

Abstract
Estrogen exerts its cardiovascular protective role at least in part by regulating endothelial hydrogen sulfide (H2S) release, but the underlying mechanisms remain to be fully elucidated. Estrogen exerts genomic effects, i.e. those involving direct binding of the estrogen receptor (ER) to gene promoters in the nucleus, and nongenomic effects, mediated by interactions of the ER with other proteins. Here, using human umbilical vein endothelial cells (HUVECs), immunological detection, MS-based analyses, and cGMP and H2S assays, we show that 17β-estradiol (E2) rapidly enhances endothelial H2S release in a nongenomic manner. We found that E2 induces phosphorylation of cystathionine γ-lyase (CSE), the key enzyme in vascular endothelial H2S generation. Mechanistically, E2 enhanced the interaction of membrane ERα with the Gα subunit Gαi-2/3, which then transactivated particulate guanylate cyclase-A (pGC-A) to produce cGMP, thereby activating protein kinase G type I (PKG-I). We also found that PKG-Iβ, but not PKG-Iα, interacts with CSE, leading to its phosphorylation, and rapidly induces endothelial H2S release. Furthermore, we report that silencing of either CSE or pGC-A in mice attenuates E2-induced aorta vasodilation. These results provide detailed mechanistic insights into estrogen's nongenomic effects on vascular endothelial H2S release and advance our current understanding of the protective activities of estrogen in the cardiovascular system.
Keywords: cyclic GMP (cGMP), protein kinase G (PKG), estrogen, hydrogen sulfide, endothelial cell, 17β-estradiol, vascular endothelial cells, cystathionine γ-lyase, vasodilation, vasorelaxation, cell signaling, gene regulation, nongenomic regulation, sulfur metabolism
Introduction
The incidence and severity of cardiovascular disease (CVD)5 are significantly increased in postmenopausal women (1). Estrogen decline during women's menopausal transition has long been thought to be the primary cause for elevated risk of CVD in postmenopausal women (2). Although epidemiological and experimental studies support the protective role of E2 in CVD, whether estrogen replacement therapy (ERT) benefits postmenopausal CVD remains controversial (3, 4). The “window of opportunity” has therefore been proposed to explain the failure of clinical trials, which supports the view that ERT could be cardiovascular protective if started early at the onset of menopause (4). This hypothesis is supported by recent randomized clinical trials showing that women receiving ERT early after menopause exhibited significantly reduced risk of CVD (5, 6). Further investigations are therefore required to fully understand estrogen's cardiovascular effects and the underlying mechanisms.
It is well-known that estrogen exerts its genomic and rapid nongenomic actions through binding to estrogen receptors (ER) ERα and ERβ, as well as the recently identified G protein–coupled ER (GPR30) (7, 8). The genomic effects of estrogen on cardiovascular function and disease have been well-defined over the past years. A number of previous studies have shown that 17β-estradiol (E2) improves lipid profiles (9), inhibits proliferation and migration of vascular smooth muscle cell (10), induces vasorelaxation by increasing endothelial nitric oxide synthase protein expression and nitric oxide (NO) release (11, 12), accelerates endothelial repair by promoting endothelial proliferation and migration (13, 14), and alleviates vascular inflammatory response by modulating functions of immune cells (15). More recently, evidence has continued to accumulate that estrogen's nongenomic actions exert numerous long-term functions in a variety of cell types. For instance, nonnuclear-initiated actions of the ER are essential for the protective effects of estrogen against vascular injury, neointima formation, and cortical bone mass (16, 17). Encouragingly, nonnuclear ER signaling promotes cardiovascular protection but does not induce an uterotrophic response or promote breast cancer tumor growth in vivo (18). Thus, the selective modulation of nonnuclear ER signaling may represent a promising avenue to protect CVD without increasing estrogen-related cancer risk.
Hydrogen sulfide (H2S) is a well-characterized gasotransmitter that is mainly synthesized by cystathionine γ-lyase (CSE) from l-cysteine in vascular tissues (19). H2S has been reported to induce vasorelaxation, stimulate endothelial cell-related angiogenic properties, and protect against atherosclerosis (20–22). We have previously reported that E2 enhances endothelial CSE gene expression through ERα-Sp1 interaction with the binding sites in CSE gene promoter and increases endothelial H2S release, which in turn promotes endothelial proliferation and migration (23). Interestingly, E2 also induces vasorelaxation by stimulating rapid endothelial H2S release within minutes, although the detailed mechanisms remain unknown (24). In view of the importance of nonnuclear ER signaling functions in CVD, it is worthwhile to fully elucidate the underlying molecular mechanisms through which E2 rapidly stimulates H2S release by vascular endothelial cells.
In the present study, we showed that E2 rapidly induced phosphorylation of CSE, the key enzyme for vascular endothelial H2S generation, and enhanced H2S release in a nongenomic manner. We also found that pGC-A/cGMP/PKG signaling was required for E2's nongenomic effects on vascular endothelial H2S release. Our findings provide novel mechanistic insights into estrogen's nongenomic effects on vascular endothelial H2S release and advances our current understanding of estrogen's cardiovascular protective actions.
Results
E2 rapidly induced H2S release by vascular endothelial cells in a nongenomic manner
We measured H2S release by HUVECs exposed to various concentrations of E2 for 15 min. E2 significantly stimulated H2S release with concentrations as low as 1 nm, and this induction peaked at 10 nm (Fig. 1A). E2 at a physiological concentration (10 nm) resulted in a significant increase in H2S release after 5 min, and this increase reached maximum after 15 min (Fig. 1B). Comparable changes in H2S release were also observed in HAECs treated with 10 nm E2 (Fig. 1C).
Figure 1.

E2 promotes vascular endothelial H2S release through its nongenomic effect. A, HUVECs were exposed to different concentrations of E2 for 15 min, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus CON, one-way ANOVA followed by the post hoc LSD test). B, HUVECs were treated with E2 (10 nm) at different time points, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). C, HAECs were treated with E2 (10 nm) at different time points, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). D, HUVECs were treated with E2 in the presence or absence of the protein synthesis inhibitor CHX (200 μm) and the transcription inhibitor Act D (10 μm). The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). E and F, HUVECs were treated with E2-BSA in different concentrations or at different time points, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). G, HUVECs were treated with E2 in the presence or absence of ER antagonist ICI182,780 (ICI, 10 μm), GPR30 antagonist (G15, 10 μm), GPR30 agonist G1 (G1, 10 μm), and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01; ***, p < 0.001 versus CON; ###, p < 0.001 versus E2; one-way ANOVA followed by the post hoc LSD test).
To confirm whether E2-induced H2S release depended on its nongenomic pathway, we incubated HUVECs with E2 and protein synthesis inhibitor cycloheximide (CHX) or transcription inhibitor actinomycin D (Act D). As expected, Act D treatment efficiently prevented the transcription of ICAM1 and PFKB3 (Fig. S1, A and B), whereas CHX treatment significantly blocked the translation of VCAM1 in HUVECs (Fig. S1C). Interestingly, both Act D and CHX treatment had no effect on E2-induced H2S release (Fig. 1D). The cell-impermeable E2-BSA compound is commonly used to identify E2's rapid actions that occur on the outer surface of the cell membrane. We also observed that E2-BSA significantly increased H2S release in a similar manner with E2 (Fig. 1, E and F).
We found that ER antagonist ICI182780 significantly reduced E2-stimulated H2S release and this effect was not inhibited by GPR30 antagonist G15 (Fig. 1G). Consistently, GPR30 agonist G1 alone had no effect on H2S release (Fig. 1G). Interestingly, G15 itself significantly induced H2S release (Fig. 1G).
E2 enhanced endothelial H2S release through rapid phosphorylation of CSE
H2S is primarily produced by CSE in vascular tissues and its phosphorylation is critical for endothelial H2S production (25). Short-term treatment with E2 did not alter CSE expression but rapidly induced phosphorylation of serine and threonine residues of CSE, which reached maximum around 15 min in HUVECs (Fig. 2A). Comparable changes in phosphorylation of serine and threonine residues of CSE were also seen in HAECs treated with 10 nm E2 (Fig. 2B). Previous reports have reported that phosphorylation of CSE Ser-377 plays an important role in regulating endothelial H2S release (26). To address whether anti-phospho-(Ser/Thr)Phe used in the present study can recognize phosphorylation of CSE Ser-377, we performed an MS analysis to examine anti-phospho-(Ser/Thr)Phe immunoprecipitated samples from HEK293 cells. Potential phosphorylation sites of CSE were predicted using NetPhos 3.1 Server (Fig. 2C). Interestingly, the residue serine 56 was found to be phosphorylated (Fig. 2D) rather than serine 377.
Figure 2.

E2 regulates endothelial H2S release through phosphorylation of CSE. A and B, HUVECs and HAECs were treated with E2 at different time points, and Thr/Ser phosphorylation of CSE and total CSE level were measured by immunoprecipitation and Western blotting. C, potential phosphorylation sites marked with yellow were predicted by NetPhos 3.1 Server. D, mass spectrometry analysis of immunoprecipitated samples anti-phospho-(Ser/Thr)Phe from HEK293 cells.
E2 stimulated phosphorylation of CSE and endothelial H2S release through PKG-Iβ
It has been previously shown that cGMP-dependent protein kinase G (PKG) phosphorylates the serine residue in CSE (26, 27). In line with this, E2-induced Thr/Ser phosphorylation of CSE was significantly reduced in the presence of 10 μm PKG inhibitor KT5823 (Fig. 3A). Moreover, E2-induced H2S production by HUVECs was also dramatically abolished by KT5823 (Fig. 3B). The specificity of KT5823 is concentration dependent, 10 μm of this inhibitor may also inhibit PKC activity. To determine whether E2-induced phosphorylation of CSE was also involved with PKC, a specific PKC inhibitor bisindolylmaleimide I (BIMI) was used. The results showed E2-induced phosphorylation of CSE and endothelial H2S release was not attenuated in the presence of 5 μm BIMI (Fig. 3, C and D). These data suggest that E2 induced phosphorylation of CSE and endothelial H2S release through activation of PKG.
Figure 3.

E2-induced endothelial H2S release and phosphorylation of CSE depends on PKG-Iβ. A, HUVECs were exposed to E2 for 15min in the presence or absence of the protein kinase G inhibitor (KT5823, 10 μm), and CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. B, HUVECs were exposed to E2 for 15 min, in the presence or absence of the protein kinase G inhibitor (KT5823, 10 μm). The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus CON; ###, p < 0.001 versus E2; Mann-Whitney test). C and D, HUVECs were exposed to E2 for 15 min in the presence or absence of the protein kinase C inhibitor (BIMI, 5 μm). CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01 versus CON; one-way ANOVA followed by the post hoc LSD test). E and F, HUVECs were transfected with PKG-Iα (20 nm) or PKG-Iβ siRNA(20 nm) for 36 h and treated with E2 for 15 min. The cell content of PKG-Iα and PKG-Iβ was detected by Western blotting; CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. G, HUVECs were transfected with PKG-Iα (20 nm) or PKG-Iβ siRNA(20 nm) for 36 h and treated with E2 for 15 min. The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus scrambled siRNA CON; ###, p, < 0.001 versus scrambled siRNA E2, one-way ANOVA followed by the post hoc LSD test). H and I, HAECs were transfected with PKG-Iβ siRNA (20 nm) for 36 h and treated with E2 for 15 min. PKG-Iβ, CSE phosphorylation level, and total CSE were detected by immunoprecipitation and Western blotting, and the medium H2S concentrations were measured.
There are two major types of PKG in vascular cells, including PKG-Iα and PKG-Iβ. siRNA-mediated silencing of PKG-Iβ, but not PKG-Iα, significantly attenuated E2-induced phosphorylation of CSE (Fig. 3, E and F). In agreement, E2-induced H2S release was significantly abolished by PKG-Iβ siRNA but remained unchanged in the presence of PKG-Iα siRNA (Fig. 3G). E2-induced phosphorylation of CSE and H2S release was also abolished by PKG-Iβ siRNA rather than PKG-Iα siRNA in HAECs (Fig. 3, H and I).
We also overexpressed PKG-Iα or -Iβ to examine their effects on CSE phosphorylation and H2S release (Fig. 4, A and B). Overexpression of PKG1β dramatically induced Thr/Ser phosphorylation of CSE with or without E2 (Fig. 4D), whereas overexpression of PKG-Iα had no effects (Fig. 4C). Basal or E2-induced H2S release was not altered by overexpression of PKG-Iα, whereas overexpression of PKG-Iβ significantly enhanced H2S production in the absence or presence of E2 (Fig. 4E).
Figure 4.

PKG-Iβ directly interacts with CSE and mediates E2-induced phosphorylation of CSE and endothelial H2S release. A and B, HUVECs were transfected with PKG-Iα (2 μg) or PKG-Iβ (2 μg) plasmid for 48 h, and treated with E2 for 15 min. The efficiency of overexpression was confirmed by Western blotting. C and D, CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. E, HUVECs were transfected with PKG-Iα (2 μg) or PKG-Iβ (2 μg) plasmid for 48 h, and treated with E2 for 15 min. The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01 versus empty vector CON, one-way ANOVA followed by the post hoc LSD test). F, the interaction between CSE protein and PKG-Iβ or PKG-Iα protein was detected by immunoprecipitation and Western blotting. Input refers to whole cell lysates, and IgG refers to normal rabbit IgG.
To test if there is a direct interaction between PKG and CSE, we performed immunoprecipitation assay. Enhanced interaction of PKG-Iβ and CSE was observed after E2 treatment, whereas the interaction of PKG-Iα and CSE remained unchanged (Fig. 4F). Taken together, these data indicate that E2 stimulated phosphorylation of CSE and H2S production through enhancing the interaction between PKG-Iβ and CSE.
E2-induced H2S release depended on elevated cGMP levels
Because PKG is a cGMP-dependent protein kinase, we investigated whether E2-induced H2S production is associated with cGMP level. E2 significantly increased cGMP levels after 5–30 min (Fig. 5A). As expected, PET-cGMP (membrane permeant analogues of cGMP) treatment induced Ser-239 phosphorylation of its major substrate vasodilator-stimulated phosphoprotein (VASP) and phosphorylation of CSE (Fig. 5B). Similar to E2, PET-cGMP rapidly induced H2S production by HUVECs after 15 min when used at a concentration of 0.1 nm and reached maximum at a concentration of 10 nm (Fig. 5C). These data revealed that E2-induced H2S release depended on elevated cGMP level.
Figure 5.

E2 induced endothelial H2S release through cGMP. A, HUVECs were treated with E2 at different time points; the level of cGMP was measured by ELISA (mean ± S.D.; n = 3 independent experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). B, HUVECs were treated with different concentrations of cGMP-PET for 15 min; the cell content of p-VSAP was detected by Western blotting. The cell content of CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. C, HUVECs were treated with different concentrations of cGMP-PET for 15 min, the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus CON, one-way ANOVA followed by the post hoc LSD test).
E2 enhanced cGMP and induced endothelial H2S production mainly through pGC-A
cGMP is generated from GTP by guanylyl cyclases (GCs). Two different forms of GCs exist in mammals, including the soluble (sGC) and the particulate GC (pGC) (28). As expected, sGC agonist BAY41-2272 significantly increased VASP phosphorylation and this effect was stronger than E2 (Fig. 6A). However, sGC inhibitor NS2028 slightly reduced E2-induced phosphorylation of VASP (Fig. 6A), indicating that sGC is not the principle enzyme for E2's effect on cGMP signaling.
Figure 6.

pGC-A is critical in E2-induced cGMP/PKG/CSE signaling pathway and endothelial H2S production. A–C, HUVECs were exposed to E2 for 15 min in the presence or absence of the sGC activator (BAY41–2272, 3*10−6 m), sGC inhibitor (NS2028, 10−5 m), donor of NO (SNP, 10−4 m), inhibitor of NO (LNNA, 10−3 m). The cell content of p-VSAP was detected by Western blotting, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; *, p < 0.05 versus CON; one-way ANOVA followed by the post hoc LSD test). D, HUVECs were transfected with pGC-A siRNA (20 nm) for 36 h and treated with E2 for 15 min; pGC-A, p-VASP, and VASP were detected by Western blotting. E, HUVECs were transfected with pGC-A siRNA (20 nm) for 36 h, and treated with E2 for 15 min; CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. F, HUVECs were transfected with pGC-A siRNA (20 nm) for 36 h and treated with E2 for 15 min. The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01 versus scrambled siRNA CON; one-way ANOVA followed by the post hoc LSD test). G and H, HAECs were transfected with pGC-A siRNA (20 nm) for 36 h, and treated with E2 for 15 min; pGC-A, CSE phosphorylation level, total CSE, and the medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.01, versus scrambled siRNA CON; one-way ANOVA followed by the post hoc LSD test). I, HUVECs were exposed to ANP (pGC-A ligand) (10−6 m) for different time; the cell content of p-VASP level was detected by Western blotting; the cell content of CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting.
sGC is the only conclusively proven receptor for NO and E2 is a well-characterized stimulator of endothelial NO (29, 30). To exclude the possible role of NO-sGC pathway in E2-induced cGMP production, NO donor sodium nitroprusside dehydrate (SNP) and NO synthase inhibitor NG-nitro-l-arginine (l-NNA) were used. SNP induced a stronger VASP phosphorylation than E2 (Fig. 6B), which manifests the activation of NO-sGC pathway is sufficient to activate PKG. Notwithstanding, l-NNA did not alter E2-induced VASP phosphorylation (Fig. 6B). As a result, E2-induced endothelial H2S release was slightly but not significantly attenuated by NS2028 and l-NNA (Fig. 6C). These data support that NO-sGC pathway is not involved in E2-induced cGMP/PKG activation.
Because pGC-A (equivalent to natriuretic peptide receptor A, NPR-A) is the main isoform of pGC expressed in vascular endothelial cells (31), we next evaluated the role of pGC-A in E2-induced cGMP signaling and H2S release. Transfection with pGC-A siRNA significantly reduced E2-induced phosphorylation of VASP and CSE, and endothelial H2S release (Fig. 6, D–F). In HEACs, silence of pGC-A also inhibited phosphorylation of VASP and CSE and endothelial H2S release that induced by E2 (Fig. 6, G and H). Conversely, pGC-A ligand atrial natriuretic peptide (ANP) induced phosphorylation of VASP and CSE in a time-dependent manner (Fig. 6I). Taken together, these data show that pGC-A is critical in E2-induced cGMP/PKG/CSE signaling pathway and H2S production.
E2 activated cGMP/PKG pathway and endothelial H2S release through the enhanced interaction of ERα with Gαi2/3 and pGC-A
Gαi is important for E2's nongenomic signaling (32). We found that at resting stage, pGC-A interacted with ERα, Gαi-2, and Gαi-3 but not Gαi-1 (Fig. 7A). E2 readily increased the interaction of ERα with Gαi-2 or Gαi-3 and pGC-A (Fig. 7A). siRNA-mediated silencing of Gαi-2 or Gαi-3 significantly reduced E2-induced phosphorylation of VASP and Thr/Ser phosphorylation of CSE (Fig. 7, B–E). As a result, E2-induced endothelial H2S release was almost completely abolished by Gαi-2 or Gαi-3 siRNA (Fig. 7, F and G). These data indicate that E2 enhanced the protein complex formation of ERα with Gαi2/3 and pGC-A.
Figure 7.

E2 activated cGMP/PKG pathway and endothelial H2S release through the enhanced interaction of ERα with Gαi2/3 and pGC-A. A, HUVECs were exposed to E2 for 15 min. The interaction between Gαi1/2/3, pGC-A, and mERα was detected by immunoprecipitation and Western blotting. B and C, HUVECs were transfected with scrambled siRNA or Gαi2/3 siRNA for 48 h, and treated with E2 for 15 min. The cell content of Gαi2/3 and p-VASP was detected by Western blotting. D and E, HUVECs were transfected with scrambled siRNA or Gαi2/3 siRNA for 48 h, and treated with E2 for 15 min. CSE phosphorylation level and total CSE were detected by immunoprecipitation and Western blotting. F and G, HUVECs were transfected with scrambled siRNA or Gαi2/3 siRNA for 48 h, and treated with E2 for 15min. The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; **, p < 0.01; ***, p < 0.001 versus scrambled siRNA CON; ###, p < 0.001, versus scrambled siRNA E2; one-way ANOVA followed by the post hoc LSD test).
To further confirm whether the interaction of Gαi with ERα is required for E2-induced nongenomic signaling and H2S production, we transfected HUVECs with ERαD258A vector, which prevents the interaction of Gαi with ERα (33). Indeed, overexpression of the ERαD258A mutant prevented E2's nongenomic activation of classical p-ERK and p-Akt pathways (Fig. 8A). In parallel, it prevented E2-induced PKG activation, as shown by the level of phosphorylated VASP (Fig. 8A). Meanwhile, ERαD258A mutant decreased E2-induced interaction of Gαi with ERα (Fig. 8B), Thr/Ser phosphorylation of CSE, and endothelial H2S release (Fig. 8, C and D). These data confirm that the interaction of Gαi with ERα is required for E2-induced cGMP/PKG activation and endothelial H2S release.
Figure 8.
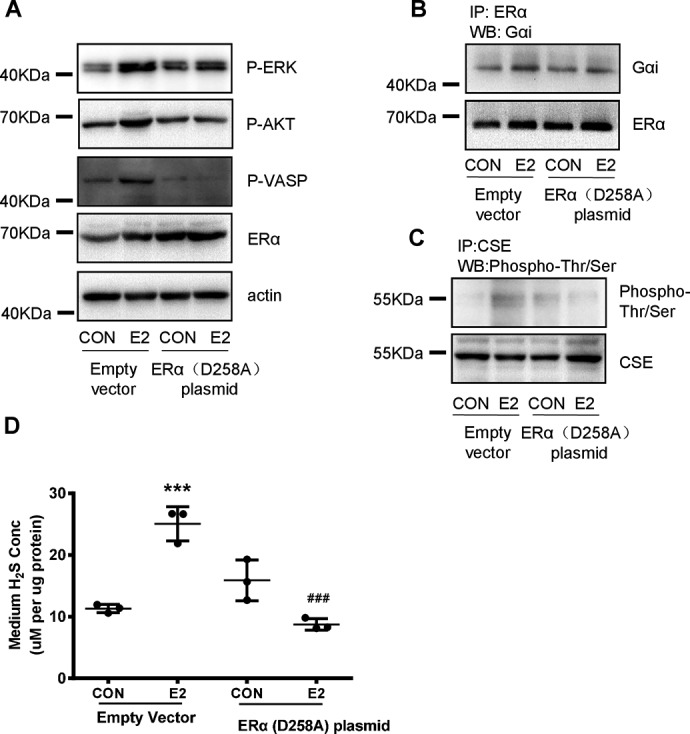
ERαD258A prevents the nongenomic effect of E2 on phosphorylation of CSE and endothelial H2S release. A, HUVECs were transfected with empty vector (2 μg) or mutant ERα (D258A, 2 μg) plasmid for 48 h and treated with E2 for 15 min. p-VSAP, VASP, p-ERK, p-AKT, and ERα were detected by Western blotting. B, HUVECs were transfected with empty vector (2 μg) or mutant ERα (D258A, 2 μg) plasmid for 48 h and treated with E2 for 15 min. The interaction between ERα and Gαi was measured by immunoprecipitation and Western blotting. C and D, HUVECs were transfected with empty vector (2 μg) or mutant ERα (D258A, 2 μg) plasmid for 48 h and treated with E2 for 15 min, CSE phosphorylation level and total CSE was detected by immunoprecipitation and Western blotting. The medium H2S concentrations were measured (mean ± S.D.; n = 3 independent experiments; ***, p < 0.001 versus empty vector CON; ###, p < 0.001 versus empty vector E2, one-way ANOVA followed by the post hoc LSD test).
Knockdown of vascular pGC-A and CSE impaired E2-induced vasodilation
Having observed the important role of CSE and pGC-A in E2-induced endothelial H2S release, we finally examined whether knockdown of CSE or pGC-A in mice can attenuate E2-induced vasodilation in vitro. Mice were received tail vein injection of control, pGC-A, or CSE AAV-shRNA for 4 weeks and aortae were dissected out to detect vascular tension. AAV-mediated shRNA was efficiently delivered to the aorta, as confirmed by GFP+ expression (Fig. S2A). Our immunostaining and Western results showed a successful silence of pGC-A and CSE protein in vascular endothelial cells (Fig. S2, B–D).
To preclude the effects of NO and PGI2, the arteries were incubated with NOS inhibitor l-NNA and COX inhibitor indomethacin for 30 min to inhibit the production of NO and prostacyclin. Then the NO- and prostacyclin-resistant responses of all three groups of aortic arteries to cumulative application of acetylcholine (Ach) were studied. Compared with control (Fig. 9A), AAV-mediated pGC-A and CSE knockdown resulted in significantly attenuated Ach-induced vasodilation (Fig. 9, B–D). In addition, we found that E2 induced vasodilation (Fig. 9E), which was significantly reduced by silencing of pGC-A and CSE expression (Fig. 9, F–H). These data suggest that activation of CSE and pGC-A is important for E2-induced vasodilation in vitro.
Figure 9.

AAV-mediated silencing of pGC-A or CSE significantly attenuated E2-induced vasodilation. Mice at 8 weeks of age were injected from the tail vein with AAV-control-shRNA (5.0*1011 v.g), AAV-pGC-A-shRNA (5.0*1011 v.g), AAV-CSE-shRNA (7.5*1011 v.g). A–D, after 4 weeks, the aortae were dissected out and pretreated for 30 min with 200 mm l-NNA (NO inhibitor) and 10 mm Indo (prostaglandin synthesis inhibitors) and preconstricted with Phe (10−6 m) before testing with Ach (10−5 m) (mean ± S.D.; n = 5 independent experiments; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test). E–H, knockdown of pGC-A or CSE expression significantly attenuated E2-induced vasodilation (mean ± S.D.; n = 5 independent experiments; ***, p < 0.001 versus CON; one-way ANOVA followed by the post hoc LSD test).
Discussion
We have previously demonstrated that E2 attenuates atherosclerosis through up-regulation of vascular endothelial CSE protein expression and H2S release (23). In addition, we found that E2 rapidly stimulates endothelial H2S release, although the mechanism remains obscure (24). The current study extends our previous studies to disclose the underlying molecular mechanisms through which E2 rapidly induces endothelial H2S release and vasodilation.
In the present study, CSE expression was not altered, but its phosphorylation was readily increased with short-term treatment of E2. A previous study has reported that vascular endothelial CSE is phosphorylated by taurolithocholic acid via Akt and cAMP-dependent PKA on serine residues, in particular Ser-377, to enhance H2S release (26, 34). Our MS results showed the anti-phospho-(Ser/Thr)Phe antibody used in the present study only recognizes phosphorylation of CSE Ser-56. However, its role in E2's rapid effect on H2S release remains to be further investigated.
In vascular endothelial cells, PKG-Iα and PKG-Iβ are predominantly expressed (35, 36). On the basis of our previous finding that PKG is critical for H2S release (24), here we further identified that E2-induced phosphorylation of CSE and H2S production depends on PKG-Iβ but not PKG-Iα. The substrate specificity of PKG-I depends on the different N termini of the two isoforms. For example, the isoform PKG-Iα specifically recognizes regulatory myosin phosphatase targeting subunit 1 or RhoA (37, 38), whereas the IP3RI-associated cGMP kinase substrate is a specific substrate for PKG-Iβ (39). Our co-immunoprecipitation result further indicated that with E2 treatment, PKG-Iβ interacted with CSE, suggesting that CSE is a new specific substrate for PKG-Iβ.
PKG-I-knockout mice show an impaired response to NO-induced vasodilatation, and vascular smooth muscle–specific PKG-Iα or PKG-Iβ expression can rescue vasodilatory response (40, 41). The mechanisms include the inhibition of intracellular Ca2+ release from sarcoplasmic/endoplasmic reticulum by PKG-Iβ or the activation of myosin light chain phosphatase by PKG-Iα (39, 42). Although most work describes a vasodilatory effect of PKG-I on vascular smooth muscles, it was recently reported that the activation of endothelial PKG also leads to vasorelaxation, and this effect is possibly related to potassium channel (43). Interestingly, H2S is able to regulate endothelial conductance (IK(Ca)) and small conductance (SK(Ca)) potassium channels (44, 45). From these points of view, our findings provide a new possible explanation for the vasodilatory effect of endothelial PKG-I, namely that PKG-Iβ interacts and activates CSE to produce H2S, which then acts on potassium channels to induce vasodilation.
We have previously reported that E2 activates PKG in vascular endothelial cells, as indicated by VASP phosphorylation at Ser-239 (46). Likewise, PKG is also found as the target of E2 in many other tissues, such as vascular smooth muscle cells, osteocytes, and breast cancer cells (47–49). sGC is the only conclusively proven receptor for NO and E2 is able to enhance endothelial NO release (29, 30). As the important regulators in cardiovascular system, H2S and NO interact with each other's biosynthesis and physiological response (50). NO increased CSE expression and H2S generation from vascular tissues (51). Likely, H2S increased endothelial nitric oxide synthase phosphorylation and NO generation through the Akt pathway (52). In our study, sGC or NOS inhibitor did not significantly alter E2-induced PKG activation and H2S release, whereas the silence of pGC-A completely abolished E2's effects. This is consistent with others' data showing that E2-enhanced cGMP level was not altered by sGC inhibitor NS2028 in hepatocyte (53). Our work indicates that NO/sGC pathway does not involve in E2-induced H2S release.
pGC-A is the trans-plasma membrane protein that is traditionally activated by ANP binding to its extracellular receptor portion. It plays an important role in cardiovascular system. For example, pGC-A knockout mice exhibited significant arterial hypertension and cardiac hypertrophy (54, 55). E2 was able to augment pGC-A signaling by increasing ANP level (55, 56). In addition to this, E2 acts at the extracellular face of the plasma membrane to stimulate pGC-A in a nongenomic manner in hepatocyte, but the detailed mechanism remains unexplored (53). Here we further revealed that membrane ERα recruited Gαi-2/3, which then transactivated pGC-A to produce cGMP. In agreement, it was shown that pGC-A activation depended on Gαi in Caenorhabditis elegans photoreceptor cells (57). Therefore, E2 can enhance pGC-A signaling via indirect regulation of its ligand ANP or direct transactivation of it in vascular endothelial cells.
We confirmed that ERα agonist PPT, but not ERβ agonist DPN, stimulated endothelial H2S release, suggesting that ERα is the key receptor in this event (24). In this study, we extended and identified that ERα interacted with Gαi-2 and Gαi-3 to stimulate PKG and H2S release. Indeed, ERα directly interacts with Gαi and mediates its nongenomic effects. The disruption of ERα-Gαi interaction prevented E2-induced nitric oxide synthase activation, re-endothelialization, and protection against vascular injury (16, 18, 32). It was reported that endothelial Gαi, in particular Gαi-2, coupled to many G protein–coupled receptors, such as α2-adrenergic receptor, endothelin B receptor, and 5-hydroxytryptamine receptor 1D, to stimulate NO release and induce vasodilation (58). Our in vitro experiments showed that knockdown of Gαi-2 or Gαi-3, but not Gαi-1, significantly reduced E2-induced cGMP signaling and H2S production. To the best of our knowledge, we for the first time reveal a molecular link between Gαi and H2S, which adds a new explanation to the mechanisms of Gαi activation and vasorelaxation.
Mutagenesis and the addition of blocking peptide revealed that amino acids 251–260 of ERα contain Gαi-binding domain that mediates Src and ERK1/2 activation (32). Furthermore, point mutation of ERαD258A prevented the interaction of ERα with Gαi and blocked nongenomic activation of its downstream signaling (33). In line with this, our data demonstrated that ERαD258A mutant prevented E2-induced ERα-Gαi interaction, nongenomic activation of Akt and ERK1/2, CSE phosphorylation, and H2S production. We also found that AAV-mediated silencing of pGC-A and CSE significantly reduced E2-induced vasodilation. These results indicated that the described nongenomic mechanism of E2-induced H2S release is essential for physiological effect of E2 on vasorelaxation. The rapid, nongenomic ER signaling contributes to long-term vascular protective actions, such as anti-atherogenesis and inhibition of neointima formation (59). Whether E2-induced rapid H2S release plays an important role in its long-term vascular protective actions remains to be investigated in our future study.
We found that E2 rapidly stimulated endothelial H2S release, and it is insensitive to transcription or protein inhibitor. In addition, the cell-impermeable E2-BSA compound mimicked E2's effect. These data suggest that E2 induced endothelial H2S release through its nongenomic action, which does not require transcription or translation of E2's target genes. Instead, E2 binds to its membrane receptors and rapidly exerts the generation of intracellular second messengers, and various signal-transduction within minutes such as phosphorylation of CSE, thereby mediating transient H2S release. Classically, three isoforms of membrane estrogen receptor, including mERα, mERβ, as well as GPR30 (also known as G protein–coupled estrogen receptor 1), exist in different tissues (60). We revealed that ERα interacted with Gαi and the point mutation of ERα blocked E2-elicited downstream signaling and H2S release. This is consistent with our previous studies showing that mERα is the critical receptor for estrogen's nongenomic effects in different type of cells (14, 61). GPR30 has no effect on H2S release. However, GPR30 antagonist G15 itself enhanced H2S release. This may be because of the low-affinity crossreactivity of the G15 to ERα (62). Hence, our data provide a new example to the concept that the modulation of different membrane estrogen receptor isoform can differ markedly in their stimulatory and/or inhibitory effects. The selective activation of these membrane ERs, therefore, is of great importance in the prevention and treatment of E2-related physiological functions and diseases.
In conclusion, we showed that E2 stimulated endothelial H2S release in a nongenomic manner. E2 enhanced the interaction of mERα and Gαi-2/3, which transactivated pGC-A to produce cGMP and subsequently activate PKG-Iβ to interact with CSE protein, leading to its phosphorylation and rapid H2S release (Fig. 10). Our study provides novel mechanistic insights into E2's nongenomic effects and also identifies the mERα signaling as a promising target for the development of novel therapeutic strategies in cardiovascular system.
Figure 10.
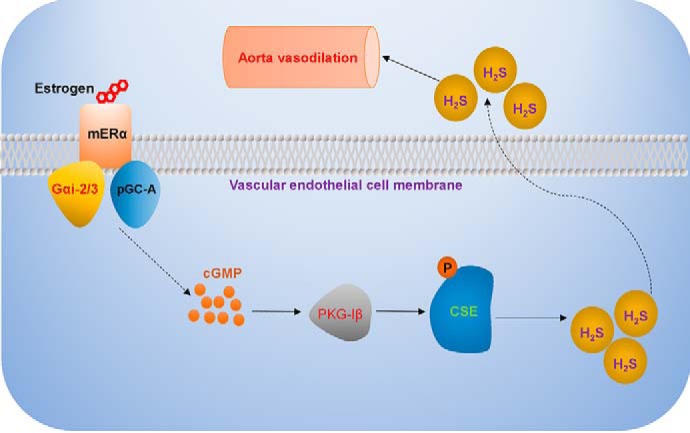
Schematic representation of nongenomic action of E2 on vascular endothelial H2S release. E2 enhances the interaction of membrane estrogen receptor α (ERα) and G α subunit Gαi-2/3, which subsequently transactivates pGC-A to produce cGMP and activates PKG-Iβ. The isoform of active PKG-Iβ interacts with CSE protein, leading to its phosphorylation and rapid endothelial H2S release.
Experimental procedures
Ethical statement
Human umbilical cords for HUVEC isolation
Human umbilical cords for isolation of HUVECs were collected from Guangzhou First Municipal People's Hospital. All participants gave informed consent. This study was approved by The Research Ethics Committee of Guangzhou First Municipal People's Hospital and Guangzhou Medical University and complied with the Declaration of Helsinki.
Mice
All animal procedures were reviewed and approved by Institutional Animal Care and Use Committee of Guangzhou Medical University. Four-week-old female mice in C57BL/6 background were obtained from the Guangdong animal experiment center (Guangdong, China), raised in the Experimental Animal Center of Guangzhou Medical University. The mice were kept under specific pathogen-free and temperature-controlled conditions on a 12 h light/dark cycle and maintained feedings and drinking water.
Reagents and antibodies
17β-estradiol (catalogue no. E2758), β-estradiol 6-(O-carboxymethyl)oxime: BSA fluorescein isothiocyanate conjugate (E2-BSA) (catalogue no. E6507), cycloheximide (catalogue no. C7698), actinomycin D (catalogue no. A9415), sodium nitroferricyanide(III) dihydrate (catalogue no. 228710), Nω-Nitro-l-arginine (catalogue no. N5501), PET-cGMP (catalogue no. P0622–1VL), and acetylcholine chloride (catalogue no. A6625) were purchased from Sigma-Aldrich. ICI 182,780 (catalogue no. ab120131) and KT5823 (catalogue no. ab120423) were obtained from Abcam (Cambridge, MA). G1 (catalogue no. G6798) and G15 (catalogue no. G6548) were supplied by Santa Cruz Biotechnology (Dallas, TX). NS2028 (catalogue no. 4517) was obtained from Tocris Bioscience (Minneapolis, MN). Endothelial Cell Medium (catalogue no. 1001-prf) was purchased from ScienCell Research Laboratories (Carlsbad, CA). Opti-MEM (catalogue no. 31985) and FBS (catalogue no. 12484) were obtained from Invitrogen. Phenylephrine (catalogue no. S2569) was obtained from Selleck Chemicals (Houston, TX). γ-cystathionine (CSE) polyclonal antibody (catalogue no. 12217–1-AP, lot no. 00050744) and NPR1 (pGC-A) polyclonal antibody (catalogue no. 55116–1-AP, lot no. 00048172) were purchased from Proteintech (Rosemont, PA). Anti-phosphotyrosine antibody (catalogue no. ab190824, lot no. GR260170–1), anti-phospho-(Ser/Thr)Phe antibody (catalogue no. ab17464, lot no. GR321281–1), and anti-ERβ antibody (catalogue no. ab3576, lot no. GR3192192–3) were obtained from Abcam. Anti-ERα antibody (catalogue no. 8644s, lot no. 4), anti-PKG-Iα antibody (catalogue no. 13511s, lot no. 1), anti-phospho-VASP (Ser239) antibody (catalogue no. 3114S, lot no. 5), anti-VASP antibody (catalogue no. 3112S, lot no. 2), anti-Gα(i) antibody (catalogue no. 5290, lot no. 3), anti-VCAM1 (catalogue no. 12367, lot no.1), and HA-Tag (Magnetic Bead Conjugate) (catalogue no. 11846, lot no. 6) were obtained from Cell Signaling Technology (Danvers, MA). Anti-PKG-Iβ antibody (catalogue no. PA527325, lot no. PA5–27325) was purchased from Thermo Fisher. Anti-Gαi-1 (catalogue no. sc-13533), anti-Gαi-2 (catalogue no. sc-7276), anti-Gαi-3 (catalogue no. sc-262) antibodies were obtained from Santa Cruz Biotechnology (Dallas, TX). All other chemicals were of analytical grade and purchased from Guangzhou Chemical Reagents (Guangzhou, China).
Cell cultures and treatments
HUVECs were isolated and cultured as described previously (13). HAECs were obtained from ScienCell Research Laboratories (catalogue no. 6100). Before treatments, HUVECs and HAECs were cultured in Endothelial Cell Medium (catalogue no. 1001-prf) containing 5% steroid-deprived FBS. Cell cultures were maintained at 37 °C in a humidified incubator in a 5% CO2 plus 95% O2 atmosphere. All plasmid transfections were performed by using Lipofectamine 3000 reagent (catalogue no. L3000015, Invitrogen) according to the manufacturer's instructions. All siRNA at 20 nm working concentration were transfected using Lipofectamine RNAiMAX Reagent following the manufacturer's instructions (catalogue no. 13778, Invitrogen). Cells (60% confluent in dishes) were serum-starved for 6–8 h followed by incubation with siRNA or plasmid for another 6 h in serum-free media. Target protein silencing or overexpression was assessed by Western blotting after 48 h of transfection.
Plasmids and siRNA and adeno-associated virus (AAV)–shRNA
ESR1 (catalogue no. H8846), EGFP-ESR1-D258A (catalogue no. H5536), EGFP-HA-empty vector (catalogue no. H7638), EGFP-HA-CSE-WT (catalogue no. H7673), EGFP-HA-CSE-S377A (catalogue no. H316), and EGFP-HA-CSE-S377D (catalogue no. H8157) expression plasmid were generated in Obio Technology Corp., Ltd. (ShangHai, China). The PKG-Iα and PKG-Iβ expression plasmid were bought from Addgene (Watertown, MA). The PKG-Iα (catalogue no. 5626772), PKG-Iβ (catalogue no. 5872111) and pGC A (catalogue no. 5625849) siRNA were purchased from Invitrogen. Gαi2 and Gαi3 siRNA were ordered from Guangzhou RiboBio Co., Ltd. (Guangzhou, China). The mouse AAV-control-shRNA, AAV-CSE-shRNA, and AAV-pGC-A-shRNA were ordered from Vigene Biosciences Inc. (ShanDong, China).
Real-time PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen; Hilden, Germany) and reverse transcribed PrimeScriptTM RT Master Mix (Takara, RR036A). Real-time PCR was performed using SYBR Green qPCR Kit (Takara, RR820A) in the QuantStudio 5 real-time system (Life Technologies). Data were expressed as relative -fold changes using the 2−ΔΔCT method. qPCR primers are summarized as follows: PFKFB3: forward, 5′-TGGCAGATGACCAGCACA-3′ and reverse, 5′-CTTCAGAGAGAGGAAGCCGA-3′; ICAM-1: forward, 5′-TCTTCCTCGGCCTTCCCATA-3′ and reverse, 5′-AGGTACCATGGCCCCAAATG-3′; β-actin: forward, 5′-CATGTACGTTGCTATCCAGGC-3′ and reverse, 5′-CTCCTTAATGTCACGCACGAT-3′.
Immunoblotting
After treatments, HUVECs were washed three times with precooling PBS before addition of the lysis buffer (100 mm Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 1 mm sodium orthovanadate, 1 mm NaF and 1 mm phenylmethylsulfonylfluoride, 3 mm PMSF) to cell culture dish on ice. Then cell lysates were scraped carefully, boiled for 18 min before being centrifuged for 7 min at 13,500 rpm. Cell lysates were separated by SDS-PAGE. The antibodies were used at the following concentrations: CSE (1:2000), pGC-A (1:1000), phosphotyrosine (1:2000), phospho-(Ser/Thr)Phe (1:2000), ERβ (1:2000), ERα (1:4000), PKG-Iα (1:1000), phospho-VASP (1:4000), VASP (1:2000), Gαi (1:2000), and PKG-Iβ (1:2000). Semiquantitative analysis of Western blotting data was performed using ImageJ software (National Institutes of Health). The relative protein levels normalized by that of intracellular β-actin were then expressed as the percentage compared with respective controls. All semiquantitative data are presented in Figs. S3 and S4.
Immunoprecipitation assay
Immunoprecipitation was performed according to the manufacturer's instructions (Dynabeads Protein G, catalogue no. 10007D, Invitrogen). In brief, an equal amount (200 μg) of cell lysate was immunoprecipitated with IP antibody–conjugated Dynabeads magnetic beads for 10 min at 4 °C under gentle agitation. The magnetic bead–Ab–Ag complex was washed with washing buffer for three times. 20 μl Elution buffer consisting of 0.2 m glycine (pH 2.0) was used to remove immunoprecipitates from the beads. The immunoprecipitates were analyzed by Western blotting.
Mass spectrometry analysis
HEK293 cells were transfected with 2 μg WT CSE plasmid for 24 h, and immunoprecipitation was performed with anti-phospho-(Ser/Thr)Phe as describe above. The anti-phospho-(Ser/Thr)Phe immunoprecipitates were subjected to trypsin digestion, and the phosphopeptides were enriched using TiO2 beads (GL Sciences) according to manufacturer's instructions. The enriched phosphopeptides separated from nanoHPLC were subjected to the tandem MS Q-EXACTIVE (Thermo Fisher Scientific) by BGI Mass Spectrometry Center (Shenzhen, China). The parameters for MS analysis are listed as follows: Electrospray voltage, 1.6 kV; precursor scan range, 350–1600 m/z at a resolution of 70,000 in Orbitrap; MS/MS fragment scan range, >100 m/z at a resolution of 17,500 in HCD mode; the number of MS/MS scans following one MS scan, 20 most abundant precursor ions above a threshold ion count of 20,000.
Measurement of cGMP levels
HUVECs were treated with 10 nm E2 at the indicated time points. Cells were lysed in 0.1 m HCl/1% Triton. cGMP was then measured by using a cGMP ELISA kit (catalogue no. 80101, New East Biosciences, King of Prussia, PA). Each time the calibration of cGMP standards are assayed at the same time as the samples. The standard wells, testing sample wells, and blank wells were set. Briefly, the testing sample and cGMP standards were added to testing sample wells and standard wells, respectively; the blank wells contained lysis buffer. HRP-conjugate reagent, chromogen solution, and stop solution were added to each well. The optical density at 450 nm was determined using a microtiter plate reader. The concentration of cGMP in the samples was then determined by comparing the optical density of the samples to the standard curve.
Measurement of H2S levels
HUVECs were treated and then the medium was collected for measuring the H2S levels as we reported previously (24). Amperometric H2S sensors (ISO-H2S-100, World Precision Instruments, Sarasota, FL) were used for the measurement of dissolved H2S concentration in the medium. In brief, the amperometric H2S sensor was calibrated with Na2S.9H2O stock solution before each experiment and a calibration curve was made. The tip of the H2S sensor was immersed into the medium for 10–15 mm and the current output was recorded. The concentration of dissolved H2S in the solution was calculated based on the calibration curve.
Animals
At 8 weeks of age, the mice were randomly divided into three groups and accepted one tail vein injection of AAV-Control-shRNA (5.0*1011 vector genomes (v. g) per mouse), AAV-pGC-A-shRNA (5.0*1011 v.g per mouse), or AAV-CSE-shRNA (7.5*1011 v.g per mouse), respectively. After 4 weeks, the aortae were dissected out and the AAV-mediated silencing of target protein was evaluated through immunofluorescence staining and Western blotting.
Artery preparation and wall tension measurement
After tail vein injection of AAV for 4 weeks, aortic blood vessels were taken to detect vascular tension. In vitro tension measurement was carried out as we reported previously (24). In brief, aortic were isolated and cut into four segments (2 mm per segment) in ice-cold physiological salt solution. The artery rings were mounted on a needle holder of DMT620 multi-channel isolated vascular tone measurement system (World Precision Instruments, Sarasota, FL) under a stereomicroscope. The organ bath was filled with oxygenated tissue buffer (PSS) and maintained at 37.8 °C. Isometric contractions were recorded by a force transducer connected to an analog-to-digital converter system. 30 min after mounting in the organ bath, all the rings were contracted using phenylephrine (Phe, 1 μm), and the functional integrity (over 90% relaxation) of the endothelial layer was determined by adding acetylcholine (10 μm). The rings were then allowed to equilibrate for an additional 60 min and the rings were contracted for a second time by the addition of Phe for 30 min and then its response to ACh or E2 was recorded.
Statistical analysis
Data are presented as mean ± S.D. All experiments were repeated at least three times. Statistical comparisons were made using one-way-ANOVA followed by the post hoc LSD test or Mann-Whitney test where the data are not normally distributed with SPSS software; the difference was considered significant if p < 0.05.
Author contributions
X. X. data curation; X. X., Q. Y., X. Liu, P. L., X. Li, Y. C., T. S., J. L., D. Z., and X. F. formal analysis; X. X. and X. F. supervision; X. X. and X. F. funding acquisition; X. X. validation; X. X., Q. Y., X. Liu, P. L., X. Li, and X. F. investigation; X. X., Q. Y., X. Liu, P. L., X. Li, Y. C., T. S., J. L., D. Z., and X. F. methodology; X. X., T. S., J. L., and D. Z. writing-review and editing; D. Z. writing-original draft; X. F. project administration.
Supplementary Material
This work was supported by National Natural Science Foundation Grants 81871137, 81471426 (to X. F.), The Project of Principal Scientists—Guangzhou Municipal Universities 'Yangcheng Scholars' Grant 1201541587 (to X. F.), The Project of Department of Education of Guangdong Province Grant 2015KTSCX109 (to X. F.), National Funds of Developing Local Colleges and Universities Grant B16056001 (to X. F.), Science and Technology Program of Guangzhou Grant 201804010376 (to X. F.), Guangzhou 121 Talent Program (to X. F.), National Natural Science Foundation for Young Scientists of China Grant 81701411 (to X. Liu), Guangdong Natural Science Foundation Grant 2017A030310158 (to X. Liu), National Natural Science Foundation Grant 81872128 (to J. L.), National Natural Science Foundation for Young Scientists of China Grant 81800428 (to D. Z.), The Innovation Project of Department of Education of Guangdong Province Grant 2017KTSCX158 (to D. Z.), Guangdong Natural Science Foundation Grant 2018A030310178 (to D. Z.), and Science and Technology Program of Guangzhou Grant 201904010289 (to D. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- CVD
- cardiovascular disease
- AAV
- adeno-associated virus
- Ach
- acetylcholine
- Act D
- actinomycin D
- ANOVA
- analysis of variance
- ANP
- atrial natriuretic peptide
- BIMI
- bisindolylmaleimide I
- CHX
- cycloheximide
- CSE
- cystathionine γ-lyase
- DPN
- 2,3-bis(4-hydroxyphenyl)-propionitrile
- E2
- 17β-estradiol
- E2-BSA
- β-Estradiol 6-(O-carboxymethyl)oxime: BSA fluorescein isothiocyanate conjugate
- ER
- estrogen receptor
- ERT
- estrogen replacement therapy
- GC
- guanylyl cyclase
- HAEC
- human aortic endothelial cell
- HUVEC
- human umbilical vein endothelial cell
- l-NNA
- NG-nitro-l-arginine
- pGC
- particulate guanylate cyclase
- Phe
- phenylephrine
- PKG
- protein kinase G
- PPT
- 4,4′,4′-(4-propyl-[1H]-pyrazole-1,3,5-triyl)trisphenol (PPT)
- sGC
- soluble GC
- SNP
- sodium nitroprusside dehydrate
- VASP
- vasodilator-stimulated phosphoprotein
- v.g
- vector genomes.
References
- 1. Mozaffarian D., Benjamin E. J., Go A. S., Arnett D. K., Blaha M. J., Cushman M., Das S. R., de Ferranti S., Despres J. P., Fullerton H. J., Howard V. J., Huffman M. D., Isasi C. R., Jimenez M. C., Judd S. E., et al. (2016) Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation 133, e38–e360 10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 2. Mendelsohn M. E., and Karas R. H. (1999) The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 340, 1801–1811 10.1056/NEJM199906103402306 [DOI] [PubMed] [Google Scholar]
- 3. Harman S. M. (2014) Menopausal hormone treatment cardiovascular disease: Another look at an unresolved conundrum. Fertil. Steril. 101, 887–897 10.1016/j.fertnstert.2014.02.042 [DOI] [PubMed] [Google Scholar]
- 4. Genazzani A. R., and Simoncini T. (2013) Pharmacotherapy: Benefits of menopausal hormone therapy—timing is key. Nat. Rev. Endocrinol. 9, 5–6 10.1038/nrendo.2012.228 [DOI] [PubMed] [Google Scholar]
- 5. Schierbeck L. L., Rejnmark L., Tofteng C. L., Stilgren L., Eiken P., Mosekilde L., Køber L., and Jensen J. E. (2012) Effect of hormone replacement therapy on cardiovascular events in recently postmenopausal women: Randomised trial. BMJ 345, e6409 10.1136/bmj.e6409 [DOI] [PubMed] [Google Scholar]
- 6. Hodis H. N., Mack W. J., Henderson V. W., Shoupe D., Budoff M. J., Hwang-Levine J., Li Y., Feng M., Dustin L., Kono N., Stanczyk F. Z., Selzer R. H., and Azen S. P. (2016) Vascular effects of early versus late postmenopausal treatment with estradiol. N. Engl. J. Med. 374, 1221–1231 10.1056/NEJMoa1505241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arnal J. F., Fontaine C., Billon-Galés A., Favre J., Laurell H., Lenfant F., and Gourdy P. (2010) Estrogen receptors and endothelium. Arterioscler. Thromb. Vasc. Biol. 30, 1506–1512 10.1161/ATVBAHA.109.191221 [DOI] [PubMed] [Google Scholar]
- 8. Meyer M. R., and Barton M. (2009) ERα, ERβ, and gpER: Novel aspects of oestrogen receptor signalling in atherosclerosis. Cardiovasc. Res. 83, 605–610 10.1093/cvr/cvp187 [DOI] [PubMed] [Google Scholar]
- 9. Bourassa P. A., Milos P. M., Gaynor B. J., Breslow J. L., and Aiello R. J. (1996) Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 93, 10022–10027 10.1073/pnas.93.19.10022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang P., Xu J., Zheng S., Huang J., Xiang Q., Fu X., and Wang T. (2010) 17β-estradiol down-regulates lipopolysaccharide-induced MCP-1 production and cell migration in vascular smooth muscle cells. J. Mol. Endocrinol. 45, 87–97 10.1677/JME-09-0166 [DOI] [PubMed] [Google Scholar]
- 11. Goglia L., Tosi V., Sanchez A. M., Flamini M. I., Fu X. D., Zullino S., Genazzani A. R., and Simoncini T. (2010) Endothelial regulation of eNOS, PAI-1 and t-PA by testosterone and dihydrotestosterone in vitro and in vivo. Mol. Hum. Reprod 16, 761–769 10.1093/molehr/gaq049 [DOI] [PubMed] [Google Scholar]
- 12. Iwakura A., Luedemann C., Shastry S., Hanley A., Kearney M., Aikawa R., Isner J. M., Asahara T., and Losordo D. W. (2003) Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation 108, 3115–3121 10.1161/01.CIR.0000106906.56972.83 [DOI] [PubMed] [Google Scholar]
- 13. Li P., Wei J., Li X., Cheng Y., Chen W., Cui Y., Simoncini T., Gu Z., Yang J., and Fu X. (2017) 17β-Estradiol enhances vascular endothelial Ets-1/miR-126–3p expression: The possible mechanism for attenuation of atherosclerosis. J. Clin. Endocrinol. Metab. 102, 594–603 10.1210/jc.2016-2974 [DOI] [PubMed] [Google Scholar]
- 14. Simoncini T., Scorticati C., Mannella P., Fadiel A., Giretti M. S., Fu X. D., Baldacci C., Garibaldi S., Caruso A., Fornari L., Naftolin F., and Genazzani A. R. (2006) Estrogen receptor α interacts with Gα13 to drive actin remodeling and endothelial cell migration via the RhoA/Rho kinase/moesin pathway. Mol. Endocrinol. 20, 1756–1771 10.1210/me.2005-0259 [DOI] [PubMed] [Google Scholar]
- 15. Arnal J. F., Laurell H., Fontaine C., Billon A., Calippe B., Lenfant F., and Gourdy P. (2009) Estrogen receptor actions on vascular biology and inflammation: Implications in vascular pathophysiology. Climacteric 12, Suppl. 1, 12–17 [DOI] [PubMed] [Google Scholar]
- 16. Bernelot Moens S. J., Schnitzler G. R., Nickerson M., Guo H., Ueda K., Lu Q., Aronovitz M. J., Nickerson H., Baur W. E., Hansen U., Iyer L. K., and Karas R. H. (2012) Rapid estrogen receptor signaling is essential for the protective effects of estrogen against vascular injury. Circulation 126, 1993–2004 10.1161/CIRCULATIONAHA.112.124529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bartell S. M., Han L., Kim H. N., Kim S. H., Katzenellenbogen J. A., Katzenellenbogen B. S., Chambliss K. L., Shaul P. W., Roberson P. K., Weinstein R. S., Jilka R. L., Almeida M., and Manolagas S. C. (2013) Non-nuclear-initiated actions of the estrogen receptor protect cortical bone mass. Mol. Endocrinol. 27, 649–656 10.1210/me.2012-1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chambliss K. L., Wu Q., Oltmann S., Konaniah E. S., Umetani M., Korach K. S., Thomas G. D., Mineo C., Yuhanna I. S., Kim S. H., Madak-Erdogan Z., Maggi A., Dineen S. P., Roland C. L., Hui D. Y., Brekken R. A., Katzenellenbogen J. A., Katzenellenbogen B. S., and Shaul P. W. (2010) Non-nuclear estrogen receptor α signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J. Clin. Invest. 120, 2319–2330 10.1172/JCI38291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., and Wang R. (2008) H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 10.1126/science.1162667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mani S., Li H., Untereiner A., Wu L., Yang G., Austin R. C., Dickhout J. G., Lhoták Š., Meng Q. H., and Wang R. (2013) Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127, 2523–2534 10.1161/CIRCULATIONAHA.113.002208 [DOI] [PubMed] [Google Scholar]
- 21. Pan L. L., Liu X. H., Gong Q. H., Yang H. B., and Zhu Y. Z. (2012) Role of cystathionine γ-lyase/hydrogen sulfide pathway in cardiovascular disease: A novel therapeutic strategy? Antioxid. Redox Signal. 17, 106–118 10.1089/ars.2011.4349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Y., Zhao X., Jin H., Wei H., Li W., Bu D., Tang X., Ren Y., Tang C., and Du J. (2009) Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179 10.1161/ATVBAHA.108.179333 [DOI] [PubMed] [Google Scholar]
- 23. Fu X., Zhou K., Gao Q., Zheng S., Chen H., Li P., Zhang Y., Suo K., Simoncini T., and Wang T. (2013) 17β-estradiol attenuates atherosclerosis development: The possible role of hydrogen sulfide. Int. J. Cardiol. 167, 1061–1063 10.1016/j.ijcard.2012.10.071 [DOI] [PubMed] [Google Scholar]
- 24. Zhou K., Gao Q., Zheng S., Pan S., Li P., Suo K., Simoncini T., Wang T., and Fu X. (2013) 17β-estradiol induces vasorelaxation by stimulating endothelial hydrogen sulfide release. Mol. Hum. Reprod. 19, 169–176 10.1093/molehr/gas044 [DOI] [PubMed] [Google Scholar]
- 25. Yuan G., Vasavda C., Peng Y. J., Makarenko V. V., Raghuraman G., Nanduri J., Gadalla M. M., Semenza G. L., Kumar G. K., Snyder S. H., and Prabhakar N. R. (2015) Protein kinase G-regulated production of H2S governs oxygen sensing. Sci. Signal. 8, ra37 10.1126/scisignal.2005846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Renga B., Bucci M., Cipriani S., Carino A., Monti M. C., Zampella A., Gargiulo A., d'Emmanuele di Villa Bianca R., Distrutti E., and Fiorucci S. (2015) Cystathionine γ-lyase, a H2S-generating enzyme, is a GPBAR1-regulated gene and contributes to vasodilation caused by secondary bile acids. Am. J. Physiol. Heart Circ. Physiol. 309, H114–H126 10.1152/ajpheart.00087.2015 [DOI] [PubMed] [Google Scholar]
- 27. Kennelly P. J., and Krebs E. G. (1991) Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. J. Biol. Chem. 266, 15555–15558 [PubMed] [Google Scholar]
- 28. Madhani M., Okorie M., Hobbs A. J., and MacAllister R. J. (2006) Reciprocal regulation of human soluble and particulate guanylate cyclases in vivo. Br. J. Pharmacol. 149, 797–801 10.1038/sj.bjp.0706920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shah R. C., Sanker S., Wood K. C., Durgin B. G., and Straub A. C. (2018) Redox regulation of soluble guanylyl cyclase. Nitric Oxide 76, 97–104 10.1016/j.niox.2018.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Simoncini T., Hafezi-Moghadam A., Brazil D. P., Ley K., Chin W. W., and Liao J. K. (2000) Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 407, 538–541 10.1038/35035131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen H., Levine Y. C., Golan D. E., Michel T., and Lin A. J. (2008) Atrial natriuretic peptide-initiated cGMP pathways regulate vasodilator-stimulated phosphoprotein phosphorylation and angiogenesis in vascular endothelium. J. Biol. Chem. 283, 4439–4447 10.1074/jbc.M709439200 [DOI] [PubMed] [Google Scholar]
- 32. Kumar P., Wu Q., Chambliss K. L., Yuhanna I. S., Mumby S. M., Mineo C., Tall G. G., and Shaul P. W. (2007) Direct interactions with Gαi and G βγ mediate nongenomic signaling by estrogen receptor α. Mol. Endocrinol. 21, 1370–1380 10.1210/me.2006-0360 [DOI] [PubMed] [Google Scholar]
- 33. Wu Q., Chambliss K., Lee W. R., Yuhanna I. S., Mineo C., and Shaul P. W. (2013) Point mutations in the ERα Gαi binding domain segregate nonnuclear from nuclear receptor function. Mol. Endocrinol. 27, 2–11 10.1210/me.2011-1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bibli S. I., Hu J., Sigala F., Wittig I., Heidler J., Zukunft S., Tsilimigras D. I., Randriamboavonjy V., Wittig J., Kojonazarov B., Schürmann C., Siragusa M., Siuda D., Luck B., Abdel Malik R., et al. (2019) Cystathionine γ lyase sulfhydrates the RNA binding protein human antigen R to preserve endothelial cell function and delay atherogenesis. Circulation 139, 101–114 10.1161/CIRCULATIONAHA.118.034757 [DOI] [PubMed] [Google Scholar]
- 35. Hofmann F., Feil R., Kleppisch T., and Schlossmann J. (2006) Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol. Rev. 86, 1–23 10.1152/physrev.00015.2005 [DOI] [PubMed] [Google Scholar]
- 36. Koika V., Zhou Z., Vasileiadis I., Roussos C., Finetti F., Monti M., Morbidelli L., and Papapetropoulos A. (2010) PKG-I inhibition attenuates vascular endothelial growth factor-stimulated angiogenesis. Vascul. Pharmacol. 53, 215–222 10.1016/j.vph.2010.08.004 [DOI] [PubMed] [Google Scholar]
- 37. Wooldridge A. A., MacDonald J. A., Erdodi F., Ma C., Borman M. A., Hartshorne D. J., and Haystead T. A. (2004) Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of serine 695 in response to cyclic nucleotides. J. Biol. Chem. 279, 34496–34504 10.1074/jbc.M405957200 [DOI] [PubMed] [Google Scholar]
- 38. Ellerbroek S. M., Wennerberg K., and Burridge K. (2003) Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 278, 19023–19031 10.1074/jbc.M213066200 [DOI] [PubMed] [Google Scholar]
- 39. Schlossmann J., Ammendola A., Ashman K., Zong X., Huber A., Neubauer G., Wang G. X., Allescher H. D., Korth M., Wilm M., Hofmann F., and Ruth P. (2000) Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Iβ. Nature 404, 197–201 10.1038/35004606 [DOI] [PubMed] [Google Scholar]
- 40. Pfeifer A., Klatt P., Massberg S., Ny L., Sausbier M., Hirneiss C., Wang G. X., Korth M., Aszódi A., Andersson K. E., Krombach F., Mayerhofer A., Ruth P., Fässler R., and Hofmann F. (1998) Defective smooth muscle regulation in cGMP kinase I-deficient mice. EMBO J. 17, 3045–3051 10.1093/emboj/17.11.3045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weber S., Bernhard D., Lukowski R., Weinmeister P., Wörner R., Wegener J. W., Valtcheva N., Feil S., Schlossmann J., Hofmann F., and Feil R. (2007) Rescue of cGMP kinase I knockout mice by smooth muscle specific expression of either isozyme. Circ. Res. 101, 1096–1103 10.1161/CIRCRESAHA.107.154351 [DOI] [PubMed] [Google Scholar]
- 42. Surks H. K., Mochizuki N., Kasai Y., Georgescu S. P., Tang K. M., Ito M., Lincoln T. M., and Mendelsohn M. E. (1999) Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Iα. Science 286, 1583–1587 10.1126/science.286.5444.1583 [DOI] [PubMed] [Google Scholar]
- 43. Kim H. Y., Lee Y. J., Han B. H., Yoon J. J., Ahn Y. M., Hong M. H., Tan R., Kang D. G., and Lee H. S. (2017) Mantidis ootheca induces vascular relaxation through PI3K/AKT-mediated nitric oxide-cyclic GMP-protein kinase G signaling in endothelial cells. J Physiol. Pharmacol. 68, 215–221 [PubMed] [Google Scholar]
- 44. Jackson-Weaver O., Osmond J. M., Riddle M. A., Naik J. S., Gonzalez Bosc L. V., Walker B. R., and Kanagy N. L. (2013) Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2+-activated K+ channels and smooth muscle Ca2+ sparks. Am. J. Physiol. Heart Circ. Physiol. 304, H1446–H1454 10.1152/ajpheart.00506.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mustafa A. K., Sikka G., Gazi S. K., Steppan J., Jung S. M., Bhunia A. K., Barodka V. M., Gazi F. K., Barrow R. K., Wang R., Amzel L. M., Berkowitz D. E., and Snyder S. H. (2011) Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 109, 1259–1268 10.1161/CIRCRESAHA.111.240242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Holt A. W., Martin D. N., Shaver P. R., Adderley S. P., Stone J. D., Joshi C. N., Francisco J. T., Lust R. M., Weidner D. A., Shewchuk B. M., and Tulis D. A. (2016) Soluble guanylyl cyclase-activated cyclic GMP-dependent protein kinase inhibits arterial smooth muscle cell migration independent of VASP-serine 239 phosphorylation. Cell. Signal. 28, 1364–1379 10.1016/j.cellsig.2016.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Marathe N., Rangaswami H., Zhuang S., Boss G. R., and Pilz R. B. (2012) Pro-survival effects of 17β-estradiol on osteocytes are mediated by nitric oxide/cGMP via differential actions of cGMP-dependent protein kinases I and II. J. Biol. Chem. 287, 978–988 10.1074/jbc.M111.294959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fallahian F., Karami-Tehrani F., Salami S., and Aghaei M. (2011) Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J. 278, 3360–3369 10.1111/j.1742-4658.2011.08260.x [DOI] [PubMed] [Google Scholar]
- 49. Keung W., Chan M. L., Ho E. Y., Vanhoutte P. M., and Man R. Y. (2011) Non-genomic activation of adenylyl cyclase and protein kinase G by 17β-estradiol in vascular smooth muscle of the rat superior mesenteric artery. Pharmacol. Res. 64, 509–516 10.1016/j.phrs.2011.05.010 [DOI] [PubMed] [Google Scholar]
- 50. Wu D., Hu Q., and Zhu D. (2018) An update on hydrogen sulfide and nitric oxide interactions in the cardiovascular system. Oxid. Med. Cell. Longev. 2018, 4579140 10.1155/2018/4579140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao W., Zhang J., Lu Y., and Wang R. (2001) The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 20, 6008–6016 10.1093/emboj/20.21.6008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Predmore B. L., Julian D., and Cardounel A. J. (2011) Hydrogen sulfide increases nitric oxide production from endothelial cells by an Akt-dependent mechanism. Front. Physiol. 2, 104 10.3389/fphys.2011.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stratton R. C., Squires P. E., and Green A. K. (2010) 17β-estradiol elevates cGMP and, via plasma membrane recruitment of protein kinase GIα, stimulates Ca2+ efflux from rat hepatocytes. J. Biol. Chem. 285, 27201–27212 10.1074/jbc.M110.103630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sabrane K., Kruse M. N., Fabritz L., Zetsche B., Mitko D., Skryabin B. V., Zwiener M., Baba H. A., Yanagisawa M., and Kuhn M. (2005) Vascular endothelium is critically involved in the hypotensive and hypovolemic actions of atrial natriuretic peptide. J. Clin. Invest. 115, 1666–1674 10.1172/JCI23360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tokudome T., Horio T., Kishimoto I., Soeki T., Mori K., Kawano Y., Kohno M., Garbers D. L., Nakao K., and Kangawa K. (2005) Calcineurin-nuclear factor of activated T cells pathway-dependent cardiac remodeling in mice deficient in guanylyl cyclase A, a receptor for atrial and brain natriuretic peptides. Circulation 111, 3095–3104 10.1161/CIRCULATIONAHA.104.510594 [DOI] [PubMed] [Google Scholar]
- 56. Belo N. O., Sairam M. R., and Dos Reis A. M. (2008) Impairment of the natriuretic peptide system in follitropin receptor knockout mice and reversal by estradiol: Implications for obesity-associated hypertension in menopause. Endocrinology 149, 1399–1406 10.1210/en.2007-0572 [DOI] [PubMed] [Google Scholar]
- 57. Liu J., Ward A., Gao J., Dong Y., Nishio N., Inada H., Kang L., Yu Y., Ma D., Xu T., Mori I., Xie Z., and Xu X. Z. (2010) C. elegans phototransduction requires a G protein-dependent cGMP pathway and a taste receptor homolog. Nat. Neurosci. 13, 715–722 10.1038/nn.2540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mombouli J. V., and Vanhoutte P. M. (1999) Endothelial dysfunction: From physiology to therapy. J. Mol. Cell. Cardiol. 31, 61–74 10.1006/jmcc.1998.0844 [DOI] [PubMed] [Google Scholar]
- 59. Kim K. H., Young B. D., and Bender J. R. (2014) Endothelial estrogen receptor isoforms and cardiovascular disease. Mol. Cell. Endocrinol. 389, 65–70 10.1016/j.mce.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Levin E. R. (2009) Plasma membrane estrogen receptors. Trends Endocrinol. Metab. 20, 477–482 10.1016/j.tem.2009.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zheng S., Huang J., Zhou K., Zhang C., Xiang Q., Tan Z., Wang T., and Fu X. (2011) 17β-Estradiol enhances breast cancer cell motility and invasion via extra-nuclear activation of actin-binding protein ezrin. PLoS One 6, e22439 10.1371/journal.pone.0022439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dennis M. K., Field A. S., Burai R., Ramesh C., Petrie W. K., Bologa C. G., Oprea T. I., Yamaguchi Y., Hayashi S., Sklar L. A., Hathaway H. J., Arterburn J. B., and Prossnitz E. R. (2011) Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 127, 358–366 10.1016/j.jsbmb.2011.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.