

To the Editor:
Recently, Bisgaard et al. [2006] detected additional chromosomal abnormalities in six patients with previously known abnormal karyotypes, such as inversions and translocations, using metaphase comparative genomic hybridization (CGH) at a resolution of 2–3 Mb. They reported previously undetected interstitial deletions with CGH in their patients including a de novo 2q33.2–q34 deletion. However, no chromosome imbalances were found using conventional cytogenetics in their six patients at the inversion or translocation breakpoints.
Deletions in the long arm of chromosome 2 are relatively rare and correlate with a broad spectrum of clinical findings, including developmental delay, mental retardation, hyperactivity with autistic traits and dysmorphic features including Pierre Robin sequence, cleft palate, temporal bone abnormalities and hypoplastic lungs [Kramer et al., 2000; Houdayer et al., 2001; Van Buggenhout et al., 2005; Pescucci et al., 2007]. Approximately one-half of the reported deletions involved a chromosome segment between bands 2q24.3 and 2q31.1 [Courtens et al., 1997] or terminal 2q37.3 deletions [Kitsiou-Tzeli et al., 2007].
In 1997, Courtens et al. reported the first 2q33.3–q34 interstitial deletion in a 5-week-old boy with phenotypic findings initially suggesting Seckel syndrome (e.g., pre- and postnatal growth retardation, microcephaly, and muscular hypotonia). Courtens et al. [1997] summarized seven cases involving at least the 2q33.3–q34 chromosome region (i.e., largest deletion 2q33–q36). Subsequently, four other cases have been reported with deletions in the 2q33–q35 region [Kramer et al., 2000; Riegel et al., 2001; Pescucci et al., 2003; Bisgaard et al., 2006].
Herein, we report a second case of a de novo 2q33.3–q34 deletion in a 6-year-old male ascertained with aCGH and referred for evaluation of a de novo balanced translocation [t(4;16)(q32.2;q24.1)]. We compared findings in our case to those reported by Courtens et al. [1997], Pescucci et al. [2003], and Bisgaard et al. [2006] involving similar 2q deletions.
Our patient was born to a para 4, gravid 4, 27-year-old mother and a 43-year-old father. The mother denied alcohol or drug exposure during pregnancy. The infant was delivered at term by an uncomplicated vaginal delivery with a birth weight of 3,182 g (25th centile) and length of 50.8 cm (50th centile). The head circumference was not known. The infant was discharged with the mother at 24 hr of age. During the first year of life, the patient was hospitalized on three occasions for respiratory infections (RSV and pneumonia). Sweat chloride testing for cystic fibrosis was negative. Asthma and severe allergies were diagnosed requiring treatment with loratadine, montelukast sodium, and a budesonide inhaler. Tympanostomy tubes were placed at 6 months and again at 18 months of age for persistent otitis media. The child had extensive dental caries requiring restoration at 3.5 years of age.
Developmental milestones were delayed (e.g., sitting alone at 1 year, first steps at 18–24 months and first words at 24 months). At 2 years of age the child developed “spacing out episodes” and was found to have an abnormal EEG although he was not placed on medication. He was subsequently seen by neurology services and diagnosed with complex partial seizures. An MRI study was normal. This child was first seen in the genetics clinic at 3 years 5 months of age for evaluation of developmental delays, retractile but palpable testes and a small appearing scrotum. The physical exam at 3 years 5 months revealed a weight of 16.8 kg (75th centile), height of 95 cm (20th centile) and head circumference of 46 cm (<3rd centile). Bilateral fifth finger clinodactyly, two posterior hair whorls, epicanthal folds, and undescended testes were noted. Chromosome analysis revealed an apparently balanced chromosome translocation, 46, XY,t(4;16)(q32.2;q24.1) at the 550 band level. Parental chromosomes were normal. Because of the possibility that the observed clinical findings may be secondary to submicroscopic deletions around the translocation breakpoints, aCGH analysis was performed on genomic DNA extracted from peripheral blood and hybridized with reference normal DNA using the Spectral Chip 2600 (1 Mb, Spectral Genomics, Inc., Waltham, MA). Two clones at 2q34 (RP11-13N10 at 208.7 Mb and RP11-90O3 at 209.8 Mb) showed copy number loss due to a 2q33.3–q34 deletion (Fig. 1) confirmed using FISH analysis. This deletion was estimated to be 1.4–4.4 Mb in size based on the clone coverage of the 1 Mb array. No abnormalities were seen on either the 4q or 16q chromosome regions. Microarray analysis of the parents showed the deletion to be de novo. To further delineate the size of the deletion, the Agilent 44K oligonucleotide microarray (Agilent Technologies, Santa Clara, CA) was similarly used for hybridization with the patient’s DNA. With this technology, the deletion was determined to be approximately 3.7 Mb in size involving 18 known genes (ADAM23, MDH1B, KIAA0971, CPO, KLF7, CREB1, FAM119A, FLJ40432, FZD5, CRYGD, CRYGC, CRYGB, CRYGA, IDH1, PIP5K3, PTHR2, MAP2, C2orf21; see Fig. 2).
Fig. 1.
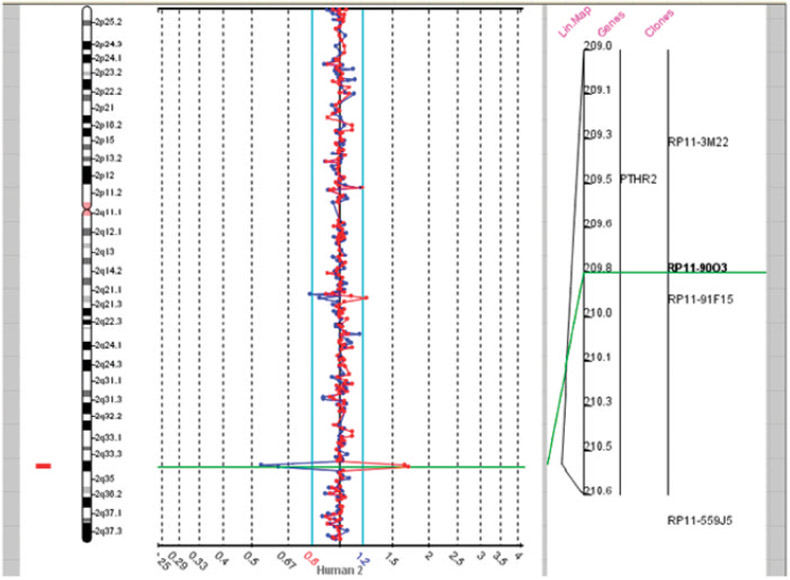
An array comparative genomic hybridization (aCGH) was carried out using a Spectral Chip 2600 (1 Mb; Spectral Genomics, Inc., Waltham, MA). Two clones at 2q33.3–2q34 (RP11-13N10 located at 208.7 Mb and RP11-90O3 at 209.8 Mb) showed DNA loss corresponding to a 1.4–4.4 Mb deletion at 2q33.3–q34.
Fig. 2.
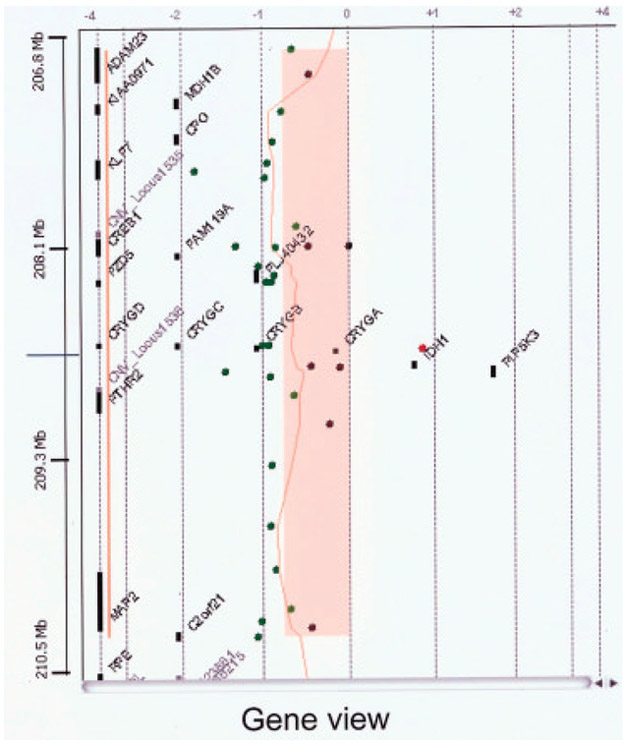
An expanded view of the 2q33.3–2q34 deletion segment (represented in salmon color) with the order of genes in the deleted region determined by the use of the Agilent 44 K oligonucleotide microarray. The approximate size of the deletion was 3.7 Mb and involves 18 known genes. Green symbols represent deleted hybridized oligonucleotide probes.
The patient was the only child born to the parents. The mother reported a history of learning disabilities but graduated from high school. She had a daughter and a son with a different partner, both having ADHD. Another son by a third partner had oppositional defiant disorder and was born with a club foot. The father reported he also had learning disabilities. There was no further family history of known genetic conditions, mental illness or birth defects.
At 6 years of age, he attended school (one half day sessions) in a special education classroom where he received occupational, speech and physical therapies. His speech was difficult to understand and he spoke in phrases without the use of complete sentences. Previous IQ testing (type unknown) revealed an IQ of 51. The parents described his behavior as “out of control” with tantrums, screaming episodes, and hyperactivity. His functional level appeared to be that of a 3- to 4-year old. He has taken clonidine, methylphenidate, and topiramate for his seizures and hyperactivity. His head circumference remained below the 3rd centile while height was at the 10–25th centile and weight was at the 10th centile. His face had a hypotonic appearance with a high forehead, mild right ptosis, a notch of his left ear and a prominent nasal tip (Fig. 3). Multiple dental caps were present on the lower teeth. There was a thin upper lip with a normal intact palate. Bilateral fifth finger clinodactyly was present as well as two to three toe syndactyly. There was mild hyperextension of the elbows. Testes were palpable in the scrotum.
Fig. 3.
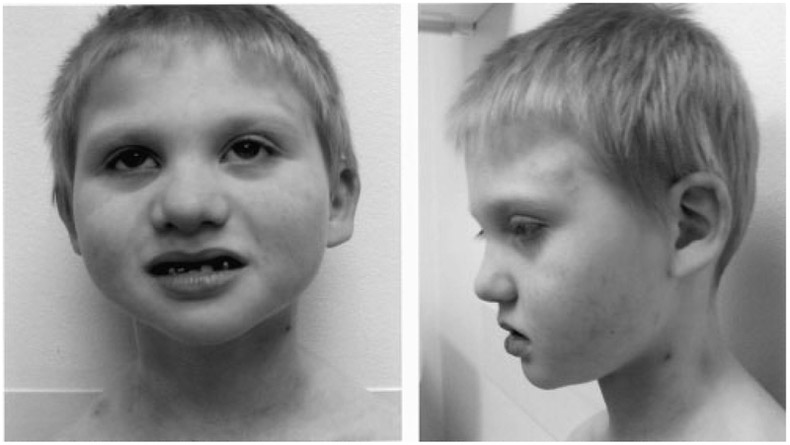
Frontal facial and profile views of our patient at 6 years of age. Note the high forehead, mild right ptosis, abnormal ears, prominent nasal tip, and extensive dental findings.
This case is the second report of a 2q33.3–q34 deletion (other subjects with similar 2q deletion are summarized in Table I). In our subject a balanced translocation, t(4:16)(q32.2;q24.1), was also present but a molecular microarray analysis (aCGH) at the 1 Mb level failed to show abnormalities in the 4q or 16q regions. However, the 2q deletion was detected and further characterized with a 44 K oligonucleotide microarray. Our patient also had severe psychomotor delays with poor eye contact, autistic behavior, microcephaly, and dysmorphic features. These features were similar to those described by Pescucci et al. [2003] and Bisgaard et al. [2006] in their patients with distal 2q deletions.
TABLE I.
Clinical Findings in Three Cases With an Interstitial Deletion at 2q33.2–q35
| Features | Courtens et al. [1997] | Pescucci et al. [2003] | Bisgaard et al. [2006] | Present case |
|---|---|---|---|---|
| Deletion segment | 2q33.3–q34 | 2q33.3–q35 | 2q33.2–q34 | 2q33.3–q34 |
| Sex | M | F | M | M |
| Age | 5 weeks | 14 years | 11 months | 3 years 5 months |
| Birth weight | 2,560 g | 1,960 g | 3,250 g | 3,182 g |
| Feeding difficulties | Yes | Yes | Yes | Yes |
| Growth failure | Yes | No | No | No |
| Hypotonia | Yes | Yes | No | Yes |
| Psychomotor retardation | Yes | Yes | Yes | Yes |
| Autistic traits | NA | Yes | Yes | Yes |
| Micrognathia | Yes | No | No | No |
| Microcephaly | Yes | Yes | 10th centile at 2 years of age | Yes |
| High forehead | Yes | No | Yes | Yes |
| Abnormal ears | Yes | Yes | No | Yes |
| Downslanting palpebral fissures | No | Yes | Yes | No |
| Other findings | Hypospadias | Enlarged cisterna magna and peripontine spaces by MRI; grand mal seizures; persistent hand clapping | Normal MRI; balanced (10;15) translocation inherited from father and grandfather | Normal MRI; complex partial seizures; de novo balanced (4;16) translocation |
The recent literature suggests that autism or autism related genes are found in chromosome regions 7q21.2–q36.2, 16p12.1–p13.3, 6q14.3–q23.2, 2q24.1–q33.1, 17q11.1–q21.2, 1q21–q44, and 3q21.3–q29 [Yang and Gill, 2007]. Our case and the reports by Pescucci et al. [2003] and Bisgaard et al. [2006] further expands the distal 2q region to include region 2q33.3–2q34 and the involvement of risk genes for autism and neurodevelopment.
REFERENCES
- Bisgaard AM, Kirchhoff M, Zeynep T, Jepsen B, Brondum-Nielsen K, Cohen M, Hamborg-Petersen B, Bryndorf T, Tommerup N, Skovby F. 2006. Additional chromosomal abnormalities in patients with a previously detected abnormal karotype, mental retardation and dysmorphic features. Am J Med Genet Part A 140A:2180–2187. [DOI] [PubMed] [Google Scholar]
- Courtens W, Speleman F, Messiaen L, Bormans J, Van Roy N, Vamos E. 1997. Interstitial deletion 2q33.3-q34 in a boy with a phenotype resembling the Seckel syndrome. Am J Med Genet 71:479–485. [PubMed] [Google Scholar]
- Houdayer C, Portnoi MF, Vialard F, Soupe V, Crumiere C, Taillemite JL, Coudere R, Vazquez MP, Bahuau M. 2001. Pierre Robin sequence and interstitial deletion 2q32.3-q33.2. Am J Med Genet 102:219–226. [DOI] [PubMed] [Google Scholar]
- Kitsiou-Tzeli S, Sismani C, Ioannides M, Bashiardes S, Ketoni A, Touliatou V, Kolialexi A, Mavrou A, Kanavakis E, Patsalis PC. 2007. Array-CGH analysis and clinical description of 2q37.3 de novo subtelomeric deletion. Eur J Med Genet 50:149–154. [DOI] [PubMed] [Google Scholar]
- Kramer BW, Martin T, Henn W, Lai S, Speer CP. 2000. Lung hypoplasia in a patient with del(2)(q33-q35) demonstrated by chromosome microdissection. Am J Med Genet 94:184–188. [PubMed] [Google Scholar]
- Pescucci C, Meloni I, Bruttini M, Ariani F, Longo I, Mari F, Canitano R, Hayek G, Zappella M, Renieri A. 2003. Chromosome 2 deletion encompassing the MAP2 gene in a patient with autism and Rett-like features. Clin Genet 64:497–501. [DOI] [PubMed] [Google Scholar]
- Pescucci C, Caselli R, Grosso S, Mencarelli MA, Mari F, Farnetani MA, Piccini B, Artuso R, Bruttini M, Priolo M, Zuffardi O, Gimelli S, Balestri P, Renieri A. 2007. 2q24-q31 deletion: Report of a case and review of the literature. Eur J Med Genet 50:21–32. [DOI] [PubMed] [Google Scholar]
- Riegel M, Morava E, Czako M, Kosztolanyi G, Schinzel A. 2001. Distal deletion, del(2)(q33.3q33.3), in a patient with severe growth deficiency and minor abnormalities. Am J Med Genet 102:227–230. [DOI] [PubMed] [Google Scholar]
- Van Buggenhout G, Van Ravenswaaij-Arts C, Mc Maas N, Thoelen R, Vogels A, Smeets D, Salden I, Matthijs G, Fryns JP, Vermeesch JR. 2005. The del(2)(q32.2-q33) deletion syndrome defined by clinical and molecular characterization of four patients. Eur J Med Genet 48:246–289. [DOI] [PubMed] [Google Scholar]
- Yang MS, Gill M. 2007. A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci 25:69–85. [DOI] [PubMed] [Google Scholar]