

Abstract
Alexander disease (AxD) is an often fatal astrogliopathy caused by dominant gain-of-function missense mutations in the glial fibrillary acidic protein (GFAP) gene. The mechanism by which the mutations produce the AxD phenotype is not known. However, the observation that features of AxD are displayed by mice that express elevated levels of GFAP from a human WT GFAP transgene has contributed to the notion that the mutations produce AxD by increasing accumulation of total GFAP above some toxic threshold rather than the mutant GFAP being inherently toxic. A possible mechanism for accumulation of GFAP in AxD patients is that the mutated GFAP variants are more stable than the WT, an attribution abetted by observations that GFAP complexes containing GFAP variants are more resistant to solvent extraction. Here we tested this hypothesis by determining the relative levels of WT and mutant GFAP in three individuals with AxD, each of whom carried a common but different GFAP mutation (R79C, R239H, or R416W). Mass spectrometry analysis identified a peptide specific to the mutant or WT GFAP in each patient, and we quantified this peptide by comparing its signal to that of an added [15N]GFAP standard. In all three individuals, the level of mutant GFAP was less than that of the WT. This finding suggests that AxD onset is due to an intrinsic toxicity of the mutant GFAP instead of it acting indirectly by being more stable than WT GFAP and thereby increasing the total GFAP level.
Keywords: astrocyte, genetic disease, intermediate filament, MS, mutant, protein stability, Alexander disease, astrogliopathy, glial fibrillary acidic protein (GFAP)
Introduction
Alexander disease (AxD)3 is an often fatal astrogliopathy caused by dominant gain-of-function coding mutations in the GFAP gene (1; reviewed in Refs. 2–4). This gene encodes glial fibrillary acidic protein, an intermediate filament protein found predominantly in astrocytes in the central nervous system. A defining feature of AxD is the accumulation in astrocytes of GFAP-rich protein aggregates called Rosenthal fibers (5, 6). Astrocytes from human patients and animal models display multiple defects (reviewed in Ref. 7), including decreased glutamate and potassium transport (8), decreased proteasome activity (9), increased production of nitric oxide and proinflammatory chemokines (10–13), defective calcium signaling (14), and aberrant mechanosensitive signaling (15). A widely held view of how the presence of mutant GFAP leads to these defects is that, rather than being toxic per se, it is more stable than the WT and thereby causes accumulation of GFAP above a critical toxic threshold (4, 8, 16, 17). The origin of this view was the observation that mice expressing elevated levels of WT GFAP from a human GFAP transgene display features of AxD (18). In addition, the presence of this WT transgene dramatically enhances the pathology displayed by AxD model mice engineered to express an endogenous mutated GFAP (8, 11, 16). Also contributing to the toxic threshold hypothesis is the analysis of transgenic mouse lines expressing different levels of the AxD-causing human R239H mutant GFAP (19). This analysis led to the conclusion that expression of the mutant GFAP does not in itself cause Rosenthal fiber formation but that these aggregates form when the level of total GFAP increases by at least some amount between 2.7% to 30%. Other contributors to this hypothesis are multiple observations that multimeric forms of GFAP are more resistant to solubilization when they contain mutant GFAP (e.g. Ref. 6, 20–23).
As a test of this toxic threshold hypothesis, we determined whether mutant GFAP is indeed more stable than the WT. Using MS, the relative levels of mutant and WT GFAP were analyzed in three AxD patients, each of whom carried one of the more prevalently occurring disease-causing GFAP mutations: R79C, R239H, or R416W. The level of mutant GFAP in all three patients was found to be less than that of the WT. This suggests that mutant GFAP is not inherently more stable than the WT and, consequently, that onset of AxD is due to the intrinsic toxicity of mutant GFAP rather than an increase in the total GFAP pool.
Results
General approach
We investigated whether mutant GFAP is more stable than WT GFAP by using MS to determine the relative amounts of mutant and WT GFAP in AxD patients harboring the R79C, R239H, or R416W mutation. A peptide unique to either the mutant or WT protein was quantified, and its molar concentration was compared to that of total GFAP obtained by quantifying several peptides common to both the mutant and WT forms.
Identification of WT- and mutant-specific peptides
The mutant and WT diagnostic peptides were selected based on their efficient detection by LC-MS/MS and sequence uniqueness to GFAP in the predicted human proteome using the PROWL algorithm ProteinInfo tool to search the NCBI database (http://prowl.rockefeller.edu).4 For the R79C case, we observed a GFAP tryptic peptide specific for WT GFAP (FASYIEK, residues 80–86). For the R416W patient, we observed a tryptic peptide specific for the mutant protein (TVEMWDGEVIK, residues 412–422). For the R239H patient, we were unable to detect either a WT-specific or mutant-specific peptide using trypsin digestion, but by using chymotrypsin, we detected the mutant-specific peptide KEIHTQY (residues 236–242). Spectra for these three diagnostic peptides are shown in Fig. 1.
Figure 1.

MS/MS spectra used for identifying peptides specific for WT or mutant GFAP. A–C, peptides are diagnostic for WT GFAP in an R79C patient (A), R416W mutant GFAP (B), and R239H mutant GFAP (C). In each panel, the first spectrum is for the unlabeled peptide and the second is for the corresponding 15N-labeled peptide. The ion m/z ratios were calculated using the Protein Prospector MS Product tool (http://prospector.ucsf.edu).4
Identification of common peptides
The total amount of GFAP in the patient samples was measured by identifying proteotypic GFAP peptides not affected by the mutations. For the R79C and R416W patients, these were the tryptic peptides FADLTDAAAR (residues 261–270), LADVYQAELR (residues 112–121), ALAAELNQLR (residues 96–105), and LLEGEENR (residues 369–376). For the R239H patient sample, these were the chymotryptic peptides RQEADEATL (residues 173–181) and ARQQVHVEL (residues 216–224). Because of difficulty in detecting ARQQVHVEL (Table 1), for R239H we also used IEKVRF (residues 84–89), which is also present in desmin and peripherin. Both of these nontarget proteins are at extremely low concentrations in the brain compared to GFAP (The Human Protein Atlas, https://www.proteinatlas.org (44)).4 The fragment ions used for quantitation of each common peptide are listed in Table S1.
Table 1.
Mutant GFAP as a percent of total GFAP in R79C, R416W, and R239H Alexander disease patients
Total GFAP is the sum of the amount of patient GFAP and 15N standard GFAP analyzed. The control peptides for trypsin digestion were FADLTDAAAR (1), LADVYQAELR (2), ALAAELNQLR (3), and LLEGEENR (4). The control peptides for chymotrypsin digestion were ARQQVHVEL (1), RQEADEATL (2), and IEKVRF (3). IEKVRF is also present in desmin and peripherin, but these proteins are not expected to be present in the brain at significant levels compared to GFAP. Were significant levels indeed present, their inclusion would lead to overestimation of the amount of WT GFAP present and, thus, underestimation of the percent mutant GFAP. With inclusion of IEKVRF, the calculated percent mutant GFAP was actually slightly higher than that obtained if it were excluded (29% versus 27%). ND, not detected in the patient sample, the 15N standard, or both; Amt, amount; Dig, digestion; MUT, mutant.
| Amt 15N Std |
Total GFAP |
Dig time |
% Mutant |
Peptide Ratios (Patient/15N Std) Intact GFAP Band |
Peptide Ratios (Patient/15N-Std) Degraded GFAP Band |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ng | ng | h | Intact | Degraded | Diagnostic | Controls | Diagnostic | Controls | ||||||
| R79C patient, trypsin digestion | WT | 1 | 2 | 3 | 4 | WT | 1 | 2 | 3 | 4 | ||||
| 50 | 120 | 40 | 21% | 25% | 0.76 | 0.99 | 1.00 | 0.96 | 0.88 | 1.41 | 1.90 | 2.09 | 1.83 | 1.72 |
| 100 | 170 | 18 | 22% | ND | 0.46 | 0.63 | 0.59 | 0.53 | 0.59 | ND | ND | ND | 0.95 | ND |
| 100 | 170 | 40 | 21% | 23% | 0.48 | 0.65 | 0.61 | 0.63 | 0.56 | 0.85 | 1.15 | 1.18 | 1.07 | 0.99 |
| 150 | 220 | 40 | 19% | 22% | 0.38 | 0.51 | 0.47 | 0.47 | 0.42 | 0.57 | 0.77 | 0.78 | 0.71 | 0.66 |
| Average ± SD | 21% ± 1% | 23% ± 2% | ||||||||||||
| R416W patient, trypsin digestion | MUT | 1 | 2 | 3 | 4 | MUT | 1 | 2 | 3 | 4 | ||||
| 13.5 | 90.5 | 40 | 45% | 19% | 2.81 | 6.13 | 6.45 | 5.89 | 6.70 | 0.25 | 1.35 | 1.53 | 1.32 | 0.96 |
| 36 | 113 | 18 | 47% | 20% | 1.11 | 2.34 | 2.55 | 2.28 | 2.32 | 0.11 | 0.54 | 0.66 | 0.56 | 0.35 |
| 36 | 113 | 40 | 45% | 22% | 1.04 | 2.27 | 2.45 | 2.16 | 2.43 | 0.11 | 0.55 | 0.56 | 0.52 | 0.38 |
| 58.5 | 135.5 | 40 | 45% | 22% | 0.67 | 1.53 | 1.54 | 1.43 | 1.41 | 0.06 | 0.32 | 0.34 | 0.30 | 0.20 |
| Average ± SD | 46% ± 1% | 21% ± 2% | ||||||||||||
| R239H patient, chymotrypsin digestion | MUT | 1 | 2 | 3 | MUT | 1 | 2 | 3 | ||||||
| 10 | 190 | 72 | 30% | ND | 10.43 | ND | 34.99 | ND | ND | ND | ND | ND | ||
| 50 | 230 | 48 | 33% | ND | 1.97 | ND | 7.17 | 4.71 | ND | ND | 7.07 | ND | ||
| 50 | 230 | 72 | 22% | ND | 1.55 | ND | 6.56 | 7.28 | ND | ND | 13.97 | ND | ||
| 100 | 280 | 72 | 33% | ND | 0.88 | 3.21 | 2.91 | 2.00 | ND | ND | 4.30 | ND | ||
| Average ± SD | 29% ± 5% | |||||||||||||
Examination of degraded GFAP
GFAP Immunoblots of the patient brain extracts displayed the typically observed pattern of a major band at the expected position for intact human GFAP of 50 kDa and a series of bands in the 35- to 45-kDa range that are presumed to be partially degraded GFAP (Fig. 2) (24–27). Because the WT and mutant GFAP composition of the degraded protein might be a sensitive indicator of their relative stabilities, this material was analyzed in addition to that in the intact region for the R79C and R416W patients (attempts to perform this analysis for the R239H patient were unsuccessful, probably because of the reduced amount of GFAP in the degraded size range coupled with decreased ionization efficiency or digestion of the chymotryptic peptides relative to the tryptic peptides used for the R79C and R416W patients).
Figure 2.
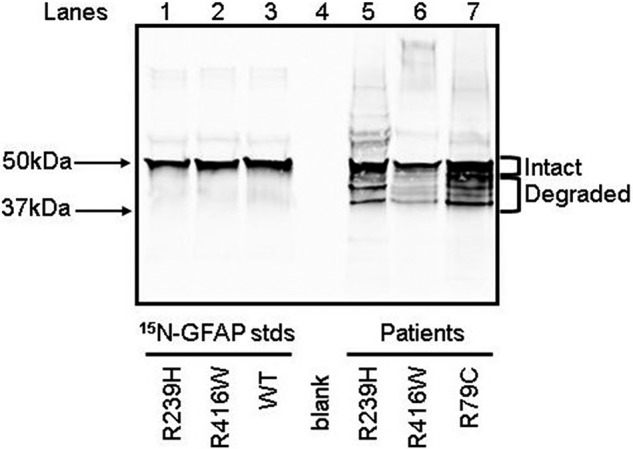
GFAP immunoblot of 15N-labeled GFAP proteins and patient total homogenate samples. For each 15N-labeled GFAP standard (lanes 1–3), 7.5 ng of purified standard was loaded on the gel. The amounts of total protein applied for the R239H, R416W, and R79C patient samples (lanes 5–7) were 200 ng, 4,600 ng, and 270 ng, respectively. These patient protein amounts reflected differences in GFAP content, which were determined by densitometric comparison of GFAP staining intensity in pilot gels with that of the 15N-labeled GFAP standards. The negative control (lane 4) had no protein loaded. The positions of relevant molecular mass markers run concurrently are indicated.
Use of an internal standard for quantitation
The signals obtained for the various peptides were quantified by comparison to signals obtained from a known amount of full-length 15N-labeled human GFAP added to each sample. The 15N-labeled GFAP was either the mutant or WT form, depending on whether the type-specific peptide used for identification was from the mutant or the WT. Thus, WT 15N-labeled GFAP was added to the R79C patient sample, and 239H and 416W mutant 15N-labeled GFAP was added to the R239H and R416W patient samples, respectively (see “Experimental procedures” for details). These 15N-labeled GFAP standards were added to the samples prior to gel electrophoresis. However, because these were full-length protein standards lacking significant degradation products, and so would not be present in the degraded bands following electrophoresis (Fig. 2), the same amount of 15N-labeled GFAP standard was run in a separate lane in the gel and then excised and combined with the excised patient degradation bands prior to extraction for analysis (see “Experimental procedures” for details).
Calculation method
The fraction of total GFAP that was mutant (or WT) was obtained by (A) determining the amount of the unique mutant (or WT) peptide in the patient sample relative to the same peptide in the 15N-labeled GFAP internal standard, (B) determining the amount of total GFAP relative to the 15N-labeled standard using GFAP peptides common to both mutant and WT forms, and (C) dividing A by B. For uniformity, results are presented in Table 1 as the percentage of total GFAP attributable to the mutant form (percent mutant); thus, for R79C, the experimentally determined WT percentage was subtracted from 100. The center and right sections of Table 1 show the data used to calculate these percentages (the signals from the diagnostic and control peptides in the patient samples normalized to those from the 15N-labeled standard). An illustration of the quantitation procedure is provided in Fig. 3 for the peptide specific for the R416W mutant GFAP (TVEMWDGEVIK) and a peptide common to both the mutant and WT GFAP (FADLTDAAAR).
Figure 3.

Illustration of the quantitation method. Data shown are from the 40-h tryptic digest of the intact GFAP band from the run using 36 ng of 15N-labeled mutant 416W protein standard and 113 ng of unlabeled R416W patient GFAP (Table 1). A and B, elution chromatograms for detection of the product ions from the R416W mutant–specific peptide TVEMWDGEVIK that were used for quantitation. A, unlabeled patient GFAP. B, [15N]GFAP internal standard. C and D, data for the common peptide FADLTDAAAR. Note that, as expected, the unlabeled and 15N-labeled peptides coelute. The table insets in B and D list the product ions, the peak areas of their signals, and the ratios of the peak area of each unlabeled ion to that of the labeled ion. These ratios provide measures of the amount of mutant GFAP and total GFAP, respectively, in the patient sample relative to the 15N standard. The ratio of these two values is the fraction of total patient GFAP that is the mutant form. The actual calculations summarized in Table 1 include additional values from other common peptides. Data for each individual peak area are provided in Table S1.
Control experiments
The calculation method used should yield the same result for any amount of 15N-labeled standard added to the samples, provided that the amount was sufficient for its peptides to be detected. The data presented in Table 1 confirm this expectation. Varying the added 15N-labeled standard up to 10-fold had no apparent effect on the percent mutant quantitation. Table 1 also shows results from testing whether the duration of proteolytic digestion was appropriate. As shown in Table 1, reducing the time of trypsin digestion from our standard of 40 h to 18 h or the time of chymotrypsin digestion from our standard of 72 h to 48 h had a marginal, if any, effect on quantitation.
Relative level of mutant GFAP in AxD patients
None of the three AxD patients had a higher percentage of mutant GFAP than WT in the intact band (Table 1), counter to expectation if mutant GFAP contributed to disease by having greater stability. The percentages of total GFAP attributed to the mutant form for the R79C patient (21%) and R239H patient (29%) were actually much lower than that of the WT, suggesting that these mutant GFAPs are less stable rather than more stable. Similarly, less mutant GFAP than WT was observed in the degraded bands. In the case of the R79C patient, the percentage of mutant GFAP in the degraded band was about the same as that in the intact band, whereas for the R416W patient, it was less than that in the intact band.
Discussion
The objective of this study was to test a proposed mechanism for AxD that posits that mutant GFAP is not directly toxic but has greater stability than WT GFAP and thus results in accumulation of GFAP to toxic levels. We used MS to determine the relative levels of mutant and WT GFAP by comparing the amount of a patient-specific peptide with that of common peptides, using recombinant 15N-labeled GFAP as an internal standard. A similar approach was used to determine the ratio of mutant to WT K-Ras proteins in clinical samples (28, 29), except that in these other studies, internal standards were 13C/15N-labeled diagnostic peptides added after trypsin digestion. Our internal standards were full-length 15N-labeled GFAP added to the initial homogenates, which provides correction for losses during sample processing (30).
Contrary to expectation for a mechanism based on greater stability of mutant GFAP, for each of the three AxD patients analyzed, the mutant GFAP was present at a similar or lower level than the WT form. This lower level of mutant GFAP could result from reduced transcription, mRNA stability, or translation rather than reduced protein stability, but this would require that each of the missense mutations acts at the nucleotide level. Evidence excluding decreased transcription or mRNA stability was obtained by Jones et al. (14), who found no difference in transcript levels of the mutant and WT GFAP mRNAs in astrocytes differentiated from induced pluripotent stem cells (iPSCs) isolated from AxD patients carrying either an R88C or R416W mutation. An effect on translation is also unlikely for these samples. In some organisms, codon use does affect translation, either by codon recognition efficiency or by dictating the secondary structure of the mRNA in the initiation region (reviewed in Ref. 31), but humans show no evidence of codon selection (32). Furthermore, the R79C and R239H mutations are to more frequently used codons rather than to ones that are rarer; the codons and their percentage of use in humans are CGC(0.79)→UGC(0.92) for R79C, and CGC(0.79)→CAC(1.15) for R239H (45). In addition, the nucleotide changes have no effect on the predicted mRNA structure in the region of the translation start site (data not shown; structures were analyzed using RNAfold via http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi).4 Thus, the most plausible explanation for the lower level of mutant GFAP is instability of the protein itself. Reduced stability provides an explanation for the otherwise puzzling observation that total GFAP levels are actually lower in the spinal cord of R236H mice than in the WT (33).
A mechanism for reduced stability of mutant GFAP arises from an analysis of the crystal structure of the human GFAP 1B rod domain (34). The structure obtained led the authors to conclude that many of the mutations in the central rod domain producing AxD would destabilize filament formation, particularly at the initial step of dimerization (a caveat, however, is that this prediction was also made for V115I, which is not believed to be disease-causing (35)). The suggested defect in polymerization of mutant GFAP would result in a greater fraction of the mutant form than WT being present as monomers and thus more available for degradation. This mechanism could explain not only the instability of R79C and R239H GFAP but also why the stability of R416W GFAP is similar to that of the WT; R79C and R239H are in the central rod domain and thus might affect dimerization, whereas R416W is in the tail region and is believed to only affect subsequent steps in polymerization (21). Several prior observations are consistent with compromised polymerization of mutant GFAP. Using a yeast two-hybrid system, Nielsen et al. (36) observed decreased dimerization for the several common AxD GFAP mutations they tested. Tian et al. (37) found that, in U251 cells expressing either GFP-tagged WT or tagged R239C GFAP, the fraction of GFAP present in the soluble fraction (monomers and short oligomers) was significantly greater for the mutant.
The possibility that analysis of the partially degraded forms of GFAP might provide a more sensitive measure of relative stabilities was not realized. Instead of containing more of the presumptively less stable R79C mutant GFAP, the percentage of R79C mutant GFAP in the partially degraded GFAP was similar to that in the intact GFAP (23% versus 21%). In the case of the R416W patient, the calculated percentage of mutant GFAP in the degraded band (21%) was even less than that in the intact band (46%) (Table 1). A likely explanation for the lower calculated percentage of mutant GFAP in the degraded R416W GFAP is that some of the cleaved GFAPs had the R416W proteotypic peptide removed. The sequences of the degradation bands present in our AxD patient samples are not known, but the series of bands observed is quite similar to those seen in a variety of other control and diseased post-mortem samples (24–27) and has been attributed to digestion by calpain (27, 38, 39). N-terminal sequencing of several of these large degradation products obtained from normal brain white matter, multiple sclerosis plaques, leucotomy scars (24) and spinal cords of patients with ALS (38), and by complete in vitro calpain digestion of GFAP (39), yielded cleavage sites after Ala-40, Asn-59, and Thr-383. Cleavage at Ala-40 or Asn-59 and/or Thr-383 could generate the series of degradation bands in our samples (Fig. 2), with Thr-383 cleavage removing the R416W diagnostic peptide. Because this peptide identifies the mutant protein, its loss would result in underestimation of the contribution of mutant protein to the total. In contrast, the R79C site is expected to be retained in the degradation products examined. Two possible explanations for the percentage of R79C GFAP being similar in the intact and degraded bands are that the clipped mutant GFAPs may also be less stable than their WT counterparts, or that calpain degradation may occur predominantly post-mortem (39) so that the composition of the degradation products reflects that of the intact GFAP at the time of death.
A remaining question is whether our finding of less mutant than WT GFAP applies to all astrocytes or only to those affected (or unaffected) by the disorder. Using Rosenthal fiber content as an indication of affected astrocytes, our data, in conjunction with that of Heaven et al. (6), suggest that the status of the astrocytes is not a critical parameter. Using urea extraction to distinguish between GFAP in normal filaments and that in Rosenthal fibers, that study estimated that about two-thirds of the GFAP in the R79C sample was associated with Rosenthal fibers, but little to none was in the R239H sample, whereas the observed percentage of mutant GFAP was similar in both samples (21% and 29%, respectively). However, a more rigorous examination of these variables with a larger sample size is required to establish these relationships.
Our finding that the levels of mutant GFAP are similar to or lower than WT levels suggests that the mutant GFAP does not initiate AxD by causing accumulation of normally functioning GFAP, but that the mutations per se produce a toxic response. This same conclusion was reached by Jones et al. (14) in their study of astrocytes derived from AxD patient iPSCs. They observed that, compared with their gene-corrected controls, these cells displayed marked differences in both their RNA transcripts and their ability to propagate intercellular calcium waves but no difference in their GFAP protein levels, prompting the comment that “mutations in GFAP are sufficient.” An example of an effect due to a GFAP mutation rather than GFAP quantity is the finding of Tang et al. (9) that proteasome activity is inhibited more strongly by soluble R239C GFAP than by WT GFAP. Another is the finding of Jany et al. (33) that, although GFAP levels in the spinal cord of R236H mice were decreased compared with the WT, the level of Gfap mRNA was increased. Thus, similar to the findings of Jones et al. (14), mutant GFAP initiates changes in gene transcription in the absence of an increase in total GFAP levels. There are also multiple reports that, at comparable levels, mutant GFAP causes significantly more physiological changes, including aggregate formation, than WT GFAP (e.g. 10, 19, 23, 40).
If mutation of GFAP per se causes disease, then how does one account for the AxD-like characteristics of mice overexpressing WT GFAP? A model proposed by Li et al. (41) provides a resolution of this seeming paradox. They hypothesized that a rate-limiting step in GFAP polymer formation is sensitive to the presence of the GFAP mutations, a suggestion now supported by the crystal structure analysis (34). Thus, either elevated production of WT GFAP or the presence of mutant GFAP would cause abnormal accumulation of precursors. Some of these precursors might then be diverted into an alternative, aberrant pathway to produce toxic complexes. Consistent with this explanation, Tang et al. (9) suggested that formation of abnormal GFAP oligomers is responsible for proteasome inhibition. In light of this kinetic bottleneck model, we suggest an alternative to the conclusion of Tanaka et al. (19) that an increase in total GFAP of between 2.7% and 30%, caused by expression of a mutant transgene, is sufficient to produce Rosenthal fiber formation. We suggest that, rather than such a modest increase in total GFAP being toxic, their observations instead define the minimum percent of total GFAP that must be the mutant form to significantly interfere with polymerization. Our data suggest that having about 20% of the total GFAP as the mutant form is sufficient to produce disease (Table 1), and a prior report of an AxD mutation that caused occasional exon skipping suggests that about 10% mutant form is sufficient (42).
The greater resistance of mutant GFAP to solubilization by various solvents was another observation that contributed to the concept that the total amount of GFAP is the critical factor for toxicity in AxD. Although our data indicate that the stability of mutant GFAP is similar to or less than that of the WT, it remains possible that GFAP complexes containing even a minority of the mutant form have increased stability. However, several observations suggest that the GFAP extraction data, which were obtained in vitro, may not reflect differences in GFAP stability in vivo. Moody et al. (43) found that GFAP in the R236H mouse actually turns over more rapidly than in the WT, and in a Drosophila AxD model, Wang et al. (23) observed no change in GFAP levels when the solvent extraction of mutant GFAP was restored to that of the WT by overexpression of αB-crystallin.
In conclusion, we found that in three AxD patients the level of mutant GFAP is lower than that of the WT, indicating that mutant GFAP is less stable rather than more stable than the WT. This observation, together with those cited above of other laboratories, suggests a reordering of events in the development of AxD. Instead of mutant GFAP having increased stability and serving simply to increase the total level of GFAP above some toxic threshold, we suggest that the initial event is toxicity of mutant GFAP per se. Although this view places toxic effects of mutant GFAP as an event occurring prior to a rise in total GFAP levels, the importance of increasing levels of total GFAP is not discounted. A number of positive feedback loops have been proposed whereby pathological changes caused by mutant GFAP, such as induction of a stress response and inhibition of proteasome activity, result in increasing GFAP production (reviewed in Ref. 2). An irreversible point in disease progression may be reached when activation of the positive feedback loops by mutant GFAP more than compensates for its relative loss because of its instability.
Experimental procedures
Tissue samples
Use of post-mortem human tissue for this study was approved by the University of Alabama at Birmingham Institutional Review Board and abided by the Declaration of Helsinki principles. The sources and characteristics of the tissues are described in Table 2. They were stored at −80 °C until use. For sample preparation, ∼20 mg of tissue was homogenized by pipetting up and down 20 times in 1 ml of a total homogenate protein buffer consisting of 2% SDS, 6.25 mm Tris-HCl (pH 7.5), 5 mm EDTA, and Roche Complete Protease Inhibitor Mixture, as described previously (9). The resulting homogenates were boiled for 30 min, followed by centrifugation at 10,000 × g for 5 min, and the supernatant was used in subsequent experiments. Protein concentrations were determined by BCA assay (Pierce).
Table 2.
Description of Alexander disease tissues used
The tissues analyzed were from typical cases of the infantile form of AxD that have been reported previously as indicated. The R79C and R239H tissues were obtained from the University of Maryland Brain and Tissue Bank (UMB BTB). The R79C sample was a 1-cm-thick coronal section of parietal cerebrum from section 12R, produced by sectioning protocol 1. The R239H sample was frontal neocortical gray matter from section 3L, produced by sectioning protocol 2. The R416W tissue was a small fragment of neocortex and underlying white matter provided by James Goldman (Columbia Medical School); its post-mortem interval (PMI) is unknown. Although the exact locations of the tissues excised for analysis are not known, information about their Rosenthal fiber content is provided in Ref. 6, which used adjacent samples in a proteomics study of Rosenthal fibers. A fraction enriched for Rosenthal fibers contained 87% of the total GFAP in the R416W sample, 73% in the R79C sample, and 3% in the R239H sample, whereas 1% to 8% was observed in controls. Thus, most of the GFAP analyzed here for the R416W and R79C samples was presumably in Rosenthal fibers, but little if any of these aggregates were present in the R239H sample.
Production of [15N]GFAP standards
pET23b plasmids containing the WT and mutant human GFAP complementary DNA sequences were provided by Roy Quinlan (Durham University) (see Ref. 21 for the method of construction). The plasmids were transfected into Bl21pLysS Escherichia coli and grown in medium having more than 98% 15N (Cambridge Isotope Laboratories, Andover, MA), and the recombinant GFAP was purified as described by Der Perng et al. (21).
Immunoblotting
Immunoblotting was used to estimate the GFAP concentrations in patient samples and to determine the gel position of the intact and degraded GFAP bands. Purified recombinant human GFAP, made as described above, was used as a quantitation standard. Protein samples were boiled in Laemmli sample buffer for 5 min and electrophoresed for 60 min in a 4%–20% SDS-PAGE gel (Pierce) at 120 V and subsequently transferred to nitrocellulose membranes for 1 h at room temperature at 100 V, followed by blocking with 5% BSA in PBS for 1 h at room temperature. The membranes were then incubated overnight with agitation in a 1:5,000 dilution of Z0334 anti-GFAP rabbit polyclonal antibody (Dako, Carpinteria, CA), washed three times in 5% nonfat dry milk/0.1% (v/v) Tween 20/PBS (diluent buffer), incubated for 1 h at room temperature in a 1:15,000 dilution of an anti-rabbit 800 CW secondary antibody (Li-Cor Biosciences, Lincoln, NE), washed three times with diluent buffer and once with PBS, and then imaged on a Li-Cor Odyssey imager.
Sample processing for LC-MS/MS
Samples were processed by gel electrophoresis, excision of bands of interest, proteolytic digestion, and analysis by LC-MS/MS. Four analyses were performed for each patient sample. The same amount of patient GFAP was used for each patient sample set, but the amount of [15N]GFAP standard or the time of proteolysis varied (Table 1). Because the [15N]GFAP standard comigrates with the intact patient GFAP, to provide an internal standard for the degraded GFAP bands, the same amount of [15N]GFAP standard for each sample was electrophoresed in separate lanes and then combined with the corresponding excised band of degraded GFAP. Electrophoresis was performed by placing the samples in Laemmli sample buffer and running on a 4%–20% SDS-PAGE gel (Pierce) for 45 min at 150 V, followed by Coomassie staining for 30 min and destaining in 15% (v/v) methanol/10% (v/v) glacial acetic for 2 h at room temperature, as described previously (6). The intact GFAP bands, corresponding to ∼45–55 kDa, and degraded GFAP bands, corresponding to ∼35–45 kDa (Fig. 2), were excised separately from the patient samples, and each gel slice containing degraded GFAP was combined with the corresponding intact GFAP band of the [15N]GFAP standard.
For trypsin digestion of the R79C and R416W patient samples, the gel slices were further destained overnight in 100 mm ammonium bicarbonate/50% acetonitrile. The following day, they were washed three times in 100 mm ammonium bicarbonate, followed by dehydration in 100% acetonitrile. The fully dried gel slices were then digested at 37 °C for 18 or 40 h (Table 1) with sequencing-grade trypsin (Promega, Madison, WI) with a 1:20 trypsin:protein ratio, determined by comparing the stained gel region with that for 1.5 μg of BSA run on the same gel.
For chymotrypsin digestion of the R239H patient samples, the gel slices were further destained overnight in 50 mm ammonium bicarbonate/50% acetonitrile, washed three times in 100 mm Tris-HCl and 10 mm CaCl2 (pH 8.0), followed by dehydration in 100% acetonitrile. The fully dried gel slices were then digested at 25 °C for 48 or 72 h with sequencing-grade chymotrypsin (Promega), starting with a 1:10 chymotrypsin:protein ratio. Fresh aliquots of chymotrypsin at the same 1:10 ratio were added every 24 h.
Following proteolysis, the digestion solutions were transferred to separate tubes, and the gel slices were extracted further in buffer consisting of 5% formic acid/50% acetonitrile for 1 h at room temperature. The extraction buffer was then combined with the corresponding peptide digest solution, vacuum-centrifuged to dryness, and analyzed after being resuspended in 0.1% formic acid as the ion-pairing agent.
Targeted LC-MS/MS acquisition
Peptides were loaded onto a 15 cm × 75 μm ChromXP C18-CL 3-μm, 300 Å cHiP Nanoflex System (Eksigent, Foster City, CA) with a 250 nl/min flow rate and eluted using a 0%–50% acetonitrile/0.1% formic acid gradient over 30 min. Data were acquired using a TripleTOF 5600 system (Sciex) with an ion spray voltage of 2.3 kV, a declustering potential of 60 V, curtain gas of 20 p.s.i., nebulizer gas of 10 p.s.i., and an interface heating temperature of 120 °C. MS survey scans were acquired for 250 ms from 300–1250 m/z, and product ion scans targeting unlabeled and 15N-labeled GFAP peptides were collected with an accumulation time of 100 ms from 100–2,000 m/z.
Author contributions
M. R. H. and M. B. conceptualization; M. R. H. and M. B. data curation; M. R. H. and M. B. formal analysis; M. R. H. and L. W. investigation; M. R. H. and M. B. methodology; M. R. H. and M. B. writing-original draft; M. R. H., S. B., and M. B. writing-review and editing; S. B. and M. B. supervision; M. B. resources; M. B. funding acquisition; M. B. project administration.
Supplementary Material
Acknowledgments
We thank Roy Quinlan for providing the GFAP WT, R79C, R239H, and R416W expression vectors; Ming Der-Perng for providing details regarding purification of the recombinant GFAP proteins; the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore and James E. Goldman (Columbia University) for providing the patient tissues; and Albee Messing for comments on the manuscript. Funding for the Targeted Metabolomics and Proteomics Laboratory came in part from National Institutes of Health Grants P30DK079337 and P30AR50948.
This work was supported by NINDS, National Institutes of Health Grant P01NS42803 (to M. B.). Funds for the Sciex 5600 TripleTOF mass spectrometer came from National Institutes of Health Shared Instrumentation Grant S10 RR027822 (to S. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- AxD
- Alexander disease
- GFAP
- glial fibrillary acidic protein
- iPSC
- induced pluripotent stem cell.
References
- 1. Brenner M., Johnson A. B., Boespflug-Tanguy O., Rodriguez D., Goldman J. E., and Messing A. (2001) Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat. Genet. 27, 117–120 10.1038/83679 [DOI] [PubMed] [Google Scholar]
- 2. Brenner M., Goldman J. E., Quinlan R. A., and Messing A. (2009) in Astrocytes in (Patho)physiology of the Nervous System (Parpura V., and Haydon P., eds.) pp. 591–648, Springer, New York [Google Scholar]
- 3. Flint D., and Brenner M. (2011) in Leukodystrophies (Raymond G. V., Eichler F., Fatemi A., and Naidu S., eds.) pp. 106–129, Mac Keith Press, London [Google Scholar]
- 4. Messing A. (2018) Alexander disease. Handb. Clin. Neurol. 148, 693–700 10.1016/B978-0-444-64076-5.00044-2 [DOI] [PubMed] [Google Scholar]
- 5. Alexander W. S. (1949) Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain 72, 373–381 10.1093/brain/72.3.373 [DOI] [PubMed] [Google Scholar]
- 6. Heaven M. R., Flint D., Randall S. M., Sosunov A. A., Wilson L., Barnes S., Goldman J. E., Muddiman D. C., and Brenner M. (2016) Composition of Rosenthal fibers, the protein aggregate hallmark of Alexander disease. J. Proteome. Res. 15, 2265–2282 10.1021/acs.jproteome.6b00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sosunov A., Olabarria M., and Goldman J. E. (2018) Alexander disease: an astrocytopathy that produces a leukodystrophy. Brain Pathol. 28, 388–398 10.1111/bpa.12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sosunov A. A., Guilfoyle E., Wu X., McKhann G. M. 2nd, and Goldman J. E. (2013) Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J. Neurosci. 33, 7439–7450 10.1523/JNEUROSCI.4506-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tang G., Perng M. D., Wilk S., Quinlan R., and Goldman J. E. (2010) Oligomers of mutant glial fibrillary acidic protein (GFAP) inhibit the proteasome system in Alexander disease astrocytes, and the small heat shock protein αB-crystallin reverses the inhibition. J. Biol. Chem. 285, 10527–10537 10.1074/jbc.M109.067975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L., Hagemann T. L., Kalwa H., Michel T., Messing A., and Feany M. B. (2015) Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat. Commun. 6, 8966 10.1038/ncomms9966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olabarria M., Putilina M., Riemer E. C., and Goldman J. E. (2015) Astrocyte pathology in Alexander disease causes a marked inflammatory environment. Acta Neuropathol. 130, 469–486 10.1007/s00401-015-1469-1 [DOI] [PubMed] [Google Scholar]
- 12. Kondo T., Funayama M., Miyake M., Tsukita K., Era T., Osaka H., Ayaki T., Takahashi R., and Inoue H. (2016) Modeling Alexander disease with patient iPSCs reveals cellular and molecular pathology of astrocytes. Acta Neuropathol. Commun. 4, 69 10.1186/s40478-016-0337-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li L., Tian E., Chen X., Chao J., Klein J., Qu Q., Sun G., Sun G., Huang Y., Warden C. D., Ye P., Feng L., Li X., Cui Q., Sultan A., et al. (2018) GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of Alexander disease. Cell Stem Cell 23, 239–251.e6 10.1016/j.stem.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones J. R., Kong L., Hanna M. G. 4th, Hoffman B., Krencik R., Bradley R., Hagemann T., Choi J., Doers M., Dubovis M., Sherafat M. A., Bhattacharyya A., Kendziorski C., Audhya A., Messing A., and Zhang S. C. (2018) Mutations in GFAP Disrupt the Distribution and Function of Organelles in Human Astrocytes. Cell Rep. 25, 947–958.e4 10.1016/j.celrep.2018.09.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang L., Xia J., Li J., Hagemann T. L., Jones J. R., Fraenkel E., Weitz D. A., Zhang S. C., Messing A., and Feany M. B. (2018) Tissue and cellular rigidity and mechanosensitive signaling activation in Alexander disease. Nat. Commun. 9, 1899 10.1038/s41467-018-04269-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hagemann T. L., Connor J. X., and Messing A. (2006) Alexander disease-associated glial fibrillary acidic protein mutations in mice induce Rosenthal fiber formation and a white matter stress response. J. Neurosci. 26, 11162–11173 10.1523/JNEUROSCI.3260-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tang G., Xu Z., and Goldman J. E. (2006) Synergistic effects of the SAPK/JNK and the proteasome pathway on glial fibrillary acidic protein (GFAP) accumulation in Alexander disease. J. Biol. Chem. 281, 38634–38643 10.1074/jbc.M604942200 [DOI] [PubMed] [Google Scholar]
- 18. Messing A., Head M. W., Galles K., Galbreath E. J., Goldman J. E., and Brenner M. (1998) Fatal encephalopathy with astrocyte inclusions in GFAP transgenic mice. Am. J. Pathol. 152, 391–398 [PMC free article] [PubMed] [Google Scholar]
- 19. Tanaka K. F., Takebayashi H., Yamazaki Y., Ono K., Naruse M., Iwasato T., Itohara S., Kato H., and Ikenaka K. (2007) Murine model of Alexander disease: analysis of GFAP aggregate formation and its pathological significance. Glia 55, 617–631 10.1002/glia.20486 [DOI] [PubMed] [Google Scholar]
- 20. Hsiao V. C., Tian R., Long H., Der Perng M., Brenner M., Quinlan R. A., and Goldman J. E. (2005) Alexander-disease mutation of GFAP causes filament disorganization and decreased solubility of GFAP. J. Cell Sci. 118, 2057–2065 10.1242/jcs.02339 [DOI] [PubMed] [Google Scholar]
- 21. Der Perng M., Su M., Wen S. F., Li R., Gibbon T., Prescott A. R., Brenner M., and Quinlan R. A. (2006) The Alexander disease-causing glial fibrillary acidic protein mutant, R416W, accumulates into Rosenthal fibers by a pathway that involves filament aggregation and the association of αB-Crystallin and HSP27. Am. J. Hum. Genet. 79, 197–213 10.1086/504411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Y. S., Lim S. C., Chen M. H., Quinlan R. A., and Perng M. D. (2011) Alexander disease causing mutations in the C-terminal domain of GFAP are deleterious both to assembly and network formation with the potential to both activate caspase 3 and decrease cell viability. Exp. Cell Res. 317, 2252–2266 10.1016/j.yexcr.2011.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L., Colodner K. J., and Feany M. B. (2011) Protein misfolding and oxidative stress promote glial-mediated neurodegeneration in an Alexander disease model. J. Neurosci. 31, 2868–2877 10.1523/JNEUROSCI.3410-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dahl D., and Bignami A. (1975) Glial fibrillary acidic protein from normal and gliosed human brain: demonstration of multiple related polypeptides. Biochim. Biophys. Acta 386, 41–51 10.1016/0005-2795(75)90244-5 [DOI] [PubMed] [Google Scholar]
- 25. Martínez A., Carmona M., Portero-Otin M., Naudí A., Pamplona R., and Ferrer I. (2008) Type-dependent oxidative damage in frontotemporal lobar degeneration: cortical astrocytes are targets of oxidative damage. J. Neuropathol. Exp. Neurol. 67, 1122–1136 10.1097/NEN.0b013e31818e06f3 [DOI] [PubMed] [Google Scholar]
- 26. Newcombe J., Woodroofe M. N., and Cuzner M. L. (1986) Distribution of glial fibrillary acidic protein in gliosed human white matter. J. Neurochem. 47, 1713–1719 10.1111/j.1471-4159.1986.tb13079.x [DOI] [PubMed] [Google Scholar]
- 27. Zoltewicz J. S., Scharf D., Yang B., Chawla A., Newsom K. J., and Fang L. (2012) Characterization of antibodies that detect human GFAP after traumatic brain injury. Biomark. Insights 7, 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Q., Chaerkady R., Wu J., Hwang H. J., Papadopoulos N., Kopelovich L., Maitra A., Matthaei H., Eshleman J. R., Hruban R. H., Kinzler K. W., Pandey A., and Vogelstein B. (2011) Mutant proteins as cancer-specific biomarkers. Proc. Natl. Acad. Sci. U.S.A. 108, 2444–2449 10.1073/pnas.1019203108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Halvey P. J., Ferrone C. R., and Liebler D. C. (2012) GeLC-MRM quantitation of mutant KRAS oncoprotein in complex biological samples. J. Proteome. Res. 11, 3908–3913 10.1021/pr300161j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brun V., Dupuis A., Adrait A., Marcellin M., Thomas D., Court M., Vandenesch F., and Garin J. (2007) Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol. Cell. Proteomics 6, 2139–2149 10.1074/mcp.M700163-MCP200 [DOI] [PubMed] [Google Scholar]
- 31. Gingold H., and Pilpel Y. (2011) Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7, 481 10.1038/msb.2011.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. dos Reis M., Savva R., and Wernisch L. (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32, 5036–5044 10.1093/nar/gkh834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jany P. L., Hagemann T. L., and Messing A. (2013) GFAP expression as an indicator of disease severity in mouse models of Alexander disease. ASN Neuro. 5, e00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim B., Kim S., and Jin M. S. (2018) Crystal structure of the human glial fibrillary acidic protein 1B domain. Biochem. Biophys. Res. Commun. 503, 2899–2905 10.1016/j.bbrc.2018.08.066 [DOI] [PubMed] [Google Scholar]
- 35. Li R., Johnson A. B., Salomons G., Goldman J. E., Naidu S., Quinlan R., Cree B., Ruyle S. Z., Banwell B., D'Hooghe M., Siebert J. R., Rolf C. M., Cox H., Reddy A., Gutiérrez-Solana L. G., et al. (2005) Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander disease. Ann. Neurol. 57, 310–326 10.1002/ana.20406 [DOI] [PubMed] [Google Scholar]
- 36. Nielsen A. L., Jørgensen P., and Jørgensen A. L. (2002) Mutations associated with a childhood leukodystrophy, Alexander disease, cause deficiency in dimerization of the cytoskeletal protein GFAP. J. Neurogenet. 16, 175–179 10.1080/01677060215305 [DOI] [PubMed] [Google Scholar]
- 37. Tian R., Gregor M., Wiche G., and Goldman J. E. (2006) Plectin regulates the organization of glial fibrillary acidic protein in Alexander disease. Am. J. Pathol. 168, 888–897 10.2353/ajpath.2006.051028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fujita K., Kato T., Yamauchi M., Ando M., Honda M., and Nagata Y. (1998) Increases in fragmented glial fibrillary acidic protein levels in the spinal cords of patients with amyotrophic lateral sclerosis. Neurochem. Res. 23, 169–174 10.1023/A:1022476724381 [DOI] [PubMed] [Google Scholar]
- 39. Zhang Z., Zoltewicz J. S., Mondello S., Newsom K. J., Yang Z., Yang B., Kobeissy F., Guingab J., Glushakova O., Robicsek S., Heaton S., Buki A., Hannay J., Gold M. S., Rubenstein R., et al. (2014) Human traumatic brain injury induces autoantibody response against glial fibrillary acidic protein and its breakdown products. PLoS ONE 9, e92698 10.1371/journal.pone.0092698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mignot C., Delarasse C., Escaich S., Della Gaspera B., Noé E., Colucci-Guyon E., Babinet C., Pekny M., Vicart P., Boespflug-Tanguy O., Dautigny A., Rodriguez D., and Pham-Dinh D. (2007) Dynamics of mutated GFAP aggregates revealed by real-time imaging of an astrocyte model of Alexander disease. Exp. Cell Res. 313, 2766–2779 10.1016/j.yexcr.2007.04.035 [DOI] [PubMed] [Google Scholar]
- 41. Li R., Messing A., Goldman J. E., and Brenner M. (2002) GFAP mutations in Alexander disease. Int. J. Dev. Neurosci. 20, 259–268 10.1016/S0736-5748(02)00019-9 [DOI] [PubMed] [Google Scholar]
- 42. Flint D., Li R., Webster L. S., Naidu S., Kolodny E., Percy A., van der Knaap M., Powers J. M., Mantovani J. F., Ekstein J., Goldman J. E., Messing A., and Brenner M. (2012) Splice site, frameshift, and chimeric GFAP mutations in Alexander disease. Hum. Mutat. 33, 1141–1148 10.1002/humu.22094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moody L. R., Barrett-Wilt G. A., Sussman M. R., and Messing A. (2017) Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease. J. Biol. Chem. 292, 5814–5824 10.1074/jbc.M116.772020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 45. Jorgensen F. G., Hobolth A., Hornshoj H., Bendixen C., Fredholm M., and Schierup M. H. (2005) Comparative analysis of protein coding sequences from human, mouse and the domesticated pig. BMC Biol. 3, 2 10.1186/1741-7007-3-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.