

Abstract
Vascular endothelial growth factor A (VEGF) signals primarily through its cognate receptor VEGF receptor-2 (VEGFR-2) to control vasculogenesis and angiogenesis, key physiological processes in cardiovascular disease and cancer. In human umbilical vein endothelial cells (HUVECs), knockdown of protein kinase D-1 (PKD1) or PKD2 down-regulates VEGFR-2 expression and inhibits VEGF-induced cell proliferation and migration. However, how PKD regulates VEGF signaling is unclear. Previous bioinformatics analyses have identified binding sites for the transcription factor activating enhancer-binding protein 2 (AP2) in the VEGFR-2 promoter. Using ChIP analyses, here we found that PKD knockdown in HUVECs increases binding of AP2β to the VEGFR-2 promoter. Luciferase reporter assays with serial deletions of AP2-binding sites within the VEGFR-2 promoter revealed that its transcriptional activity negatively correlates with the number of these sites. Next we demonstrated that AP2β up-regulation decreases VEGFR-2 expression and that loss of AP2β enhances VEGFR-2 expression in HUVECs. In vivo experiments confirmed increased VEGFR-2 immunostaining in the spinal cord of AP2β knockout mouse embryos. Mechanistically, we observed that PKD phosphorylates AP2β at Ser258 and Ser277 and suppresses its nuclear accumulation. Inhibition of PKD activity with a pan-PKD inhibitor increased AP2β nuclear localization, and overexpression of both WT and constitutively active PKD1 or PKD2 reduced AP2β nuclear localization through a Ser258- and Ser277-dependent mechanism. Furthermore, substitution of Ser277 in AP2β increased its binding to the VEGFR-2 promoter. Our findings uncover evidence of a molecular pathway that regulates VEGFR-2 expression, insights that may shed light on the etiology of diseases associated with aberrant VEGF/VEGFR signaling.
Keywords: vascular endothelial growth factor (VEGF), angiogenesis, protein kinase D (PKD), signal transduction, endothelial cell, vascular endothelial growth factor receptor-2
Introduction
Vascular endothelial growth factor (VEGF)5 is the principal angiogenic growth factor that modulates physiological and pathological angiogenesis and plays a key role in cancer, ischemic and inflammatory diseases, and wound repair (1, 2). VEGF receptor 2 (VEGFR-2) is recognized as the prominent receptor mediating VEGF-stimulated angiogenesis, vascular permeability, and remodeling (1). Upon binding of VEGF, VEGFR-2 undergoes dimerization and autophosphorylation to initiate activation of downstream signaling pathways (2, 3). Numerous previous studies have contributed to a remarkable knowledge of VEGFR-2-mediated signaling cascades (4); however, the molecular mechanism controlling the expression levels of VEGFR-2 in endothelial cells remains unclear.
Protein kinase D (PKD), a family of serine/threonine kinases, regulates a variety of cellular functions, including proliferation, migration, and protein transport (5, 6). In humans, three isoforms of PKD (PKD1, PKD2, and PKD3) have been described, of which PKD1 and PKD2 are predominantly expressed in endothelial cells (7). PKD is activated mostly by cell growth–promoting substances such as phorbol ester, platelet-derived growth factor, and G protein–coupled receptor ligands (8). PKD1 and PKD2 have been shown to function downstream of VEGFR-2 to directly phosphorylate heat shock protein 27 (HSP27), histone deacetylases (HDACs), and cAMP response element–binding protein (CREB) to modulate endothelial cell proliferation and migration (7, 9, 10), but the role of PKD1 and PKD2 in the regulation of VEGFR-2 expression remains poorly understood.
Transcription factor activator protein-2 (AP2, TFAP2) transcription factors constitute a family of closely related and evolutionarily conserved sequence-specific DNA-binding proteins. The five murine ap2 genes (AP2α, AP2β, AP2γ, AP2δ, and AP2ϵ) are expressed in developing limbs, epithelia, and neuroectoderm, including neural crest–derived tissues such as facial mesenchyme (11, 12). The importance of AP2 family members is highlighted by the embryonic or perinatal lethality of AP2 knockout mice (13–17). AP2 family members also play an integral role in developmentally controlled sleep behavior (18). AP2 proteins form homo- and heterodimers with other AP2 family members and can function as either transcriptional activators or repressors of genes involved in mammalian development, differentiation, and carcinogenesis (19). The transcriptional activity of AP2 is regulated by several interaction partners, subcellular localization, and posttranslational modifications (19, 20). Although AP2 binding sites on the VEGFR-2 promoter have been predicted by computational modeling approaches (21), the specific AP2 family member (AP2α, AP2β, or AP2γ) that interacts with the VEGFR-2 promoter and its mode of transcriptional regulation has not been identified in endothelial cells.
In this study, we demonstrate that PKD1 and PKD2 directly regulate VEGFR-2 transcription through transcription factor AP2β in endothelial cells. We demonstrate that silencing PKD1 or PKD2 enhances association of AP2β with the VEGFR-2 promoter, decreasing VEGFR-2 transcription. The repressive effect of AP2β on VEGFR-2 expression was confirmed in AP2β-overexpressing and silenced endothelial cells as well as AP2β knockout mouse embryos. Furthermore, we identified that PKD directly phosphorylates AP2β at serine 258 and 277 and that PKD-mediated phosphorylation controls subcellular localization and association with the VEGFR-2 promoter of AP2β.
Results
PKD regulates VEGFR-2 expression, proliferation, and migration of endothelial cells and angiogenesis
To evaluate the role of PKD in VEGFR-2 expression in endothelial cells, human umbilical vein endothelial cells (HUVECs) were transfected with PKD1 siRNA or PKD2 siRNA. Knockdown of PKD1 or PKD2 down-regulated VEGFR-2 protein and transcript (Fig. 1, A and B). The inhibitive effect of PKD on VEGFR-2 was also observed in human blood outgrowth endothelial cells (BOECs) (Fig. 1). Administration of a pan-PKD inhibitor, CRT0066101 (CRT), reduced the phosphorylation of HSP27, a well-established downstream target of PKD1 and PKD2, and significantly inhibited the expression of VEGFR-2 (Fig. 1C), suggesting that activation of PKD suppresses the expression control of VEGFR-2. Indeed, overexpression of WT PKD1, a constitutively active form of PKD1 (PKD1.CA), and PKD2 in HUVECs increased the expression of VEGFR-2, among which PKD1.CA exhibited the greatest effect on VEGFR-2 up-regulation (Fig. 1D). Furthermore, in accordance with a published report (7), down-regulation of PKD1 or PKD2 significantly inhibited VEGF-induced endothelial proliferation and migration (Fig. 1, E and F). The effect of PKD on angiogenesis in vivo was further examined in a Matrigel plug assay (Fig. 1G). Matrigel containing PBS vehicle, VEGF alone, or a combination of VEGF and CRT, was subcutaneously implanted in mice. Our results showed that inhibition of PKD activity with CRT suppressed VEGF-induced angiogenesis in vivo. Taken together, these results suggest that endogenous PKD1 and PKD2 control expression of VEGFR-2 and positively regulate the proliferation and migration of endothelial cells as well as angiogenesis in vivo.
Figure 1.

PKD regulates VEGFR-2 expression, proliferation, and migration of endothelial cells and angiogenesis. A and B, BOECs (A) and HUVECs (A and B) were transfected with control siRNA or PKD1 or PKD2 siRNA for 48 h. VEGFR-2 levels were determined by Western blotting (A) and qPCR (B). C, HUVECs were incubated with CRT (2.5 μm) or DMSO as a control for 24 h, and then protein was collected and analyzed by Western blotting. D, HUVECs were infected with a lentivirus expressing LacZ as a control, WT PKD1, PKD1.CA, and WT PKD2 for 24 h, and then protein was collected for Western blotting. E and F, siRNA-transfected HUVECs as described in A were serum-starved and stimulated with VEGF (10 ng/ml). Migration was examined with a Boyden chamber assay 4 h after VEGF stimulation (E). Proliferation was determined using a [3H]thymidine incorporation assay 20 h after VEGF stimulation (F). G, the Matrigel plug assay was performed by diluting Matrigel in EBM-2 medium containing heparin in the presence of PBS vehicle, VEGF (10 ng/ml), or a combination of VEGF and CRT0066101 (10 μm). Matrigel was injected into 8- to 10-week-old mice subcutaneously. Fourteen days after implantation, mice were euthanized, and Matrigel plugs were removed, photographed, imaged, and quantitated for vessels as a measure of angiogenesis. Western blot images are representative of three independent experiments. *, p < 0.05; ***, p < 0.001. Scale bar for the Matrigel plug in G = 5 mm; scale bar for H&E in G = 50 μm.
Knockdown of PKD2 enhances binding of AP2β to the VEGFR-2 promoter
We sought to determine the molecular mechanism through which PKD modulates VEGFR-2 expression. Based on bioinformatics data, three AP2 binding sites exist within the VEGFR-2 promoter between bp −143 to −134, −102 to −89, and −69 to −60 (21). Thus, we hypothesized that PKD may down-regulate VEGFR-2 through AP2-mediated transcriptional repression of the VEGFR-2 promoter. The results of our ChIP assay showed that AP2α bound the VEGFR-2 promoter but did not show differences among control, PKD1 knockdown, and PKD2 knockdown groups (Fig. 2A). However, increased AP2β bound the VEGFR-2 promoter upon PKD1 knockdown, and this effect was most pronounced in PKD2-ablated HUVECs, as evidenced by the CHIP assay (Fig. 2A).
Figure 2.
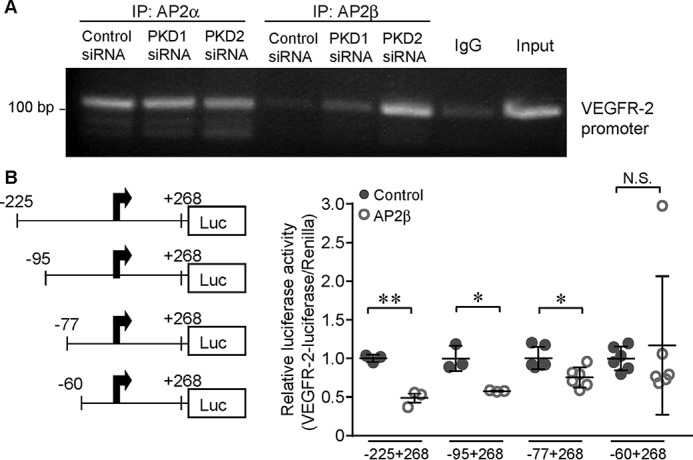
AP2β negatively regulates the transcriptional activity of VEGFR-2. A, HUVECs were transfected with control siRNA or PKD1 or PKD2 siRNA for 48 h, and then cross-linked chromatin–protein complexes were immunoprecipitated (IP) using AP2α or AP2β antibodies as well as control IgG. After reverse cross-linking, DNA fragments were isolated, and the VEGFR-2 promoter was amplified by PCR using promoter-specific primers and run on a 2% agarose gel. 5% of the input was run as a positive control. B, HUVECs were transfected with VEGFR-2 firefly luciferase (Luc) promoter constructs as indicated, with or without AP2β-GFP and an internal Renilla luciferase control vector. After 48 h, luciferase activity was determined using the DualGlo luciferase kit, and data were normalized with respect to Renilla activity. *, p < 0.05; **, p < 0.01. N.S., not significant.
AP2β negatively regulates the transcriptional activity of VEGFR-2
To demonstrate AP2β-specific inhibition of the VEGFR-2 promoter, serial deletion luciferase constructs of the VEGFR-2 promoter containing three, two, one, or no AP2β binding sites were transfected into HUVECs with or without an AP2β-GFP expression plasmid. Maximum inhibition of luciferase activity was observed with the construct containing three AP2β binding sites, and the number of AP2β binding sites positively correlated with increasing inhibition, confirming negative regulation of VEGFR-2 transcription by AP2β (Fig. 2B).
AP-2β controls the expression of VEGFR-2 in vitro and in vivo
Next, to confirm the regulatory role of AP2 in expression VEGFR-2, we demonstrated that siRNA-mediated knockdown of AP2β enhanced VEGFR-2 expression (Fig. 3A). Conversely, overexpression of AP2β decreased VEGFR-2 protein expression (Fig. 3B). We also performed qPCR and confirmed that knockdown of AP2β increased but overexpression of AP2β decreased the mRNA levels of VEGFR-2 (Fig. 3C). Furthermore, immunohistochemistry was performed to examine the expression of VEGFR-2 in genomic AP-2β knockout mouse embryos. Increased VEGFR-2 protein expression was observed in the spinal cord of AP-2β knockout mice at embryonic day 13 compared with similarly aged WT control embryos (Fig. 3, D–G). These results suggest that endogenous AP-2β suppresses VEGFR-2 expression in vitro and in vivo.
Figure 3.

AP2β decreases the expression of VEGFR-2 in vitro and in vivo. A–C, HUVECs were transfected with AP2β siRNA to knock down AP2β (A and C) or transfected with the AP2β-GFP plasmid to overexpress AP2β (B and C). VEGFR-2 levels were determined by Western blotting (A and B) and qPCR (C), respectively. D–G, photomicrographs showing VEGFR-2 immunolabeling in the dorsal horn of the spinal cord of AP2-β+/+ (D and E) or AP2-β−/− (F and G) mice at embryonic day 13. The area of neuroepithelium shown in D and F is depicted at high power using oil immersion in E and G. Scale bars in E and G = 25 μm; scale bars in D and F = 100 μm. *, p < 0.05; **, p < 0.01.
PKD activates AP2β at Ser258 and Ser277 and suppresses nuclear accumulation of AP2
We hypothesized that PKDs could directly regulate AP2β function by serine phosphorylation at positions 258 and 277. Previous studies have suggested Ser258 as a phosphorylation site in AP2β using structural analysis (22). However, attempts to reliably detect serine phosphorylation of AP2β using phosphoserine-specific antibodies have proven challenging. Consequently, the effects of PKD1 and PKD2 on AP2β phosphorylation have not been examined. Therefore, to determine whether PKD1 is the upstream kinase for these sites, we performed Phos-tagTM assays with lysates from HEK293 cells that were cotransfected with vector, WT AP2β, or mutated AP2β-S258A or AP2β-S277A together with PKD1.CA or vector control. Expression of PKD1.CA led to a shift of WT AP2β, indicating its phosphorylation by PKD1. This was decreased when Ser258 or Ser277 were blunted by serine-to-alanine substitutions, identifying both as PKD1 phosphorylation sites (Fig. 4A). To further examine the effect of endogenous PKD on AP2β phosphorylation, we collected lysates of HUVECs, performed immunoprecipitation with an AP2β antibody, and probed the immunoprecipitates with a pMOTIF antibody that specifically recognizes PKD-phosphorylated substrates. Our results confirmed that endogenous PKD phosphorylates AP2β and that this phosphorylation is reduced when the pan-PKD inhibitor CRT is present in the medium (Fig. 4B).
Figure 4.

PKD activates AP-2β at S258 and Ser277 and controls the cellular localization of AP2β. A, HEK293 cells were cotransfected with vector, AP2β, AP2β-S258A, or AP2β-S277A together with PKD1.CA or a vector control. Lysates were subjected to Phos-tagTM analysis to determine potential phosphorylation as indicated by shifting. The bottom arrow indicates the AP2β bands, and the top arrow indicates the shift of the phosphorylated AP2β bands. B, lysates from control (Con) HUVECs and HUVECs pretreated with CRT (2.5 μm) for 30 min were collected and subjected to immunoprecipitation (IP) followed by Western blotting. C and D, 293T cells were transfected with AP2β-GFP for 24 h and then treated with CRT (2.5 μm) or DMSO as a control for 30 min. Cellular localization of GFP was examined. The images were acquired using a confocal microscope (LSM 880, Carl Zeiss) and are representative of three independent experiments. The nuclear localization (N ≥ C) of AP2β was counted blindly and quantified. 303 GFP-positive cells in the control groups and 390 GFP positive cells in the CRT group were counted. E and F, 293T cells were transfected with AP2β-GFP, AP2β-S258A-GFP, AP2β-S277A-GFP, or AP2β-S258A/S277A-GFP together with mCherry-PKD1.WT and mCherry-PKD1.CA for 24 h. Nuclear localization (N ≥ C) of AP2β was counted blindly in cells double-positive for both GFP and mCherry. The image is representative of three independent experiments. An average of 237 double-positive cells was counted in each group. G, 293T cells were transfected with AP2β-GFP, AP2β-S258A-GFP, AP2β-S277A-GFP, or AP2β-S258A/S277A-GFP together with PKD2- WT and PKD2.CA for 24 h. Cellular localization of GFP was visualized. The images are representative of three independent experiments. Nuclear localization of AP2β was counted blindly and quantified. An average of 210 GFP-positive cells was counted in each group, and the quantification is presented in Fig. S1. H, nuclear lysates from control HUVECs and HUVECs pretreated with CRT (2.5 μm) for 2 h were collected and subjected to Western blotting. I, nuclear lysates were collected from HUVECs infected with a lentivirus overexpressing PKD1.WT or PKD1.CA for 24 h and then analyzed by Western blotting. J, 293T cells were transfected with PKD2.CA together with GFP, AP2β-GFP, AP2β-S258A-GFP, AP2β-S277A-GFP, or AP2β-S258A/S277A-GFP. A ChIP assay was performed 24 h after transfection. n = 6/group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, p < 0.05. Scale bars = 10 μm.
To define the functional consequence of PKD-mediated phosphorylation of AP2β, the cellular localization of AP2β was examined using a fusion protein, AP2β-GFP, in 293T cells. As shown in Fig. 4, C and D, administration of the pan-PKD inhibitor CRT significantly increased nuclear accumulation of AP2β. The effect of PKD1 on AP2β nuclear localization was further examined in 293T cells transfected with mCherry-PKD1 or mCherry-PKD1.CA (Fig. 4, E and F). In cells transfected with PKD1, mutation of either Ser258 or Ser277 increased AP2β nuclear accumulation, as shown by enriched GFP signals in nuclei. Notably, overexpression of PKD1.CA decreased the nuclear localization of WT AP2β but not AP2β with mutations. Similar results were observed in PKD2-overexpressing 293T cells (Fig. 4G and Fig. S1). In these experiments, substitution of both Ser258 and Ser277 did not significantly increase the nuclear localization of AP2β compared with single Ser258 or Ser277 substitution, suggesting that Ser258 and Ser277 play redundant roles in the AP2β nuclear localization. Nuclear protein was also collected from HUVECs stimulated with CRT and exhibited increased nuclear accumulation of AP2β (Fig. 4H). Overexpression of PKD1.CA in HUVECs was shown to decrease the nuclear AP2β protein level (Fig. 4I), suggesting that active PKD promotes cytoplasmic retention of AP2β. Correspondingly, AP2β with a Ser277 substitution showed significantly increased binding to the VEGFR-2 promoter (Fig. 4J). These results indicate that PKD-mediated phosphorylation of AP2β, especially Ser277, controls the nuclear localization and DNA binding of AP2β.
Discussion
In this study, we report that PKD regulates VEGFR-2 expression through AP2β in endothelial cells. It has been well-established that VEGF activity is controlled autonomously by its own expression levels through various circulating sequestering proteins and via VEGFR-2, its cognate tyrosine kinase receptor, predominantly responsible for transducing the angiogenic effects of VEGF (23). Several small-molecule VEGFR-2 inhibitors and VEGFR-2 monoclonal antibodies (24) have been developed to therapeutically target tumor angiogenesis in cancer patients. Although VEGF is secreted by a variety of cell types, the expression of VEGFR-2 is mainly restricted to vascular endothelial cells. Here our results identify a novel mechanism through which PKD phosphorylates AP2β and regulates the cytoplasmic localization and DNA binding of AP2β to control transcription of VEGFR-2 in endothelial cells.
Because of the important role of VEGFR-2 in endothelial cells, multiple mechanisms have been reported to control the expression of VEGFR-2. Although endocytosis of VEGFR-2 was originally thought to induce its degradation and down-regulation, recent studies have indicated that VEGFR-2 endocytosis and trafficking in endosomal compartments are essential for VEGF-stimulated extracellular signal-regulated kinase activation but not down-regulation of VEGFR-2 (25). Previous studies identified that c-Cbl and activated PKC-mediated protein modification are important for down-regulation of VEGFR-2 levels (26, 27). The promoter of VEGFR-2 was cloned, and positive regulatory elements were identified between bp −225 to +127 (21). Important subsequent studies identified several molecules or protein complexes capable of increasing the transcriptional activity of VEGFR-2, including nuclear focal adhesion kinase, Sp1, and complexes consisting of mutant p53 bound to switching/sucrose non-fermenting (28–31). AP2 was predicted to bind the promoter region of VEGFR-2 between bp −143 to −134, −102 to −89, and −69 to −60 (21); our promoter luciferase assay (Fig. 2) confirmed that all three binding regions are involved in AP2β-suppressed VEGFR-2 expression. Although AP2 is generally considered a transcriptional activator, it has been shown to negatively regulate the transcription of several genes through interaction or competition with other positive transcription factors, such as Sp1 (32–37). Indeed, the three identified AP2-binding elements in the VEGFR-2 promoter are either near or overlapping with Sp1-binding sites (21). Thus, it is possible that AP2β may compete or interact with Sp1 to repress transcription of VEGFR-2. Notably, although VEGFR-2 is often up-regulated during oncogenesis and recognized as a promising marker of several types of cancer, VEGFR-2 expression is negatively controlled by the inflammatory cytokines tumor necrosis factor α and interleukin-1β (38), β-amyloid (39), and oxidized low-density lipoprotein (40), all of which induce endothelial cell dysfunction. Our results raise the possibility that AP2β-mediated suppression of VEGFR-2 might be involved in endothelial cell dysfunction.
Positive feedback in the regulation of VEGFR-2 expression has been supported by several previous studies. VEGFR-2 translocates to nuclei and interacts with several nuclear proteins, including Sp1, to directly regulate its transcription (31). VEGF binding to membrane-bound VEGFR-2 induces an increase in gene transcription and protein expression of VEGFR-2 (41). Endogenous VEGF also activates and maintains the expression of VEGFR-2 in endothelial cells (42, 43). Previous studies have indicated that PKD1 and PKD2 are required for VEGF-stimulated endothelial cell migration, proliferation, and tubulogenesis by mediating phosphorylation of HSP27, CREB-binding protein/p300, and HDACs (9, 10, 44). Our results demonstrate that genetic knockdown or pharmacological inhibition of PKD decreases VEGFR-2 expression in endothelial cells, suggesting that PKD1 and PKD2 act as a positive feedback mechanism to maintain the expression of VEGFR-2 upon stimulation of VEGF.
Previous studies have shown that knockdown of PKD1 phosphorylates AP2α at Ser258 to negatively regulate expression of the ABC transporter in THP-1 cells (22). A follow-up study reported that knockdown of PKD1 leads to decreased phosphorylation levels of both AP2α and AP2β in 3T3-L1 preadipocytes (45). The Ser258 in the basic domain of AP2, which is conserved among species of all five subtypes of AP2, exists within an LXRXXS/T sequence, a phosphorylation motif of PKD1 (45). The LXRXXS (Ser277) motif of the AP2β basic domain is completely identical to that of AP2α (45). Given that Ser258 and Ser277 are functionally important and well-conserved phosphorylation sites among AP2 family members, it stands to reason that PKD activates AP2β at Ser258 and Ser277 to regulate nuclear localization and DNA binding of AP2β.
Indeed, our results showed that PKD controlled the cytoplasmic retention of AP2β, as shown by the increased nuclear localization of AP2β upon inhibition of PKD (Fig. 4). Typically, PKD1 and PDK2 reside in the cytosol as well as in intracellular compartments such as the Golgi and mitochondria. A recent report demonstrated that stimulation of cholecystokinin-2 promotes nuclear PKD2 localization in epithelial cells (46). PKD has also been shown to directly interact with and control the nuclear export of HDAC5 (47). Our results showed that two phosphorylated sites, Ser258 and Ser277, are required for PKD-controlled cytoplasmic retention of AP2β. Furthermore, our results showed that mutation of AP2β Ser277 increased the association of AP2β with the VEGFR-2 promoter. However, the molecular mechanism by which PKD-mediated phosphorylation controls cellular localization of AP2β and how the two phosphorylated sites of AP2β synergistically control the biological function of AP2β still requires further investigation. Our study does not exclude the possibility that PKD phosphorylates AP2β in both the cytosol and nucleus. Our results (data not shown) do not support the hypothesis that PKD forms complexes with AP2β in endothelial cells. Although PKD phosphorylates multiple downstream targets to control their cellular localization, not all phosphorylated substrates form complexes with PKD (5). Thus, we anticipate that PKD phosphorylates AP2β and rapidly dissociates from it. In future studies, we will examine the association of AP2β and other candidate molecules, such as 14-3-3 protein, which have been shown to associate with PKD-phosphorylated substrates and sequester them to the cytosol (49, 50).
In conclusion, our results identified a novel signaling cascade of PKD–AP2β that controls the transcription of VEGFR-2 in endothelial cells. Our results provide insights for therapeutic strategies targeting VEGFR-2 in several diseases, including cancer, diabetic retinopathy, and ischemic diseases.
Experimental procedures
Cells and reagents
HUVECs (Lonza) and BOECs were passaged in endothelial cell basal medium supplemented with EGM-MV SingleQuots (Lonza). 293T cells (ATCC) were cultured in DMEM with 10% FBS. VEGF was purchased from R&D Systems. Antibodies against AP2α (SC184) and AP2β (SC8976) were from Santa Cruz Biotechnology. Antibodies against VEGFR-2 (2479) and phosphorylated HSP27 (2405) were from Cell Signaling Technology. The PKD2 antibody (07-488) was from EMD Millipore, and the HSP27 antibody (18284-1-AP) was from Proteintech. The β-actin antibody (A2228) was purchased from Sigma-Aldrich. The GFP antibody (A-11120) was purchased from Thermo Fisher and used for the ChIP assay. The pMOTIF antibody was from Cell Signaling Technology (51). The PKD1/2 antibody (2052, Cell Signaling Technology) that was initially used for immunoblotting (Fig. 1A) to examine PKD1 protein levels has been discontinued. Subsequently, the mouse monoclonal PKD1 antibody developed by Storz and co-workers (52) was used in the Western blot depicted in Fig. 1D. Light-chain-specific secondary antibodies (211-032-171, Jackson ImmunoResearch Laboratories) were used in the Western blots of immunoprecipitation. Nuclear proteins were collected as described previously (53).
The human AP2β (TFAP2b) retrovirus and plasmid were generated from the pEGFP-C1 vector and generously shared by Dr. T. Sargent (NICHD, National Institutes of Health, Bethesda, MD) as reported previously (54, 55). The AP2β Ser258 and Ser277 mutants were generated using the QuikChange II site-directed mutagenesis (Aligent) kit according the manufacturer's instructions. The expression plasmids for constitutively active FLAG-tagged PKD2 (PKD2.CA, PKD2.S706E/S710E mutation) and FLAG-tagged PKD2 were generated as described previously (56, 57). The expression plasmids for mCherry-tagged WT PKD1 and mCherry-tagged PKD1.CA (PKD1.S738E.S742E) were generated as described previously (52). Lentivirus vectors of PKD1, PKD1.CA (PKD1.S738E.S742E), and PKD2 were used as described previously (58). The previously described PKD inhibitor CRT0066101 (48) was obtained from Sigma-Aldrich.
siRNA transfection
1 × 105 HUVECs were seeded in 60-mm plates and cultured for 24 h. The next day, cells were transfected with 100 nm siRNA using Oligofectamine (Invitrogen) in Opti-MEM serum-reduced medium. After 4 h, antibiotic-free medium was added, and cell lysates were prepared 24 or 48 h after transfection. PKD1 siRNA (SI00042371, Qiagen) target sequences was 5′-AACAAAGCTGTTAAACTGTTA-3′. The PKD2 siRNA (SI02224768, Qiagen) target sequences was 5′-CACGACCAACAGATACTATAA-3′. The AP2β siRNA (SI00049266, Qiagen) target sequence was 5′-TTCGAGTTTAGTAATACTGAT-3′. Allstars negative control siRNA (Qiagen) was used in the control group.
Phos-tagTM SDS-PAGE
Phos-tagTM SDS-PAGE was performed according to the manufacturer's instructions (Wako Chemicals USA Inc., Richmond, VA). In brief, 2 μg of protein per sample was separated by electrophoresis at 50 V per gel on an 8% SDS-PAGE gel containing Phos-tagTM reagent (50 μmol/liter) and MnCl2 (50 μmol/liter). Prior to protein transfer, gels were incubated in transfer buffer containing 1 mm EDTA for 10 min (twice) and then incubated in transfer buffer for 10 min. Proteins were transferred to a PVDF membrane and detected by standard immunoblotting techniques using Odyssey (LI-COR, Lincoln, NE).
Proliferation assay
Control, PKD1, or PKD2 siRNA–treated, serum-starved HUVECs (4 × 104/ml) were seeded in 24-well plates. After 24 h, the cells were serum-starved (0.2% FBS) and stimulated with VEGF (10 ng/ml). The next day, 1 μCi of [H3]thymidine was added to each well. 4 h later, cells were washed with chilled PBS, fixed with 100% cold methanol, and collected for measurement of TCA-precipitable radioactivity.
Migration assay
5 × 104 HUVECs were transfected with the indicated siRNA, serum-starved, and treated with or without 10 ng/ml VEGF. Following 4-h incubation at 37 °C, cells that remained in the upper chamber were gently removed. Cells that had migrated through the filter were fixed in 4% paraformaldehyde and stained with 0.2% crystal violet dissolved in 2% ethanol. Migration was quantitated by counting the number of cells in four separate fields on the filter using bright-field optics with a Nikon Diaphot microscope.
Real-time PCR
Total RNA was isolated from cells with the RNeasy Mini Kit (Qiagen). RNA was reverse-transcribed by using oligo(dT) priming using the iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed with a TaqMan SYBR Green Master Mix (Applied Biosystems). The comparative cycle threshold method was used to calculate the relative abundance of mRNA compared with that of β-actin expression.
Immunohistochemistry
The spinal cord of embryonic day 13 AP2β knockout mice (17) was harvested and fixed in neutral buffered 10% formalin at room temperature for 24 h before processing, embedding in paraffin, and sectioning. Sections were deparaffinized and then subjected to VEGFR-2 immunohistochemistry staining. Stable diaminobenzidine was used as a chromogen substrate, and the sections were counterstained with a hematoxylin solution.
ChIP assay
Cells were cross-linked with 1% formaldehyde followed by sonication. The sheared chromatin was immunoprecipitated with Dynabeads conjugated with control IgG or GFP primary antibodies. The eluted immunoprecipitates were incubated at 65 °C for 6 h for reverse formaldehyde cross-linking. DNA was extracted with a Qiagen PCR purification kit and subjected to PCR with specific primers (Table S1).
Matrigel plug assay
The Matrigel plug assay was performed by diluting Matrigel in EBM-2 medium containing heparin in the presence of PBS vehicle, VEGF (10 ng/ml), or a combination of VEGF and CRT0066101 (10 μm). Matrigel was injected into 8- to 10-week-old mice subcutaneously. Fourteen days after implantation, mice were euthanized, and the Matrigel plugs were removed, photographed, imaged, and quantitated for vessels as a measure of angiogenesis. All animals were maintained in accordance with Institutional Animal Care and Use Committee guidelines.
Statistics
Statistics was performed using Kruskal–Wallis ANOVA with Dunn's multiple comparisons test and Student's t test. Data are represented as mean ± S.D. and considered statistically significantly different at p < 0.05.
Author contributions
Y. W., L. H. H., P. S., and D. M. funding acquisition; Y. W. validation; Y. W., L. H. H., R. S. A., E. W., S. D., H. R. D., F. W., T. S., I. A. S., S. G., and R. B. investigation; Y. W. writing-original draft; Y. W., P. S., and D. M. project administration; L. H. H., R. B., and D. M. conceptualization; H. R. D., I. A. S., and S. G. methodology; I. A. S., S. G., P. S., and D. M. resources; P. S. and D. M. writing-review and editing; R. B. formal analysis; D. M. supervision.
Supplementary Material
Acknowledgment
We thank Victoria Pham for assisting with HUVEC culture.
This work was supported by NHLBI, National Institutes of Health Grant HL140411 (to D. M.); NCI, National Institutes of Health Grants CA78383-20 (to D. M.), CA187035 (to L. H. H.), and CA200572 (to P. S.); Florida Department of Health Cancer Research Chair Fund Florida Grant 3J-02 (to D. M.); American Heart Association Grants 13POST14510025 (to L. H. H.) and 19CDA34700013 (to Y. W.); and Mayo Clinic Ted and Loretta Rogers Cardiovascular Career Development Award Honoring Hugh C. Smith (to Y. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1 and Table S1.
- VEGF
- vascular endothelial growth factor
- VEGFR
- vascular endothelial growth factor receptor
- PKD
- protein kinase D
- HDAC
- histone deacetylase
- CREB
- cAMP response element–binding protein
- HUVEC
- human umbilical vein endothelial cell
- BOEC
- blood outgrowth endothelial cell
- CRT
- CRT0066101
- CA
- constitutively active
- qPCR
- quantitative PCR.
References
- 1. Ferrara N. (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev. 25, 581–611 10.1210/er.2003-0027 [DOI] [PubMed] [Google Scholar]
- 2. Welti J., Loges S., Dimmeler S., and Carmeliet P. (2013) Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Invest. 123, 3190–3200 10.1172/JCI70212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Abhinand C. S., Raju R., Soumya S. J., Arya P. S., and Sudhakaran P. R. (2016) VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal. 10, 347–354 10.1007/s12079-016-0352-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simons M., Gordon E., and Claesson-Welsh L. (2016) Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–625 10.1038/nrm.2016.87 [DOI] [PubMed] [Google Scholar]
- 5. Rozengurt E. (2011) Protein kinase D signaling: multiple biological functions in health and disease. Physiology 26, 23–33 10.1152/physiol.00037.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Durand N., Borges S., and Storz P. (2015) Functional and therapeutic significance of protein kinase D enzymes in invasive breast cancer. Cell Mol. Life Sci. 72, 4369–4382 10.1007/s00018-015-2011-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evans I. M., and Zachary I. C. (2011) Protein kinase D in vascular biology and angiogenesis. IUBMB Life 63, 258–263 10.1002/iub.456 [DOI] [PubMed] [Google Scholar]
- 8. Rozengurt E., Rey O., and Waldron R. T. (2005) Protein kinase D signaling. J. Biol. Chem. 280, 13205–13208 10.1074/jbc.R500002200 [DOI] [PubMed] [Google Scholar]
- 9. Evans I. M., Britton G., and Zachary I. C. (2008) Vascular endothelial growth factor induces heat shock protein (HSP) 27 serine 82 phosphorylation and endothelial tubulogenesis via protein kinase D and independent of p38 kinase. Cell Signal. 20, 1375–1384 10.1016/j.cellsig.2008.03.002 [DOI] [PubMed] [Google Scholar]
- 10. Ha C. H., Wang W., Jhun B. S., Wong C., Hausser A., Pfizenmaier K., McKinsey T. A., Olson E. N., and Jin Z. G. (2008) Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 283, 14590–14599 10.1074/jbc.M800264200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moser M., Imhof A., Pscherer A., Bauer R., Amselgruber W., Sinowatz F., Hofstädter F., Schüle R., and Buettner R. (1995) Cloning and characterization of a second AP-2 transcription factor: AP-2β. Development 121, 2779–2788 [DOI] [PubMed] [Google Scholar]
- 12. Mitchell P. J., Timmons P. M., Hébert J. M., Rigby P. W., and Tjian R. (1991) Transcription factor AP-2 is expressed in neural crest cell lineages during mouse embryogenesis. Genes Dev. 5, 105–119 10.1101/gad.5.1.105 [DOI] [PubMed] [Google Scholar]
- 13. Schorle H., Meier P., Buchert M., Jaenisch R., and Mitchell P. J. (1996) Transcription factor AP-2 essential for cranial closure and craniofacial development. Nature 381, 235–238 10.1038/381235a0 [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., Hagopian-Donaldson S., Serbedzija G., Elsemore J., Plehn-Dujowich D., McMahon A. P., Flavell R. A., and Williams T. (1996) Neural tube, skeletal and body wall defects in mice lacking transcription factor AP-2. Nature 381, 238–241 10.1038/381238a0 [DOI] [PubMed] [Google Scholar]
- 15. Nottoli T., Hagopian-Donaldson S., Zhang J., Perkins A., and Williams T. (1998) AP-2-null cells disrupt morphogenesis of the eye, face, and limbs in chimeric mice. Proc. Natl. Acad. Sci. U.S.A. 95, 13714–13719 10.1073/pnas.95.23.13714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. West-Mays J. A., Zhang J., Nottoli T., Hagopian-Donaldson S., Libby D., Strissel K. J., and Williams T. (1999) AP-2α transcription factor is required for early morphogenesis of the lens vesicle. Dev. Biol. 206, 46–62 10.1006/dbio.1998.9132 [DOI] [PubMed] [Google Scholar]
- 17. Moser M., Pscherer A., Roth C., Becker J., Mücher G., Zerres K., Dixkens C., Weis J., Guay-Woodford L., Buettner R., and Fässler R. (1997) Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2β. Genes Dev. 11, 1938–1948 10.1101/gad.11.15.1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bringmann H. (2018) Sleep-active neurons: conserved motors of sleep. Genetics 208, 1279–1289 10.1534/genetics.117.300521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckert D., Buhl S., Weber S., Jäger R., and Schorle H. (2005) The AP-2 family of transcription factors. Genome Biol. 6, 246 10.1186/gb-2005-6-13-246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ding X., Fan C., Zhou J., Zhong Y., Liu R., Ren K., Hu X., Luo C., Xiao S., Wang Y., Feng D., and Zhang J. (2006) GAS41 interacts with transcription factor AP-2β and stimulates AP-2β-mediated transactivation. Nucleic Acids Res. 34, 2570–2578 10.1093/nar/gkl319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patterson C., Perrella M. A., Hsieh C. M., Yoshizumi M., Lee M. E., and Haber E. (1995) Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J. Biol. Chem. 270, 23111–23118 10.1074/jbc.270.39.23111 [DOI] [PubMed] [Google Scholar]
- 22. Iwamoto N., Abe-Dohmae S., Lu R., and Yokoyama S. (2008) Involvement of protein kinase D in phosphorylation and increase of DNA binding of activator protein 2α to downregulate ATP-binding cassette transporter A1. Arterioscler. Thromb. Vasc. Biol. 28, 2282–2287 10.1161/ATVBAHA.108.174714 [DOI] [PubMed] [Google Scholar]
- 23. Ferrara N., Gerber H. P., and LeCouter J. (2003) The biology of VEGF and its receptors. Nat. Med. 9, 669–676 10.1038/nm0603-669 [DOI] [PubMed] [Google Scholar]
- 24. Javle M., Smyth E. C., and Chau I. (2014) Ramucirumab: successfully targeting angiogenesis in gastric cancer. Clin. Cancer Res. 20, 5875–5881 10.1158/1078-0432.CCR-14-1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Simons M. (2012) An inside view: VEGF receptor trafficking and signaling. Physiology 27, 213–222 10.1152/physiol.00016.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murdaca J., Treins C., Monthouel-Kartmann M. N., Monthouël-Kartmann M. N., Pontier-Bres R., Kumar S., Van Obberghen E., Giorgetti-Peraldi S. (2004) Grb10 prevents Nedd4-mediated vascular endothelial growth factor receptor-2 degradation. J. Biol. Chem. 279, 26754–26761 10.1074/jbc.M311802200 [DOI] [PubMed] [Google Scholar]
- 27. Singh A. J., Meyer R. D., Band H., and Rahimi N. (2005) The carboxyl terminus of VEGFR-2 is required for PKC-mediated down-regulation. Mol. Biol. Cell 16, 2106–2118 10.1091/mbc.e04-08-0749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patterson C., Wu Y., Lee M. E., DeVault J. D., Runge M. S., and Haber E. (1997) Nuclear protein interactions with the human KDR/flk-1 promoter in vivo: regulation of Sp1 binding is associated with cell type-specific expression. J. Biol. Chem. 272, 8410–8416 10.1074/jbc.272.13.8410 [DOI] [PubMed] [Google Scholar]
- 29. Pfister N. T., Fomin V., Regunath K., Zhou J. Y., Zhou W., Silwal-Pandit L., Freed-Pastor W. A., Laptenko O., Neo S. P., Bargonetti J., Hoque M., Tian B., Gunaratne J., Engebraaten O., Manley J. L., et al. (2015) Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes Dev. 29, 1298–1315 10.1101/gad.263202.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun S., Wu H. J., and Guan J. L. (2018) Nuclear FAK and its kinase activity regulate VEGFR2 transcription in angiogenesis of adult mice. Sci. Rep. 8, 2550 10.1038/s41598-018-20930-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Domingues I., Rino J., Demmers J. A., de Lanerolle P., and Santos S. C. (2011) VEGFR2 translocates to the nucleus to regulate its own transcription. PLoS ONE 6, e25668 10.1371/journal.pone.0025668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen A., Beno D. W., and Davis B. H. (1996) Suppression of stellate cell type I collagen gene expression involves AP-2 transmodulation of nuclear factor-1-dependent gene transcription. J. Biol. Chem. 271, 25994–25998 10.1074/jbc.271.42.25994 [DOI] [PubMed] [Google Scholar]
- 33. Chen T. T., Wu R. L., Castro-Munozledo F., and Sun T. T. (1997) Regulation of K3 keratin gene transcription by Sp1 and AP-2 in differentiating rabbit corneal epithelial cells. Mol. Cell Biol. 17, 3056–3064 10.1128/MCB.17.6.3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Getman D. K., Mutero A., Inoue K., and Taylor P. (1995) Transcription factor repression and activation of the human acetylcholinesterase gene. J. Biol. Chem. 270, 23511–23519 10.1074/jbc.270.40.23511 [DOI] [PubMed] [Google Scholar]
- 35. Jiang M. S., Tang Q. Q., McLenithan J., Geiman D., Shillinglaw W., Henzel W. J., and Lane M. D. (1998) Derepression of the C/EBPα gene during adipogenesis: identification of AP-2α as a repressor. Proc. Natl. Acad. Sci. U.S.A. 95, 3467–3471 10.1073/pnas.95.7.3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhu C. H., Huang Y., Oberley L. W., and Domann F. E. (2001) A family of AP-2 proteins down-regulate manganese superoxide dismutase expression. J. Biol. Chem. 276, 14407–14413 10.1074/jbc.M009708200 [DOI] [PubMed] [Google Scholar]
- 37. Yeh C. C., Wan X. S., and St Clair D. K. (1998) Transcriptional regulation of the 5′ proximal promoter of the human manganese superoxide dismutase gene. DNA Cell Biol. 17, 921–930 10.1089/dna.1998.17.921 [DOI] [PubMed] [Google Scholar]
- 38. Giraudo E., Primo L., Audero E., Gerber H. P., Koolwijk P., Soker S., Klagsbrun M., Ferrara N., and Bussolino F. (1998) Tumor necrosis factor-α regulates expression of vascular endothelial growth factor receptor-2 and of its co-receptor neuropilin-1 in human vascular endothelial cells. J. Biol. Chem. 273, 22128–22135 10.1074/jbc.273.34.22128 [DOI] [PubMed] [Google Scholar]
- 39. Cho S. J., Park M. H., Han C., Yoon K., and Koh Y. H. (2017) VEGFR2 alteration in Alzheimer's disease. Sci. Rep. 7, 17713 10.1038/s41598-017-18042-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang M., and Jiang L. (2016) Oxidized low-density lipoprotein decreases VEGFR2 expression in HUVECs and impairs angiogenesis. Exp. Ther. Med. 12, 3742–3748 10.3892/etm.2016.3823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen B. Q., Lee D. Y., Gerber H. P., Keyt B. A., Ferrara N., and Zioncheck T. F. (1998) Homologous up-regulation of KDR/Flk-1 receptor expression by vascular endothelial growth factor in vitro. J. Biol. Chem. 273, 29979–29985 10.1074/jbc.273.45.29979 [DOI] [PubMed] [Google Scholar]
- 42. Lee S., Chen T. T., Barber C. L., Jordan M. C., Murdock J., Desai S., Ferrara N., Nagy A., Roos K. P., and Iruela-Arispe M. L. (2007) Autocrine VEGF signaling is required for vascular homeostasis. Cell 130, 691–703 10.1016/j.cell.2007.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. E G, Cao Y., Bhattacharya S., Dutta S., Wang E., and Mukhopadhyay D. (2012) Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J. Biol. Chem. 287, 3029–3041 10.1074/jbc.M111.293985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang S., Li X., Parra M., Verdin E., Bassel-Duby R., and Olson E. N. (2008) Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. U.S.A. 105, 7738–7743 10.1073/pnas.0802857105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iwamoto N., and Yokoyama S. (2011) Protein kinase D regulates the adiponectin gene expression through phosphorylation of AP-2: a common pathway to the ABCA1 gene regulation. Atherosclerosis 216, 90–96 10.1016/j.atherosclerosis.2011.01.043 [DOI] [PubMed] [Google Scholar]
- 46. von Blume J., Knippschild U., Dequiedt F., Giamas G., Beck A., Auer A., Van Lint J., Adler G., and Seufferlein T. (2007) Phosphorylation at Ser244 by CK1 determines nuclear localization and substrate targeting of PKD2. EMBO J. 26, 4619–4633 10.1038/sj.emboj.7601891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., and McKinsey T. A. (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell Biol. 24, 8374–8385 10.1128/MCB.24.19.8374-8385.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harikumar K. B., Kunnumakkara A. B., Ochi N., Tong Z., Deorukhkar A., Sung B., Kelland L., Jamieson S., Sutherland R., Raynham T., Charles M., Bagherzadeh A., Bagherazadeh A., Foxton C., Boakes A., et al. (2010) A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 9, 1136–1146 10.1158/1535-7163.MCT-09-1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Scholz R. P., Regner J., Theil A., Erlmann P., Holeiter G., Jähne R., Schmid S., Hausser A., and Olayioye M. A. (2009) DLC1 interacts with 14-3-3 proteins to inhibit RhoGAP activity and block nucleocytoplasmic shuttling. J. Cell Sci. 122, 92–102 10.1242/jcs.036251 [DOI] [PubMed] [Google Scholar]
- 50. Wang Y., Waldron R. T., Dhaka A., Patel A, Riley M. M., Rozengurt E., and Colicelli J. (2002) The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol. Cell Biol. 22, 916–926 10.1128/MCB.22.3.916-926.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Döppler H., Storz P., Li J., Comb M. J., and Toker A. (2005) A phosphorylation state-specific antibody recognizes Hsp27, a novel substrate of protein kinase D. J. Biol. Chem. 280, 15013–15019 10.1074/jbc.C400575200 [DOI] [PubMed] [Google Scholar]
- 52. Borges S., Döppler H., Perez E. A., Andorfer C. A., Sun Z., Anastasiadis P. Z., Thompson E., Geiger X. J., and Storz P. (2013) Pharmacologic reversion of epigenetic silencing of the PRKD1 promoter blocks breast tumor cell invasion and metastasis. Breast Cancer Res. 15, R66 10.1186/bcr3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Y., Zhao B., Zhang Y., Tang Z., Shen Q., Zhang Y., Zhang W., Du J., Chien S., and Wang N. (2012) Kruppel-like factor 4 is induced by rapamycin and mediates the anti-proliferative effect of rapamycin in rat carotid arteries after balloon injury. Br. J. Pharmacol. 165, 2378–2388 10.1111/j.1476-5381.2011.01734.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zarelli V. E., and Dawid I. B. (2013) Inhibition of neural crest formation by Kctd15 involves regulation of transcription factor AP-2. Proc. Natl. Acad. Sci. U.S.A. 110, 2870–2875 10.1073/pnas.1300203110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang L., Zeng H., Wang P., Soker S., and Mukhopadhyay D. (2003) Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J. Biol. Chem. 278, 48848–48860 10.1074/jbc.M310047200 [DOI] [PubMed] [Google Scholar]
- 56. Spratley S. J., Bastea L. I., Döppler H., Mizuno K., and Storz P. (2011) Protein kinase D regulates cofilin activity through p21-activated kinase 4. J. Biol. Chem. 286, 34254–34261 10.1074/jbc.M111.259424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sturany S., Van Lint J., Muller F., Wilda M., Hameister H., Hocker M., Brey A., Gern U., Vandenheede J., Gress T., Adler G., and Seufferlein T. (2001) Molecular cloning and characterization of the human protein kinase D2: a novel member of the protein kinase D family of serine threonine kinases. J. Biol. Chem. 276, 3310–3318 10.1074/jbc.M008719200 [DOI] [PubMed] [Google Scholar]
- 58. Liou G. Y., Doppler H., Braun U. B., Döppler H., Braun U. B., Panayiotou R., Scotti Buzhardt M., Radisky D. C., Crawford H. C., Fields A. P., Murray N. R., Wang Q. J., Leitges M., and Storz P. (2015) Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat. Commun. 6, 6200 10.1038/ncomms7200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.