

Abstract
Virulent strains of the bacterial pathogen Vibrio cholerae cause the diarrheal disease cholera by releasing cholera toxin into the small intestine. V. cholerae acquired its cholera toxin genes by lysogenic infection with the filamentous bacteriophage CTXφ. CTXφ uses its minor coat protein pIII, located in multiple copies at the phage tip, to bind to the V. cholerae toxin-coregulated pilus (TCP). However, the molecular details of this interaction and the mechanism of phage internalization are not well-understood. The TCP filament is a polymer of major pilins, TcpA, and one or more minor pilin, TcpB. TCP are retractile, with both retraction and assembly initiated by TcpB. Consistent with these roles in pilus dynamics, we hypothesized that TcpB controls both binding and internalization of CTXφ. To test this hypothesis, we determined the crystal structure of the C-terminal half of TcpB and characterized its interactions with CTXφ pIII. We show that TcpB is a homotrimer in its crystallographic form as well as in solution and is present in multiple copies at the pilus tip, which likely facilitates polyvalent binding to pIII proteins at the phage tip. We further show that recombinant forms of TcpB and pIII interact in vitro, and both TcpB and anti-TcpB antibodies block CTXφ infection of V. cholerae. Finally, we show that CTXφ uptake requires TcpB-mediated retraction. Our data support a model whereby CTXφ and TCP bind in a tip-to-tip orientation, allowing the phage to be drawn into the V. cholerae periplasm as an extension of the pilus filament.
Keywords: bacteriophage, Vibrio cholerae, type IV pili, X-ray crystallography, protein structure, CTXφ, minor pilins, pIII, TcpB, toxin coregulated pilus
Introduction
The Gram-negative bacterium Vibrio cholerae is the causative agent of the diarrheal disease cholera. V. cholerae is ingested in contaminated water and food and colonizes the small intestine. V. cholerae use toxin-coregulated pili (TCP),4 long thin filaments belonging to the type IV pilus (T4P) family, to self-associate, drawing the bacteria into protective microcolonies (1). V. cholerae releases cholera toxin, an ADP-ribosylating toxin that acts on epithelial cells of the small intestine, resulting in profuse watery diarrhea that is the hallmark of cholera disease (2). Pandemic V. cholerae acquired the genes encoding the cholera toxin subunits by infection with the filamentous lysogenic bacteriophage CTXφ, which integrates its genome in tandem copies into the bacterial genome (3). CTXφ infects V. cholerae by first binding to its primary receptor, the surface-displayed TCP (3, 4), and then to its secondary receptor, the periplasmic protein TolA (4, 5). Expression of the tcp operon is coregulated with that of the cholera toxin genes by the master regulator ToxT (6). CTXφ binds to both TCP and TolA via its minor coat protein, pIII, which is present in three to five copies at the tip of the phage filament (3, 4). Both interactions are critical for phage infection. Once in the periplasm, the pVIII major coat proteins of CTXφ are shed into the inner membrane, and the circular single-stranded phage genome is delivered into the cytoplasm (7).
TCP are retractile polymers of thousands of copies of the major pilin protein, TcpA (1, 8), plus a low-abundance minor pilin, TcpB (9). TCP and other T4P are assembled by a macromolecular protein machine that spans both membranes and the intervening periplasm of Gram-negative bacteria (10–13). Major pilins are small globular proteins with an extended N-terminal α-helical spine, α1. The N-terminal half of α1, α1N, is conserved and hydrophobic, with the exception of an invariant Glu-5. α1N anchors pilin subunits in the inner membrane prior to pilus assembly and forms a helical array in the core of the assembled pilus filament, displaying its globular C-terminal domains on the pilus surface (8, 14, 15). Pilus assembly occurs at the inner membrane, with subunits docking into the base of the growing pilus, attracted in part by charge complementarity between Glu-5 of each incoming subunit and the positively charged N-terminal amine on the terminal subunit in the growing filament (16). The pilus grows across the periplasm and through a gated multimeric secretin channel in the outer membrane for display on the bacterial surface where it performs a multitude of functions that are critical to pathogenesis (13, 17). Pilus assembly can involve dozens of components, including four or five distinct minor pilins, low-abundance pilin-like proteins that resemble the major pilins and are thought to form a priming complex in the inner membrane that recruits the major pilins (9, 18–21). Presumably, the minor pilin-priming complex forms the tip of the pilus, but this has not been demonstrated directly. V. cholerae and enterotoxigenic Escherichia coli (ETEC) possess comparatively simple T4P systems, with fewer than a dozen assembly components and a single minor pilin that initiates pilus assembly (9, 22). X-ray crystal structures of the ETEC minor pilin CofB show a pilin-like protein connected to an additional C-terminal “effector” domain via a flexible linker (22, 23). The linker and C-terminal domains twist around each other in the crystal lattice, forming a trimer. Kawahara et al. (23) proposed that the CofB homotrimer represents a priming complex that initiates ETEC T4P assembly.
T4P functions include self-association and microcolony formation, DNA uptake, twitching motility, substrate secretion, and signaling of host cells. Like V. cholerae TCP, other T4P are receptors for bacteriophage (24). T4P functions, including DNA and phage uptake, require pilus retraction, a process whereby the pili disassemble, with the major pilins melting back into the inner membrane one subunit at a time (10). Rapid assembly and retraction, facilitated by dedicated ATPases, allow T4P to bind to substrates, surfaces, or nearby bacteria and, upon retraction, draw substrates into the periplasm, pull bacteria along surfaces, and bring bacteria together in aggregates or into intimate contact with host cells. V. cholerae TCP are also retractile; however, they lack a retraction ATPase. Instead, TCP retraction, like assembly, is initiated by the minor pilin TcpB, which signals retraction by mechanical rather than enzymatic means (9). By analogy with ETEC CofB, to which TcpB has very limited sequence similarity, monomeric TcpB was proposed to dock into the growing pilus via its pilin domain and block passage of the pilus through the secretin channel via its effector domain. Stalling pilus assembly would trigger the iterative collapse of major pilins one subunit at a time into the inner membrane, retracting the pilus (9). TcpB, like TcpA and other major pilins, has a glutamate at position 5. Glu-5 is required to initiate retraction but not to prime pilus assembly (9). Consistent with this finding, a V. cholerae tcpB-E5V mutant makes abundant pili that are not retractile (9).
The role of the minor pilin TcpB in CTXφ transduction has not been established. CTXφ infection of V. cholerae has been studied using pCTX-Kmφ, a double-stranded replicative fragment of the CTXφ genome in which a kanamycin resistance cassette has been inserted into the gene encoding the cholera toxin catalytic subunit, CtxA (3). The single-stranded form of pCTX-Kmφ is packaged by V. cholerae into infectious particles that confer kanamycin resistance onto recipient V. cholerae, allowing facile selection in growth media containing this antibiotic. The V. cholerae ΔtcpB mutant makes low levels of pili (9) but are completely resistant to CTX-Kmφ transduction (25). Furthermore, transduction efficiency is reduced 1–2 orders of magnitude for the tcpB-E5V mutant (25) despite having WT levels of pili (9). These results suggest that TcpB-mediated pilus retraction is necessary for phage uptake. However, we wondered whether TcpB might also play a direct role in CTXφ binding. We hypothesize that V. cholerae TcpB is located at the tip of TCP and acts as the primary receptor for CTXφ. This would allow the bound filamentous phage to be drawn into the cell upon pilus retraction as if it were an extension of the pilus. We show here that recombinant TcpB binds to the CTXφ minor coat protein pIII and that the presence of either TcpB or anti-TcpB antibodies substantially reduces V. cholerae transduction. Using immunogold labeling, we provide direct evidence that TcpB is indeed located at the pilus tips in multiple copies and show that TcpB-mediated pilus retraction is required for CTXφ infection. Finally, we present an X-ray crystal structure of the C-terminal half of TcpB, which is a trimer both in the crystal lattice and in solution. Our results lead us to a molecular mechanism for CTXφ binding and uptake, with implications for understanding pilus dynamics and substrate uptake in other T4P systems.
Results
Crystal structure of the C-terminal portion of TcpB
To understand how the V. cholerae minor pilin TcpB performs its dual functions in initiating both pilus assembly and retraction, we sought its crystal structure. Most type IV pilin structures have been obtained by recombinantly expressing an N-terminally truncated form of the protein lacking its first ∼25 amino acids (α1N), which are hydrophobic and anchor the C-terminal domain into the inner membrane and the pilus filament. ΔN-TcpB (residues 25–423; Fig. 1A) (22) was expressed and purified, but efforts to crystallize this TcpB form were unsuccessful. Thus, smaller portions of TcpB were expressed based on its sequence alignment with ETEC CofB (Fig. 1, A and B) and in light of the ΔN-CofB structure (22) (Fig. 1B). TcpB-pilin (residues 25–228) corresponds to the globular “pilin domain” of CofB without α1N, and TcpB-C (residues 243–423) represents the C-terminal half of TcpB and corresponds to the effector domain of CofB and the β-repeats connecting it to the pilin domain (Fig. 1, A and B). Soluble protein was produced for both forms, and the TcpB-C structure was solved to 1.53-Å resolution (Table 1 and Fig. 1C).
Figure 1.

Structure of the C-terminal half of V. cholerae TcpB and comparison with ETEC CofB. A, domain organization of TcpB and CofB and their recombinantly expressed forms. Domain boundaries and cysteines are indicated. B, amino acid sequences of TcpB and CofB aligned based on their structures and colored as in A. Identical residues are shown in boldface text. TcpB and CofB are 20% identical in their C-terminal halves (β-repeat and effector domain) and 16% identical overall. Disulfide bonds seen in the crystal structures are indicated with solid lines. The disulfide bond predicted between TcpB Cys-250 and Cys-261 is shown with a dashed line. The peptide used to generate anti-TcpB(64–78) antibodies is indicated. C, crystal structure of TcpB-C (residues 243–423) shown in ribbon representation, colored and labeled as in A and B. Residues 243–259 are not resolved in the crystal structure and are illustrated as the first strand of the three-stranded βR based on their alignment with βR2 of CofB. D, crystal structure of ΔN-CofB (22). Only the C-terminal half of ΔN-CofB (residues 261–518) is shown, omitting the pilin domain.
Table 1.
X-ray data collection and refinement statistics
| TcpB-C(243–423) | |
|---|---|
| Data collection | |
| X-ray source | SSRLa 9-2 |
| Wavelength (Å) | 1.0 |
| Space group | R32:H |
| No. molecules/AUb | 1 |
| Unit cell parameters (Å; °) | a = b = 63.29, c = 236.29; α = β = 90, γ = 120 |
| Resolution (Å) | 39.4–1.53 (1.80–1.76) |
| Completeness (%) | 99.9 (100.0) |
| Total observations | 144,061 (7,938) |
| Mean I/σ(I) | 26.9 (12.4) |
| Multiplicity | 3.8 (3.8) |
| Rpimc | 0.016 (0.053) |
| Structure refinement | |
| No. unique reflections refined against | 26,883 |
| Reflections in Rfree set | 1,342 |
| Rworkd (%) | 17.8 |
| Rfreee (%) | 19.8 |
| r.m.s.d.,f bond lengths (Å) | 0.008 |
| r.m.s.d., bond angles (°) | 0.99 |
| No. atoms | |
| Protein atoms (no H) | 1,384 |
| O atoms from waters | 141 |
| Ligand/ion atoms | 20 |
| B values | |
| Protein atoms (Å2) | 25.2 |
| Water oxygen atoms (Å2) | 30.8 |
| Ligand atoms (Å2) | 35.9 |
| Ramachandran outliers (%) | 0 |
| Ramachandran favored (%) | 100 |
| PDB code | 6MIC |
Note—Values in parentheses are for the highest resolution shell (1.8 Å - 1.76 Å).
a Stanford Synchrotron Radiation Lightsource.
b Asymmetric unit.
cRpim = Σhkl{1/(Nhkl − 1)}1/2Σi |Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl).
d Rwork = Σhkl‖Fobs| − |Fcalc‖/Σhkl|Fobs|.
e Rfree is the cross-validation R factor for 5% of the reflections against which the model was not refined.
f r.m.s.d., root mean square deviation.
TcpB-C is an elongated protein with an N-terminal β-hairpin connected by an extended 14-residue loop to a second β-hairpin at the proximal end of a C-terminal β-sandwich motif (Fig. 1C). The β-sandwich has three β-strands per β-sheet, with long irregular loops at its distal end. The second β-hairpin and β-sandwich together comprise the effector domain of TcpB. TcpB-C measures ∼74 Å in length and ∼30 Å in width. Despite limited sequence homology, the TcpB-C structure is strikingly similar to its corresponding region in CofB (Fig. 1D): both proteins share the same fold and superimpose with a root mean square deviation of 1.9 Å (main-chain atoms). The N-terminal β-hairpin of TcpB-C corresponds in position and sequence to the second of two homologous three-stranded β-sheets in CofB, referred to as β-repeat 1 (βR1) and βR2, that connect the pilin domain to the effector domain. TcpB has only one such β-sheet (βR), based on its sequence alignment with CofB, but its first strand is not resolved in the crystal structure. This is despite the presence of both Cys-250 and Cys-261, which correspond to one of two disulfide-bonded cysteine pairs that stabilize the β-repeats of CofB. Three of the four disulfide-bonded cysteine pairs in CofB are present at equivalent positions in the TcpB sequence, one in the pilin domain, one in βR, and one in the effector domain. Although all three cysteine pairs are likely to be disulfide-bonded, only the Cys-321–Cys-421 bond is apparent in the TcpB-C crystal structure, linking a proximal loop in the effector domain with the end of the terminal β-strand, as is seen for CofB.
The extended structure of TcpB-C is stabilized in the crystal lattice by its trimeric arrangement (Fig. 2A), which is identical to the trimer found in the ΔN-CofB crystal (22, 23). In both crystallographic trimers, the C-terminal effector domains align along their long axes, coming closer together at their distal ends. The β-repeats wrap around the trimer axis and hydrogen-bond to βRs of adjacent monomers, creating three interconnected β-sheets that together form a pillar. In the TcpB trimer, each of these three β-sheets comprises βR from one monomer and effector domain β-hairpin of a second monomer, joined by a central β-strand of the third monomer. This central β-strand is the extended loop that connects βR with the effector domain. In CofB, both βR1 and βR2 contribute to the β-sheets that form the pillar of the trimer. Although the pillar is stabilized by backbone hydrogen bonds, the effector domains are held together by van der Waals interactions among hydrophobic side chains, burying ∼3500 Å2 of surface area for a single monomer.
Figure 2.

TcpB-C forms a homotrimer. A, TcpB-C and ΔN-CofB crystallographic trimers, with each subunit, A, B, and C, colored differently. Only the β-repeats and effector domains of the ΔN-CofB structure are shown. B, chain C of TcpB-C, shown in the same orientation as in A, at left in cartoon representation and at right with electrostatic surface potential, calculated using DelPhi (54). Red represents negative charge, blue is positive charge, and white is neutral (scale from −5 to +5 kT). The trimerization surface is mostly uncharged. C, Coomassie-stained nonreducing SDS-PAGE of recombinant TcpB forms in the absence and presence of cross-linker DSP. Fragments containing the C-terminal half of TcpB (residues 243–423) appear at approximately triple their mass in the presence of DSP, indicating the presence of trimers, whereas the TcpB fragment comprising the pilin domain remains as a monomer. The theoretical masses of monomeric and trimeric forms are shown in the table on the right. MM, mass markers (kDa).
The extensive hydrophobic surface area of the trimerization interface, evident in the electrostatic surface representation of the monomer (Fig. 2B), may provide the driving force for trimerization as a mechanism to nucleate pilus assembly, as suggested for ETEC CofB (23). To assess whether soluble TcpB exists as a trimer in solution, where it is present in much lower concentration than in the crystal, we performed cross-linking on all three TcpB forms: ΔN-TcpB, TcpB-pilin, and TcpB-C. All protein forms migrate to their predicted masses on SDS-PAGE in the absence of cross-linker, but bands appear at ∼3-fold higher masses for both ΔN-TcpB and TcpB-C in the presence of the amine-reactive cross-linker dithiobis(succinimidyl propionate) (DSP), consistent with the trimeric form (Fig. 2C). In contrast, TcpB-pilin, which lacks the C-terminal trimerization domains, has a slightly lower apparent mass in the presence of cross-linker, which likely represents internally cross-linked protein that is not fully denatured and is thus more compact.
TcpB is present at the ends of the V. cholerae toxin-coregulated pilus
TcpB, like core minor pilins in other T4P systems, is predicted to localize to the pilus tip (9), consistent with its role in initiating pilus assembly. However, tip association has not been directly demonstrated for any minor pilin. To investigate TcpB localization on the pilus, purified TCP were applied to carbon-coated copper grids and probed with chicken antibody raised against ΔN-TcpB (residues 25–423; Table 2) followed by gold-labeled secondary antibody. The grids were stained with uranyl acetate and imaged by transmission EM. Gold clusters were observed at the ends of a small proportion of the pilus filaments (Fig. 3A), consistent with tip localization for TcpB. In some cases, two or three gold clusters were observed at the pilus ends, which presumably represent their distal tips (Fig. 3B).
Table 2.
Antibodies
| Antibody | Description | Source/Ref. |
|---|---|---|
| Anti-TcpB(64–78) | Rabbit polyclonal antibody against TcpB peptide (residues 64–78) | 9 |
| Anti-TcpB | Chicken polyclonal antibodies raised against soluble TcpB (ΔN-TcpB, residues 25–423) | This study, Pacific Immunology (Ramona, CA) |
| Anti-pIII(186–202) | Rabbit polyclonal antibody against CTXφ pIII peptide (residues 186–202) | This study, Pacific Immunology (Ramona, CA) |
| Gold-labeled donkey anti-chicken | Donkey polyclonal antibody against chicken antibodies, conjugated to 12-nm gold particles | Jackson ImmunoResearch Laboratories (West Grove, PA) |
| Anti-CofJ | Rabbit polyclonal antibody against CofJ peptide (residues 164–185) | 32 |
Figure 3.

Immunogold labeling of TcpB at pilus ends. Purified TCP were adhered to carbon-coated copper grids, treated with chicken anti-TcpB antibody raised against ΔN-TcpB (Table 2) and gold-labeled secondary antibody, then stained with uranyl acetate, and imaged by transmission EM. Single (A) and multiple (B) gold labels are seen at ends of TCP. Magnification of insets is indicated.
V. cholerae TcpB binds to the CTXφ tip-associated protein pIII
The filamentous phage CTXφ infects V. cholerae by binding to TCP via pIII, which is located in multiple copies at the phage tip. Because TcpB appears to be present in multiple copies at the tips of TCP, we reasoned that TcpB might interact directly with pIII. A pIII:TcpB interaction would mean end-to-end binding between the phage and the pilus. Because both filaments are ∼8 nm in diameter, such an interaction would allow phage uptake through the outer membrane secretion channel without increasing the diameter of the pilus. To test this idea, recombinant pIII was produced in a soluble form lacking its C-terminal 26 amino acids, which are predicted to anchor the protein in the bacterial inner membrane prior to incorporation into CTXφ (pIII-ΔTM; residues 1–355). pIII was expressed and purified, and its interaction with TcpB was assessed by capture ELISAs. First, pIII was immobilized on microtiter plates, and ΔN-TcpB was added in increasing concentrations. Unbound TcpB was washed off, and bound TcpB was detected using rabbit anti-TcpB antibody raised against a peptide corresponding to residues 64–78 on the pilin domain of TcpB. The amount of bound TcpB increases with increasing TcpB concentrations for pIII-coated wells, whereas only background TcpB binding was observed in wells coated with the major pilin TcpA or with bovine serum albumin (BSA) (Fig. 4A). Next, the inverse experiment was performed in which wells were coated with TcpB, and pIII was added in solution. Unbound pIII was washed off the plates, and anti-pIII antibody (Table 2) was added to detect bound pIII. The amount of antibody bound in the wells increased with increasing amounts of pIII for wells coated with TcpB, whereas no binding was observed for wells coated with TcpA or BSA (Fig. 4B). These data indicate a specific interaction between V. cholerae TcpB and CTXφ pIII. Interestingly, pIII binding to TcpB became saturated at a 10-fold lower concentration than that of the immobilized antigen. This may be because TcpB has a tendency to bind to the wells in a manner in which the pIII-binding site is inaccessible.
Figure 4.
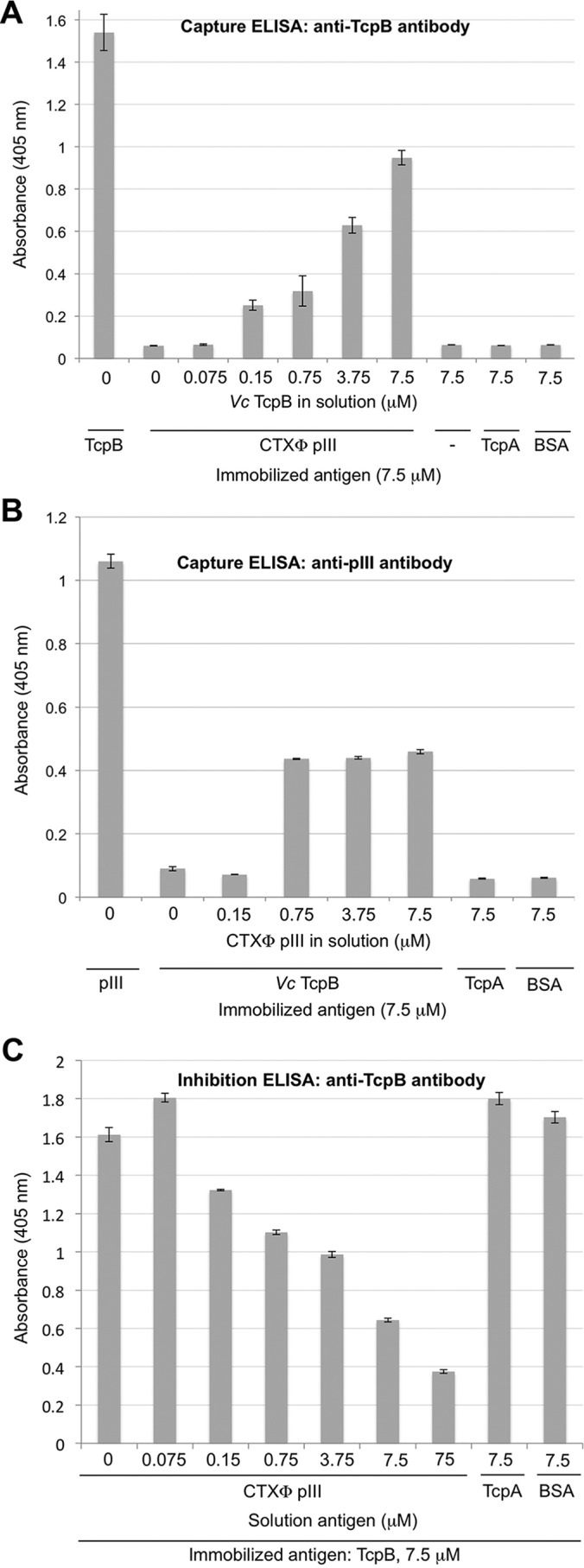
V. cholerae TcpB binds to CTXφ pIII in ELISAs. A, CTXφ pIII protein was immobilized on ELISA microplate wells, and V. cholerae TcpB was added in increasing amounts in solution. Captured TcpB was detected with rabbit anti-TcpB(64–78) antibody and enzyme-linked secondary antibody. TcpB was immobilized as a positive control; negative controls were blocked wells lacking immobilized antigen (−) and immobilized antigens TcpA and BSA to show the specificity of the TcpB:pIII interaction. B, TcpB was immobilized on ELISA microplate wells, and pIII was added in increasing amounts in solution. Captured pIII was detected with anti-pIII antibody and enzyme-linked secondary antibody. pIII was immobilized as a positive control, and TcpA and BSA were immobilized as negative controls for capture. C, TcpB was immobilized, and pIII was added in increasing amounts in solution to inhibit anti-TcpB(64–78) antibody binding. Bound antibody was detected with enzyme-linked secondary antibody. TcpA and BSA were added in solution as negative controls for inhibition. For all experiments, absorbance was read at 405 nm. Values represents the average of three separate experiments; error bars represent the S.E.
To further examine the interaction between TcpB and pIII, inhibition ELISAs were performed. TcpB was immobilized on microtiter plates, and varying amounts of soluble pIII were added to the wells to block the TcpB epitopes. After washing off unbound pIII, rabbit anti-TcpB(64–78) antibodies were added, and their binding to immobilized TcpB was detected using enzyme-linked secondary antibody. The amount of bound anti-TcpB antibody decreased with increasing amounts of pIII (Fig. 4C), indicating that pIII inhibits anti-TcpB binding to immobilized TcpB. In contrast, no reduction in anti-TcpB binding to immobilized TcpB was observed when either BSA or TcpA was added as an inhibitor. These data further support an interaction between TcpB and pIII. The reciprocal experiment in which TcpB was added to immobilized pIII showed no inhibition of anti-pIII antibody binding. It may be that the anti-pIII antibody binds to pIII with a higher affinity than does TcpB. Although we observed specific pIII:TcpB interactions by ELISA, we were not able to show such interactions using pulldown assays, suggesting that binding between these proteins is relatively weak.
Anti-TcpB antibodies and soluble TcpB both block CTXφ infection of V. cholerae
To further investigate the role of TcpB in CTXφ uptake, we performed transduction assays in the presence or absence of polyclonal anti-TcpB antibody (Table 2) or ΔN-TcpB (Fig. 5A). For these assays, we used CTX-Kmφ, in which the ctxA gene is inactivated by insertion of kanamycin resistance gene (KmR) (3). CTX-Kmφ were isolated from the culture supernatant of V. cholerae CL101 (3, 26) and incubated with V. cholerae WT strain O395 in the absence or presence of anti-TcpB antibody or ΔN-TcpB. Bacteria from the infection mixtures were grown on Luria-Bertani (LB) broth agar plates containing kanamycin to identify transductants. The presence of anti-TcpB antibodies or ΔN-TcpB in the infection mixture reduced the number of CTX-Kmφ cells by 90–95% relative to untreated cells (Fig. 5A). In contrast, addition of an unrelated antibody, anti-CofJ, or BSA had little to no effect on transduction (Fig. 5A). These results are consistent with CTXφ binding to TcpB as a prelude to phage uptake.
Figure 5.

CTX-Kmφ infection of V. cholerae is inhibited by recombinant TcpB and anti-TcpB antibodies. A, V. cholerae WT (O395) and ΔtcpA and ΔtcpB strains were incubated with buffer or recombinant TcpB (ΔN-TcpB), rabbit anti-TcpB antibody(64–78), BSA, or anti-CofJ antibody; then transduced with CTX-Kmφ; and plated on LB-Km plates. The number of cfu/ml for each infection is reported (n = 3); error bars represent the S.E. B, V. cholerae WT was incubated with three different forms of recombinant TcpB: ΔN-TcpB (residues 25–423), TcpB-C (residues 243–423), and TcpB-pilin (residues 25–228); then transduced with CTX-Kmφ; and plated on LB-Km plates. cfu/ml was calculated for each infection as for A.
As controls, infection of V. cholerae strains lacking the major pilin, ΔtcpA, and the minor pilin, ΔtcpB, were also tested. As shown previously (3, 25), no transductants were observed for either strain, whereas WT levels of transduction were obtained in a ΔtcpB mutant complemented with ectopically expressed TcpB (+ptcpB). Because some pili are present in the ΔtcpB strain (9), its complete lack of transduction is likely due to the absence of TcpB on the pilus.
To map the region of TcpB that interacts with CTXφ, phage transduction assays were performed in the presence of the additional recombinant forms of TcpB, TcpB-C (residues 243–423) and TcpB-pilin (residues 25–228). TcpB-C reduced transduction levels by 73%, comparable with that of the larger ΔN-TcpB, whereas TcpB-pilin did not show significant reduction in transduction (Fig. 5B), suggesting that CTXφ binds to the C-terminal half of TcpB. Given this, it is notable that pIII competes with anti-TcpB(64–78) antibody (Fig. 4C), despite their binding sites being located on different domains. This result may mean these sites are in close proximity.
TcpB-mediated retraction is necessary for CTXφ infection of V. cholerae
We hypothesized that CTXφ infection of V. cholerae involves both binding (adsorption) to TcpB at the tip of the pilus and uptake of CTXφ into the periplasm by TcpB-mediated pilus retraction. To test this hypothesis, we utilized a panel of V. cholerae tcpB Glu-5 substitution mutants. Glu-5 is necessary for efficient pilus retraction but not for assembly; hence, substitution of Glu-5 for valine (tcpB-E5V) results in production of abundant pili that are nonretractile and therefore nonfunctional with respect to autoagglutination, secretion of the colonization factor TcpF (9), and CTXφ uptake (25). More conserved Glu-5 substitutions to glutamine (tcpB-E5Q) and aspartate (tcpB-E5D) also impact pilus functions but to a lesser degree than for valine (9). CTXφ infection of the ΔtcpB strain complemented with tcpB Glu-5 mutants was tested relative to WT V. cholerae. The tcpB-E5D and tcpB-E5Q strains showed ∼80% transduction compared with the WT strain, whereas V. cholerae tcpB-E5V transduction showed only 5% (Fig. 6). These data suggest that TCP retraction is required to bring tip-associated CTXφ into the V. cholerae periplasm.
Figure 6.
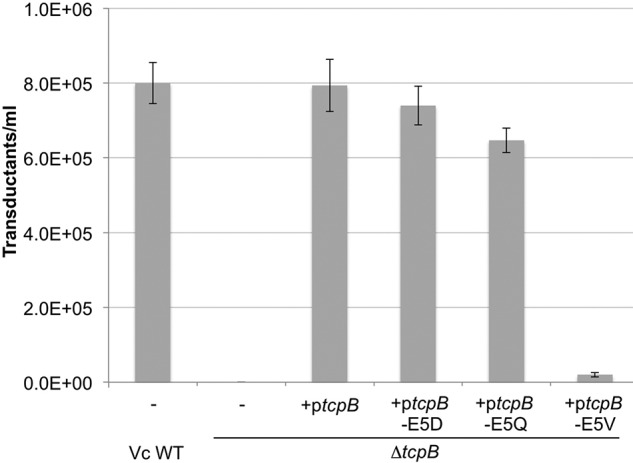
TcpB-mediated pilus retraction is required for CTX-Kmφ infection of V. cholerae. V. cholerae ΔtcpB was transformed with ptcpB encoding WT or Glu-5 substitutions. Cells were grown overnight in pilus-inducing conditions with 0.001 mm rhamnose to induce ectopic expression of TcpB. Cells from overnight culture were infected with CTX-Kmφ and plated on LB-Km. The number of cfu/ml for each infection is reported (n = 3); error bars represent the S.E.
Discussion
Our results show that the minor pilin TcpB is present at the ends of V. cholerae TCP, that CTXφ pIII binds specifically to TcpB during phage infection, and that TcpB-mediated pilus retraction is necessary for efficient phage uptake. Together, these results are consistent with a model in which CTXφ binds via pIII to TcpB at the pilus tip and is drawn through the outer membrane secretin channel by retraction as if it were an extension of the pilus (Fig. 7). Such a mechanism would avoid distorting the secretin channel, as would be required for a pilus with a laterally associated phage filament. Nonetheless, lateral interactions between CTXφ and the pili have been observed by EM (27). Lateral interactions between the TCP major pilin TcpA and the phage major coat protein pVIII are not critical for CTXφ infection of V. cholerae in vitro, as an Ff family coliphage expressing CTXφ pIII fused to the fd pIII is capable of infecting E. coli (4). Lateral interactions may, however, be important for recruiting phage in the environment or in the intestinal tract where concentrations of both are much lower as they would provide a much larger interface than that of TcpB:pIII. Pilus retraction may even force laterally associated phage toward TcpB at the pilus tip as the pilus is pulled into the bacterium.
Figure 7.

Model for T4P-mediated uptake of CTXφ. A, CTXφ binds via its tip-associated minor coat protein pIII to the minor pilin TcpB trimer at the tip of a growing V. cholerae TCP filament. B–D, random incorporation of TcpB monomer at the base of the growing pilus stalls pilus assembly (B), triggering retraction (C), which draws the phage into the periplasm as if it were an extension of the pilus (D and E). TcpB is shown as a trimer, with its pilin domain colored blue and its effector domain colored green. IM, inner membrane; OM, outer membrane.
Our immunogold labeling images represent the most definitive localization of a minor pilin at the tip of a pilus to date. Here, we show both singly and multiply labeled tips, consistent with a TcpB homotrimer at the pilus tip, as seen in the TcpB crystal structure. Trimerization via periplasmic minor pilin C-terminal domains may serve to nucleate assembly for both V. cholerae TCP and ETEC T4P (23), similar to the minor pilin-priming complex that initiates assembly of the complex T4P like Pseudomonas aeruginosa and Neisseria gonorrhoeae and the type II secretion (T2S) system pseudopili (28–30). Physical evidence for a priming complex for the T2S system was obtained with a heterotrimeric crystal structure of the ETEC minor pilins GspI, GspJ, and GspK. Whereas the TcpB and CofB homotrimers form compact structures that when present at the pilus tip could pass easily through the secretin channel, the GspIJK heterotrimers form a bulky mass that has been proposed to block their passage through the secretin channel (28). This mechanism explains a fundamental difference between the T4P, which are displayed on the bacterial surface, and T2S pseudopili, which are restricted to the bacterial periplasm where they extrude substrates across the outer membrane. Blocking passage of the pseudopilus through the secretin channel could, in theory, stall its growth and trigger disassembly, as TcpB has been proposed to do for V. cholerae TCP (9). Similar to TCP, T2S systems lack a retraction ATPase. Although pseudopilus retraction has not been demonstrated, the minor pilin–mediated assemble/stall/retract model is consistent with a piston-like mechanism to extrude substrates through the secretin channel (31).
Biological functions of tip-associated minor pilins beyond their roles in priming assembly have recently been recognized for other T4P. Oki et al. (21) showed that the trimeric form of CofB binds to the N terminus of CofJ, a protein secreted by the ETEC T4P machinery (32). The authors propose that secreted CofJ may bind to intestinal epithelial cells and recruit ETEC by binding to trimeric CofB at the tip of its pilus. Similarly, the V. cholerae competence pilus, a complex T4P, was shown recently to bind to DNA via its tip and draw the DNA to the cell surface by retraction (33). Point mutations in genes encoding two of the minor pilins necessary for pilus assembly reduce DNA uptake without affecting pilus assembly. We show here an additional biological role for tip-associated minor pilins as receptors for CTXφ. Although we were able to demonstrate interactions between TcpB and pIII directly by ELISA and indirectly by transduction, we were not able to pull down one protein with the other or to demonstrate binding by immunogold labeling, suggesting that these interactions are weak. CTXφ binding may be facilitated by avidity between the three TcpB molecules in the homotrimer at the pilus tip and the three to five pIII proteins at the tip of CTXφ.
In addition to initiating TCP assembly and binding to pIII, we show here a third role of TcpB in CTXφ infection, inducing pilus retraction, which brings the phage tip into the periplasm where pIII binds to the C-terminal domain of TolA (4, 5, 27, 34). TCP retraction was predicted previously as an uptake mechanism for CTXφ (3) based on the similarities between CTXφ and the Ff family of filamentous phage, including M13. CTXφ resembles the Ff coliphage in gene organization and structure; both phage types have three to five copies of a minor coat protein, pIII, at their tip and thousands of copies of pVIII, which forms a cylindrical shell around the single-stranded circular phage genome. Similar to CTXφ, Ff phage use pIII to bind to the F pilus on E. coli, which spontaneously retracts to bring the phage into the periplasm (35–37) where pIII binds to TolA (4, 5, 38, 39). The relatively weak interaction between individual pIII and TcpB molecules may facilitate the handoff to TolA as a first step in phage uncoating. We note that this process is analogous to DNA uptake by the competence pilus. After being drawn into the periplasm by pilus retraction, DNA interacts with ComEA, the periplasmic component of the bacterial competence system, which is thought to ratchet the DNA into the cell (40–42).
An outstanding question is whether TcpB-mediated retraction is indeed spontaneous, triggered by random incorporation of TcpB monomers into the growing pilus (Fig. 7), or whether phage binding somehow signals retraction. TcpB is produced in much lower quantities than TcpA (9); hence its designation as a “minor” pilin. Most likely, TcpB is incorporated randomly and infrequently into the growing pilus to trigger retraction, which would draw any bound phage into the cell. Alternatively, phage binding to the pilus may provide a mechanosensory signal that enhances incorporation of TcpB to trigger retraction. For instance, tension on the pilus from phage binding might enlarge the gap at the base of the pilus where the major and minor pilins dock, enhancing the ability of TcpB to dock. The crystal structure of ETEC CofB (22, 23) contains the pilin domain, which is similar in structure but slightly larger than that of the corresponding major pilin CofA (43). Thus, CofB may be less likely than CofA to incorporate into the base of a growing pilus, except when the pilus is pulled, opening up the gap at the base of the pilus enough for CofB to dock. Although it is well-established that retraction generates force on the pilus (44, 45), it remains to be seen whether force on the pilus triggers retraction. Interestingly, retraction events for the V. cholerae competence T4P occur with the same frequency and rate in both the absence and presence of DNA, suggesting that retraction is spontaneous in this system and not triggered by DNA binding (33). However, the mechanism that drives retraction in the competence T4P differs from that of TCP, utilizing a retraction ATPase rather than a single minor pilin. It remains to be seen whether these processes are directly comparable.
The results described here advance our understanding of an important biological process driving the evolution of bacterial pathogens and help to clarify the role of the minor pilin in pilus dynamics and functions. A detailed understanding of the phage:host interaction and phage uptake mechanism may inform strategies to block this process to control transfer of mobile genetic elements and to exploit the process as a mechanism to deliver antibiotics into the bacterial periplasm.
Experimental procedures
Strains, plasmids, and antibodies
V. cholerae strains and plasmids used for this study are listed in Table 3. V. cholerae cells used as recipients for CTXφ infections were grown without antibiotics. V. cholerae RT4225 was grown in 100 μg/ml ampicillin (Ap); CL101 and phage transductants were grown with 50 μg/ml Km. The V. cholerae TcpB Glu-5 mutants were prepared by transforming V. cholerae ΔtcpB with ptcpB with mutations encoding the Glu-5 substitutions described elsewhere (9). Antibodies are listed in Table 2.
Table 3.
Bacterial strains, plasmids and primers
| Reagent | Description | Source/Ref. |
|---|---|---|
| Strains | ||
| V. cholerae O395 | Wild-type classical O1, Ogawa, SmR | 1 |
| V. cholerae CL101 | O395, pCTX-Kmφ plasmid | 3 |
| V. cholerae ΔtcpA (RT4031) | O395 ΔtcpA | 52 |
| V. cholerae ΔtcpB (RT4368) | O395 ΔtcpB | 52 |
| V. cholerae RT4225 | O395, tcpA (H181A), pMT5:toxT | 26 |
| E. coli (DH5α) | F − endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80dlacZΔM15 Δ (lacZYA-argF)U169, hsdR17 (rK − mK+), λ– | Life Technologies |
| E. coli Origami (DE3) | Δ(ara-leu)7697 ΔlacX74 ΔphoA PvuII phoR araD139 ahpC galE galK rpsL F′[lac+ lacI q pro] (DE3) gor522::Tn10 trxB (KanR, StrR, TetR)4 | |
| E. coli SHuffle® T7 Express lysY Competent E. coli cells | MiniF lysY (CamR)/fhuA2 lacZ::T7 gene1 [lon] ompT ahpC gal λatt::pNEB3-r1-cDsbC (SpecR, lacIq) ΔtrxB sulA11 R(mcr-73::miniTn10–TetS)2 [dcm] R(zgb-210::Tn10–TetS) endA1 Δgor Δ(mcrC-mrr)114::IS10 | New England Biolabs |
| Plasmids | ||
| pJMA10.1 | pBAD22 derivative; araC replaced with PrhaB, Bla, ApR; NcoI site removed | 9 |
| ptcpB | pJMA10.1 containing the tcpB gene | 9 |
| ptcpB-E5Q | ptcpB, E5Q | 9 |
| ptcpB-E5D | ptcpB, E5D | 9 |
| ptcpB-E5V | ptcpB, E5V | 9 |
| pET-15b | Encodes an N-terminal His·Tag® sequence followed by a thrombin site, ApR, T7lac | Novagen |
| pET-15b-tcpB | pET:15b with the gene fragment encoding ΔN-TcpB (residues 25–423) inserted into the NdeI and BamHI sites downstream of the His·Tag | 9 |
| pET-15b-tcpB-pilin | pET:15b with the gene fragment encoding TcpB-pilin (residues 25–228) inserted into the NdeI and BamHI sites downstream of the His·Tag | This study |
| pET-15b-tcpB-C | pET:15b with the gene fragment encoding TcpB-C (residues 243–423) inserted into the NdeI and BamHI sites downstream of the His·Tag | This study |
| pET-15b-pIII | pET:15b with the gene fragment encoding pIII residues 1–355 (pIII-ΔTM) inserted into the NdeI and BamHI sites downstream of the His·Tag | This study |
| pET-15b-tcpA | pET:15b with the gene fragment encoding ΔN-TcpA (residues 29–199) inserted into the NdeI and XhoI sites downstream of the His·Tag | 53 |
| Primers | ||
| TcpB-pilin-for | CGGTCAGTCTTGCTTAAACCGGAGAAATAGCAAT | |
| TcpB-pilin-rev | ATTGCTATTTCTCCGGTTTAAGCAAGACTG ACCGCCA | |
| TcpB-C-for | GGAATTCCATATGTTTTTAGAAGATAGTGAGTTGTGTTGGGA | |
| TcpB-C-rev | TCGCGGATCCTTAATTTTCACACCATTGAAACGCTATAAA | |
| pIII-ΔTM-for | GGAATTCAATATGTCCGCCATCAATTGTG | |
| pIII-ΔTM-rev | CGTAGGATCCTTAGTGCAGGTTTTCAGAAAAGAGGGAG |
Expression and purification of TcpB
TcpB-pilin (residues 25–228) was expressed from plasmid pET-15b-tcpB encoding TcpB with a His tag in place of α1N, residues 1–24 of the mature pilin (ΔN-TcpB; residues 25–423 (9)). A stop codon was inserted into the tcpB gene fragment at codon 229 using primers listed in Table 3 to produce pET-15b-tcpB-pilin. The gene fragment for TcpB-C (residues 243–423) was PCR-amplified from pET-15b-tcpB using TcpB-C forward and reverse primers (Table 3). The PCR products were digested with NdeI and BamHI, cloned into these restriction sites in pET-15b (Novagen) downstream of a sequence encoding a His tag and linker with a thrombin cleavage site, and ligated using T4 DNA ligase. All three plasmids, pET-15b-tcpB, pET-15b-tcpB-pilin, and pET-15b-tcpB-C, were used to transform SHuffle® T7 Express lysY Competent E. coli cells (New England Biolabs). Cells were grown overnight in LB broth supplemented with ampicillin (100 μg/ml) to an optical density at 600 nm (OD600) of 0.1 in a shaking incubator at 37 °C, and protein expression was induced by isopropyl β-d-thiogalactopyranoside (IPTG; final concentration 0.4 mm). Cells were incubated a further 20 h at 20 °C and harvested by centrifugation (5000 × g for 1 h at 4 °C). The cell pellet was suspended in PBS (137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.4; 20 ml/liter of cell pellet), cryocooled in liquid nitrogen and thawed three times. EDTA-free protease inhibitor mixture tablets (Roche Applied Science) and lysozyme (final concentration, 1 mg/ml) were added, and the suspension was incubated for 2–3 h at room temperature. Cell suspensions were clarified by sonication, and cell debris was removed by centrifugation at 40,000 × g for 1 h at 4 °C. Cell lysates were filtered and loaded onto a nickel-nitrilotriacetic acid column pre-equilibrated with wash buffer (30 mm imidazole, 20 mm Tris-HCl, pH 7.4, 500 mm NaCl). The column was washed with 30 column volumes of the same buffer, and His-tagged TcpB proteins were eluted with 300 mm imidazole, 20 mm Tris-HCl, pH 7.4, 500 mm NaCl. Thrombin (GE Healthcare) was added at ∼1 unit/mg of TcpB to cleave off the His tag and linker, and the protein mixture was dialyzed against a buffer of 20 mm Tris-HCl, pH 7.4, 100 mm NaCl. Digested TcpB was loaded onto a Sephacryl S-100 HR size-exclusion column (GE Healthcare). Fractions were collected and analyzed by SDS-PAGE, and those containing the TcpB of the correct size were combined and concentrated on an Amicon stirred-cell concentrator (Millipore) to 20–25 mg/ml and stored with 1 mm benzamidine at −80 °C.
Crystallization and structure determination of TcpB-C
TcpB-pilin and TcpB-C were screened for initial crystallization conditions at the Hauptman-Woodward Medical Research Institute. Small crystals were obtained for TcpB-C only, and conditions were optimized in-house. Diffraction-quality crystals were obtained in 200 mm NaCl, 100 mm sodium acetate, pH 4.6, 20% (±)-2-methyl-2,4-pentanediol. The crystals were cryocooled in mother liquor with 30% glycerol as cryoprotectant. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource using Blu-Ice (46) (Table 1). Raw data were processed with iMosflm (47) and the CCP4 program POINTLESS (48). The data were scaled by AIMLESS, and structure factor amplitudes were calculated and unique data were generated by CTRUNCATE and UNIQUE, respectively (48). The Matthews coefficient indicates 47% solvent content, consistent with a single molecule in the asymmetric unit. The structure was solved by molecular replacement using Phaser (49) with ETEC CofB (Protein Data Bank code 4QS4; residues 357–514) as a model. The TcpB-C model spanning residues 260–423 was completed and refined using the PHENIX suite (50). Residues 260–423 were resolved in the electron density map. Data and refinement statistics are listed in Table 1.
Cross-linking of TcpB fragments
The various forms of TcpB (25 μm) were incubated at 37 °C for 2 h in the absence or presence of 0.5 mm DSP in 20-μl reactions. Laemmli sample buffer (51) lacking reducing agent was added, and each sample was loaded onto an SDS-polyacrylamide gel, electrophoresed, and stained with Coomassie dye.
Immunogold labeling of TcpB on TCP
V. cholerae RT4225 cells (26) were grown for 17 h in LB broth, pH 6.5, with 1 mm IPTG and 100 mg/ml Ap on a Ferris wheel rotator spinning at 60 rpm at 30 °C to induce pilus production. Cells were removed by centrifugation (15 min at 7000 × g), and pili were isolated from the culture supernatant by ammonium sulfate precipitation (30% saturation). The ammonium sulfate was removed by dialysis, and pilus samples were concentrated to 11 mg/ml. Diluted samples of 50 μg/ml pilus were applied in 25-μl drops to carbon-coated copper grids (CF-300, Electron Microscopy Sciences, Hatfield, PA) and incubated for 10 min at 20 °C in a humidified chamber. Grids were washed with TBS containing 0.1% (v/v) Tween 20 (TBST) and blocked for 1 h in TBST containing 2% BSA. Samples were incubated with chicken anti-TcpB antibody raised against ΔN-TcpB (residues 25–423; Table 2), diluted 1:50 in TBST, 1% BSA, for 1 h. Grids were washed in three drops of TBST and incubated for 30 min with anti-chicken secondary antibody conjugated to 12-nm colloidal gold (Jackson ImmunoResearch Laboratories, West Grove, PA) diluted 1:60 in TBST, 1% BSA. Grids were washed in TBST and stained with 25 μl of 3% uranyl acetate for 60 s (Electron Microscopy Sciences) and imaged on a Tecnai F20 FEG scanning transmission electron microscope operating at 200 keV.
Cloning, expression, and purification of CTXφ pIII
CTXφ pIII was expressed and purified in a soluble form lacking its C-terminal 21 amino acids, which represent the transmembrane segment. The gene fragment encoding residues 1–355 of CTXφ pIII (pIII-ΔTM) was PCR-amplified from V. cholerae CL101 using primers listed in Table 3. The PCR product was digested with NdeI and BamHI, and the product was ligated into pET-15b-pIII, which was transformed into SHuffle T7 Express lysY Competent E. coli cells. Cells were grown overnight in 200 ml of LB broth containing 100 μg/ml ampicillin (LB-Ap) at 37 °C shaking at 200 rpm. A 10-ml aliquot of overnight culture was used to inoculate 1 liter of LB-Ap and grown at 37 °C to an OD600 of ∼0.2. Protein expression was induced by addition of IPTG to 0.4 mm, and cells were grown overnight at 19 °C shaking. Cells were pelleted by centrifugation at 4000 × g for 30 min, the supernatant was discarded, and the pellet was resuspended in lysis buffer 2 (20 mm Bis-Tris, pH 6.5, 100 mm NaCl, 10% glycerol, 0.1% Tween, 5 mm imidazole) with 10 μg/ml lysozyme and a cOmpleteTM protease inhibitor mixture tablet (EDTA-free; Sigma-Aldrich). Cells were incubated in lysis buffer 2 with stirring at room temperature for 1 h and then lysed by sonication. The cell lysate was centrifuged at 10,000 × g for 15 min at 4 °C. The supernatant and resuspended cell pellet were analyzed by SDS-PAGE. pIII was found to be in the insoluble pellet fraction. Thus, this fraction was denatured in 6 m guanidine hydrochloride at room temperature for 2 h. Cellular debris was removed by centrifugation at 40,000 × g for 40 min, and the supernatant was passed through a 0.45-μm membrane filter and loaded onto a gravity column containing nickel-nitrilotriacetic acid beads (Qiagen) for affinity purification using the N-terminal His tag. The column was washed using 30 column volumes of wash buffer (20 mm Bis-Tris, pH 6.5, 100 mm NaCl). The column was next washed with 20 column volumes of 6 m urea followed by 4 and 2 m urea. pIII was eluted with 300 mm imidazole. pIII was concentrated using a stirred-cell concentrator (Millipore) with a 10,000-Da molecular-mass-cutoff membrane and loaded onto a size-exclusion column (HiPrep 26/60 Sephacryl S-100 HR, GE Healthcare). Fractions containing pIII were pooled and concentrated to 1.5–2 mg/ml, flash frozen in liquid nitrogen, and stored at −80 °C.
ELISAs
ΔN-TcpB or pIII (35 μl of a 7.5 μm solution in TBS) was added to the wells of polystyrene microtiter plates and incubated overnight at 4 °C. Plates were washed three times with TBST and once with TBS and then blocked with 200 μl/well 5% blocking buffer (TBST, 5% (w/v) skim milk powder) for 1 h at 37 °C. Plates were washed three times with TBST. Varying concentrations of protein (pIII, ΔN-TcpB, ΔN-TcpA, or BSA) were added and incubated for 30 min at 20 °C. Plates were washed three times before adding 35 μl of primary antibody raised against peptides on TcpB and pIII (Table 2) and diluted 1:1000 in blocking buffer. After 2-h incubation at 20 °C, plates were washed five times with TBST, and 35 μl of alkaline phosphatase–conjugated secondary antibody diluted (1:500) in blocking buffer was added to each well and incubated for 1 h at 20 °C. Plates were washed five times with TBS and incubated for 30 min at 20 °C with 35 μl of para-nitrophenyl phosphate substrate diluted in alkaline phosphatase buffer (TBS, 1 m MgCl2, 0.1% Tween 20). The absorbance was measured at 405 nm after 30 min at 20 °C. Error bars represent S.E.
CTXφ transduction assays
V. cholerae strains used as recipients in transduction assays were grown in LB broth, pH 6.5, at 30 °C on a Ferris wheel rotator spinning at 60 rpm for 17 h. ΔtcpB strains ectopically expressing TcpB and TcpB Glu-5 mutants were induced with 0.001% rhamnose. CTX-Kmφ were isolated from V. cholerae CL101 after overnight growth at 37 °C with shaking at 200 rpm. The CL101 strain carries a replicative form of the CTXφ genome, pCTX-Kmφ, in which a kanamycin (Km) resistance cassette has been inserted into the ctxA gene, disrupting it (3). Thus, CTX-Kmφ infection of V. cholerae imparts Km resistance. CL101 cells were removed from the overnight culture by centrifugation at 16,000 × g for 30 s, and the supernatant containing the phage was passed twice through a 0.2-μm-pore filter to remove residual cells. Phage transduction assays were performed by mixing 100 μl of the filtered supernatant with 100 μl of the recipient V. cholerae overnight cultures and shaking for 30 min at 20 °C. Where indicated, 100 μl of CTXφ was mixed with 10 μl of 4.7 mm TcpB, rabbit anti-TcpB(64–78) antibody (Table 2), anti-CofJ antibody, or BSA and incubated at 30 °C for 30 min prior to infection of V. cholerae cells. Serial dilutions of the infection mixture were plated on LB-Km agar plates to enumerate the transductants. Cells were grown overnight at 37 °C, and colony-forming units (cfu) were counted. Values are averages of three separate experiments. Error bars represent S.E.
Author contributions
M. G.-R. and L. C. conceptualization; M. G.-R. and L. C. data curation; M. G.-R., S. K., B. A. B., and L. C. formal analysis; M. G.-R., S. K., B. A. B., and L. C. investigation; M. G.-R. methodology; L. C. funding acquisition; L. C. project administration.
Acknowledgments
This work made use of the 4D LABS shared facilities supported by the Canada Foundation for Innovation (CFI), British Columbia Knowledge Development Fund (BCKDF), Western Economic Diversification Canada (WD), and Simon Fraser Uiversity (SFU).
This work was supported by Canadian Institutes for Health Research (CIHR) Research Grant MOP125959 and by a Simon Fraser University VPR RPE Grant. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 6MIC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TCP
- toxin-coregulated pilus/pili
- T4P
- type IV pilus/pili
- ETEC
- enterotoxigenic E. coli
- βR
- β-repeat
- DSP
- dithiobis(succinimidyl propionate)
- Km
- kanamycin
- T2S
- type II secretion
- Ap
- ampicillin
- IPTG
- isopropyl β-d-thiogalactopyranoside
- TBS
- Tris-buffered saline
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- cfu
- colony-forming units.
References
- 1. Taylor R. K., Miller V. L., Furlong D. B., and Mekalanos J. J. (1987) Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U.S.A. 84, 2833–2837 10.1073/pnas.84.9.2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaper J. B., Morris J. G. Jr., and Levine M. M. (1995) Cholera. Clin. Microbiol. Rev. 8, 48–86 10.1128/CMR.8.1.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Waldor M. K., and Mekalanos J. J. (1996) Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 272, 1910–1914 10.1126/science.272.5270.1910 [DOI] [PubMed] [Google Scholar]
- 4. Heilpern A. J., and Waldor M. K. (2003) pIIICTX, a predicted CTXφ minor coat protein, can expand the host range of coliphage fd to include Vibrio cholerae. J. Bacteriol. 185, 1037–1044 10.1128/JB.185.3.1037-1044.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heilpern A. J., and Waldor M. K. (2000) CTXφ infection of Vibrio cholerae requires the tolQRA gene products. J. Bacteriol. 182, 1739–1747 10.1128/JB.182.6.1739-1747.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DiRita V. J., Parsot C., Jander G., and Mekalanos J. J. (1991) Regulatory cascade controls virulence in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A. 88, 5403–5407 10.1073/pnas.88.12.5403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLeod S. M., Kimsey H. H., Davis B. M., and Waldor M. K. (2005) CTXφ and Vibrio cholerae: exploring a newly recognized type of phage-host cell relationship. Mol. Microbiol 57, 347–356 10.1111/j.1365-2958.2005.04676.x [DOI] [PubMed] [Google Scholar]
- 8. Li J., Egelman E. H., and Craig L. (2012) Structure of the Vibrio cholerae type IVb pilus and stability comparison with the Neisseria gonorrhoeae type IVa pilus. J. Mol. Biol. 418, 47–64 10.1016/j.jmb.2012.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ng D., Harn T., Altindal T., Kolappan S., Marles J. M., Lala R., Spielman I., Gao Y., Hauke C. A., Kovacikova G., Verjee Z., Taylor R. K., Biais N., and Craig L. (2016) The Vibrio cholerae minor pilin TcpB initiates assembly and retraction of the toxin-coregulated pilus. PLoS Pathog. 12, e1006109 10.1371/journal.ppat.1006109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Craig L., Forest K. T., and Maier B. (2019) Type IV pili: dynamics, biophysics and functional consequences. Nat. Rev. Microbiol. 17, 429–440 10.1038/s41579-019-0195-4 [DOI] [PubMed] [Google Scholar]
- 11. McCallum M., Burrows L. L., and Howell P. L. (2019) The dynamic structures of the type IV pilus. Microbiol. Spectr. 7, PSIB-0006-2018 10.1128/microbiolspec.PSIB-0006-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang Y. W., Kjær A., Ortega D. R., Kovacikova G., Sutherland J. A., Rettberg L. A., Taylor R. K., and Jensen G. J. (2017) Architecture of the Vibrio cholerae toxin-coregulated pilus machine revealed by electron cryotomography. Nat. Microbiol. 2, 16269 10.1038/nmicrobiol.2016.269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berry J. L., and Pelicic V. (2015) Exceptionally widespread nanomachines composed of type IV pilins: the prokaryotic Swiss Army knives. FEMS Microbiol. Rev. 39, 134–154 10.1093/femsre/fuu001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kolappan S., Coureuil M., Yu X., Nassif X., Egelman E. H., and Craig L. (2016) Structure of the Neisseria meningitidis type IV pilus. Nat. Commun. 7, 13015 10.1038/ncomms13015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F., Coureuil M., Osinski T., Orlova A., Altindal T., Gesbert G., Nassif X., Egelman E. H., and Craig L. (2017) Cryoelectron microscopy reconstructions of the Pseudomonas aeruginosa and Neisseria gonorrhoeae type IV pili at sub-nanometer resolution. Structure 25, 1423–1435.e4 10.1016/j.str.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Craig L., Volkmann N., Arvai A. S., Pique M. E., Yeager M., Egelman E. H., and Tainer J. A. (2006) Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol. Cell 23, 651–662 10.1016/j.molcel.2006.07.004 [DOI] [PubMed] [Google Scholar]
- 17. Melville S., and Craig L. (2013) Type IV pili in Gram-positive bacteria. Microbiol. Mol. Biol. Rev. 77, 323–341 10.1128/MMBR.00063-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Giltner C. L., Habash M., and Burrows L. L. (2010) Pseudomonas aeruginosa minor pilins are incorporated into type IV pili. J. Mol. Biol. 398, 444–461 10.1016/j.jmb.2010.03.028 [DOI] [PubMed] [Google Scholar]
- 19. Helaine S., Dyer D. H., Nassif X., Pelicic V., and Forest K. T. (2007) 3D structure/function analysis of PilX reveals how minor pilins can modulate the virulence properties of type IV pili. Proc. Natl. Acad. Sci. U.S.A. 104, 15888–15893 10.1073/pnas.0707581104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Winther-Larsen H. C., Wolfgang M. C., van Putten J. P., Roos N., Aas F. E., Egge-Jacobsen W. M., Maier B., and Koomey M. (2007) Pseudomonas aeruginosa type IV pilus expression in Neisseria gonorrhoeae: effects of pilin subunit composition on function and organelle dynamics. J. Bacteriol. 189, 6676–6685 10.1128/JB.00407-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oki H., Kawahara K., Maruno T., Imai T., Muroga Y., Fukakusa S., Iwashita T., Kobayashi Y., Matsuda S., Kodama T., Iida T., Yoshida T., Ohkubo T., and Nakamura S. (2018) Interplay of a secreted protein with type IVb pilus for efficient enterotoxigenic Escherichia coli colonization. Proc. Natl. Acad. Sci. U.S.A. 115, 7422–7427 10.1073/pnas.1805671115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kolappan S., Ng D., Yang G., Harn T., and Craig L. (2015) Crystal structure of the minor pilin CofB, the initiator of CFA/III pilus assembly in enterotoxigenic Escherichia coli. J. Biol. Chem. 290, 25805–25818 10.1074/jbc.M115.676106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawahara K., Oki H., Fukakusa S., Yoshida T., Imai T., Maruno T., Kobayashi Y., Motooka D., Iida T., Ohkubo T., and Nakamura S. (2016) Homo-trimeric structure of the type IVb minor pilin CofB suggests mechanism of CFA/III pilus assembly in human enterotoxigenic Escherichia coli. J. Mol. Biol. 428, 1209–1226 10.1016/j.jmb.2016.02.003 [DOI] [PubMed] [Google Scholar]
- 24. Bradley D. E. (1973) A pilus-dependent Pseudomonas aeruginosa bacteriophage with a long noncontractile tail. Virology 51, 489–492 10.1016/0042-6822(73)90448-0 [DOI] [PubMed] [Google Scholar]
- 25. Gao Y., Hauke C. A., Marles J. M., and Taylor R. K. (2016) Effects of tcpB mutations on biogenesis and function of TCP, the type IVb pilus of Vibrio cholerae. J. Bacteriol. 198, 2818–2828 10.1128/JB.00309-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirn T. J., Lafferty M. J., Sandoe C. M., and Taylor R. K. (2000) Delineation of pilin domains required for bacterial association into microcolonies and intestinal colonization by Vibrio cholerae. Mol. Microbiol. 35, 896–910 10.1046/j.1365-2958.2000.01764.x [DOI] [PubMed] [Google Scholar]
- 27. Ford C. G., Kolappan S., Phan H. T., Waldor M. K., Winther-Larsen H. C., and Craig L. (2012) Crystal structures of a CTXφ pIII domain unbound and in complex with a Vibrio cholerae TolA domain reveal novel interaction interfaces. J. Biol. Chem. 287, 36258–36272 10.1074/jbc.M112.403386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Korotkov K. V., and Hol W. G. (2008) Structure of the GspK-GspI-GspJ complex from the enterotoxigenic Escherichia coli type 2 secretion system. Nat. Struct. Mol. Biol. 15, 462–468 10.1038/nsmb.1426 [DOI] [PubMed] [Google Scholar]
- 29. Nguyen Y., Sugiman-Marangos S., Harvey H., Bell S. D., Charlton C. L., Junop M. S., and Burrows L. L. (2015) Pseudomonas aeruginosa minor pilins prime type IVa pilus assembly and promote surface display of the PilY1 adhesin. J. Biol. Chem. 290, 601–611 10.1074/jbc.M114.616904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cisneros D. A., Bond P. J., Pugsley A. P., Campos M., and Francetic O. (2012) Minor pseudopilin self-assembly primes type II secretion pseudopilus elongation. EMBO J. 31, 1041–1053 10.1038/emboj.2011.454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sandkvist M. (2001) Type II secretion and pathogenesis. Infect. Immun. 69, 3523–3535 10.1128/IAI.69.6.3523-3535.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yuen A. S., Kolappan S., Ng D., and Craig L. (2013) Structure and secretion of CofJ, a putative colonization factor of enterotoxigenic Escherichia coli. Mol. Microbiol. 90, 898–918 10.1111/mmi.12407 [DOI] [PubMed] [Google Scholar]
- 33. Ellison C. K., Dalia T. N., Vidal Ceballos A., Wang J. C., Biais N., Brun Y. V., and Dalia A. B. (2018) Retraction of DNA-bound type IV competence pili initiates DNA uptake during natural transformation in Vibrio cholerae. Nat. Microbiol. 3, 773–780 10.1038/s41564-018-0174-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Houot L., Navarro R., Nouailler M., Duché D., Guerlesquin F., and Lloubes R. (2017) Electrostatic interactions between the CTX phage minor coat protein and the bacterial host receptor TolA drive the pathogenic conversion of Vibrio cholerae. J. Biol. Chem. 292, 13584–13598 10.1074/jbc.M117.786061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clarke M., Maddera L., Harris R. L., and Silverman P. M. (2008) F-pili dynamics by live-cell imaging. Proc. Natl. Acad. Sci. U.S.A. 105, 17978–17981 10.1073/pnas.0806786105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jacobson A. (1972) Role of F pili in the penetration of bacteriophage f1. J. Virol. 10, 835–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marvin D. A. (1998) Filamentous phage structure, infection and assembly. Curr. Opin Struct. Biol. 8, 150–158 10.1016/S0959-440X(98)80032-8 [DOI] [PubMed] [Google Scholar]
- 38. Riechmann L., and Holliger P. (1997) The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli. Cell 90, 351–360 10.1016/S0092-8674(00)80342-6 [DOI] [PubMed] [Google Scholar]
- 39. Lubkowski J., Hennecke F., Plückthun A., and Wlodawer A. (1999) Filamentous phage infection: crystal structure of g3p in complex with its coreceptor, the C-terminal domain of TolA. Structure 7, 711–722 10.1016/S0969-2126(99)80092-6 [DOI] [PubMed] [Google Scholar]
- 40. Chen I., and Gotschlich E. C. (2001) ComE, a competence protein from Neisseria gonorrhoeae with DNA-binding activity. J. Bacteriol. 183, 3160–3168 10.1128/JB.183.10.3160-3168.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salzer R., Kern T., Joos F., and Averhoff B. (2016) The Thermus thermophilus comEA/comEC operon is associated with DNA binding and regulation of the DNA translocator and type IV pili. Environ. Microbiol. 18, 65–74 10.1111/1462-2920.12820 [DOI] [PubMed] [Google Scholar]
- 42. Seitz P., Pezeshgi Modarres H., Borgeaud S., Bulushev R. D., Steinbock L. J., Radenovic A., Dal Peraro M., and Blokesch M. (2014) ComEA is essential for the transfer of external DNA into the periplasm in naturally transformable Vibrio cholerae cells. PLoS Genet. 10, e1004066 10.1371/journal.pgen.1004066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kolappan S., Roos J., Yuen A. S., Pierce O. M., and Craig L. (2012) Structure of CFA/III and longus type IV pili from enterotoxigenic Escherichia coli. J. Bacteriol. 194, 2725–2735 10.1128/JB.00282-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Clausen M., Jakovljevic V., Søgaard-Andersen L., and Maier B. (2009) High-force generation is a conserved property of type IV pilus systems. J. Bacteriol. 191, 4633–4638 10.1128/JB.00396-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maier B., Koomey M., and Sheetz M. P. (2004) A force-dependent switch reverses type IV pilus retraction. Proc. Natl. Acad. Sci. U.S.A. 101, 10961–10966 10.1073/pnas.0402305101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez A., Moorhead P., McPhillips S. E., Song J., Sharp K., Taylor J. R., Adams P. D., Sauter N. K., and Soltis S. M. (2008) Web-Ice: integrated data collection and analysis for macromolecular crystallography. J. Appl. Crystallogr. 41, 176–184 10.1107/S0021889807057822 [DOI] [Google Scholar]
- 47. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., and Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 52. Kirn T. J., Bose N., and Taylor R. K. (2003) Secretion of a soluble colonization factor by the TCP type 4 pilus biogenesis pathway in Vibrio cholerae. Mol. Microbiol. 49, 81–92 10.1046/j.1365-2958.2003.03546.x [DOI] [PubMed] [Google Scholar]
- 53. Craig L., Taylor R. K., Pique M. E., Adair B. D., Arvai A. S., Singh M., Lloyd S. J., Shin D. S., Getzoff E. D., Yeager M., Forest K. T., and Tainer J. A. (2003) Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol. Cell 11, 1139–1150 10.1016/S1097-2765(03)00170-9 [DOI] [PubMed] [Google Scholar]
- 54. Oron A., Wolfson H., Gunasekaran K., and Nussinov R. (2003) Using DelPhi to compute electrostatic potentials and assess their contribution to interactions. Curr. Protoc. Bioinformatics Chapter 8, Unit 8.4 10.1002/0471250953.bi0804s02 [DOI] [PubMed] [Google Scholar]