

Abstract
Chemokines play diverse roles in human pathophysiology, ranging from trafficking leukocytes and immunosurveillance to the regulation of metabolism and neural function. Chemokine function is intimately coupled to binding tissue glycosaminoglycans (GAGs), heparan sulfate (HS), chondroitin sulfate (CS), and dermatan sulfate (DS). Currently, very little is known about how the structural features and sequences of a given chemokine, the structure and sulfation pattern of a given GAG, and structural differences among GAGs and among chemokines impact binding interactions. In this study, we used solution NMR spectroscopy to characterize the binding interactions of two related neutrophil-activating chemokines, CXCL1 and CXCL5, with HS, CS, and DS. For both chemokines, the dimer bound all three GAGs with higher affinity than did the monomer, and affinities of the chemokines for CS and DS were lower than for HS. NMR-based structural models reveal diverse binding geometries and show that the binding surfaces for each of the three GAGs were different between the two chemokines. However, a given chemokine had similar binding interactions with CS and DS that were different from HS. Considering the fact that CXCL1 and CXCL5 activate the same CXCR2 receptor, we conclude that GAG interactions play a role in determining the nature of chemokine gradients, levels of free chemokine available for receptor activation, how chemokines bind their receptors, and that differences in these interactions determine chemokine-specific function.
Keywords: nuclear magnetic resonance (NMR), heparan sulfate, chondroitin sulfate, dermatan sulfate, chemokine, glycosaminoglycan
Introduction
Humans express ∼50 chemokines that show a remarkable repertoire of functions from combating microbial infection to homeostasis and developmental biology. Fundamental to these diverse functions is the migration of immune and nonimmune cells to proximal and remote locations. Most, if not all, chemokines share the following properties: they exist as monomers and dimers, activate receptors that belong to the GPCR class, and interact with tissue glycosaminoglycans (GAGs) (1–3).2 Biophysical, cellular, and animal model studies have shown that GAG interactions orchestrate leukocyte trafficking by regulating chemokine gradients, availability, how GAG-bound chemokines are presented to their receptors, and endothelial activation (4–12).
GAGs heparan sulfate (HS), chondroitin sulfate (CS), and dermatan sulfate (DS) are linear sulfated polysaccharides, which are covalently attached to core proteins and are the glycan part of a family of proteins called proteoglycans (PGs). Proteoglycan structures are quite diverse, are expressed by many cell types, and are also an integral component of the connective tissue and extracellular matrix (12–15). For instance, syndecans, a major class of membrane-spanning PGs, are of varying molecular weight, are expressed in different cell types, and carry varying number of HS and CS chains (15). Whereas syndecan-1 (Sdc-1) and Sdc-3 contain HS and CS, Sdc-2 and Sdc-4 contain only HS. Sdc-1 and Sdc-4 are expressed on the endothelium and epithelium, and both syndecans have been implicated in chemokine-mediated neutrophil recruitment and are essential for successful resolution of inflammation (16–18). HS chains are located close to the N terminus of the ectodomains and so more accessible for interacting with the receptors on the blood leukocytes, whereas CS chains are closer to the membrane surface and less accessible. Extracellular matrix (ECM) PGs such as perlecan, versican, and decorin contain varying levels of HS, CS, or DS and exist as noncovalent macromolecular complexes with ECM proteins such as collagen and laminin. GAGs and ectodomain of PGs also exist in the free form because they are cleaved by sheddases during neutrophil recruitment. These observations collectively suggest that the chemokine functional response depends on GAG structural features, their location within a PG, and the nature and location of PG in the tissue.
The basic building blocks of HS consist of repeating disaccharide units of d-glucuronic acid (GlcA) and GlcNAc (Fig. 1). HS has a modular structure with sulfated sequences separated by nonsulfated regions and is also more diverse because of differential N-sulfation and O-sulfation. In addition to N-sulfation, glucosamine can have 6-O-sulfation, and GlcA can have 2-O-sulfation and epimerize at C5 to l-iduronic acid (IdoA). The basic building blocks of CS and DS consist of d-GlcA and GalNAc, and on average, the disaccharide unit has one sulfate with O-sulfation either at C4 or C6 of GalNAc (Fig. 1). DS can be considered a variant of CS with the difference arising from C5 epimerization of GlcA to IdoA.
Figure 1.

A, schematic of heparin/HS, CS, and DS structures. R stands for potential sulfation sites. B, sequences of CXCL1 and CXCL5. Basic residues implicated in GAG binding that are also conserved in other neutrophil-activating chemokine are highlighted in blue and shaded in gray, those that are involved in GAG binding in CXCL1 alone are in bold and underlined, and corresponding residues that are not conserved in CXCL5 are in red. C, structures of CXCL1 dimer and monomer. All CXCR2-activating chemokines share high sequence similarity and have the same structural fold. The individual monomers in the dimer are shown in dark and light blue for clarity. Different structural and functional regions are labeled in both the monomer and dimer.
Chemokines are classified into two major (CXC and CC) families. A subset of seven CXC chemokines, including CXCL1 and CXCL5, characterized by the N-terminal ELR residues, function as agonists for the CXCR2 receptor (19). These chemokines recruit neutrophils across the endothelium and ECM to the infected and injury site and have also been implicated in nonimmune physiological functions (20–22). CXCL1 and CXCL5 are differentially expressed in different tissues and clinical symptoms, indicating nonredundant roles that are highly context-dependent (23–25). CXCL1 and CXCL5 share 51% sequence similarity, possess the same structural fold, and reversibly exist as monomers and dimers (Fig. 1) (26–29). Very little is known regarding how structural features and sequence of a chemokine, structure and sulfation pattern of a GAG, differences between GAGs, and differences between chemokines impact binding interactions and function.
In this study, we characterized the binding of CXCL1 and CXCL5 to HS, CS, and DS using solution NMR spectroscopy. We observe pronounced differences in binding geometries that could be attributed to differences in participation of a few select conserved and chemokine-specific residues and differences in GAG structures. Shared properties include higher affinity of the dimer for all three GAGs and higher affinity for HS compared with CS and DS. Considering that CXCL1 and CXCL5 activate the same CXCR2 receptor, we propose that diversity of GAG interactions play a role in determining the nature of chemokine gradients, levels of free chemokine available for receptor activation, how chemokines bind their receptors, and thereby impact function in a chemokine-specific manner.
Results
NMR chemical shifts are exquisitely sensitive to local structural changes, and so the binding surfaces were defined from chemical shift perturbation (CSP) on titrating GAGs to 15N-labeled proteins. Considering that CXCL1 and CXCL5 exist as monomers and dimers (monomer–dimer equilibrium constant, 1–4 μm) (27, 29), we initially characterized binding at low concentrations where both monomers and dimers are present. During GAG titration, four species will exist in solution: both monomer and dimer in the free and GAG-bound forms. Because of coupled equilibria, peak intensity changes reflect relative populations and binding constants.
For both chemokines and all three GAGs, the peaks corresponding to the monomer disappear on GAG binding (Fig. 2), indicating that the dimer is the high-affinity ligand. Therefore, all subsequent studies were carried out at 100–120 μm, where CXCL1 and CXCL5 exist as dimers. The chemical shifts in the GAG-bound form were assigned by collecting a series of spectra on adding small increments of GAG until essentially there were no changes in chemical shifts. We observe only one set of peaks during the titration, indicating that the free and bound-forms are in the fast exchange regime in the NMR time scale.
Figure 2.

Sections of the 1H-15N HSQC spectra showing the overlay of CXCL1 (A–D) and CXCL5 (E–H) in the free (black) and GAG-bound forms (red, heparin; blue, HS; green, CS; orange, DS). Dimer and monomer peaks are indicated as d and m. The monomer peaks disappear on GAG binding, indicating that the dimer is the high-affinity ligand. The spectra were collected using a ∼10 μm chemokine sample in 50 mm sodium phosphate, pH 6.0.
We had previously shown that the length of a 14-mer (dp14) is more than sufficient to cover all of the chemokine-binding interface from heparin-binding studies (29–31). For our current studies, we used CS and DS 14-mers. HS has a modular structure with sulfated domain separated by nonsulfated regions, and so we used a HS 30-mer to capture the modular HS structure. We also characterized binding to a heparin 26-mer and observed that the CSP profiles of backbone amides on binding the 14-mer and 26-mer were no different. The CSP profiles on binding HS and heparin were essentially identical, indicating that binding interactions of HS sulfated domain is similar to heparin. For both chemokines, the CSP profiles indicate that basic residues located in the N-loop, 40s loop, and C-terminal helix mediate binding. However, the profiles vary between the chemokines, and also profiles on CS and DS binding were quite different from HS for a given chemokine.
CXCL1–HS interactions
HS binding resulted in perturbation of basic residues Arg-8, His-19, Lys-21, Lys-45, Arg-48, Lys-49, Lys-60, Lys-61, and Lys-65. The perturbation profile on HS binding is very similar to what was previously observed for heparin (Figs. 3 and 4). Lys-29 showed negligible perturbation but is implicated in binding from mutagenesis, functional, and NMR studies tailored to detect lysine side chain chemical shifts (9, 31). Mapping these residues reveals that His-19, Lys-21, Lys-45, Lys-60, Lys-61, and Lys-65 constitute one binding domain (defined as the α-domain), and Arg-8, Lys-29, Arg-48, and Lys-49 constitute a second binding domain (defined as the β-domain) (Fig. 5). The CXCL1 structure reveals that α- and β-domains are located on opposite faces of the protein and that a single GAG chain cannot simultaneously bind both domains. Isothermal titration calorimetry studies also show a stoichiometry of two heparins binding one CXCL1 dimer (Fig. S1). Some neighboring and buried residues also show significant CSP, but these are unlikely to be involved in direct binding and are likely due to indirect binding-induced structural changes.
Figure 3.

Sections of 1H-15N HSQC spectra showing the overlay of CXCL1 in the free (black) and heparin-bound (red, A), HS-bound (blue, B), CS-bound (green, C), and DS-bound (orange, D) forms. Arrows indicate the direction of the peak movement.
Figure 4.
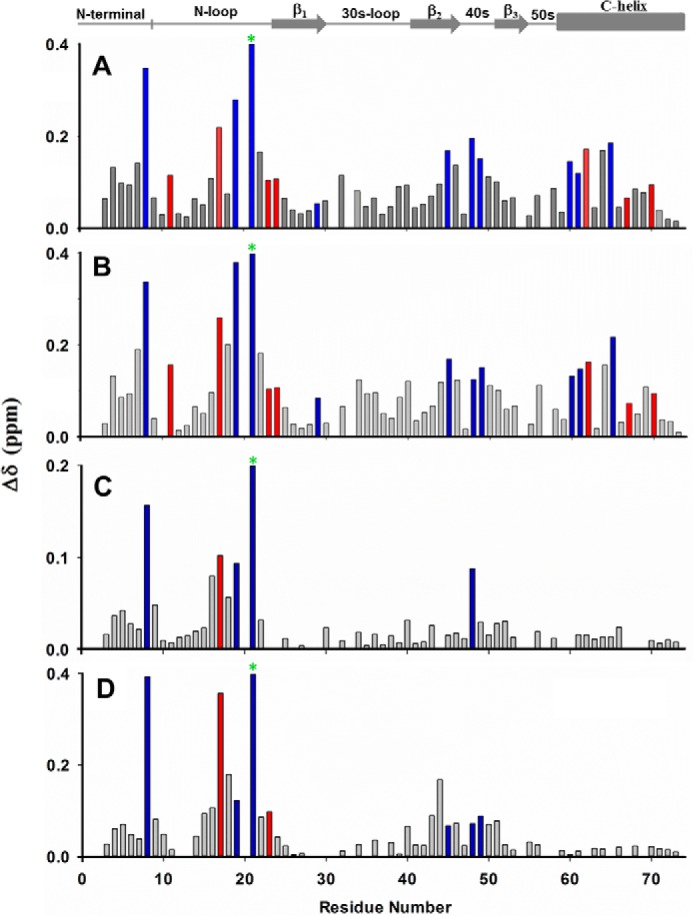
NMR backbone amide CSP profiles of CXCL1 on adding heparin (A), HS (B), CS (C), and DS (D). Basic and buried residues that show significant perturbation are shown in blue and red, respectively. The chemical shift of Lys-21 (green asterisks) is perturbed by 1.20, 0.95, 0.26, and 0.75 ppm for heparin, HS, CS, and DS, respectively.
Figure 5.

GAG binding surfaces in CXCL1. Heparin and HS bind both α- and β-domains, and CS and DS bind the γ-domain. GAG-binding basic residues are painted in dark and light blue to distinguish the two monomers of the dimer, and α-domain and β-domain residues are labeled in yellow and white, respectively.
CXCL1–CS and CXCL1–DS interactions
The CSP profiles for CS binding were dramatically different than that for HS. CS binding resulted in selective perturbation of the basic residues Arg-8, His-19, Lys-21, and Arg-48. Lack of perturbation of the helical residues Lys-60, Lys-61, and Lys-65 is striking (Figs. 3 and 4). Of the perturbed residues, His-19 and Lys-21 are from the α-domain, and Arg-8 and Arg-48 are from the β-domain. We define this new interface as the γ-domain. Whereas both α- and β-domains span the dimer interface, the γ-domain exists within a monomer (Fig. 5). The CSP profile for DS was similar but for some differences (Figs. 3 and 4). Compared with CS, perturbation of Arg-48 was less profound, and in addition, the CSP profile indicates that Lys-45 and Lys-49 could be involved in binding. Mapping these residues also suggests that DS binds the γ-domain. For both CS and DS, like in HS, some neighboring and buried residues show significant CSP that can be attributed to indirect interactions. Dissociation constants were determined by fitting the binding-induced chemical shift changes as described previously (32). Affinities of CXCL1 for CS and DS were weaker than that for HS (Kd 50 ± 10 versus 4 ± 2 μm). We show sections of the spectra that highlight GAG binding-induced chemical shift changes and also Kd plots for a select set of peaks (Fig. S2).
CXCL5–HS interactions
HS binding resulted in perturbation of the basic residues His-23, Lys-25, Lys-49, Lys-52, Lys-64, Lys-65, Lys-69, and Lys-76. The perturbation profile on HS binding is very similar to that for heparin (Figs. 6 and 7). Modeling and NMR studies that detect lysine side chain chemical shifts have shown that Lys-76 is not involved in binding, indicating that its chemical shift change must be due to indirect structural changes (30, 31). The remaining residues form a contiguous surface within one monomer of the dimer (Fig. 8A). Chemical shift changes were also observed for a few buried residues that can be attributed to indirect interactions.
Figure 6.

Sections of 1H-15N HSQC spectra showing the overlay of CXCL5 in the free (black) and heparin-bound (red, A), HS (blue, B), CS (green, C), and DS (orange, D) forms. Arrows indicate the direction of the peak movement. Gln-38 and Lys-64 that are differentially perturbed between heparin/HS and CS/DS are highlighted.
Figure 7.
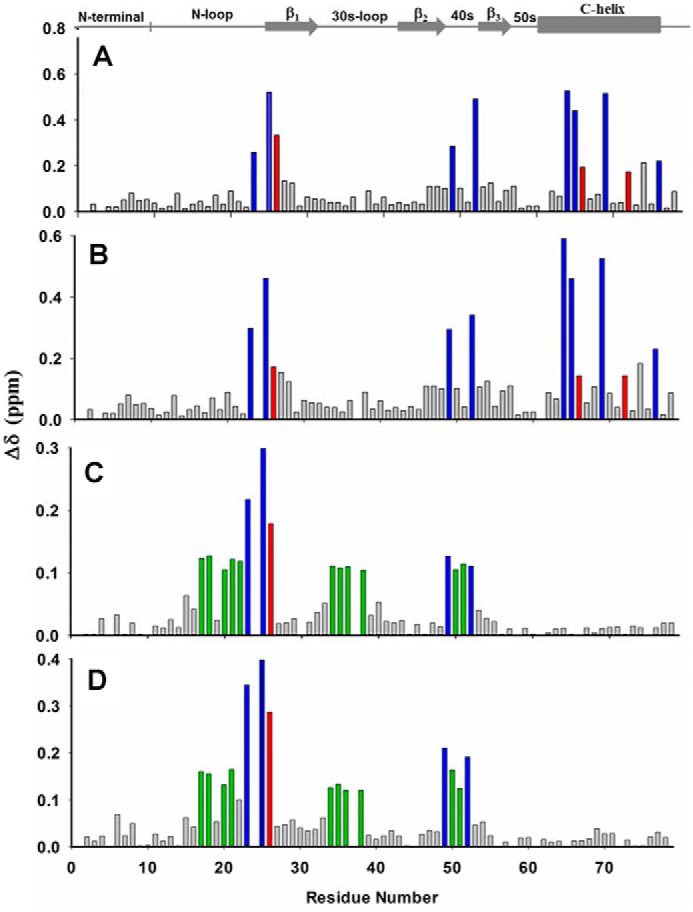
NMR backbone amide CSP profiles of CXCL5 on adding heparin (A), HS (B), CS (C), and DS (D). Basic and buried residues that show significant perturbation are shown in blue and red, respectively. In the cases of CS and DS, hydrophobic/polar residues that show significant perturbation are shown in green.
Figure 8.

GAG binding surfaces in CXCL5. Heparin/HS and CS/DS binding surfaces are shown in A and B. Basic residues are painted in blue, and nonbasic residues for CS/DS binding are painted in light and dark green to distinguish their location in the two monomers. In the case of HS/heparin, the binding surface is located within the monomer.
Sequences reveal that the same residues that mediate HS binding in CXCL5 also mediate binding in CXCL1 (Fig. 1). However, in CXCL1, additional interactions are observed for Arg-8, Lys-29, and Lys-49. Arg-8 is absolutely conserved among all seven CXCR2-activating chemokines and is essential for receptor activation. Lys-29 is unique to CXCL1, and Lys-49 is quasiconserved and observed only in four of the seven chemokines (3). In CXCL5, the residues corresponding to Lys-49 and Lys-29 are Glu and Phe, respectively, indicating that the strategic location and participation of a few conserved and chemokine-specific basic residues determine very different HS binding geometries.
CXCL5–CS and CXCL5–DS interactions
Perturbation profiles for CS and DS are similar but very different compared with HS (Figs. 6 and 7). Chemical shift changes of basic residues were observed only for His-23, Lys-25, Lys-49, and Lys-52. Similar to CXCL1, C-terminal helical basic residues are not perturbed. However, perturbation of several N-loop and 30s loop nonbasic residues that are not in the vicinity of a basic residue is striking. Mapping the perturbed residues suggests a binding interface that spans the dimer that is very different than in CXCL1 (Fig. 8B). Binding affinities of CXCL5 for CS and DS are also weaker compared with that for HS (Kd = 30 ± 10 versus 3 ± 2 μm).
Structural models of GAG-bound CXCL1/CXCL5 complexes
CS/DS-bound CXCL1 complexes
We carried out HADDOCK-based calculations that use CSP data as ambiguous interaction restraints to drive the docking process. We modeled binding of CS-4S and CS-6S, which contain a sulfate at either the 4-O or 6-O position, and DS, which contains a sulfate at the 4-O position.
For all three GAGs, we performed three different HADDOCK runs to ensure that the input constraints did not bias specific structural models and that all possible binding geometries within a monomer and across the dimer interface were considered. We modeled binding of one GAG chain with constraints to only one monomer of the dimer, binding of two GAG chains with constraints to both monomers of the dimer, and binding of one GAG chain with constraints to both monomers of the dimer. For all three GAGs, all runs resulted in only one major cluster with the GAG binding to a monomer of a dimer (Fig. 9).
Figure 9.

Models of CS/DS-bound CXCL1 and CXCL5 complexes. The structural models of CXCL1 bound to CS-6S (A and B), CS-4S (C), and DS-4S (D), and CXCL5 bound to CS-6S (E and F), CS-4S (G), and DS-4S (H) are shown. In A and E, CXCL1 and CXCL5 dimer structures are shown in ribbon presentation, and CS-6S is shown as sticks. Two monomers of the dimer are shown in blue and cyan, and CS-6S is shown to bind one monomer of the dimer. B–D and F–H highlight the GAG-binding residues, with basic residues shown in blue and polar residues in green.
Structural models reveal that all three GAGs adopt the same geometry. Ionic interactions were mediated by basic residues Arg-8, His-19, Lys-21, Lys-45, Arg-48, and Lys-49, and H-bonding and packing interactions by polar residues Gln-10, Asn-22, and Asn-46. For all three GAGs, CXCL1 engages two sulfates, two carboxylates, and two N-acetyl groups. Differences in GAG backbone structure and topology of the acidic groups did impact binding interactions. For instance, His-19 is involved in binding to CS6S and not CS4S or DS, Arg-8 engages a sulfate in CS6S and a carboxylate in CS4S and DS, and Lys-21 engages a sulfate in CS4S and DS and a carboxylate in CS6S.
NMR studies, which used naturally occurring CS that is a mixture of CS-4S and CS-6S, showed significant perturbation for Arg-8, His-19, Lys-21, and Arg-48. Lack of chemical shift changes for Lys-45 and Lys-49 can be attributed to chemical shift changes from direct and indirect interactions that are of opposite sign and similar magnitude and cancel out. A similar observation has been made for one of the lysines in heparin binding to CXCL1 and CXCL8 (29, 33).
CS/DS-bound CXCL5 complexes
The rationale and the docking strategy were the same as described for CXCL1 complexes. We initially modeled binding of one GAG chain with constraints to both monomers of the dimer (run 1) and two GAG chains with constraints to both monomers of the dimer (run 2). For all three GAGs, both runs resulted in similar structures with one GAG binding across the dimer interface in run 1 and two GAG chains across the dimer interface on opposite faces of the protein in run 2 (Fig. 9). Structural models reveal that all three GAGs adopt the same geometry. Electrostatic interactions were mediated by basic residues His-23, Lys-25 Lys-49, and Lys-52 and H-bonding and packing interactions by nonbasic residues Gln-17, Thr-18, Gln-20, Met-26, Leu-48, Asn-50, Ile-35′, and Pro-37′ (where ′ corresponds to residues from the second monomer). For all three GAGs, CXCL5 engages two sulfates, two carboxylates, one N-acetyl, and one hydroxyl group. However, differences in GAG structure result in some differences in residue-specific interactions. For instance, whereas Lys-49 binds a sulfate in CS-6S and a carboxylate in CS-4S and DS, Lys-25 and Lys-52 bind a sulfate in all three GAGs. NMR studies show significant CSP for all residues identified from modeling studies except for Pro-37, which is to be expected because prolines do not have a backbone amide proton. We also modeled binding of GAG chains with constraints given to only one monomer of the dimer (run 3). Run 3 resulted in two major clusters: one similar to that observed in run 1 despite the absence of explicit constraints to the second monomer, and a second cluster that involved binding only to the N-loop and 40s loop residues within a monomer. The latter model can be ruled out because it is inconsistent with the NMR data.
HS-bound CXCL1/CXCL5 complexes
NMR studies reveal that the CSP profiles of HS and heparin binding to both CXCL1 and CXCL5 are very similar. To mimic the diversity in HS structures, we modeled HS that is either missing N-sulfate, 2-O-sulfate, or 6-O-sulfate. For both CXCL1 and CXCL5, the binding geometries of all three variants were similar to heparin, suggesting that the binding interface is plastic, and the structural scaffold and distribution of sulfates and carboxylates dictate a similar binding geometry (Fig. 10).
Figure 10.

Models of heparin/HS-bound CXCL1 and CXCL5 complexes. The structural models of heparin (A and B) and HS-NS, 2OS (C) bound to CXCL1 α-domain, heparin (D and E) and HS-NS, 2OS (F) bound to CXCL1 β-domain, and heparin (G and H) and HS-NS, 2OS (I) bound to CXCL5 are shown. In A, D, and G, CXCL1 and CXCL5 dimer structures are shown in ribbon presentation, and GAG is shown as sticks. Two monomers of the dimer are shown in blue and cyan, and basic residues implicated in binding are highlighted. Heparin and HS-NS, 2OS bind across the dimer interface in CXCL1 and within the monomer in CXCL5. Binding interactions of HS-NS, 6OS and HS-2OS, 6OS were similar to HS-NS, 2OS and are not shown.
Discussion
GAGs bind hundreds of proteins with diverse structural scaffolds and functions from growth factors and enzymes to chemokines and cytokines. Describing the molecular basis of GAG interactions has been challenging because structures of GAG-bound protein complexes have been hard to come by. Therefore, many studies have reported structural models of proteins bound to heparin on the basis of NMR backbone CSP-based methods. In recent years, lysine, arginine, and histidine side chain chemical shifts have also been shown to be quite sensitive and useful for identifying GAG-binding residues (31, 34, 35). Heparin has been and continues to be the GAG of choice for structural and biophysical studies because it is more uniformly sulfated and because of availability of size-defined oligosaccharides. Both HS and heparin share a repeating disaccharide unit composed of glucosamine and hexuronic acid, and it is assumed that heparin is a good surrogate for capturing HS interactions. Our studies for two related chemokines validate this premise by providing experimental evidence that the same basic residues mediate binding heparin and HS. We point out native HS polymer used in our studies is intrinsically heterogeneous because of differential N- and O-sulfation and that the NMR chemical shift changes are a sum of interactions to all of the different structures present in the polymer. To our knowledge, ours is the first NMR study that has characterized residue-specific binding interactions to heparin and HS polymers. Whether our observation holds for other HS-binding proteins needs to be determined. Very few studies have characterized the structural basis of CS and DS interactions or how the structural features of the same protein mediate binding different GAGs. We show that a given chemokine can have different binding surfaces for different GAGs, that related chemokines can have different binding surfaces for a given GAG, and that binding affinities can vary between GAGs, indicating that differences in chemokine sequences and GAG structure result in different binding interactions.
Considering GAGs bind diverse protein classes (36–39), chemokine sequence must play a major role in determining selectivity, affinity, and geometry for a given GAG and between GAGs. We observe for two related chemokines, a cluster of basic residues located in the N-loop, 40s loop, and C-terminal helix mediate HS binding via ionic interactions. Participation of additional residues from elsewhere in the sequence in CXCL1 results in a very different binding geometry. For both chemokines, binding interactions of CS and DS did not involve helical residues. This observation is quite striking, because helical residues in CXCR2-activating chemokines have long been assumed as important in GAG binding. NMR structural models for both chemokines suggest that N- and 2-O-sulfation in HS interact with helical lysines. Collectively, these data indicate that distribution of basic residues in the context of chemokine structure, differences in participation of a few select basic residues, and differences in GAG sulfation pattern can result in very different binding interactions. Knowledge of structures and molecular dynamics simulation studies are essential to fully appreciate how and which GAG sulfate/carboxylate groups mediate binding and how differences in GAG structures and chemokine sequence determine the large diversity of binding interactions. Previous NMR studies indicate that the binding interactions of CS, DS, and heparin hexasaccharide binding to CXCL8 are similar (40), which is different from our observations for CXCL1 and CXCL5.
Animal model studies have shown that the ability of chemokines to reversibly exist as monomers and dimers and GAG interactions regulate neutrophil recruitment (9, 41, 42). During active neutrophil trafficking, chemokine concentration can vary by orders of magnitude, and so it may exist as a monomer, dimer, or both at different times and locations. Our results indicate that both CXCL1 and CXCL5 dimers bind all GAGs with higher affinity. NMR studies for a number of chemokines have demonstrated that GAG binding and dimerization are coupled (43–49), suggesting that this is a shared property of all chemokines for all GAGs. CXCL1 and CXCL5 are selectively expressed and/or under specific disease conditions and are also co-expressed in other conditions (22–25, 50–52). Further, CXCL1 and CXCL5 KO mice show distinct differences in disease models, indicating that the unique phenotype of each chemokine is critical for successful resolution (23, 24). CXCL1 and CXCL5 have also been implicated as major players in housekeeping functions including in developmental biology, suggesting that GAG interactions must play an important role in these highly spatiotemporally regulated processes (53, 54). Additional studies similar to those reported here for other chemokines and other GAG-binding proteins with different structural scaffolds are essential to fully appreciate how differences in GAG interactions regulate and dictate functional response in health and disease.
Materials and methods
Recombinant 15N-labeled CXCL1 and CXCL5 were expressed and purified as described previously (27). Heparin (dp26), HS (dp30), CS (dp14), and DS (dp14) were purchased from Iduron. According to the manufacturer, the oligosaccharides were purified using high-resolution gel filtration chromatography. The major disaccharide unit in heparin is IdoA, 2S-GlcNS,6S (∼75%), in chondroitin sulfate is GlcA-GalNAc sulfated at C6 or C4, and in dermatan sulfate is IdoA-GalNAc,4S (∼88%). The heparan sulfate polymer on an average has 1.75 sulfate groups (0.65 N-sulfate and 1.1 O-sulfate groups) per disaccharide. All GAGs contain uronic acid at the nonreducing end and a C4–C5 double bond as a result of the endolytic action of heparinase or chondroitin ABC lyase.
NMR titration experiments
GAG titrations were carried out in 50 mm sodium phosphate, pH 5.7, containing 1 mm 2,2-dimethyl-2-silapentanesulfonic acid, 1 mm sodium azide, and 10% D2O (v/v). NMR spectra for CXCL1 were acquired at 40 °C and for CXCL5 at 25 °C on Bruker Avance III 800 MHz (equipped with a TXI cryoprobe) or 600 MHz (with QCI probe) spectrometers. For all GAGs, a series of HSQC spectra was collected essentially until no change in chemical shifts was observed. The chemical shifts of the WT CXCL1 dimer and CXCL5 dimer are available (27, 29). 1H-15N HSQC spectra were collected by adding aliquots of GAGs (3–8 mm) prepared in the same buffer to 100–120 μm chemokine. The final chemokine:GAG molar ratio was 1:4. The chemical shift perturbation (Δδobs) was calculated as a weighted average of 1H (ΔδH) and 15N(ΔδN) chemical shift changes (Δδobs = [(ΔδH)2 + (ΔδN/5)2]1/2). To determine relative GAG affinities of the monomer and dimer, GAGs were titrated to 8–15 μm chemokine.
Structural models of GAG-bound CXCL1 and CXCL5 complexes
Molecular docking of GAGs to CXCL5/CXCL1 dimer was carried out using high ambiguity driven biomolecular DOCKing (HADDOCK) using NMR CSP as structural constraints described previously (29, 30, 55, 56). Docking simulations were carried out using CXCL1 and CXCL5 dimer NMR and various GAGs. HS, CS, and DS oligosaccharide structures generated using the GLYCAM web server (http://glycam.org/).3 The topology and parameter files for all oligosaccharides were generated using the PRODRG server (57). After an initial rigid body docking, the top 1000 structures that had the best intermolecular energies were then sequentially subjected to semiflexible simulated annealing and explicit solvent refinement. The pair-wise ligand interface root-mean-square-deviation matrix over all structures was calculated, and final structures were clustered using a cut-off value of 7.5 Å. The clusters were sorted using root-mean-square deviation and HADDOCK score (weighted sum of a combination of energy terms). The structures with best HADDOCK score were used for representation.
Author contributions
K. M. S. and K. R. conceptualization; K. M. S. data curation; K. M. S. software; K. M. S. formal analysis; K. M. S. and K. R. supervision; K. M. S. validation; K. M. S. and K. R. investigation; K. M. S. visualization; K. M. S. and K. R. methodology; K. M. S. and K. R. writing-original draft; K. M. S. project administration; K. M. S. and K. R. writing-review and editing; K. R. resources; K. R. funding acquisition.
Supplementary Material
Acknowledgment
We thank Dr. Tianzhi Wang for assistance with the NMR experiments.
This work was supported in part by National Institutes of Health Grants P01HL107152 and R21AI124681 and a Sealy and Smith Foundation grant to the Sealy Center for Structural Biology and Molecular Biophysics. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- GAG
- glycosaminoglycan
- HS
- heparan sulfate
- CS
- chondroitin sulfate
- DS
- dermatan sulfate
- CSP
- chemical shift perturbation
- PG
- proteoglycan
- ECM
- extracellular matrix
- GlcA
- d-glucuronic acid
- IdoA
- l-iduronic acid.
References
- 1. Stone M. J., Hayward J. A., Huang C., Huma E. Z., and Sanchez J. (2017) Mechanisms of regulation of the chemokine-receptor network. Int. J. Mol. Sci. 18, E342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salanga C. L., and Handel T. M. (2011) Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp. Cell Res. 317, 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rajarathnam K., Sepuru K. M., Joseph P. R. B., Sawant K. V., and Brown A. J. (2018) Glycosaminoglycan interactions finetune chemokine-mediated neutrophil trafficking: structural insights and molecular mechanisms. J. Histochem. Cytochem. 66, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Massena S., Christoffersson G., Hjertström E., Zcharia E., Vlodavsky I., Ausmees N., Rolny C., Li J. P., and Phillipson M. (2010) A chemotactic gradient sequestered on endothelial heparan sulfate induces directional intraluminal crawling of neutrophils. Blood 116, 1924–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanino Y., Coombe D. R., Gill S. E., Kett W. C., Kajikawa O., Proudfoot A. E., Wells T. N., Parks W. C., Wight T. N., Martin T. R., and Frevert C. W. (2010) Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J. Immunol. 184, 2677–2685 10.4049/jimmunol.0903274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sarris M., Masson J. B., Maurin D., Van der Aa L. M., Boudinot P., Lortat-Jacob H., and Herbomel P. (2012) Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr. Biol. 22, 2375–2382 10.1016/j.cub.2012.11.018 [DOI] [PubMed] [Google Scholar]
- 7. Wang L., Fuster M., Sriramarao P., and Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 10.1038/ni1233 [DOI] [PubMed] [Google Scholar]
- 8. Frevert C. W., Kinsella M. G., Vathanaprida C., Goodman R. B., Baskin D. G., Proudfoot A., Wells T. N., Wight T. N., and Martin T. R. (2003) Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am. J. Respir. Cell Mol. Biol. 28, 464–472 10.1165/rcmb.2002-0084OC [DOI] [PubMed] [Google Scholar]
- 9. Sawant K. V., Poluri K. M., Dutta A. K., Sepuru K. M., Troshkina A., Garofalo R. P., and Rajarathnam K. (2016) Chemokine CXCL1 mediated neutrophil recruitment: role of glycosaminoglycan interactions. Sci. Rep. 6, 33123 10.1038/srep33123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Monneau Y., Arenzana-Seisdedos F., and Lortat-Jacob H. (2016) The sweet spot: how GAGS help chemokines guide migrating cells. J. Leukoc. Biol. 99, 935–953 10.1189/jlb.3MR0915-440R [DOI] [PubMed] [Google Scholar]
- 11. Derler R., Gesslbauer B., Weber C., Strutzmann E., Miller I., and Kungl A. (2017) Glycosaminoglycan-mediated downstream signaling of CXCL8 binding to endothelial cells. Int. J. Mol. Sci. 18, E2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Celie J. W., Beelen R. H., and van den Born J. (2009) Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front. Biosci. (Landmark Ed.) 14, 4932–4949 [DOI] [PubMed] [Google Scholar]
- 13. Schaefer L., and Schaefer R. M. (2010) Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 339, 237–246 10.1007/s00441-009-0821-y [DOI] [PubMed] [Google Scholar]
- 14. Djerbal L., Lortat-Jacob H., and Kwok J. (2017) Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj. J. 34, 363–376 10.1007/s10719-017-9761-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Couchman J. R. (2010) Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 26, 89–114 10.1146/annurev-cellbio-100109-104126 [DOI] [PubMed] [Google Scholar]
- 16. Ishiguro K., Kadomatsu K., Kojima T., Muramatsu H., Iwase M., Yoshikai Y., Yanada M., Yamamoto K., Matsushita T., Nishimura M., Kusugami K., Saito H., and Muramatsu T. (2001) Syndecan-4 deficiency leads to high mortality of lipopolysaccharide-injected mice. J. Biol. Chem. 276, 47483–47488 10.1074/jbc.M106268200 [DOI] [PubMed] [Google Scholar]
- 17. Tanino Y., Chang M. Y., Wang X., Gill S. E., Skerrett S., McGuire J. K., Sato S., Nikaido T., Kojima T., Munakata M., Mongovin S., Parks W. C., Martin T. R., Wight T. N., and Frevert C. W. (2012) Syndecan-4 regulates early neutrophil migration and pulmonary inflammation in response to lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 47, 196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gill S. E., Nadler S. T., Li Q., Frevert C. W., Park P. W., Chen P., and Parks W. C. (2016) Shedding of syndecan-1/CXCL1 complexes by matrix metalloproteinase 7 functions as an epithelial checkpoint of neutrophil activation. Am. J. Respir. Cell Mol. Biol. 55, 243–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rajarathnam K., Schnoor M., Richardson R. M., and Rajagopal S. (2019) How do chemokines navigate neutrophils to the target site: dissecting the signaling pathways. Cell Signal. 54, 69–80 10.1016/j.cellsig.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi Y. (2008) The role of chemokines in neutrophil biology. Front. Biosci. 13, 2400–2407 10.2741/2853 [DOI] [PubMed] [Google Scholar]
- 21. Cardona A. E., Li M., Liu L., Savarin C., and Ransohoff R. M. (2008) Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J. Leukoc. Biol. 84, 587–594 10.1189/jlb.1107763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z. J., Cao D. L., Zhang X., Ji R. R., and Gao Y. J. (2013) Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain 154, 2185–2197 10.1016/j.pain.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cai S., Batra S., Lira S. A., Kolls J. K., and Jeyaseelan S. (2010) CXCL1 regulates pulmonary host defense to Klebsiella Infection via CXCL2, CXCL5, NF-κB, and MAPKs. J. Immunol. 185, 6214–6225 10.4049/jimmunol.0903843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mei J., Liu Y., Dai N., Favara M., Greene T., Jeyaseelan S., Poncz M., Lee J. S., and Worthen G. S. (2010) CXCL5 regulates chemokine scavenging and pulmonary host defense to bacterial infection. Immunity 33, 106–117 10.1016/j.immuni.2010.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Z'Graggen K., Walz A., Mazzucchelli L., Strieter R. M., and Mueller C. (1997) The C-X-C chemokine ENA-78 is preferentially expressed in intestinal epithelium in inflammatory bowel disease. Gastroenterology 113, 808–816 10.1016/S0016-5085(97)70175-6 [DOI] [PubMed] [Google Scholar]
- 26. Fairbrother W. J., Reilly D., Colby T. J., Hesselgesser J., and Horuk R. (1994) The solution structure of melanoma growth stimulating activity. J. Mol. Biol. 242, 252–270 10.1006/jmbi.1994.1577 [DOI] [PubMed] [Google Scholar]
- 27. Sepuru K. M., Poluri K. M., and Rajarathnam K. (2014) Solution structure of CXCL5: a novel chemokine and adipokine implicated in inflammation and obesity. PLoS One 9, e93228 10.1371/journal.pone.0093228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ravindran A., Sawant K. V., Sarmiento J., Navarro J., and Rajarathnam K. (2013) Chemokine CXCL1 dimer is a potent agonist for the CXCR2 receptor. J. Biol. Chem. 288, 12244–12252 10.1074/jbc.M112.443762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sepuru K. M., and Rajarathnam K. (2016) CXCL1/MGSA is a novel glycosaminoglycan (GAG)-binding chemokine: structural evidence for two distinct non-overlapping binding domains. J. Biol. Chem. 291, 4247–4255 10.1074/jbc.M115.697888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sepuru K. M., Nagarajan B., Desai U. R., and Rajarathnam K. (2016) Molecular basis of chemokine CXCL5–glycosaminoglycan interactions. J. Biol. Chem. 291, 20539–20550 10.1074/jbc.M116.745265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sepuru K. M., Iwahara J., and Rajarathnam K. (2018) Direct detection and characterization of lysine side chain NH3+ in protein-heparin complexes using NMR spectroscopy. Analyst 143, 635–638 10.1039/C7AN01406F [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ravindran A., Joseph P. R., and Rajarathnam K. (2009) Structural basis for differential binding of the interleukin-8 monomer and dimer to the CXCR1 N-domain: role of coupled interactions and dynamics. Biochemistry 48, 8795–8805 10.1021/bi901194p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joseph P. R., Mosier P. D., Desai U. R., and Rajarathnam K. (2015) Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: structural plasticity mediates differential binding interactions. Biochem. J. 472, 121–133 10.1042/BJ20150059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joseph P. R. B., Sawant K. V., Iwahara J., Garofalo R. P., Desai U. R., and Rajarathnam K. (2018) Lysines and arginines play non-redundant roles in mediating chemokine-glycosaminoglycan interactions. Sci. Rep. 8, 12289 10.1038/s41598-018-30697-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sepuru K. M., and Rajarathnam K. (2018) Distinct differences in structural states of conserved histidines in two related proteins: NMR studies of chemokines CXCL1 and CXCL8 in the free form and macromolecular complexes. Biochemistry 57, 5969–5977 10.1021/acs.biochem.8b00756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meneghetti M. C., Hughes A. J., Rudd T. R., Nader H. B., Powell A. K., Yates E. A., and Lima M. A. (2015) Heparan sulfate and heparin interactions with proteins. J. R. Soc. Interface 12, 0589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu D., and Esko J. D. (2014) Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 83, 129–157 10.1146/annurev-biochem-060713-035314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pomin V. H., and Mulloy B. (2015) Current structural biology of the heparin interactome. Curr. Opin. Struct. Biol. 34, 17–25 10.1016/j.sbi.2015.05.007 [DOI] [PubMed] [Google Scholar]
- 39. Gandhi N. S., and Mancera R. L. (2008) The structure of glycosaminoglycans and their interactions with proteins. Chem. Biol. Drug Des 72, 455–482 10.1111/j.1747-0285.2008.00741.x [DOI] [PubMed] [Google Scholar]
- 40. Pichert A., Samsonov S. A., Theisgen S., Thomas L., Baumann L., Schiller J., Beck-Sickinger A. G., Huster D., and Pisabarro M. T. (2012) Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 22, 134–145 10.1093/glycob/cwr120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gangavarapu P., Rajagopalan L., Kolli D., Guerrero-Plata A., Garofalo R. P., and Rajarathnam K. (2012) The monomer-dimer equilibrium and glycosaminoglycan interactions of chemokine CXCL8 regulate tissue-specific neutrophil recruitment. J. Leuko. Biol. 91, 259–265 10.1189/jlb.0511239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sawant K., Xu R., Cox R., Hawkins H., Kolli D., Garofalo R. P., and Rajarathnam K. (2015) Chemokine CXCL1 mediated neutrophil recruitment in the mouse lung: role CXCR2 activation. J. Innate Immun. 7, 647–658 10.1159/000430914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deshauer C., Morgan A. M., Ryan E. O., Handel T. M., Prestegard J. H., and Wang X. (2015) Interactions of the chemokine CCL5/RANTES with medium-sized chondroitin sulfate ligands. Structure 23, 1066–1077 10.1016/j.str.2015.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monneau Y. R., Luo L., Sankaranarayanan N. V., Nagarajan B., Vivès R. R., Baleux F., Desai U. R., Arenzana-Seidedos F., and Lortat-Jacob H. (2017) Solution structure of CXCL13 and heparan sulfate binding show that GAG binding site and cellular signalling rely on distinct domains. Open Biol. 7, 170133 10.1098/rsob.170133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brown A. J., Sepuru K. M., Sawant K. V., and Rajarathnam K. (2017) Platelet-derived chemokine CXCL7 dimer preferentially exists in the glycosaminoglycan-bound form: implications for neutrophil–platelet crosstalk. Front. Immunol. 8, 1248 10.3389/fimmu.2017.01248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joseph P. R. B., Sawant K. V., and Rajarathnam K. (2017) Heparin-bound chemokine CXCL8 monomer and dimer are impaired for CXCR1 and CXCR2 activation: implications for gradients and neutrophil trafficking. Open Biol. 7, 170168 10.1098/rsob.170168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sepuru K. M., Nagarajan B., Desai U. R., and Rajarathnam K. (2018) Structural basis, stoichiometry, and thermodynamics of chemokines KC/mCXCL1 and MIP2/mCXCL2 binding to glycosaminoglycan heparin. J. Biol. Chem. 293, 17817–17828 10.1074/jbc.RA118.004866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ziarek J. J., Veldkamp C. T., Zhang F., Murray N. J., Kartz G. A., Liang X., Su J., Baker J. E., Linhardt R. J., and Volkman B. F. (2013) Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardioprotection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 288, 737–746 10.1074/jbc.M112.394064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jansma A. L., Kirkpatrick J. P., Hsu A. R., Handel T. M., and Nietlispach D. (2010) NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J. Biol. Chem. 285, 14424–14437 10.1074/jbc.M109.091108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Disteldorf E. M., Krebs C. F., Paust H. J., Turner J. E., Nouailles G., Tittel A., Meyer-Schwesinger C., Stege G., Brix S., Velden J., Wiech T., Helmchen U., Steinmetz O. M., Peters A., Bennstein S. B., et al. (2015) CXCL5 drives neutrophil recruitment in Th17-mediated GN. J. Am. Soc. Nephrol. 26, 55–66 10.1681/ASN.2013101061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin M., Carlson E., Diaconu E., and Pearlman E. (2007) CXCL1/KC and CXCL5/LIX are selectively produced by corneal fibroblasts and mediate neutrophil infiltration to the corneal stroma in LPS keratitis. J. Leukoc. Biol. 81, 786–792 10.1189/jlb.0806502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nouailles G., Dorhoi A., Koch M., Zerrahn J., Weiner J. 3rd, Faé K. C., Arrey F., Kuhlmann S., Bandermann S., Loewe D., Mollenkopf H. J., Vogelzang A., Meyer-Schwesinger C., Mittrücker H. W., McEwen G., et al. (2014) CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Invest. 124, 1268–1282 10.1172/JCI72030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mei J., Liu Y., Dai N., Hoffmann C., Hudock K. M., Zhang P., Guttentag S. H., Kolls J. K., Oliver P. M., Bushman F. D., and Worthen G. S. (2012) Cxcr2 and Cxcl5 regulate the IL-17/G-CSF axis and neutrophil homeostasis in mice. J. Clin. Invest. 122, 974–986 10.1172/JCI60588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsai H. H., Frost E., To V., Robinson S., Ffrench-Constant C., Geertman R., Ransohoff R. M., and Miller R. H. (2002) The chemokine receptor CXCR2 controls positioning of oligodendrocyte precursors in developing spinal cord by arresting their migration. Cell 110, 373–383 10.1016/S0092-8674(02)00838-3 [DOI] [PubMed] [Google Scholar]
- 55. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein–protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 10.1021/ja026939x [DOI] [PubMed] [Google Scholar]
- 56. de Vries S. J., van Dijk A. D., Krzeminski M., van Dijk M., Thureau A., Hsu V., Wassenaar T., and Bonvin A. M. (2007) HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins 69, 726–733 10.1002/prot.21723 [DOI] [PubMed] [Google Scholar]
- 57. Schüttelkopf A. W., and van Aalten D. M. F. (2004) PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr D Biol Crystallogr. 60, 1355–1363 10.1107/S0907444904011679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.