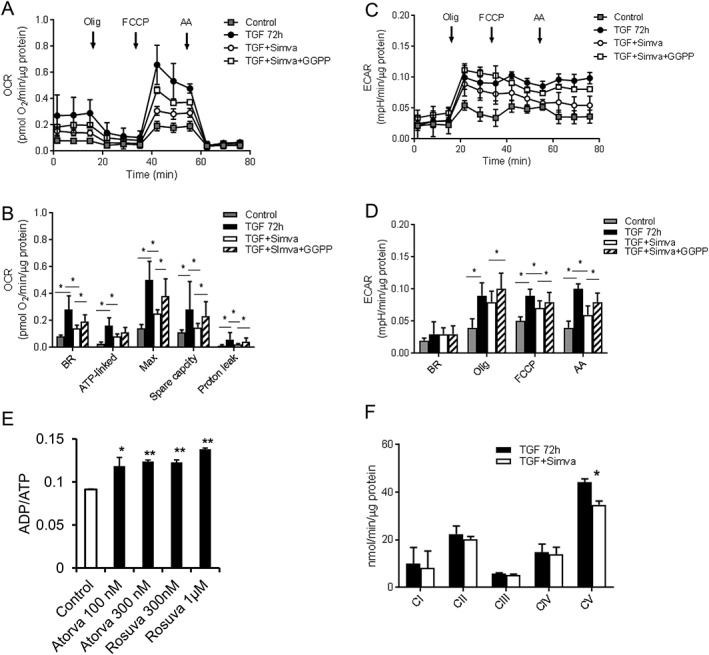
Effect of statins on cellular respiration. (A) Graphical representation of pooled data of cellular respiration (OCR) from Seahorse assays of hVFs in the control, differentiated (TGF), the simvastatin‐treated (TGF + Sim), and GGPP/simvastatin co‐administration (TGF + Sim + GGPP) groups. (B) Bar graphs depict that simvastatin reduced the ATP OCR, the proton leak‐linked OCR, and the maximal OCR without any significant effect on spare capacity OCR or non‐mitochondrial OCR. The inhibitory effect of simvastatin on the mitochondrial OCR was reversed by GGPP. (C) Graphical representation of pooled data of extracellular acidification rate (ECAR) from Seahorse assays of hVFs in all the three groups. (D) Simvastatin treatment of the differentiated hVFs (TGF + Sim) in vitro reduced ECAR, after addition of FCCP or AA. GGPP reversed ECAR to a similar level as control differentiated hVFs upon addition of FCCP and AA (TGF + Sim + GGPP); n = 6. Data are mean ± SD. (E) Bar graph depicting that both lipophilic atorvastatin (100 and 300 nmol/L) and hydrophilic rosuvastatin (300 nmol/L and 1 μmol/L) increased the ADP/ATP ratio. (F) Enzymatic activities of mitochondrial OXPHOS complexes in lysates from differentiated (TGF) and simvastatin‐treated hVFs showed significant reduction in Complex V activity by simvastatin (TGF + Simva). n = 3. Data are mean ± SEM. Atorva, atorvastatin; GGPP, geranylgeranyl pyrophosphate; Simva, simvastatin; Rosuva, rosuvastatin.
3.4. Inhibition of geranylgeranyl pyrophosphate synthase unlike Rho‐kinase promotes de‐differentiation
Digeranyl bisphosphonate (DGBP, 30 μmol/L), a GGPP synthase inhibitor, induced significant de‐differentiation as evident from the reduced expression of α‐SMA compared with the differentiated control (Figure 4A ). However, inhibition of Rho‐kinase by Y27632 (10 μmol/L) did not affect the differentiation in the same period of 72 h. Similarly, both basal OCR and ATP‐linked OCR were significantly inhibited by DGBP (Figure 4B ) unlike the Rho‐kinase inhibitor, Y27632 or GGPP (30 nmol/L), itself (Figure 4C ).
Figure 4.
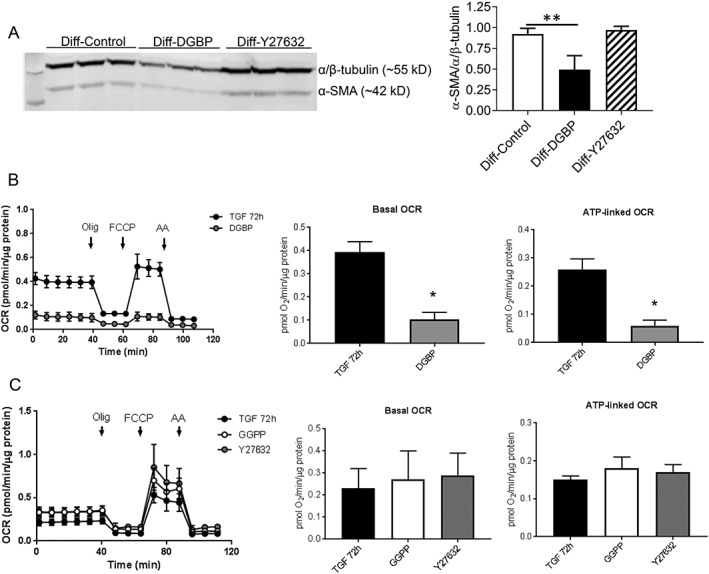
Effect of either GGPP synthase or Rho kinase inhibition on hVF differentiation and cellular respiration. (A) Representative immunoblot showing the in vitro effect of either GGPP synthase inhibitor digerenyl bisphonate (DGBP, 30 μmol/L) or ROCK inhibitor Y27632 (10 μmol/L) for 72 h on already‐differentiated myofibroblasts (Diff), and corresponding bar graph shows significant de‐differentiation by DGBP without any effect by Y27632. (B) Graphical representation of pooled data of cellular respiration (OCR) from seahorse assays of differentiated hVFs (TGF) and the GGPP synthase‐inhibited (DGBP) groups. Both basal and ATP‐linked OCRs were significantly inhibited by DGBP. (C) Graphical representation of pooled data of OCR showing no significant effect by GGPP or Y27632 on both basal and ATP‐linked respiration. Data are mean ± SD. * P < 0.05; one‐way ANOVA.
3.5. Time‐dependent effect of simvastatin on fibroblast de‐differentiation and oxygen consumption rate
To determine the time‐dependent relationship between cellular respiration and de‐differentiation, in vitro, simvastatin (200 nmol/L) was administered for various time periods at the interval of 12 h up to 72 h on differentiated hVFs followed by immunoblotting (α‐SMA) and Seahorse assay. Simvastatin significantly (P < 0.05) induced de‐differentiation from 24 h (Figure 5A,B ) onwards while significant (P < 0.05) reduction in both basal OCR and ATP‐linked OCR from 18 h onwards (Figure 5C–E ). In addition to α‐SMA expression, we also confirmed the time‐dependent statin‐induced de‐differentiation of myofibroblasts with another differentiation marker, paxillin (Figure S5 ).
Figure 5.
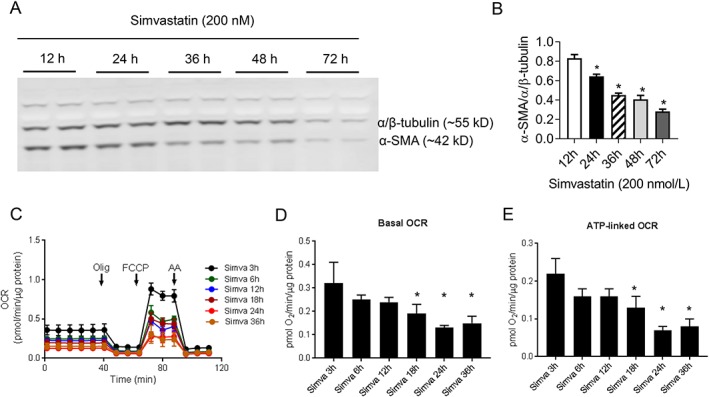
Time‐course effect of statins in human ventricular myofibroblasts de‐differentiation and cellular respiration. (A) Representative immunoblots showing the time‐dependent effect of in vitro treatment of simvastatin (200 nmol/L), and corresponding bar graph depicts significant de‐differentiation from 24 h onwards. (B) Graphical representation of pooled data of time‐dependent effect of simvastatin (200 nmol/L) on OCR of differentiated hVFs. Respective bar graphs depict significant reduction in both basal and ATP‐linked OCRs from 18 h onwards. Data mean ± SD. * P < 0.05; one‐way ANOVA.
3.6. Decreased pro‐fibrotic myofibroblasts in heart failure patients on statin therapy
In the context of in vitro statin‐induced de‐differentiation of cardiac myofibroblasts, we determined the impact of statin therapy in HF patients. hVFs, isolated from HF patients, displayed a high α‐SMA expression, suggesting predominant composition of differentiated myofibroblasts (Figures 6A , S3 ), whereas hVFs from HF + statin patients showed significantly (P = 0.01) reduced expression of α‐SMA (Figures 6A,B , S3 ) (α‐SMA/GAPDH ratios: 1 ± 0.19 [HF, n = 4] and 0.23 ± 0.06 [HF + statin, n = 4]). Immunocytochemistry showed a greater population of α‐SMA+ cells in the HF group than in the HF + statin group (Figure 6C , S3 ). Corresponding to the differentiation status of hVFs in terms of α‐SMA expression between the two groups, a reciprocal expression of SPRY1, a negative regulator of fibrosis,20, 21 existed with lower expression in the HF group (0.15 ± 0.01, n = 4) and significantly (P = 0.01) greater expression in the HF + statin group (0.51 ± 0.12, n = 4) (Figure 6D,E ), confirming the effect of statin therapy in HF patients leading to mitigated fibrotic phenotype of hVFs.
Figure 6.
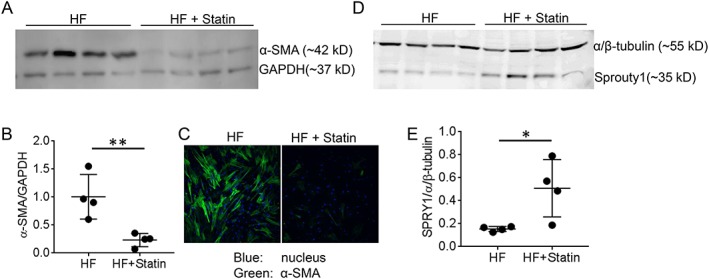
Effect of statin therapy on differentiation of ventricular fibroblasts in heart failure patients. (A) Representative immunoblot of human ventricular fibroblasts (hVFs) isolated from HF patients displayed a high α‐SMA expression while hVFs from HF + statin showed significantly reduced α‐SMA expression. (B) The individual data points column graph displays each sample value of α‐SMA/GAPDH density ratio with lines depicting mean ± SD; n = 4. (C) Representative immunocytochemistry showed a higher population of α‐SMA+ (green) cells in the HF group vs. HF + statin group (n = 3). (D) Immunoblot shows the expression of SPRY1, a negative regulator of fibrosis, in lysates of hVFs isolated from failing heart patients. (E) The individual data points column graph displays each sample value of SPRY1/GAPDH density ratio with lines depicting mean ± SD, with lower expression in the HF group and significantly higher expression in HF + statin therapy; n = 4. * P < 0.05, ** P < 0.01 considered significant vs. HF; unpaired t‐test.
3.7. Decreased cellular respiration in human ventricular fibroblasts from heart failure patients on statin therapy
The cellular respiration in hVFs isolated from HF patients was determined to assess whether in vitro statin‐induced changes in fibroblasts bioenergetics also occur in vivo. The basal OCR (0.137 ± 0.056 vs. 0.232 ± 0.025, P = 0.021), the ATP‐linked OCR (0.093 ± 0.039 vs. 0.154 ± 0.024, P = 0.047), maximal OCR (0.258 ± 0.102 vs. 0.456 ± 0.058, P = 0.02), and spare capacity OCR (0.148 ± 0.056 vs. 0.287 ± 0.058, P = 0.025) were significantly reduced in hVFs isolated from HF + statin patients, without any effect on the proton leak‐linked‐related OCR (0.0195 ± 0.008 vs. 0.0146 ± 0.004, P = 0.322) or the non‐mitochondrial‐related e_k; OCR (0.031 ± 0.017 vs. 0.046 ± 0.03, P = 0.400) (Figure 7 A,B). Long‐term in vivo statin therapy did not significantly affect the ECAR (Figure 7C,D ). Immunoblotting of hVF lysates with total OXPHOS human antibody cocktail showed significantly (P < 0.05) reduced Complex V subunit expression following statin therapy, while other complexes (I to IV) did not change significantly (Figure 7E,F ).
Figure 7.
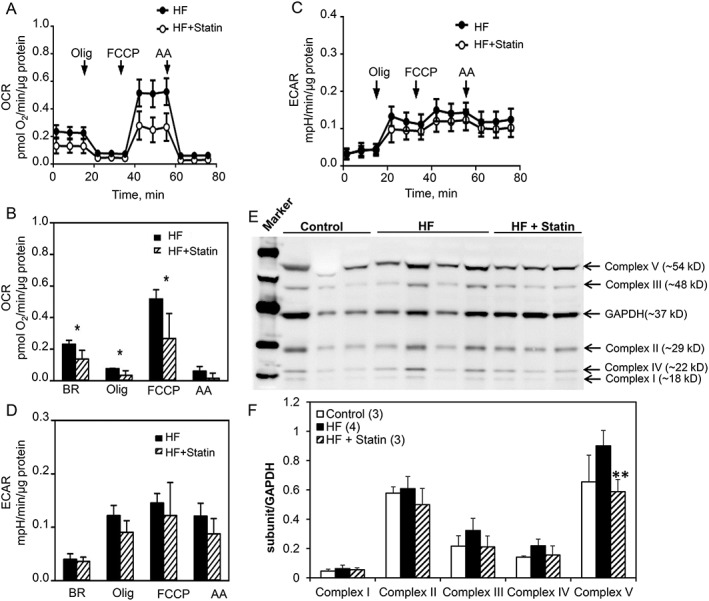
Reduced mitochondrial respiration in hVFs from HF patients under statin therapy. (A) Graphical representation of pooled OCR data from Seahorse assay of hVFs isolated from failing heart patients. (B) Bar graphs depicting the mean OCR for each group at basal, following oligomycin, FCCP, and antimycin A. The basal, ATP‐linked, maximal, and spare capacity OCRs were significantly reduced in hVFs from HF + statin vs. HF group, without significant effect on the Olig‐insensitive proton leak‐linked‐related OCR or non‐mitochondrial OCR. (C) Graphical representation of pooled data of extracellular acidification rate (ECAR) from Seahorse assays of hVFs isolated from failing heart patients. (D) Bar graph shows ECAR data (mean ± SD) with no significant difference in ECAR between the two groups, following addition of either oligomycin, FCCP, or antimycin A; n = 6; analysed by unpaired t‐test. Data are mean ± SD. (E) Representative immunoblot with total OXPHOS human antibody coScktail and corresponding bar graph (F) showed significantly reduced Complex V subunit following statin therapy; n = 4, analysed by one‐way ANOVA.
4. Discussion
The salient findings from this study are (i) in vitro, both lipophilic and hydrophilic statins concentration‐dependently induced myofibroblast de‐differentiation and reduced cellular respiration with increased ADP/ATP ratio; (ii) statin‐induced myofibroblast de‐differentiation was sensitive to MVA, GGPP, and glibenclamide; (iii) statin‐induced myofibroblast de‐differentiation was mimicked by direct GGPP synthase inhibition; (iv) simulating the in vitro effect, statin therapy in HF patients significantly reduced cardiac myofibroblast differentiation and lowered hVF cellular respiration including basal, ATP‐linked, maximal, and spare capacity respirations. Excessive cardiac fibrosis is considered to be one of the major predisposing factors leading to mechanical and electrical dysfunctions in HF.1 Accumulation of myofibroblasts within pathologic lesions is a feature of various fibrotic disorders,5 including progressive HF and cardiac hypertrophy.6 Therefore, reversing myofibroblasts back to fibroblasts might provide a new therapeutic approach in attenuating fibrosis and cardiac dysfunctions.22 Several strategies have been attempted in non‐cardiac myofibroblast reversal, including reduced strain, inhibition of SMAD3, TGF‐β receptor blockade, prostaglandin E2, and nitrated fatty acids.22, 23, 24, 25 However, studies on de‐differentiation of human cardiac myofibroblasts are scarce.
4.1. Concentration‐dependent de‐differentiation effect of statins
Our study, for the first time, reports that human ventricular myofibroblasts can be de‐differentiated by statins. Statins are inhibitors of HMG‐CoA reductase commonly prescribed for their hypolipidaemic effects in patients at risk of cardiac dysfunctions. In vitro application of both hydrophilic and lipophilic statins on already‐differentiated hVFs induced myofibroblast de‐differentiation in a concentration‐dependent manner as evident from the decrease in α‐SMA expression, a marker of myofibroblasts. While both atorvastatin and rosuvastatin effects were displayed in a logarithmic range of doses, simvastatin evinced a very narrow window of effective concentrations between 100 and 500 nmol/L without causing cytotoxicity. This difference could be due to the varied proportion by which the binding enthalpy contributes to the binding affinity that is not the same for all statins, indicating that the balance among hydrogen bonding, van der Waals, and hydrophobic interactions is not the same for all of them.26 Simvastatin is high in potency followed by atorvastatin and rosuvastatin, which may be due to their differences in affinity and lipophilicity.27 Rosuvastatin, being hydrophilic, requires active transportation via organic anion transporting polypeptide 1B1 while lipophilic statins can passively diffuse into the cells.28 Although several prior in vitro studies have demonstrated statin‐induced prevention of differentiation to myofibroblasts,7, 8, 9 their ability to de‐differentiate already‐differentiated myofibroblasts is hitherto largely unknown. In addition to α‐SMA, the reduced expression of the negative regulator of fibrosis, Sprouty1, was reversed by simvastatin (Supporting Information, Figure S1 ).
4.2. Mechanisms of statin‐induced de‐differentiation
Statin‐induced de‐differentiation appears to be HMG‐CoA reductase‐dependent because MVA, the reduced product of HMG‐CoA that is common to both the cholesterol‐synthesis and cholesterol‐independent pathways, prevented the statin‐induced de‐differentiation. The signalling pathway downstream to MVA in the myofibroblast de‐differentiation process is independent of cholesterol synthesis but involves the isoprenoid GGPP, a predominant signalling intermediate molecule in the cholesterol synthesis‐independent pathway13, 14 as supplementation of GGPP completely prevented the statin‐induced de‐differentiation. The association between altered bioenergetics and statin‐induced de‐differentiation seems to involve KATP channels, the molecular sensors of cellular metabolism.15 Co‐incubation of statins with glibenclamide, a KATP channel inhibitor, attenuated the statin‐induced myofibroblast de‐differentiation, suggesting that activation of KATP channels by increased ADP/ATP ratio, caused by statins, may play a role in statin‐induced myofibroblast de‐differentiation. In conformity, recently, nicorandil, a KATP channel agonist, was demonstrated to attenuate myocardial fibrosis and improve cardiac function after myocardial infarction in rats.29
4.3. Effect of statins on cellular respiration
Previously, altered cellular respiration has been reported to be associated with myofibroblast differentiation.10 In this study, statins decreased the cellular respiration of hVFs via cholesterol synthesis‐independent pathway. Our study is the first to show that GGPP can improve statin‐induced compromised cellular respiration in hVFs and prevent de‐differentiation; nevertheless, the mechanism is unclear. It probably involves maintenance of the glycolytic activity mitigated by statins, as evident from the reversal of ECAR by GGPP (Figure 3C,D ). Direct inhibition of GGPP synthase by DGBP is enough to affect the cellular respiration and induce de‐differentiation (Figure 4), suggesting GGPP as an important intermediate signalling molecule influencing OCR; however, GGPP supplementation alone does not enhance the OCR (Figure 4C ). In muscle cells, statin‐induced deficiency of Coenzyme Q10 (CoQ10)/Ubiquinone is believed to be the aetiology of mitochondrial dysfunction.30 However, it becomes complicated across different tissue types; unlike in skeletal muscles, statins seem to protect mitochondria in cardiac myocytes from oxidative stress,31 and hence, statin effects on cellular respiration cannot be generalized across various cell types/tissues/organs. The possibility of statin‐induced decrease in mitochondrial respiration as the underlying mechanism of statin‐induced myofibroblast de‐differentiation is supported by the evidence of a similar de‐differentiation effect (Figure 2D ) following direct inhibition of mitochondrial respiration by oligomycin. The role of compromised Complex V in the statin effect is also supported by the reduced functional activity of Complex V following simvastatin (Figure 3F ). Further evidence comes from the time‐dependent effect of simvastatin on de‐differentiation vs. OCR where reduction in OCR occurs earlier (Figure 5).
4.4. Impact of statin therapy on ventricular fibroblasts in heart failure patients
The high population of myofibroblasts in HF patients not on statin therapy was significantly attenuated in patients on statin therapy. The isolated hVFs from HF + statin patients showed significantly reduced expression of α‐SMA and significantly increased expression of Spry1, an intrinsic negative regulator of fibrosis the downregulation of which is reported in cardiac pathologies to activate connective tissue growth factor, lysyl oxidase, fibroblast growth factor, and fibrosis.20, 21 The major effect of statins on reducing the myofibroblast population in HF patients could be due to either statin‐induced prevention of differentiation or promotion of de‐differentiation of the myofibroblasts. In addition to a reduced population of myofibroblasts, the hVFs isolated from HF + statin patients also showed significantly reduced cellular respiration, including basal, ATP‐linked, maximal, proton leak‐linked, and spare capacity respirations, without any change in ECAR. Statins induced regression of cardiac hypertrophy and fibrosis in a transgenic rabbit model of human hypertrophic cardiomyopathy32 and reversed myocardial fibrosis through activation of AMP‐activated protein kinase in a mouse model of metabolic syndrome.33 The reversal effects of statins on fibrosis in these animal models might also be due to statin‐induced de‐differentiation of myofibroblasts, as found in this study. We also observed reduced fibrosis in HF patients under statin therapy (Supporting Information, Figure S4 ). The overall observations from this study are illustrated for easy understanding in a simple schematic model (Figure 8).
Figure 8.
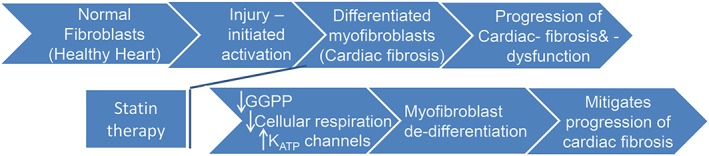
Schematic model depicting the potential relationship between statin therapy, mitochondrial energetics, and de‐differentiation of hVFs. Cardiac fibrosis with persistent myofibroblasts is one of the major underlying factors in heart failure (HF) progression. Statin therapy significantly reduced the high level of differentiated myofibroblasts in HF patients. The underlying mechanisms of statin‐induced de‐differentiation of myofibroblasts involve GGPP‐dependent signalling mechanisms, lowered cellular respiration, and KATP channels. GGPP, geranylgeranyl pyrophosphate; KATP, adenosine 5′‐triphosphate‐sensitive potassium channels.
4.5. Study limitations
Ethical consideration and stringent exclusion/inclusion criteria to prevent potential confounding factors limited our study. hVFs, used in this study, are from late‐stage failing heart of left ventricular assist device and heart transplant patients while the statin therapy duration was at least 1 year. The significant differences between hVF from HF and HF + Statin remain to be tested on large cohorts that how duration of statin therapy or degree of cardiac damage influences cellular remodelling. Moreover, this study could not have excluded unknown co‐morbidities in HF patients that had a possible effect on the results. However, in vitro studies corroborate the novel observations from the HF patients' samples. The in vivo mechanisms of de‐differentiation need to be determined using an animal model. Nevertheless, this pioneer study demonstrates a significant in vivo effect of statin therapy on cellular respiration and differentiation of cardiac fibroblasts in human.
5. Conclusions
We demonstrate that (i) statins can induce de‐differentiation of myofibroblasts isolated from human failing hearts; (ii) statin therapy lowers the left ventricular myofibroblast population in human HF; and (iii) the underlying mechanisms in statin‐induced de‐differentiation involve GGPP‐sensitive signalling, lowered bioenergetics, and KATP channels. As myofibroblasts' persistence results in chronic/excessive fibrosis, causing cardiac dysfunction,34 strategies that can de‐differentiate myofibroblasts back to fibroblasts provide a novel therapeutic approach to mitigate fibrosis and HF progression.
Conflict of interest
E.G.S. and A.N.R. have a financial interest in Cellular Logistics, Inc. No other authors have any conflicts of interest to report.
Funding
This work was supported by Aurora Health Care, Cardiovascular Surgical Research Award to G.R.R. (570–5028).
Supporting information
Figure S1. Effect of simvastatin on Sprouty 1 expression in hVF. Immunoblot showing the in vitro effect of simvastatin (200 nmol/L) for 72 h on Sprouty 1 expression in differentiated hVFs (TGF) and corresponding bar graph shows reversal of reduced sprouty 1 expression by simvastatin.
Figure S2. Full Gels corresponding to Figure 2C.
Figure S3. Immunoblot corresponding to Figure 6A‐C after including control (non HF) hVFs, in addition to HF and HF + statin.
Figure S4. Representative photographs of Masson's trichrome staining of ventricle histology sections display severe fibrosis (blue) in the failing heart (top) compared with the failing heart patients under statin therapy (bottom).
Figure S5. Simvastatin reduces paxillin expression. Representative immunoblots showing the time‐dependent effect of in vitro treatment of simvastatin (200 nmol/L) and corresponding bar graph depicts significant decrease in paxillin expression normalized to α/β‐tubulin.
Table S1. Materials used.
Table S2. Clinical characteristics of patients under study: Both HF (No statin) and HF + Statin groups were well‐matched with no significant difference between the groups.
Table S3. Antibodies used for immunoblotting and immuno‐localization.
Acknowledgements
The authors acknowledge Catherine Warner for immunoblotting and cell‐based assays; Ismail Ahmad for histology; and Steven Komas and Monika Zielonka from Medical College of Wisconsin, Cancer Center Redox and Bioenergetics Shared Resource (CCRBSR) for Seahorse expertise.
Emelyanova, L. , Sra, A. , Schmuck, E. G. , Raval, A. N. , Downey, F. X. , Jahangir, A. , Rizvi, F. , and Ross, G. R. (2019) Impact of statins on cellular respiration and de‐differentiation of myofibroblasts in human failing hearts. ESC Heart Failure, 6, 1027–1040. 10.1002/ehf2.12509.
References
- 1. Ichiki T, Schirger JA, Huntley BK, Brozovich FV, Maleszewski JJ, Sandberg SM, Sangaralingham SJ, Park SJ, Burnett JC Jr. Cardiac fibrosis in end‐stage human heart failure and the cardiac natriuretic peptide guanylyl cyclase system: regulation and therapeutic implications. J Mol Cell Cardiol 2014; 75: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rosenbloom J, Mendoza FA, Jimenez SA. Strategies for anti‐fibrotic therapies. Biochim Biophys Acta 2013; 1832: 1088–1103. [DOI] [PubMed] [Google Scholar]
- 3. Hinz B. Myofibroblasts. Exp Eye Res 2016. Jan; 142: 56–70. [DOI] [PubMed] [Google Scholar]
- 4. Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol 2012; 180: 1340–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryu JH, Daniels CE. Advances in the management of idiopathic pulmonary fibrosis. F1000. Med Rep 2010; 2: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 1995; 91: 161–170. [DOI] [PubMed] [Google Scholar]
- 7. Meyer‐Ter‐Vehn T, Katzenberger B, Han H, Grehn F, Schlunck G. Lovastatin inhibits TGF‐β‐induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci 2008; 49: 3955–3960. [DOI] [PubMed] [Google Scholar]
- 8. Watts KL, Sampson EM, Schultz GS, Spiteri MA. Simvastatin inhibits growth factor expression and modulates profibrogenic markers in lung fibroblasts. Am J Respir Cell Mol Biol 2005; 32: 290–300. [DOI] [PubMed] [Google Scholar]
- 9. Rizvi F, Siddiqui R, DeFranco A, Homar P, Emelyanova L, Holmuhamedov E, Ross G, Tajik AJ, Jahangir A. Simvastatin reduces TGF‐β1‐induced SMAD2/3‐dependent human ventricular fibroblasts differentiation: role of protein phosphatase activation. Int J Cardiol 2018; 270: 228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Negmadjanov U, Godic Z, Rizvi F, Emelyanova L, Ross G, Richards J, Holmuhamedov EL, Jahangir A. TGF‐β1‐mediated differentiation of fibroblasts is associated with increased mitochondrial content and cellular respiration. PLoS One 2015; 10: e0123046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. LaRosa JC, Deedwania PC, Shepherd J, Wenger NK, Greten H, DeMicco DA, Breazna A, Investigators TNT. Comparison of 80 versus 10 mg of atorvastatin on occurrence of cardiovascular events after the first event (from the Treating to New Targets [TNT] trial). Am J Cardiol 2010; 105: 283–287. [DOI] [PubMed] [Google Scholar]
- 12. Porter KE, Turner NA, O'Regan DJ, Balmforth AJ, Ball SG. Simvastatin reduces human atrial myofibroblast proliferation independently of cholesterol lowering via inhibition of RhoA. Cardiovasc Res 2004; 61: 745–755. [DOI] [PubMed] [Google Scholar]
- 13. Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res 2005; 97: 1232–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res 2017; 120: 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature 2006; 440: 470–476. [DOI] [PubMed] [Google Scholar]
- 16. Ross GR, Bajwa T Jr, Edwards S, Emelyanova L, Rizvi F, Holmuhamedov EL, Werner P, Downey FX, Tajik AJ, Jahangir A. Enhanced store‐operated Ca2+ influx and ORAI1 expression in ventricular fibroblasts from human failing heart. Biol Open 2017; 6: 326–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shchepina LA, Pletjushkina OY, Avetisyan AV, Bakeeva LE, Fetisova EK, Izyumov DS, Saprunova VB, Vyssokikh MY, Chernyak BV, Skulachev VP. Oligomycin, inhibitor of the F0 part of H+‐ATP‐synthase, suppresses the TNF‐induced apoptosis. Oncogene 2002; 21: 8149–8157. [DOI] [PubMed] [Google Scholar]
- 18. Divakaruni AS, Paradyse A, Ferrick DA, Murphy AN, Jastroch M. Analysis and interpretation of microplate‐based oxygen consumption and pH data. Methods Enzymol 2014; 547: 309–354. [DOI] [PubMed] [Google Scholar]
- 19. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 2012; 7: 1235–1246. [DOI] [PubMed] [Google Scholar]
- 20. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008; 456: 980–984. [DOI] [PubMed] [Google Scholar]
- 21. Adam O, Lohfelm B, Thum T, Gupta SK, Puhl SL, Schafers HJ, Bohm M, Laufs U. Role of miR‐21 in the pathogenesis of atrial fibrosis. Basic Res Cardiol 2012; 107: 278. [DOI] [PubMed] [Google Scholar]
- 22. Driesen RB, Nagaraju CK, Abi‐Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res 2014; 101: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wettlaufer SH, Scott JP, McEachin RC, Peters‐Golden M, Huang SK. Reversal of the transcriptome by prostaglandin E2 during myofibroblast dedifferentiation. Am J Respir Cell Mol Biol 2016; 54: 114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reddy AT, Lakshmi SP, Zhang Y, Reddy RC. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J 2014; 28: 5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kosla J, Dvorakova M, Dvorak M, Cermak V. Effective myofibroblast dedifferentiation by concomitant inhibition of TGF‐β signaling and perturbation of MAPK signaling. Eur J Cell Biol 2013; 92: 363–373. [DOI] [PubMed] [Google Scholar]
- 26. Carbonell T, Freire E. Binding thermodynamics of statins to HMG‐CoA reductase. Biochemistry 2005; 44: 11741–11748. [DOI] [PubMed] [Google Scholar]
- 27. Rageh AH, Atia NN, Abdel‐Rahman HM. Lipophilicity estimation of statins as a decisive physicochemical parameter for their hepato‐selectivity using reversed‐phase thin layer chromatography. J Pharm Biomed Anal 2017; 142: 7–14. [DOI] [PubMed] [Google Scholar]
- 28. Gazzerro P, Proto MC, Gangemi G, Malfitano AM, Ciaglia E, Pisanti S, Santoro A, Laezza C, Bifulco M. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev 2012; 64: 102–146. [DOI] [PubMed] [Google Scholar]
- 29. Lee TM, Lin SZ, Chang NC. Nicorandil regulates the macrophage skewing and ameliorates myofibroblasts by inhibition of RhoA/Rho‐kinase signalling in infarcted rats. J Cell Mol Med 2018; 22: 1056–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ramachandran R, Wierzbicki AS. Statins, muscle disease and mitochondria. J Clin Med 2017; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jones SP, Teshima Y, Akao M, Marban E. Simvastatin attenuates oxidant‐induced mitochondrial dysfunction in cardiac myocytes. Circ Res 2003; 93: 697–699. [DOI] [PubMed] [Google Scholar]
- 32. Patel R, Nagueh SF, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen HA, Quinones MA, Zoghbi WA, Entman ML, Roberts R, Marian AJ. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circulation 2001; 104: 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hermida N, Markl A, Hamelet J, Van Assche T, Vanderper A, Herijgers P, van Bilsen M, Hilfiker‐Kleiner D, Noppe G, Beauloye C, Horman S, Balligand JL. HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP‐activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovasc Res 2013; 99: 44–54. [DOI] [PubMed] [Google Scholar]
- 34. Hinz B. The extracellular matrix and transforming growth factor‐β1: tale of a strained relationship. Matrix Biol 2015; 47: 54–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effect of simvastatin on Sprouty 1 expression in hVF. Immunoblot showing the in vitro effect of simvastatin (200 nmol/L) for 72 h on Sprouty 1 expression in differentiated hVFs (TGF) and corresponding bar graph shows reversal of reduced sprouty 1 expression by simvastatin.
Figure S2. Full Gels corresponding to Figure 2C.
Figure S3. Immunoblot corresponding to Figure 6A‐C after including control (non HF) hVFs, in addition to HF and HF + statin.
Figure S4. Representative photographs of Masson's trichrome staining of ventricle histology sections display severe fibrosis (blue) in the failing heart (top) compared with the failing heart patients under statin therapy (bottom).
Figure S5. Simvastatin reduces paxillin expression. Representative immunoblots showing the time‐dependent effect of in vitro treatment of simvastatin (200 nmol/L) and corresponding bar graph depicts significant decrease in paxillin expression normalized to α/β‐tubulin.
Table S1. Materials used.
Table S2. Clinical characteristics of patients under study: Both HF (No statin) and HF + Statin groups were well‐matched with no significant difference between the groups.
Table S3. Antibodies used for immunoblotting and immuno‐localization.