

Abstract
Cholesterol esters are a key ingredient of foamy cells in atherosclerotic lesions; their formation is catalyzed by two enzymes: acyl-CoA:cholesterol acyltransferases (ACATs; also called sterol O-acyltransferases, or SOATs) ACAT1 and ACAT2. ACAT1 is present in all body cells and is the major isoenzyme in macrophages. Whether blocking ACAT1 benefits atherosclerosis has been under debate for more than a decade. Previously, our laboratory developed a myeloid-specific Acat1 knockout (KO) mouse (Acat1−M/−M), devoid of ACAT1 only in macrophages, microglia, and neutrophils. In previous work using the ApoE KO (ApoE−/−) mouse model for early lesions, Acat1−M/−M significantly reduced lesion macrophage content and suppressed atherosclerosis progression. In advanced lesions, cholesterol crystals become a prominent feature. Here we evaluated the effects of Acat1−M/−M in the ApoE KO mouse model for more advanced lesions and found that mice lacking myeloid Acat1 had significantly reduced lesion cholesterol crystal contents. Acat1−M/−M also significantly reduced lesion size and macrophage content without increasing apoptotic cell death. Cell culture studies showed that inhibiting ACAT1 in macrophages caused cells to produce less proinflammatory responses upon cholesterol loading by acetyl low-density lipoprotein. In advanced lesions, Acat1−M/−M reduced but did not eliminate foamy cells. In advanced plaques isolated from ApoE−/− mice, immunostainings showed that both ACAT1 and ACAT2 are present. In cell culture, both enzymes are present in macrophages and smooth muscle cells and contribute to cholesterol ester biosynthesis. Overall, our results support the notion that targeting ACAT1 or targeting both ACAT1 and ACAT2 in macrophages is a novel strategy to treat advanced lesions.
Keywords: cholesterol, atherosclerosis, macrophage, vascular smooth muscle cells, lipid metabolism, ACAT1/SOAT1, Inflammation
Introduction
Atherosclerotic cardiovascular disease remains a major contributor to human morbidity and mortality worldwide. As reviewed in Refs. 1–3, during the early stages of atherogenesis, chronic inflammation causes monocytes to adhere to activated endothelial cells, infiltrate into the subendothelial space of the arterial wall, and transform into macrophages. Under sustained hyperlipidemic condition, macrophages that are retained in the intimal layer of the artery continue to take up cholesterol and other lipids, mostly in the form of denatured lipoproteins. Upon entering the cell interior, lipoprotein-derived cholesterols are converted to cholesterol esters (CEs)3 for storage, causing the cholesterol-loaded cells to become foamy. In advanced atherosclerotic lesions, in addition to cholesterol, several oxysterols, either synthesized enzymatically or produced through auto-oxidation, etc., also accumulate, as reviewed in Ref. 4. The unesterified forms of oxysterols at high concentrations can cause various toxic effects to cells (5). As the lesion continues to develop, foamy macrophages secrete various inflammatory cytokines, which stimulate the smooth muscle cells to migrate from the medial layer to the intimal layer of the artery and to proliferate. In advanced lesions, both macrophages and smooth muscle cells become foamy cells; they secrete matrix metalloproteinases, which degrade collagen and other extracellular matrix proteins, cause plaque instability, and eventually lead to plaque rupture.
In macrophages and other cell types, CEs accumulated in the cytosolic compartment appear as lipid droplets and cause the cells to look foamy in appearance. These lipid droplets exist in a dynamic state: re-esterification by the enzyme acyl-CoA:cholesterol acyltransferase (ACAT; also called sterol O-acyltransferase (SOAT)) and hydrolysis by neutral cholesterol ester hydrolases constitute the cholesterol/CE cycle, causing CEs to undergo continuous turnover, as reviewed in Ref. 6. For esterification, there are two ACAT genes, SOAT1 and SOAT2, encoding two distinct enzymes: ACAT1 (7) and ACAT2 (8–10). Both enzymes use long-chain fatty acyl-CoAs, and sterols with 3-β-OH, including cholesterol and various oxysterols as their substrates (11, 12). ACAT1 and ACAT2 are integral membrane proteins located in the ER region, and they are allosterically activated by cholesterol or oxysterols. In humans, ACAT1 is ubiquitously expressed in essentially all body cells; high levels of ACAT2 are mainly found in intestinal enterocytes (13, 14). Low levels of ACAT2 are also detectable in many other cell types, and their expression levels can become much elevated under various disease states (15). For example, in human and in mouse macrophages, ACAT1 is the major isoenzyme (16); however, in human and mouse advanced atherosclerotic lesions, a significant amount of ACAT2 becomes clearly detectable (17). In normal adult human hepatocytes, the ACAT2 protein expression is low (13); however, its expression becomes highly elevated in a certain subset of human hepatocarcinoma (12), as well as in patients with gallstones (18). In mouse, under normal conditions and in a mouse model for diet induced insulin resistance, ACAT2 is the major isoenzyme in hepatocytes (19). Both ACAT1 and ACAT2 are present in mouse adipocytes and play important roles in adipogenesis (20). For CE hydrolysis, there are a number of candidate hydrolytic enzymes, all capable of hydrolyzing CEs. The two major hydrolytic enzymes identified in macrophages are neutral cholesteryl ester hydrolase 1 (21, 22) and lipoprotein lipase, as reviewed in Ref. 23. Lipoprotein lipase also catalyzes the hydrolysis of retinyl esters and diacylglycerol (24).
Whether ACAT should be considered as a target for atherosclerosis treatment has been under debate for many years, as reviewed in Ref. 15. Regarding the pharmacological approach, two different ACAT inhibitors (CI 1011 and pactimibe) tested safe in humans, reached clinical trial phase 3 stage but were both abandoned at the end, in part because neither one was found to augment the actions of statin, which is the clinically approved drug for treating atherosclerosis. Because the small molecule inhibitor approach always stands the chance of producing off-target side effect(s), it is important that the pros and cons of ACAT blockage be carefully evaluated by using a different approach, the gene knockout approach in animals. This is an area where much disagreement occurred among different groups of investigators. Early studies by Yagyu yet al. (25) showed that in mouse models for atherosclerosis progression, whole body Acat1 knockout (KO) (Acat1−/−) attenuated atherosclerosis lesions and diminished lesion macrophage content. Accad et al. (26) acquired similar results but concluded that Acat1−/−) actually enlarged atherosclerotic lesions. In the same mouse models, Acat1−/− also caused dry eye syndrome, as well as severe cutaneous xanthomatosis, which is a rare skin disease not associated with diet-induced atherosclerosis in humans. Acat1−/− also drastically shortened the life spans of the hyperlipidemic mice (26). Whole-body Acat1−/− also affected the proliferation rates of stem cells within the bone marrow and caused a significant increase in leukocyte counts in the blood (27, 28). To minimize the side effects of whole body Acat1−/−, a milder approach was undertaken (29), by performing transplant experiments using bone marrow cells from either WT mice or from Acat1−/− mice as the donor cells and the irradiated hyperlipidemic mice as the recipient mice. The results showed that when compared with bone marrow cells from the WT mice, those from Acat1−/− mice altered lesion composition and significantly increased lesion development. In contrast, very recently, Yamazaki et al. (22) performed similar experiments and received outcomes that are at variance with those reported by Fazio et al. (29). It is known that bone marrow contains myeloid cells and lymphoid cells; both myeloid cells and lymphoid cells affect atherosclerosis at various stages. The variable effects observed by different groups using bone marrow isolated from the Acat1−/− mice in transplant experiments might be caused by the lack of ACAT1 in macrophages as well as by the lack of ACAT1 in lymphoid cells (i.e. T cells and B cells). To focus on studying the roles of ACAT1 in macrophages, Huang et al. (28) developed a myeloid-specific Acat1 KO mouse (Acat1−M/−M); these mice lack Acat1 only in the myeloid cell lineage, which includes macrophages, microglia, and neutrophils. The myeloid-specific Acat1 KO mice do not exhibit dry eye syndrome nor leukocytosis (28). When crossed with the ApoE KO (ApoE−/−) mouse model for atherosclerosis, myeloid Acat1 KO significantly reduced lesion macrophage content and suppressed atherosclerosis progression; mechanistic studies showed that ACAT1 deficiency in macrophages slows down the entry of these macrophages into the subendothelial layer of the arterial vessel (28).
During various stages of atherosclerosis, in addition to CEs, cholesterol crystals, often referred to as cholesterol clefts, also develop and accumulate within lesions. These crystals cause damage to lysosomal membranes, trigger the formation of inflammasomes, and cause severe adverse immune responses (30, 31). Studies in macrophage cell culture showed that cholesterol crystal formations are initiated within the late endo/lysosomes (32, 33). Cell culture studies showed that without cholesterol acceptors present in the cell exterior, ACAT blockage in macrophages caused a significant increase in the cholesterol crystal content that led to detrimental consequences; however, when external cholesterol acceptors were present, ACAT blockage did not induce cholesterol crystal formation, and no detrimental consequence could be observed (34). At the in vivo level, whether ACAT blockage affects cholesterol crystal formation in atherosclerotic lesions had not been closely examined, because of one important technical limitation: in mouse models, cholesterol crystals become a prominent feature within the necrotic core in advanced atherosclerotic lesions, and it takes continuous feedings of a atherosclerosis diet for 15–20 weeks to develop advanced lesions in mouse. Both the total Acat1 KO mouse and the myeloid-specific Acat1 KO (Acat1−M/−M) mouse exhibit normal life spans. However, in the ApoE KO genetic background, presumably because of exaggerated xanthomatosis, the whole-body Acat1 KO mouse had a much-shortened life span and began to die 8–10 weeks after continuous high-cholesterol diet feeding. The much-shortened life span of the Acat1−/−/ApoE−/− mouse prohibited the investigators from pursuing the effect of whole body Acat1 KO in advanced atherosclerotic lesions. Recently, Huang et al. (28) showed that when fed continuously with a high-cholesterol diet, the Acat1−M/−M/ApoE−/− mouse fared much better in terms of survival rate. Here we took advantage of this opportunity and investigated the effects of myeloid-specific Acat1 KO in the ApoE KO mouse model for advanced and reported our findings.
Results
Silencing myeloid Acat1 reduces the development of advanced atherosclerosis in ApoE−/− mouse
To assess the effect of a prolonged modified Paigen diet treatment on the overall health of the ApoE−/− and ApoE−/−/Acat1−M/−M mice, we monitored the mouse weights and survival rates over a 20-week period. As shown in Fig. 1A, mice from all groups (ApoE−/−, ApoE−/−/Acat1−M/−M, and ApoE−/−/Acat1−/−) consistently gained weight after week 4. By week 20 the ApoE−/−/Acat1−M/−M mouse exhibited 16.6% higher weight gains than the ApoE−/− control mice. We suspect that this is because the ApoE−/− mouse exhibits subcutaneous xanthomas, which has been demonstrated by multiple groups. As detailed in our previous work, this skin disease is exacerbated in the ApoE−/−/Acat1−M/−M mouse and may contribute to the weight gain throughout the duration of the diet. Interestingly, we found that myeloid-specific Acat1 knockout was protected from the occurrence of early death associated with the total Acat1 knockout mouse. On average, 76% of ApoE−/−/Acat1−M/−M mice survived the extended diet study, unlike the ApoE−/−/Acat1−/− mice, which only survived to week 9. Thus, the ApoE−/−/Acat1−M/−M mouse provides a unique opportunity to study the effects of knocking out macrophage Acat1 in late stage atherosclerosis. To compare the atherosclerotic burden in 20-week modified Paigen diet–fed ApoE−/− and ApoE−/−/Acat1−M/−M mice, we isolated whole aortic tissues from these mice and performed Enface analysis by staining them with Sudan IV, a red β-naphthol diazo dye that stains neutral lipids and other lipid materials. The quantification of total Sudan IV staining revealed that the ApoE−/−/Acat1−M/−M male mice exhibited 42.7% less area percentage of atherosclerotic plaques than those of the ApoE−/− control, indicating that knocking out myeloid Acat1 slowed the progression of advanced lesions (Fig. 2B). We next performed H&E staining of aortic sections taken from both mouse types and found that the atheroma lesion areas, measured in the ascending and descending aortas, were significantly reduced by 36.7 and 49.3%, respectfully, in the ApoE−/−/Acat1−M/−M mice (Fig. 2, D and E). These results extend our 12-week study reported previously, which demonstrated that ApoE−/−/Acat1−M/−M mice are protected from early to mid-stage atherosclerosis (28).
Figure 1.

Silencing myeloid Acat1 rather than silencing Acat1 globally extends the life span of modified Paigen diet–fed ApoE−/− mice. A, body weights of male ApoE−/−, ApoE−/−/Acat1−M/−M, and ApoE−/−/Acat1−/− mice fed a modified Paigen diet for 20 weeks. B, survival curves of ApoE−/−, ApoE−/−/Acat1−M/−M, and ApoE−/−/Acat1−/− mice.
Figure 2.

Deleting myeloid Acat1 reduces the atherosclerotic burden in ApoE−/− mice fed a modified Paigen diet for an extended period of time. A, enface staining of whole aortas isolated from ApoE−/− and ApoE−/−/Acat1−M/−M mice treated with modified Paigen diet for 20 weeks. B and D, quantitation of lesion area detected in male (B) and female (C) aortas. D and E, H&E staining of cross-sections of ascending (D) and descending (E) aortas from male ApoE−/− and ApoE−/−/Acat1−M/−M mice. The data are reported as means ± S.E. *, p ≤ 0.05; **, p ≤ 0.01.
Previously, we showed that the mechanism by which myeloid Acat1 deletion prevents early lesions likely involves the reduction of macrophage content within the ascending atherosclerotic plaques (28). Here, by performing fluorescent confocal microscopic analysis using CD68 to monitor macrophage presence, we again observed a marked reduction of macrophage content, in the intimal layer of both the ascending plaques and the descending plaques from ApoE−/−/Acat1−M/−M mice, relative to those from the ApoE−/− control mice (Fig. 3). In addition to macrophages, the atherosclerotic lesions contain other immune cell types, especially the T lymphocytes. During atherosclerosis development and progression stages, T cells exist in several subtypes that play both pro-inflammatory and anti-inflammatory roles (35, 36). CD45 is a pan-leukocyte marker that recognizes all T cells and other white blood cells but does not recognize myeloid cells or smooth muscle cells. Here we used CD45 to examine the leukocyte contents within the advanced lesions by performing confocal microscopy. The results (Fig. 4) showed that in advanced lesions, silencing myeloid Acat1 in the ApoE−/− mice tends to reduce plaque leukocyte contents, suggesting that Acat1−M/−M may indirectly impact leukocyte abundance in atherosclerotic lesion. The significance of this finding is not clear at present and requires further investigation.
Figure 3.
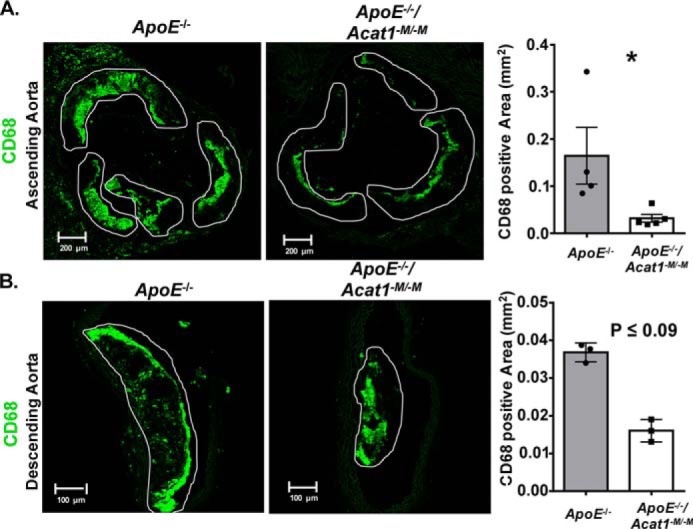
Deleting myeloid Acat1 in the ApoE−/− mice reduces macrophage presence in advanced aortic lesions. A, confocal microscopy analysis of macrophage content (CD68 stain in green) in aortic sections of ascending lesions. B, CD68-positive area found in the aortic sections of descending lesions of ApoE−/− and ApoE−/−/Acat1−M/−M mice. Areas outlined in white represent atherosclerotic plaques. The data are reported as means ± S.E. *, p ≤ 0.05.
Figure 4.
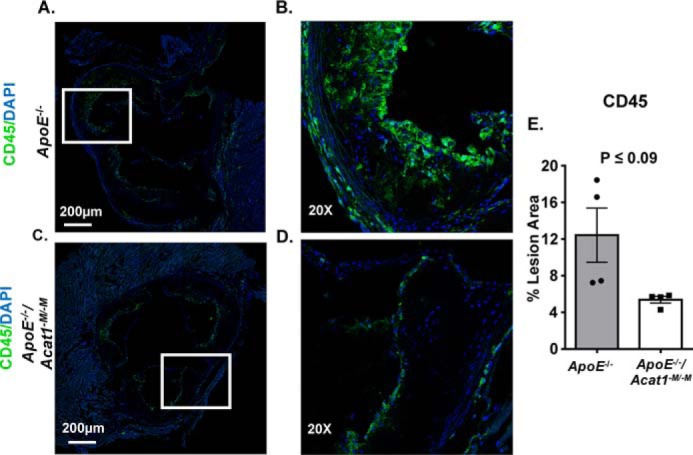
Deleting myeloid Acat1 in the ApoE−/− mice tends to reduce leukocyte contents in advanced lesions. A and C, confocal microscopy analysis of leukocyte contents based on the pan-leukocyte marker CD45 in aortic sections of ascending lesions from ApoE−/− (A) and ApoE−/−/Acat1−M/−M mice (C). The original magnification of confocal microscopy in A and C is 10×. B and D, zoomed-in views of CD45 (green) and 4′,6′-diamino-2-phenylindole (DAPI, blue) signals. E, percentage area positive for CD45 was calculated per plaque area. The data are reported as means ± S.E.
Inhibiting ACAT1 in macrophages attenuated the proinflammatory responses to acetyl LDL-mediated cholesterol loading
Macrophages exhibit plastic phenotypes. Their abilities to participate in various immune responses is controlled by the environment. In atherosclerosis, chronic inflammation that occurs at the surface areas of the aortic vessel attract monocytes and macrophages to migrate and adhere to the activated endothelium and become resident cells within the intimal region of the aortic tissue. Under hypercholesterolemia, cholesterol loading causes macrophages to become more inflammatory, in part by up-regulating the expressions of several proinflammatory response genes (reviewed in Ref. 37). In our current study, the results described in Fig. 3 showed that in the ApoE−/− mouse model for advanced lesion, myeloid Acat1 deletion caused a significant decrease in macrophage content in the lesion. To provide a plausible cellular basis for this finding, we hypothesize that ACAT1 blockage may cause macrophages to produce less proinflammatory responses upon cholesterol loading. To test this possibility, we isolated peritoneal macrophages from ApoE−/− mice and preincubated the cells without or with a potent and specific small molecule ACAT1 inhibitor K604 at 0.5 μm (38) for 18 h. For cholesterol loading, we treated the cells with high concentration (100 μg/ml) of acetylated low-density lipoprotein (AcLDL), a denatured form of LDL that produces cholesterol loading in macrophages (39, 40), for 1 day. Afterward, we harvested the cells to monitor their inflammatory responses by performing Western blotting analysis (Fig. 5A) and mRNA analysis (Fig. 5D). For Western blotting analysis, we chose to monitor two proteins: iNOS and COX2. These are two of the nonsecretory proteins in macrophages that are known to respond positively to pro-inflammatory signals, including lipopolysaccharides (41). The results showed that in peritoneal ApoE−/− macrophages, loading AcLDL significantly induced iNOS expression; under this condition, inhibiting ACAT1 with K604 significantly attenuated the induced expression of iNOS (Fig. 5C). Additional results showed that loading AcLDL also significantly induced COX2 expression; inhibiting ACAT1 with K604 tended to attenuate the induced expression of COX2, but the effect of K604 did not reach statistical significance (Fig. 5B). For mRNA analyses, we monitored nine pro-inflammatory response genes, including the genes that encode iNos and Cox2. The results showed (Fig. 5D) that loading AcLDL significantly induced the expressions of all nine genes; inhibiting ACAT1 with K604 significantly attenuated the induced gene expressions of iNos, Ccl5, Cox2, and Tnfα, but did not significantly attenuate the induced gene expressions of Mcp1, Ccl3, Ccl7, and Cxcl10. The peritoneal macrophages employed in Fig. 5 were isolated from mice lacking ApoE. To test the generality of findings described in Fig. 5, we repeated the experiment by using the mouse RAW 264.7 cell line. This cell line has been employed as a model cell for macrophages by many investigators. The results showed that loading AcLDL in RAW macrophages significantly up-regulated the expressions of both COX2 and iNOS proteins, and inhibiting ACAT1 with K604 significantly attenuated the induced expressions of both proteins (Fig. 6, A and C). Additional results (Fig. 6E) showed that loading AcLDL in RAW macrophages significantly induced the expressions of seven of the nine genes tested; inhibiting ACAT1 with K604 significantly attenuated the induced gene expressions of iNos, Cox2, and Tnfα but did not attenuate the induced gene expressions of Mcp1, Ccl3, Ccl5, and Ccl7. The result presented in Fig. 6 largely corroborated with the result presented in Fig. 5 and supports the hypothesis that under hypercholesterolemia, ACAT1 blockage in macrophages causes these cells to exhibit a less inflammatory phenotype; the less inflammatory phenotype causes their sparse presence within the plaque.
Figure 5.

Inhibiting ACAT1 ameliorates AcLDL-induced iNOS expression in peritoneal macrophages isolated from ApoE−/− mice. Peritoneal macrophages isolated from ApoE−/− mice were treated without or with the small molecule ACAT1 inhibitor K604 at 0.5 μm for 18 h and then treated without or with 100 μg/ml of AcLDL for 24 h. A, Western blotting analysis of COX2 and iNOS protein expressions. B and C, densitometry analysis of COX2 (B) and iNOS (C) protein expressions. D, QPCR analysis of various pro-inflammatory response genes. The data are reported as means ± S.E. *, p ≤ 0.05.
Figure 6.

Blocking ACAT1 ameliorates AcLDL induced iNOS and COX2 expressions in RAW macrophages. RAW macrophages were treated without or with K604 at 0.5 μm for 18 h and then treated without or with 100 μg/ml of AcLDL for 24 h. A and C, Western blotting analysis of COX2 and iNOS protein expression. B and D, densitometry analysis of COX2 (B) and iNOS (D) protein expression. E, mRNA levels of pro-inflammatory response genes. The data are reported as means ± S.E. *, p ≤ 0.05; **, p ≤ 0.01.
We next monitored cholesterol crystal contents in the lesions after the H&E staining of aortic cross-sections isolated from the ApoE−/− and ApoE−/−/Acat1−M/−M mice. As demonstrated in Fig. 7, when compared with the control ApoE−/− mice, the ApoE−/−/Acat1−M/−M mice possess significantly fewer cholesterol crystals in the necrotic core (depicted by space devoid of H&E stain). Quantitation using National Institutes of Health ImageJ software revealed that reductions in cholesterol crystals in the ascending and descending aortas were by 88 and by 41%, respectfully. Results of a TUNEL analysis revealed that there was no difference in the number of apoptotic cells present within the plaques of ApoE−/− and ApoE−/−/Acat1−M/−M mice (Fig. 8).
Figure 7.

Silencing myeloid Acat1 in ApoE−/− mice reduces cholesterol cleft development in advanced atheromas. H&E staining of cross-sections isolated from the ascending (A) and descending (B) aortas of 20-week fed ApoE−/− and ApoE−/−/Acat1−M/−M mice. The area devoid of H&E stain was quantified as positive cholesterol cleft area (examples depicted by the arrows). The data are reported as means ± S.E. *, p ≤ 0.05.
Figure 8.
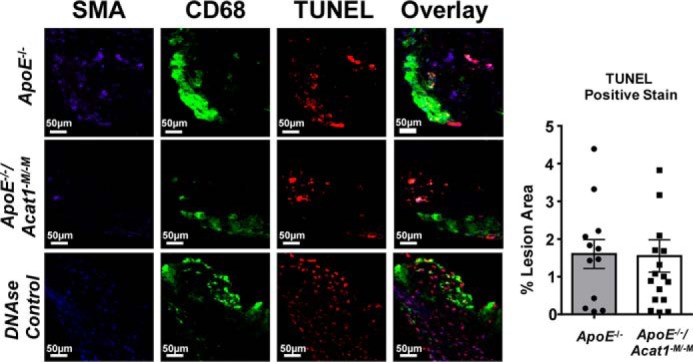
TUNEL analysis of apoptosis in aortic lesions. Cell death was detected by TUNEL analysis in ApoE−/− (top panel) and ApoE−/−/Acat1−M/−M mice (middle panel) fed a modified Paigen diet for 20 weeks. Aortic lesions were stained with markers for TUNEL (red) CD68 (green) and SMA (blue). DNase-treated aortic sections (bottom panel) were used as a positive control for TUNEL staining. The number of TUNEL-positive cells were quantified per lesion area. The data are reported as means ± S.E. *, p ≤ 0.05.
Both ACAT1 and ACAT2 are expressed in advanced atherosclerotic lesions
Given that ACAT1 is a major contributor to foam cell formation in macrophages (16), we next wanted to compare the percentage of foam cells within the advanced lesions of 20-week fed ApoE−/− and ApoE−/−/Acat1−M/−M mice. We stained aortic sections with Oil Red O to detect neutral lipids in foam cells (Fig. 9). We found that surprisingly, although there were significant reductions in the overall plaque burden as demonstrated in Fig. 2, the level of neutral lipid containing areas within the ascending aorta of the ApoE−/−/Acat1−M/−M mouse was actually comparable with that of the ApoE−/− mouse. We speculated that in advanced lesions, in addition to ACAT1 in macrophages, ACAT2 and/or nonmacrophage ACAT1 may play active roles in CE formation within the atherosclerosis plaques. To address this question, we used immunofluorescence microscopy to visualize ACAT1 and ACAT2 protein expression within the ascending lesions of ApoE−/− and ApoE−/−/Acat1−M/−M mice (Fig. 10). Our results showed that ACAT2 was present in aortic lesions of both mouse lines. These findings support and extend the early results by Sakashita et al. (17). Our results also show that ACAT1-positive cells were observed within the medial layers of plaques from both mouse groups. Together, these results suggest that in the advanced lesions, in addition to ACAT1 in macrophages, ACAT2 in macrophages and smooth muscle cells as well as nonmacrophage ACAT1 play active roles in neutral lipid content build up, by contributing to cholesteryl ester formation through their cholesterol acyl-transferase activity.
Figure 9.
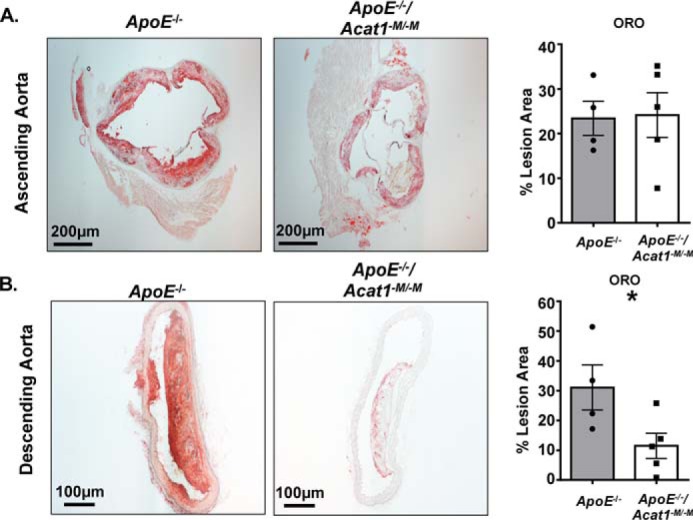
Neutral lipid content is diminished in the descending aortic plaques but remains unchanged in plaques located in the aortic arch of ApoE−/−/Acat1−M/−M mice. Oil Red O staining of cross-sections of advanced lesions found in the ascending (A) and descending (B) aortas of ApoE−/− and ApoE−/−/Acat1−M/−M mice. The data are reported as means ± S.E. *, p ≤ 0.05.
Figure 10.
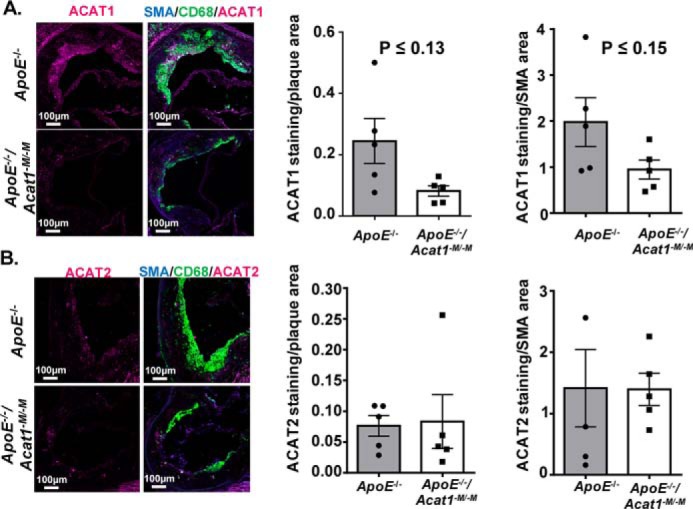
Advanced atherosclerotic lesions from ApoE−/−and ApoE−/−/Acat1−M/−M mice express ACAT2 protein. A, ACAT1 expression (depicted in fuchsia) detected by confocal microscopy analysis of ascending aorta cross-sections from ApoE−/− and ApoE−/−/Acat1−M/−M mice. B, immunofluorescence staining of ACAT2 (fuchsia), CD68 (green), and SMA (blue) in late stage lesions from ApoE−/− and ApoE−/−/Acat1−M/−M mice. The data are reported as means ± S.E. *, p ≤ 0.05.
25-Hydroxycholesterol induces ACAT1 and ACAT2 protein expression in vascular smooth muscle cells and induces ACAT2 protein expression in macrophages
In advanced lesions, oxysterols are known to build up at high concentrations. To examine whether ACAT2 can play an active role in CE formation when vascular smooth muscle cells and macrophages are activated by oxysterols, we took an in vitro approach to monitor its expression in response to 25-hydroxycholesterol (25HC). We chose 25HC because its ample presence in the atherosclerotic plaques had been demonstrated (4). In addition, Wang et al. (19) demonstrated that in cells of hepatic and intestinal origin, saturated fatty acids or sterols/oxysterols including 25HC added to culture media protects the ACAT2 protein, but not the ACAT1 protein, against proteasome mediated degradation.
We treated RAW macrophages and primary mouse vascular smooth muscle cells (VSMCs) with various concentrations of 25HC (0.5, 1, 5 μm) for 48 h and then monitored ACAT1 and ACAT2 expression by performing Western blotting and QPCR analyses. The results of Western blotting analysis showed that 25HC increased the expression of both ACAT1 and ACAT2 proteins in VSMCs, yet in macrophages only ACAT2 proteins responded to 25HC (Figs. 11 and 12). QPCR analyses showed that in VSMCs, mRNA levels of ACAT2 were not impacted by 25HC, whereas higher concentrations (5 μm) did induce Acat1 mRNA levels. In macrophages, mRNA levels of nether the Acat1 nor the Acat2 were impacted by 25HC. Together, these results show that in VSMCs, 25HC induces both ACAT1 and ACAT2 protein. Regarding the mechanism of actions of 25HC on ACATs, for ACAT1, the mechanism may involve transcription activation, perhaps in a smooth muscle cell–specific manner, because in macrophages, 25HC does not increase ACAT1 protein expression in macrophages. For ACAT2, the mechanism may involve stabilizing against its degradation, in both smooth muscle cells and macrophages, similar to the one recently demonstrated in hepatocytes and intestinal enterocytes (19).
Figure 11.

25-Hydroxycholesterol stimulates both ACAT1 and ACAT2 protein expression in vascular smooth muscle cells. A and D, Western blotting analysis of primary VSMC ACAT2 (A) and ACAT1 (D) expression in response to (0.5, 1, and 5 μm) 25HC treatment (48 h). B and E, quantitation of protein expression of ACAT2 (B) and ACAT1 (E). C and F, expression levels of ACAT2 (C) and ACAT1 (F) mRNA in 25HC-treated VSMCs detected by QPCR. The data are reported as means ± S.E. *, p ≤ 0.05.
Figure 12.

A and D, 25HC-treated RAW 264.7 macrophages exhibit elevated ACAT2 protein expression Western blotting analysis of ACAT2 (A) and ACAT1 (D) expression in RAW macrophages treated with 25HC (0.5, 1, and 5 μm) for 48 h. B and E, quantitation of protein expression of ACAT2 (B) and ACAT1 (E). C and E, QPCR analysis of ACAT2 (C) and ACAT1 (F) mRNA levels in 25HC-treated RAW macrophages cells. The data are reported as means ± S.E. *, p ≤ 0.05.
To test the effect of 25HC on overall CE formation in RAW macrophages and in VSMCs, we measured CE formation by using a radioactive assay in intact cells, in which we added radiolabeled oleate (complexed to BSA) to cells in culture for a given amount of time, and then measured the level of radiolabeled CEs formed after performing TLC analysis of the lipid extractions. The results showed that at 5 μm 25HC significantly induced CE formation in both RAW macrophages and VSMCs (Fig. 13); upon treatment with K604, an ACAT1-specific small molecule inhibitor (38), the CE formation was reduced by 80%, providing evidence that in cells treated with 25HC, ACAT1 remains as the major CE-forming enzyme in both macrophages and VSMCs. To determine the contribution of ACAT2 to the overall CE formation in 25HC-treated RAW macrophages and in VSMCs, we added pyripyroprene A (PPPA), an ACAT2-specific small molecule inhibitor at 1 and 5 μm to determine whether certain percentage of total ACAT activity can be inhibited by PPPA in these cells (43). The results indicate that although not statistically significant, there is a trend toward a reduction in CE formation, in response to PPPA in both RAW macrophages and in VSMCs (by 35 and 27% reduction, respectfully). These results indicate that upon stimulation by 25HC, ACAT1 is a major isoform in macrophages and VSMCs, and ACAT2 may play a secondary role, contributing to ∼30% of overall CE formation activity. These results provide a plausible explanation for our observation made in vivo (described in Fig. 9); namely, in the absence of ACAT1 in macrophages, plenty of foam cells are present in advanced lesions from the ApoE−/−/Acat1−M/−M mice. CEs present in these foam cells can be attributed to the nonmacrophage ACAT1 (VSMC-specific), as well as ACAT2 present in both macrophage and VSMCs within the plaques.
Figure 13.

25HC enhances cholesterol ester formation in RAW macrophages and VSMCs. Shown is cholesterol ester formation (measured by a radioactive oleate pulse assay) in RAW macrophages (A) and VSMCs (B) in response to 25HC with or without ACAT1 inhibitor K604 or ACAT2 inhibitor PPPA. The data are reported as means ± S.E. *, p ≤ 0.05.
Discussion
Whether blocking ACAT1 in macrophages is pro-atherosclerotic or anti-atherosclerotic has been under debate for many years. The results presented in the current work show that in a mouse model for advanced atherosclerotic lesions, lack of myeloid ACAT1 caused less macrophage presence, significantly reduced lesion size and the cholesterol crystals, without a detectable increase in apoptotic index within the lesion. These results confirm and extend our previous results reporting the effects of myeloid ACAT1 deficiency in the same mouse model for early atherosclerotic lesions (28) and show that in advanced lesions, myeloid selective silencing of ACAT1 is atheroprotective.
The results of the cell culture study presented here showed that inhibiting ACAT1 causes macrophages to produce less inflammatory responses upon cholesterol loading by denatured LDL (AcLDL). These results provide a plausible explanation for the sparse presence of lesion macrophages in the ApoE−/−/Acat1−M/−M mice: in atherosclerosis, ACAT1 blockage in macrophages causes these cells to be less responsive to cholesterol loading and exhibit a less inflammatory phenotype; this less inflammatory characteristic slows down their presence within the plaque. At present, it is not clear why ACAT1 blockage causes macrophages to become less inflammatory. Interestingly, in a previous study, we had found that (41) inhibiting ACAT1 causes macrophages grown in culture to produce less inflammatory responses upon treatment with lipopolysaccharide, the key bacterial endotoxin that drives acute inflammation in various immune cells. Lipopolysaccharide enters the cell interior and acts mainly via the Toll-like receptor (TLRs) signaling. It is possible that in macrophages, cholesterol loading activates the TLRs to mediate the proinflammatory responses (44–46); ACAT1 blockage may directly or indirectly attenuate the signaling activities of the TLRs to suppress the proinflammatory responses. This and other possibilities require further investigation.
In our current study, we also show that, in the advanced lesions isolated from the ApoE−/−/Acat1−M/−M mice, a significant amount of residual foamy cells, marked by the neutral lipid staining dye Oil Red O, are still present (Fig. 6). These findings correlate well with the results demonstrating the significant presence of nonmacrophage ACAT1, in the medial layer of the plaques as well as the presence of ACAT2 present in both the intimal and the medial layer of the plaques (Fig. 7). Earlier, Rong et al. (47) performed studies in cell culture and demonstrated that ACAT1 in smooth muscle cells contributes to foam cell formation. Our current study provides the in vivo proof that indeed, there is ample ACAT1 present in foamy smooth muscle cells. Regarding the presence of ACAT2 in atherosclerotic lesions: Sakashita et al. (17) demonstrated that in the advanced human and mouse atherosclerotic lesions, ACAT2 is present; most of the ACAT2 signal was localized to the macrophage-rich region. Here we confirmed and extended the findings by Sakashita et al. by showing that in a mouse model for advanced lesion, a significant amount of ACAT2 signals are present in regions of the lesion in which both macrophages and smooth muscle cells reside. To evaluate the relative contribution of ACAT1 versus ACAT2 in cholesterol esterification in macrophages and in smooth muscle cells, we performed experiments in cell culture, using the ACAT1-specific inhibitor K604 and the ACAT2-specific inhibitor PPPA as tools. The results show that in both macrophages and smooth muscle cells, either under basal conditions or after cells were treated with 25HC, ACAT1 remains as the major isoenzyme in cholesterol esterification in these cells. The physical presence of ACAT2 in these cells could be demonstrated by Western blotting, and radioactive oleate pulse assays showed that the contribution of ACAT2 is perhaps up to 20–30% of the overall cholesterol esterification process. Still, ACAT2 may play even more significant roles in foam cell formation in vivo. This question can only be addressed by further experimentation. Our current work shows that, in advanced lesions, both ACAT1 and ACAT2 are present in smooth muscle cells and contribute to foam cell formation. In the future, it would be interesting to test whether blocking ACATs in smooth muscle cells only would benefit atherosclerosis development and progression.
The system employed in our current study has several limitations. First, the myeloid cell lineage includes neutrophils, dendritic cells, and eosinophils, in addition to monocytes/macrophages. We cannot rule out the possibility that the beneficial phenotype observed in the myeloid-specific Acat1 KO was due to the lack of ACAT1 in monocytes/macrophages only. The other limitation is that only one mouse model, the ApoE−/− KO mouse, for atherosclerosis was used. The validity of this interpretation needs to be further validated by using different animal models for advanced atherosclerosis.
Our work supports the work of other investigators, who took the small molecule ACAT inhibitors approach and showed that either by using an ACAT1-specific inhibitor K604 (38, 48) or by using the isotype-nonspecific ACAT inhibitors including CI 1011 (49, 50), F1394 (47, 51), or pactimibe (52), all of which demonstrated reductions in atherosclerotic lesions in preclinical models, without overt systemic or plaque toxicity. Based on these previous results and our current work, we recommend the use of ACAT inhibitors to target advanced lesions. For efficacy, an inhibitor that inhibits both ACAT1 and ACAT2 would be more efficient. For safety reasons, an inhibitor that blocks ACAT1 with higher potency than inhibiting ACAT2 would be more appropriate.
When macrophages were treated with isotype nonspecific ACAT inhibitors under the condition in which cellular lipid efflux was minimal, the inhibitors caused excess free cholesterol built up inside the cells and produced detrimental consequences (33, 53). Because the actions of the ACAT inhibitors depend on the lipid efflux system to produce efficient cholesterol removal from the cell interior, we recommend that in the future, patients to be selected for ACAT inhibitor anti-atherosclerosis clinical trials should first be screened to carry no monogenic mutations in genes involved in the cellular lipid efflux process.
Experimental procedures
Mice
Homozygous ApoE−/− knockout mice (C57BL/6) were purchased from The Jackson Laboratory. The ApoE−/−/Acat1−/− and ApoE−/−/Acat1−M/−M mice (generated by crossing the LyzMCre, ApoE−/−, and Acat1Flox/Flox mice) were created as previously described (28). For atherosclerosis studies, the mice at postnatal day 21 were fed a standard chow diet for 8 weeks and then treated with an atherogenic diet (designated here as modified Paigen diet; 1.25% cholesterol, 35 kCal from fat) purchased from Research Diet (catalog no. 99020201) for an additional 20 weeks. The body weights and rates of survival were recorded once every week to monitor the overall health of the animals. Mice that exhibited extreme weight loss, extensively large xanthomas, skin ulcers, or loss of mobility were euthanized, and the dates of death were noted. All mouse work was approved by the Dartmouth College Institutional Animal Care and Use Committee and followed the guidelines of the National Institutes of Health.
Histological characterization of atherosclerotic plaques
Most of the procedures employed here were based on procedures described by Huang et al. (28) with minor modifications. To characterize atherosclerosis in ApoE−/− and ApoE−/−/Acat1−M/−M mice, we euthanized the mice and isolated whole aortas using a dissecting microscope. The aortic tissues were fixed in 10% formalin and preserved in paraffin or OCT blocks for sectioning. For Enface staining, we incubated whole aortas with Sudan IV (a neutral lipid stain) for 10 min and then washed them with 70% ethanol and PBS. The Sudan IV–positive area was determined using ImageJ software, by first converting the images to 8 bit, then setting the threshold for positive stain, and finally measuring the percentage area using the analyze/measure function. For histological analysis of plaque lesion size, we stained paraffin sections of ascending and descending plaques with H&E staining. To quantify in ImageJ, the selection tool was used to encircle the plaques within the aortic sections. The total lesion area was calculated by summing the areas calculated for each identified plaque. Cholesterol crystal area was determined by measuring the areas devoid of H&E staining within the necrotic cores of the lesions (51). Cross-sectional slices of aortas were also stained with Oil Red O (ORO) to detect the neutral lipid content within the plaques using techniques described in our previous work (28). In short, sections were washed with PBS, incubated with 0.3% ORO (made in isopropanol) for 15 min, and washed 20 times with H2O before being mounted with glycerol and a coverslip. H&E- and ORO-stained sections were visualized with an Olympus IX73 inverted fluorescence microscope.
Immunofluorescence microscopy
The immunofluorescence microscopic methods described in Ref. 28 were used to quantify the macrophage contents, leukocyte contents, apoptotic cells, ACAT1, and ACAT2 protein expressions within the aortic plaques isolated from ApoE−/− and ApoE−/−/Acat1−M/−M mice (28). Frozen OCT sections were thawed, blocked with 3% BSA, washed with TBST, and then stained with one of the primary antibodies: anti-CD68, anti–smooth muscle actin (SMA), anti-ACAT1 DM102, anti-ACAT2 DM56 (13), rabbit polyclonal anti-ACAT2 antibody (19), or rat anti-CD45 (Thermofisher) overnight at 4 °C. The sections were then washed with TBST and incubated with Alexa Fluor–conjugated secondaries. For TUNEL staining, we followed the protocol provided by the manufacturer (Roche). The OCT aortic sections were washed with TBST, fixed in 4% paraformaldehyde, permeabilized with 2% Triton X-100 and 1% sodium citrate, and then incubated with the TUNEL substrate mixture (5 μl of terminal deoxynucleotidyltransferase enzyme + 45 μl of fluorescein label solution) for 1 h at 37 °C. The sections were then stained with CD68 and SMA antibodies overnight. The sections were also treated with 100 units DNase for 15 min to serve as a positive control for TUNEL staining. After incubating with secondary antibodies, the sections were washed and mounted with anti-fade medium. We utilized Zeiss LS510 and LSM 880 confocal microscopes to visualize the images and take pictures. To quantify the fluorescence signal of positively stained cells, we used ImageJ software. We changed the image to 8-bit, set the threshold, drew a circle around the lesion area with the selection tool, and then measured the positive signal within the encircled region. The fluorescence within the regions was then summed to quantify the total area positive for each stain.
Peritoneal macrophages and RAW 264.7 macrophages
To isolate peritoneal macrophages we injected 8-week-old ApoE−/− mice with thioglycollate using a sterile 27-gauge needle. The 4% thioglycollate (Sigma T9032) media were prepared in H2O and autoclaved and then aged for several weeks in the dark at room temperature. until it turned brown. Four days later, the mice were sacrificed, cleaned with 70% ethanol, and injected with 4–5 ml of Hanks buffer into the intraperitoneal space. The mice were put on an orbital shaker for 2 min to wash the peritoneal space. A small opening near the abdomen was cut to expose the peritoneal cavity; a 19-gauge needle was used to retrieve the fluid in the cavity. The cells were spun down at 1200 rpm and then plated in a 6-well dish in media containing DMEM + 10% fetal calf serum + penicillin/streptomycin. Usually cells isolated from one mouse were used for a 6-well plate. 2.5 h after cells plating, the cells were washed with PBS to eliminate nonadherent cells and replenished with fresh growth medium. Experimental treatments were performed 24 h post seeding. The cells were treated without or with 0.5 μm K604, with 0.1% DMSO for 18 h, and then cells were fed without or with AcLDL at a concentration of 100 μg/ml for an additional 24 h. AcLDL was prepared as described in Ref. 54. RAW 264.7 macrophages were cultured in DMEM containing 10% heat-inactivated bovine calf serum and penicillin/streptomycin. The cells were plated in a 12-well dish at a density of 2.5 × 105 cells/well.
Primary vascular smooth muscle cells
To isolate primary vascular smooth muscle cells, we used a procedure described by Kuang et al. (55). In short, the aortas were collected from WT mice; forceps were used to remove the adventitia and endothelial layer from the aortic tissue. The remaining tissue was cut into small pieces and digested with collagenase and elastase for 1 h. The cells were then collected and plated in a dish with DMEM/Ham's F-12 medium + 10% bovine calf serum for several days. VSMCs that migrated from the tissue were trypsinized, replated, and allowed to propagate. The primary VSMC culture was maintained in DMEM/Ham's F-12 medium + 10% calf serum + penicillin/streptomycin and split into a 6-well dish at 4 × 10 5 cells/well. To study how RAW macrophages and WT VSMCs respond to 25HC, when cells reached ∼80% confluent, we treated these cells with various concentrations of 25HC (0, 0.5, 1.0, 5.0 μm) for 48 h and then harvested the cells for Western blotting or RT-PCR analysis. 25HC was solubilized in ethanol to prepare a working stock of 12.4 mm.
Western blotting analysis
For Western blotting analysis of ACAT1 and ACAT2 protein expression, the procedure described in Ref. 13 was used. RAW macrophages and WT VSMCs cells were harvested in 10% SDS and lysed with a 21-gauge syringe. The protein concentration of the supernatant was determined by the Lowry protein assay. The lysates were run on a 10% gel and transferred to a nitrocellulose membrane for 4 h. After blocking in 5% milk in TBST for 1 h, the membranes were incubated with a polyclonal anti-ACAT1 antibody (DM102), an antibody against ACAT2 (DM56) (13), or the anti-ACAT2 antibody prepared by Dr. Bo-Liang Li and Dr. Bao-Liang Song (19). β-Tubulin (from GenScript) was used as a loading control. To detect COX2 and iNOS protein expression, antibodies against COX 2 used were from Santa Cruz Biotechnology; antibodies against iNOS used were from Abcam. Lysates were prepared in radioimmune precipitation assay buffer (plus protease inhibitor 1:100) and briefly sonicated for 5 s on ice. After centrifugation the lysates were run on a 10% gel and transferred to nitrocellulose membrane in Towbin buffer. The intensities were normalized to β-tubulin expression.
RT-PCR analysis
To isolate RNA from cells, we added TRIzol reagent directly to the cells and followed the manufacturer's instructions (Thermo Fisher Scientific). The RNA was dissolved in water and then treated with DNaseI (Ambion). 1 μg of RNA was used to make cDNA using the BioBad iscript cDNA synthesis kit. QPCR was performed with Bio-Rad Syber green mix using an Applied Biosystems Step One RT-PCR system. The following primers were used to detect the genes of interest: ACAT1 PF, 5′-AGCCCAGAAAAATTTCATGGACACATACAG-3′; ACAT1 PR, 5′-CCCTTGTTCTGGAGGTGCTCTCAGATCTTT-3′; ACAT2 PF, 5′-TTTGCTCTATGCCTGCTTCA-3′; ACAT2 PR, 5′-CCATGAAGAGAAAGGTCCACA-3′; β-actin PF, 5′-CAACGAGCGGTTCCGAT-3′; and β-actin PR, 5′-GCCACAGGATTCCATACCCA-3′. The ΔΔCt values were calculated and normalized to the control group (cells treated with solvent only). Primers used to monitor genes that respond to proinflammatory signals were as described in Ref. 41.
[3H]Oleate pulse to measure cholesterol ester biosynthesis in intact cells
The procedures described in Ref. 42 were employed to measure ACAT activity in intact cells. RAW Macrophages and WT VSMCs were plated into a 12-well dish and treated with or without K604 (ACAT1-specific inhibitor (38) or PPPA, an ACAT2-specific inhibitor (43). To initiate the pulse, the cells were incubated with radiolabeled C18:1 conjugated to BSA at 37 °C. 20 min after the substrate was added, we washed the cells three times with PBS. The cells were then lysed in NaOH, and 10 μl of the lysate was used for protein determination. Total lipids were extracted using the Folch method. The lipids were then dried down with N2, resuspended in ethyl acetate, and loaded onto a TLC plate. The [3H]cholesterol ester band (identified by iodine staining) was scraped off the plate and read in a scintillation counter.
Statistical analysis
For mouse studies five to eight mice per mouse line were used to compare atherosclerosis progression. Unless stated, only male mice were employed. For in vitro studies, all experiments were performed two or three times, each with replicates. The results were quantified using Microsoft Excel and Prism GraphPad. The results are expressed as means ± S.E. Two tailed t tests were performed to evaluate the significance among the experimental groups.
Author contributions
E. M. M., L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. conceptualization; E. M. M., H. L., J. B., P. S., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. data curation; E. M. M., H. L., J. B., P. S., L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. formal analysis; E. M. M., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. supervision; E. M. M., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. validation; E. M. M., H. L., J. B., P. S., L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. investigation; E. M. M., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. visualization; E. M. M., H. L., J. B., L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. methodology; E. M. M., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. writing-original draft; E. M. M., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. project administration; E. M. M., H. L., J. B., L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. writing-review and editing; L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. resources; L.-H. H., B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. funding acquisition; B.-L. S., B.-L. L., C. C. Y. C., and T.-Y. C. software.
Acknowledgments
We thank Dr. Brent Berwin at Geisel School of Medicine at Dartmouth for advice and Irene Cofie, Zachary Panton, and Matthew Tso for contributions to this work. We acknowledge the shared resources facilities of the Pre-clinical Imaging and Microscopy Resource at the Norris Cotton Cancer Center at Dartmouth with NCI Cancer Center Support Grant 5P30 CA023108–37.
This work was supported by grants R01AG037609 and R01AG063544 from the NIH National Institute on Aging (to T.-Y. C. and C. C. Y. C.), a Ruth L. Kirschstein NRSA Postdoctoral Fellowship 1F32HL124953 from the National Heart, Lung, and Blood Institute (to E. M. M.), and a supplement (3R01AG037609–09S1) from the National Institute on Aging (to E. M. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CE
- cholesterol ester
- ACAT
- acyl-CoA:cholesterol acyltransferase
- SOAT
- sterol O-acyltransferase
- LDL
- low-density lipoprotein
- AcLDL
- acetylated LDL
- KO
- knockout
- H&E
- hematoxylin and eosin
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- 25HC
- 25-hydroxycholesterol
- VSMC
- vascular smooth muscle cell
- QPCR
- quantitative PCR
- PPPA
- pyripyroprene A
- TLR
- Toll-like receptor
- ORO
- Oil Red O
- SMA
- smooth muscle actin
- DMEM
- Dulbecco's modified Eagle's medium
- iNOS
- inducible nitric-oxide synthase.
References
- 1. Lusis A. J. (2000) Atherosclerosis. Nature 407, 233–241 10.1038/35025203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Libby P. (2008) The molecular mechanisms of the thrombotic complications of atherosclerosis. J. Intern. Med. 263, 517–527 10.1111/j.1365-2796.2008.01965.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Choudhury R. P., and Fisher E. A. (2009) Molecular imaging in atherosclerosis, thrombosis, and vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 29, 983–991 10.1161/ATVBAHA.108.165498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown A. J., and Jessup W. (1999) Oxysterols and atherosclerosis. Atherosclerosis 142, 1–28 10.1016/S0021-9150(98)00196-8 [DOI] [PubMed] [Google Scholar]
- 5. Yamanaka K., Urano Y., Takabe W., Saito Y., and Noguchi N. (2014) Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis. 5, e990 10.1038/cddis.2013.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown M. S., and Goldstein J. L. (1983) Lipoprotein metabolism in the macrophage: Implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 52, 223–261 10.1146/annurev.bi.52.070183.001255 [DOI] [PubMed] [Google Scholar]
- 7. Chang C. C., Huh H. Y., Cadigan K. M., and Chang T. Y. (1993) Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 268, 20747–20755 [PubMed] [Google Scholar]
- 8. Cases S., Novak S., Zheng Y. W., Myers H. M., Lear S. R., Sande E., Welch C. B., Lusis A. J., Spencer T. A., Krause B. R., Erickson S. K., and Farese R. V. Jr. (1998) ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase: its cloning, expression, and characterization. J. Biol. Chem. 273, 26755–26764 10.1074/jbc.273.41.26755 [DOI] [PubMed] [Google Scholar]
- 9. Anderson R. A., Joyce C., Davis M., Reagan J. W., Clark M., Shelness G. S., and Rudel L. L. (1998) Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 273, 26747–26754 10.1074/jbc.273.41.26747 [DOI] [PubMed] [Google Scholar]
- 10. Oelkers P., Behari A., Cromley D., Billheimer J. T., and Sturley S. L. (1998) Characterization of two human genes encoding acyl coenzyme A:cholesterol acyltransferase-related enzymes. J. Biol. Chem. 273, 26765–26771 10.1074/jbc.273.41.26765 [DOI] [PubMed] [Google Scholar]
- 11. Liu J., Chang C. C., Westover E. J., Covey D. F., and Chang T. Y. (2005) Investigating the allosterism of acyl coenzyme a: cholesterol acyltransferase (ACAT) by using various sterols: in vitro and intact cell studies. Biochem. J. 391, 389–397 10.1042/BJ20050428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu M., Hu X. H., Li Q., Xiong Y., Hu G. J., Xu J. J., Zhao X. N., Wei X. X., Chang C. C., Liu Y. K., Nan F. J., Li J., Chang T. Y., Song B. L., and Li B. L. (2013) A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol. Cell Biol. 5, 404–415 10.1093/jmcb/mjt039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang C. C., Sakashita N., Ornvold K., Lee O., Chang E. T., Dong R., Lin S., Lee C. Y., Strom S. C., Kashyap R., Fung J. J., Farese R. V. Jr., Patoiseau J. F., Delhon A., and Chang T. Y. (2000) Immunological quantitation and localization of ACAT-1 and ACAT-2 in human liver and small intestine. J. Biol. Chem. 275, 28083–28092 [DOI] [PubMed] [Google Scholar]
- 14. Sakashita N., Miyazaki A., Takeya M., Horiuchi S., Chang C. C., Chang T. Y., and Takahashi K. (2000) Localization of human acyl-coenzyme A: cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. Am. J. Pathol. 156, 227–236 10.1016/S0002-9440(10)64723-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang T. Y., Li B. L., Chang C. C., and Urano Y. (2009) Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 297, E1–E9 10.1152/ajpendo.90926.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miyazaki A., Sakashita N., Lee O., Takahashi K., Horiuchi S., Hakamata H., Morganelli P. M., Chang C. C., and Chang T. Y. (1998) Expression of ACAT-1 protein in human atherosclerotic lesions and cultured human monocytes-macrophages. Arterioscler. Thromb. Vasc. Biol. 18, 1568–1574 10.1161/01.ATV.18.10.1568 [DOI] [PubMed] [Google Scholar]
- 17. Sakashita N., Miyazaki A., Chang C. C., Chang T. Y., Kiyota E., Satoh M., Komohara Y., Morganelli P. M., Horiuchi S., and Takeya M. (2003) Acyl-coenzyme A:cholesterol acyltransferase 2 (ACAT2) is induced in monocyte-derived macrophages: in vivo and in vitro studies. Lab. Invest. 83, 1569–1581 10.1097/01.LAB.0000095687.17383.39 [DOI] [PubMed] [Google Scholar]
- 18. Parini P., Davis M., Lada A. T., Erickson S. K., Wright T. L., Gustafsson U., Sahlin S., Einarsson C., Eriksson M., Angelin B., Tomoda H., Omura S., Willingham M. C., and Rudel L. L. (2004) ACAT2 is localized to hepatocytes and is the major cholesterol-esterifying enzyme in human liver. Circulation 110, 2017–2023 10.1161/01.CIR.0000143163.76212.0B [DOI] [PubMed] [Google Scholar]
- 19. Wang Y. J., Bian Y., Luo J., Lu M., Xiong Y., Guo S. Y., Yin H. Y., Lin X., Li Q., Chang C. C. Y., Chang T. Y., Li B. L., and Song B. L. (2017) Cholesterol and fatty acids regulate cysteine ubiquitylation of ACAT2 through competitive oxidation. Nat. Cell Biol. 19, 808–819 10.1038/ncb3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu Y., Chen C. Y., Li J., Cheng J. X., Jang M., and Kim K. H. (2018) In vitro exploration of ACAT contributions to lipid droplet formation during adipogenesis. J. Lipid Res. 59, 820–829 10.1194/jlr.M081745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Igarashi M., Osuga J., Uozaki H., Sekiya M., Nagashima S., Takahashi M., Takase S., Takanashi M., Li Y., Ohta K., Kumagai M., Nishi M., Hosokawa M., Fledelius C., Jacobsen P., et al. (2010) The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ. Res. 107, 1387–1395 10.1161/CIRCRESAHA.110.226613 [DOI] [PubMed] [Google Scholar]
- 22. Yamazaki H., Takahashi M., Wakabayashi T., Sakai K., Yamamuro D., Takei A., Takei S., Nagashima S., Yagyu H., Sekiya M., Ebihara K., and Ishibashi S. (2019) Loss of ACAT1 attenuates atherosclerosis aggravated by loss of NCEH1 in bone marrow–derived cells. J. Atheroscler. Thromb. 26, 246–259 10.5551/jat.44040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sekiya M., Osuga J., Igarashi M., Okazaki H., and Ishibashi S. (2011) The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J. Atheroscler. Thromb. 18, 359–364 10.5551/jat.7013 [DOI] [PubMed] [Google Scholar]
- 24. Albert J. S., Yerges-Armstrong L. M., Horenstein R. B., Pollin T. I., Sreenivasan U. T., Chai S., Blaner W. S., Snitker S., O'Connell J. R., Gong D. W., Breyer R. J. 3rd, Ryan A. S., McLenithan J. C., Shuldiner A. R., Sztalryd C., et al. (2014) Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315 10.1056/NEJMoa1315496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yagyu H., Kitamine T., Osuga J., Tozawa R., Chen Z., Kaji Y., Oka T., Perrey S., Tamura Y., Ohashi K., Okazaki H., Yahagi N., Shionoiri F., Iizuka Y., Harada K., et al. (2000) Absence of ACAT-1 attenuates atherosclerosis but causes dry eye and cutaneous xanthomatosis in mice with congenital hyperlipidemia. J. Biol. Chem. 275, 21324–21330 10.1074/jbc.M002541200 [DOI] [PubMed] [Google Scholar]
- 26. Accad M., Smith S. J., Newland D. L., Sanan D. A., King L. E. Jr., Linton M. F., Fazio S., and Farese R. V. Jr. (2000) Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Invest. 105, 711–719 10.1172/JCI9021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang L. H., Gui J., Artinger E., Craig R., Berwin B. L., Ernst P. A., Chang C. C., and Chang T. Y. (2013) Acat1 gene ablation in mice increases hematopoietic progenitor cell proliferation in bone marrow and causes leukocytosis. Arterioscler. Thromb. Vasc. Biol. 33, 2081–2087 10.1161/ATVBAHA.112.301080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang L. H., Melton E. M., Li H., Sohn P., Rogers M. A., Mulligan-Kehoe M. J., Fiering S. N., Hickey W. F., Chang C. C., and Chang T. Y. (2016) Myeloid acyl-CoA:cholesterol acyltransferase 1 deficiency reduces lesion macrophage content and suppresses atherosclerosis progression. J. Biol. Chem. 291, 6232–6244 10.1074/jbc.M116.713818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fazio S., Major A. S., Swift L. L., Gleaves L. A., Accad M., Linton M. F., and Farese R. V. Jr. (2001) Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Invest. 107, 163–171 10.1172/JCI10310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nuñez G., Schnurr M., Espevik T., Lien E., Fitzgerald K. A., Rock K. L., Moore K. J., et al. (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 10.1038/nature08938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rajamaki K., Lappalainen J., Oörni K., Välimäki E., Matikainen S., Kovanen P. T., and Eklund K. K. (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5, e11765 10.1371/journal.pone.0011765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kruth H. S., Skarlatos S. I., Lilly K., Chang J., and Ifrim I. (1995) Sequestration of acetylated LDL and cholesterol crystals by human monocyte-derived macrophages. J. Cell Biol. 129, 133–145 10.1083/jcb.129.1.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kellner-Weibel G., Yancey P. G., Jerome W. G., Walser T., Mason R. P., Phillips M. C., and Rothblat G. H. (1999) Crystallization of free cholesterol in model macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 19, 1891–1898 10.1161/01.ATV.19.8.1891 [DOI] [PubMed] [Google Scholar]
- 34. Kellner-Weibel G., Luke S. J., and Rothblat G. H. (2003) Cytotoxic cellular cholesterol is selectively removed by apoA-I via ABCA1. Atherosclerosis 171, 235–243 10.1016/j.atherosclerosis.2003.08.024 [DOI] [PubMed] [Google Scholar]
- 35. Spitz C., Winkels H., Bürger C., Weber C., Lutgens E., Hansson G. K., and Gerdes N. (2016) Regulatory T cells in atherosclerosis: critical immune regulatory function and therapeutic potential. Cell Mol. Life Sci. 73, 901–922 10.1007/s00018-015-2080-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mailer R. K. W., Gisterå A., Polyzos K. A., Ketelhuth D. F. J., and Hansson G. K. (2017) Hypercholesterolemia enhances T cell receptor signaling and increases the regulatory T cell population. Sci. Rep. 7, 15655 10.1038/s41598-017-15546-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tall A. R., and Yvan-Charvet L. (2015) Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 15, 104–116 10.1038/nri3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ikenoya M., Yoshinaka Y., Kobayashi H., Kawamine K., Shibuya K., Sato F., Sawanobori K., Watanabe T., and Miyazaki A. (2007) A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 191, 290–297 10.1016/j.atherosclerosis.2006.05.048 [DOI] [PubMed] [Google Scholar]
- 39. Goldstein J. L., Ho Y. K., Basu S. K., and Brown M. S. (1979) Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. U.S.A. 76, 333–337 10.1073/pnas.76.1.333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brown M. S., Goldstein J. L., Krieger M., Ho Y. K., and Anderson R. G. (1979) Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J. Cell Biol. 82, 597–613 10.1083/jcb.82.3.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang L. H., Melton E. M., Li H., Sohn P., Jung D., Tsai C. Y., Ma T., Sano H., Ha H., Friedline R. H., Kim J. K., Usherwood E., Chang C. C. Y., and Chang T. Y. (2018) Myeloid-specific Acat1 ablation attenuates inflammatory responses in macrophages, improves insulin sensitivity, and suppresses diet-induced obesity. Am. J. Physiol. Endocrinol. Metab. 315, E340–E356 10.1152/ajpendo.00174.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang C. C., Doolittle G. M., and Chang T. Y. (1986) Cycloheximide sensitivity in regulation of acyl coenzyme A:cholesterol acyltransferase activity in Chinese hamster ovary cells: 1. Effect of exogenous sterols. Biochemistry 25, 1693–1699 10.1021/bi00355a038 [DOI] [PubMed] [Google Scholar]
- 43. Lada A. T., Davis M., Kent C., Chapman J., Tomoda H., Omura S., and Rudel L. L. (2004) Identification of ACAT1- and ACAT2-specific inhibitors using a novel, cell-based fluorescence assay: individual ACAT uniqueness. J. Lipid Res. 45, 378–386 10.1194/jlr.D300037-JLR200 [DOI] [PubMed] [Google Scholar]
- 44. Zhu X., Lee J. Y., Timmins J. M., Brown J. M., Boudyguina E., Mulya A., Gebre A. K., Willingham M. C., Hiltbold E. M., Mishra N., Maeda N., and Parks J. S. (2008) Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 283, 22930–22941 10.1074/jbc.M801408200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhu X., Owen J. S., Wilson M. D., Li H., Griffiths G. L., Thomas M. J., Hiltbold E. M., Fessler M. B., and Parks J. S. (2010) Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 51, 3196–3206 10.1194/jlr.M006486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ito A., Hong C., Rong X., Zhu X., Tarling E. J., Hedde P. N., Gratton E., Parks J., and Tontonoz P. (2015) LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. eLife 4, e08009 10.7554/eLife.08009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rong J. X., Kusunoki J., Oelkers P., Sturley S. L., and Fisher E. A. (2005) Acyl-coenzymeA (CoA):cholesterol acyltransferase inhibition in rat and human aortic smooth muscle cells is nontoxic and retards foam cell formation. Arterioscler. Thromb. Vasc. Biol. 25, 122–127 10.1161/01.ATV.0000148202.49842.3b [DOI] [PubMed] [Google Scholar]
- 48. Yoshinaka Y., Shibata H., Kobayashi H., Kuriyama H., Shibuya K., Tanabe S., Watanabe T., and Miyazaki A. (2010) A selective ACAT-1 inhibitor, K-604, stimulates collagen production in cultured smooth muscle cells and alters plaque phenotype in apolipoprotein E-knockout mice. Atherosclerosis 213, 85–91 10.1016/j.atherosclerosis.2010.08.048 [DOI] [PubMed] [Google Scholar]
- 49. Nicolosi R. J., Wilson T. A., and Krause B. R. (1998) The ACAT inhibitor, CI-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters. Atherosclerosis 137, 77–85 10.1016/S0021-9150(97)00279-7 [DOI] [PubMed] [Google Scholar]
- 50. Bocan T. M., Krause B. R., Rosebury W. S., Mueller S. B., Lu X., Dagle C., Major T., Lathia C., and Lee H. (2000) The ACAT inhibitor avasimibe reduces macrophages and matrix metalloproteinase expression in atherosclerotic lesions of hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 20, 70–79 10.1161/01.ATV.20.1.70 [DOI] [PubMed] [Google Scholar]
- 51. Rong J. X., Blachford C., Feig J. E., Bander I., Mayne J., Kusunoki J., Miller C., Davis M., Wilson M., Dehn S., Thorp E., Tabas I., Taubman M. B., Rudel L. L., and Fisher E. A. (2013) ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler. Thromb. Vasc. Biol. 33, 4–12 10.1161/ATVBAHA.112.252056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Terasaka N., Miyazaki A., Kasanuki N., Ito K., Ubukata N., Koieyama T., Kitayama K., Tanimoto T., Maeda N., and Inaba T. (2007) ACAT inhibitor pactimibe sulfate (CS-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein E-deficient mice. Atherosclerosis 190, 239–247 10.1016/j.atherosclerosis.2006.03.007 [DOI] [PubMed] [Google Scholar]
- 53. Tabas I. (2002) Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 110, 905–911 10.1172/JCI0216452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Basu S. K., Goldstein J. L., Anderson G. W., and Brown M. S. (1976) Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 73, 3178–3182 10.1073/pnas.73.9.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuang S. Q., Kwartler C. S., Byanova K. L., Pham J., Gong L., Prakash S. K., Huang J., Kamm K. E., Stull J. T., Sweeney H. L., and Milewicz D. M. (2012) Rare, nonsynonymous variant in the smooth muscle-specific isoform of myosin heavy chain, MYH11, R247C, alters force generation in the aorta and phenotype of smooth muscle cells. Circulation research 110, 1411–1422 10.1161/CIRCRESAHA.111.261743 [DOI] [PMC free article] [PubMed] [Google Scholar]