

Abstract
During the last 40 years, understanding of bone biology and the pathogenesis of osteoporosis, the most common and impactful bone disease of old age, has improved dramatically thanks to basic and clinical research advances, genetic insights from humans and rodents, and newer imaging technologies. Culprits of osteoporosis are no longer a matter of speculation based on in vitro observations. Instead, they can be identified and dissected at the cellular and molecular level using genetic approaches; and their effect on distinct bone envelopes and anatomic regions can be functionally assessed in vivo. The landscape of pharmacotherapies for osteoporosis has also changed profoundly with the emergence of several potent antiresorptive drugs as well as anabolic agents, displacing estrogen replacement as the treatment of choice. In spite of these major positive developments, the optimal duration of the available therapies and their long-term safety remain matters of conjecture and some concern. Moreover, antiresorptive therapies are used indiscriminately for patients of all ages on the assumption that suppressing remodeling is always beneficial for bone, but rebound remodeling upon their discontinuation suggests otherwise. In this invited perspective, I highlight the latest state of knowledge of bone-intrinsic and extrinsic mechanisms responsible for the development of osteoporosis in both sexes; differences between the mechanisms responsible for the effects of aging and estrogen deficiency; and the role of old osteocytes in the development of cortical porosity. In addition, I highlight advances toward the goal of developing drugs for several degenerative diseases of old age at once, including osteoporosis, by targeting shared mechanisms of aging.
Keywords: OSTEOPOROSIS, DISEASES AND DISORDERS OF/RELATED TO BONE, SEX STEROIDS, CELL/TISSUE SIGNALING, ENDOCRINE PATHWAYS, AGING, MENOPAUSE
And so, from hour to hour, we ripe and ripe, And then, from hour to hour, we rot and rot..
—William Shakespeare, As You Like It, Act 2:7
Introduction
One of the most consequential facts of modern biomedicine is that life expectancy has increased dramatically in developed countries over the past two centuries.(1) If the pace continues through this century, most babies born since 2000 will celebrate their 100th birthday. However, it remains unclear whether the increases in life expectancy are accompanied by postponement of functional limitations and disability. In view of this situation, it is imperative for the bone and mineral research community to elucidate whether skeletal involution and osteoporosis—one of the most common diseases of old age—are inexorable accompaniments of longevity, or whether they can be prevented, or at least slowed, by targeting molecular pathways and mechanisms of aging, so that “bone health span” can increase in tandem with lifespan. In this review, I summarize the last 40 years of progress toward meeting the challenge and provide some answers to these fundamental questions.
In keeping with the playwright’s wisdom, both women and men of all races start losing bone immediately after they reach peak bone mass, around the third decade (Fig. 1). At the proximal femur, bone loss is approximately linear over life.(2) The incidence of osteoporotic fractures, however, does not begin to rise until the sixth decade in the case of radiologic vertebral fractures, and the middle of the seventh decade for wrist and hip fractures. The risk of hip fractures in a man is, on average, the same as a woman 5 to 10 years younger.(3)
Fig. 1.
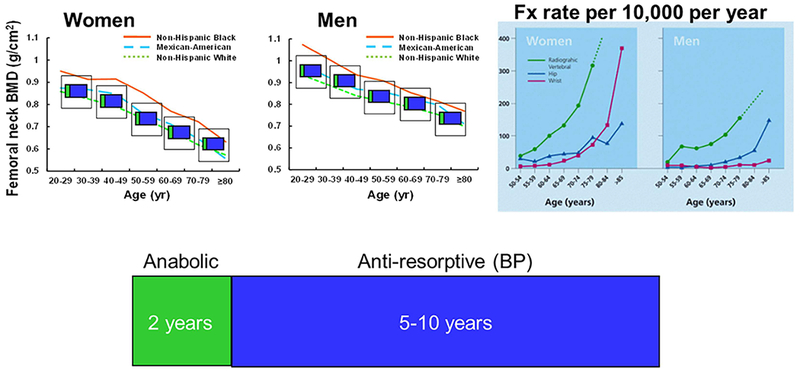
Bone loss, fractures, and “symptomatic” therapies. Femoral BMD and fracture rates are reproduced with permission from Looker and colleagues(2) and Sambrook and Cooper.(3) The duration of anabolic and BP treatment are per the latest ASBMR recommendations. BP = bisphosphonate; Fx = fracture.
In the last 40 years, the landscape of pharmacotherapies used to prevent such fractures has changed profoundly with the emergence of several potent antiresorptive drugs as well as anabolic agents, displacing estrogen replacement as the treatment of choice. In spite of these major positive developments, the optimal duration of these therapies and their long-term safety remain matters of conjecture and some concern. More important, their effects are short-lived and upon their discontinuation, bone mass returns to baseline. This clearly shows that although these therapies counter downstream changes, ie, increased resorption or decreased formation, they do not affect the upstream disease-specific pathogenic molecular mechanisms, which is the subject of the following sections.
In the case of the most potent and shorter acting antiresorptive denosumab, discontinuation causes rebound bone loss.(4,5) This indicates that powerful inherent homeostatic mechanisms force return to a predetermined baseline. Fora lack of a better understanding, we call these homeostatic mechanisms by the Frostian term “mechanostat.” Currently, the maximum recommended duration for an anabolic therapy is 2 years, and for the most commonly used antiresorptive class, bisphosphonates, 5 to 10 years (Fig. 1). This clearly poses a dilemma of when to use them for a maximal benefit, and whether it is safe to repeat them and how many times.
Aging Versus Estrogen Deficiency
Most of the changes of old age are, in the final analysis, due to atrophy; in terms of the skeleton atrophy is osteoporosis. How much of the atrophy of old age is due to under-function of the steroid-producing glands, and how much is due to old age per se are, as yet, unanswered questions.
—Albright and Reifenstein, Parathyroid Glands and Metabolic Bone Disease, Baltimore, MD: Williams & Wilkins Co.; 1948
The abrupt decline of estrogen levels at menopause undoubtedly causes loss of bone and contribute to the development of osteoporosis. However, the effects of estrogen deficiency and aging inexorably overlap, making it very difficult, if not impossible, to dissect their independent contributions to the cumulative bone anatomic deficit.
Almost 40 years ago, and drawing from earlier work of others, Michael Parfitt proposed that the stage of bone loss that begins at menopause is short-lived.(6–9) In a few years, it is followed by a later stage of loss that continues throughout life. He suggested that the two stages differ not only in timing and rate, but in structural and mechanical effects, remodeling abnormalities, cellular defects, and pathogenesis. The first stage is caused by estrogen deficiency and the mechanism is increased osteoclastic resorption. The second stage is a consequence of aging and the mechanism is failure of osteoblasts to refill cavities of normal or reduced size.
Some years later, Larry Riggs and colleagues proposed a unitary model of involutional osteoporosis according to which estrogen deficiency is the predominant cause of both the early and the late phases of bone loss in postmenopausal women, and also a contributing cause of the continuous phase of bone loss in aging men.(10) In 2010, I reviewed evidence on the effects of aging on bone, which had emerged in the interim, and suggested that it provides a paradigm shift from the “estrogen-centric” account of the pathogenesis of osteoporosis to one in which age-related mechanisms intrinsic to bone are protagonists and age-related changes in other organs and tissues, such as the ovaries, accentuate them.(11)
A search of the Web of Science website for articles published from the mid-20th century to the present with the word “estrogen” or “aging” AND “osteoporosis” in the title is depicted in Fig. 2.
Fig. 2.
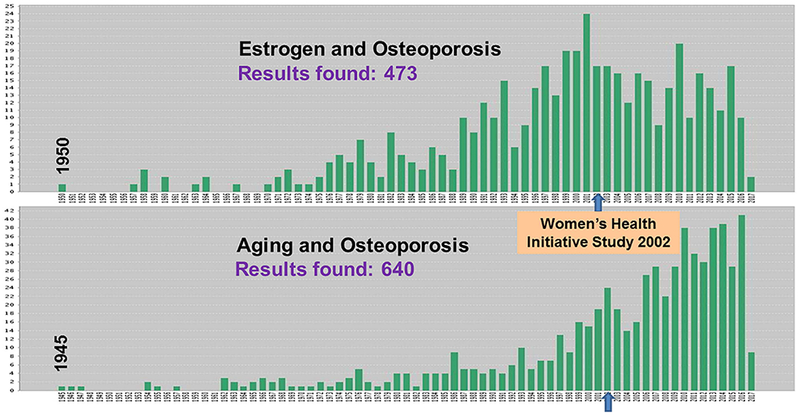
Articles with “estrogen” or “aging” AND “osteoporosis” in the title. The figure depicts a search of the Web of Science website for articles published from the mid twentieth century to the present with the word “estrogen” or “aging” AND “osteoporosis” in the title.
The results of this search clearly show a diverging trend starting around 2002. This coincides with the publication of the seminal Women’s Health Initiative Study which revealed that the health risks from the use of combined estrogen plus progestin in healthy postmenopausal women exceed the benefits.(12) This observation suggests that: (i) the role of aging in the pathogenesis of osteoporosis has been understudied until recently; and (ii) the pharmaceutical industry and the ups and downs of drug popularity has a major influence in the shaping of research priorities.
The Evolutionary Perspective
A much wider perspective of the contribution of aging versus sex steroid deficiency, however, can be gained from the evolutionary record (Fig. 3).
Fig. 3.
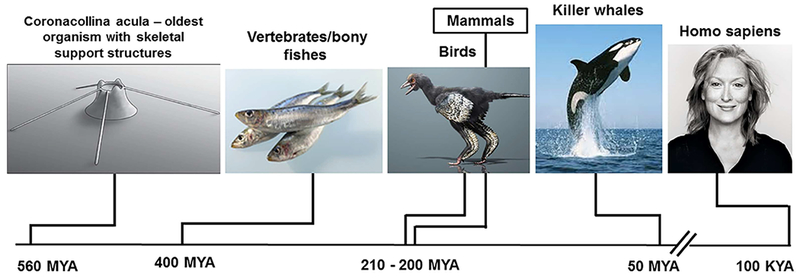
Evolutionary history of the bony skeleton. MYA = millions of years ago; KYA = thousands of years ago.
According to the latest fossil evidence, the oldest organism with individual skeletal parts is Coronacollina acula, a pre-Cambrian thimble-like creature with at least four needle-like bony “spicules” attached to its main body.(13) Vertebrates and bony fishes appeared 400 million years ago and birds and mammals 200 million years later. Killer whales and Homo sapiens are much later arrivals, with Homo sapiens appearing on the stage no more than 100,000 years ago. Aging is a shared characteristic of all birds, mammals, other vertebrates, and invertebrates alike(14,15); but, in natural populations, only women, killer whales, and short-fin pilot whales live long past menopause.
In subsequent sections, I review evidence that the overarching cause of the involution of the mammalian skeleton and the diseases that accompany it is, indeed, aging. Age-related mechanisms intrinsic to bone that are conserved through hundreds of millions of years of evolution are the primary culprits.
The Validity of the Mouse in the Study of Osteoporosis Mechanisms
Current knowledge of osteoporosis mechanisms relies heavily on experimental evidence from mice.(11,16,17) The rationale for using the mouse model is straightforward. In humans, one can mostly make associations. In mice, one can do experiments that functionally interrogate the effects of genes and test specific hypotheses about cellular and molecular mechanisms. The relatively short lifespan of mice makes the model particularly useful in the search for mechanisms that are relevant to aging in other mammals.
It has been well documented that sex steroid–sufficient mice and rats lose bone mass and strength with age.(18–22) As shown in Fig. 4, work from the Center for Osteoporosis and Metabolic Bone Diseases (hereafter “our center”) has documented that both female and male C57BL6/J mice exhibit a progressive loss of bone mineral density and strength in the spine and femur between the ages of 4 and 31 months. These findings have been confirmed more recently with μCT(22) (Fig. 5, Table 1). Indeed, as shown in, both female and male mice show a decrease of cortical thickness in the femur (mid-diaphysis) (Fig. 5A) and the spine (L4) (Fig. 5B). Both male and female mice also lose trabecular bone (BV/TV) in the distal femur and the vertebra with age (Table 1). Notably, in agreement with studies from others showing rapid trabecular bone loss in the femur between 2 and 6 months in both sexes,(20) BV/TV is very low (approximately 6%) at this site in females; but twofold to threefold higher in males.
Fig. 4.
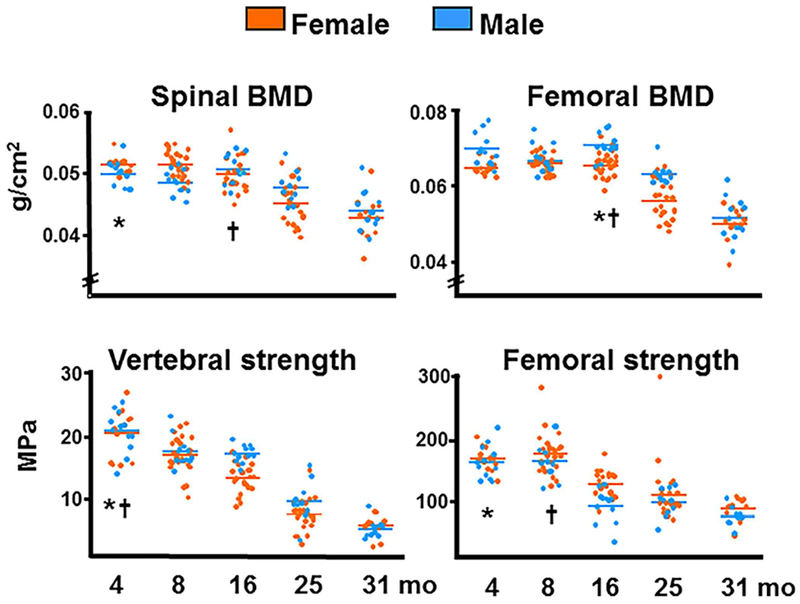
BMD and bone strength decrease with age in C57BL6/J mice. Spinal and femoral BMD was assessed by dual-energy X-ray absorptiometry; strength, by compression testing of the sixth lumbar vertebra and by three-point bending of the left femur. Colored horizontal lines indicate the mean values for each sex. * and † indicate the age after which a time-dependent decline began in females and males, respectively. Reproduced with permission from Almeida and colleagues.(21)
Fig. 5.
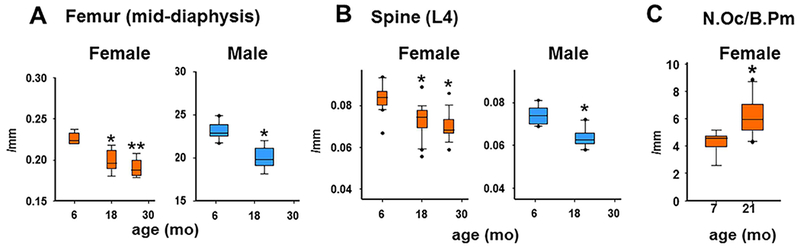
Cortical thickness decreases with age in female and male C57BL6 mice. Cortical thickness was determined at the mid-diaphysis (A) and the anterior lumbar vertebra 4 (B). Number of mice analyzed, ordered by increasing age: female (8, 8, and 7 months), male (10 and 10 months). Box-plots represent the 25th to 75th percentile values and the line denotes the median value. Whiskers represent the 95% CI, and dots represent data falling outside this CI. *p < 0.05 versus 6-month-old to 7-month-old sex-matched animals; **p < 0.05 versus 6-month-old to 7-month-old and 18-month-old to 19-month-old sex-matched animals. (C) Histomorphometric determination of N.Oc/B.Pm. For 7-month-old mice, n = 9. For 21-month-old mice, n = 10. *p < 0.05 versus 7-month-old. Reproduced with permission from Piemontese and colleagues.(22) N.Oc/B.Pm = number of osteoclasts per bone perimeter.
Table 1.
Age-Related Changes in Trabecular Bone in C57BL6 Mice
| Females | Males | ||||
|---|---|---|---|---|---|
| Vertebra (L4) | 6 months (n = 20) | 18 months (n = 20) | 26 months (n = 18) | 7 months (n = 10) | 21 months (n = 10) |
| BV/TV (%) | 25.8 ± 3.5 | 20.7 ± 5.1* | 16.2 ± 6.1* | 32.4 ± 2.6 | 23.3 ± 3.8* |
| Femur | 6 months (n = 8) | 18 months (n = 8) | 26 months (n = 7) | 7 months (n = 10) | 21 months (n = 10) |
| BV/TV (%) | 6.2 ± 2.0 | 1.1 ± 0.6* | 0.6 ± 0.6* | 21.3 ± 4.2 | 6.5 ± 1.6* |
Data were analyzed by ANOVA or Student’s t test. Animal numbers are shown in parentheses. Reproduced with permission from Piemontese and colleagues.(22)
p < 0.05 versus 6-month-old or 7-month-old mice of the same sex and strain.
At the endocortical surface of the femur, the age-dependent changes in females are accompanied by an increase in osteoclast number (Fig. 5C).(22) Wall width, a measure of decreased osteoblast vigor, decreased with age in that study, but osteoblast number and bone formation rate was no different between young and old female mice. The age-dependent decrease in wall width in mice is in full agreement with the evidence that decreased wall width in humans is the histologic hallmark of osteoporosis in the elderly.(23)
In addition to the age-dependent decline in bone mass, female C57BL/6J mice, like humans, exhibit a progressive increase in cortical porosity occurring primarily near the proximal and distal metaphyses (Fig. 6A).(22) However, this phenomenon is highly variable, with porosity values ranging from 1% to 14% in 26-month-old female mice (Fig. 6B). Cortical porosity, nonetheless, is not significantly affected by age in male C57BL/6J mice. Practically identical results, including the sex difference, are found in a second strain of mice, F1 hybrids of a C57BL/6J and BALB/c C57BL/6J C57BL/6J.(22) Notably, similar to the findings in mice, HRpQCT measurements of the radius and tibia show that postmenopausal women have greater cortical porosity than men over the age of 50 years.(24,25) At this stage the reason for the difference in cortical porosity between the sexes remains a matter of speculation. Albeit, a greater decrease in sex steroid levels in female than male mice with age cannot account for the greater porosity of cortical bone in female mice. Alternative reasons may be the higher level of androgens in male mice and humans; or that the female skeleton is genetically more sensitive to the development of age-dependent porosity than the male skeleton, analogous to the influence of sex-specific loci on BMD in children.(26)
Fig. 6.
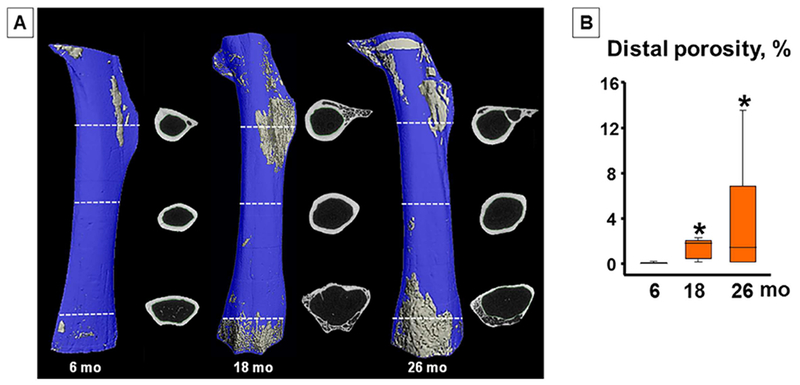
Cortical porosity increases with age in female mice. (A) Inverse μCT images of femurs (proximal at top) from female C57BL6 mice, excluding epiphyses. Cortical voids are depicted in gray within a blue bone matrix. Lines show location of single μCT slices (before binarization). (B) Quantification of porosity of the distal half of the femur. Number of mice analyzed (ordered by increasing age in months): (8,8,7). Box-plots represent the 25th to 75th percentile values and the line denotes the median value. Whiskers represent the 95% CI. *p < 0.05 versus 6-month B6 females. Reproduced with permission from Piemontese and colleagues.(22)
Furthermore, cross-sections of the femur from 6-month-old and 11-month-old adult mice show no evidence of intracortical remodeling (Fig. 7A and B), but ample endocortical remodeling as indicated by the fluorochrome labeling (Fig. 7A). Additional results from this work have established that this is BMU-based endocortical remodeling, because it can be blocked by stopping osteoclastic resorption using osteoprotegerin (OPG).(22) In contrast to the younger mice, 21-month-old mice show extensive intracortical remodeling. Indeed, scalloped cement lines marked by the green arrows in Fig. 7C form boundaries around both small and large pores containing a central capillary. These intracortical pores are lined with osteoclasts, not shown in Fig. 7C, as well as osteoblasts indicated by the fluorochrome labeling present in adjacent sections (Fig. 7D). These histologic features identify the cortical pores of aged mice as osteons, many of which contain BMUs engaged in intracortical remodeling. Albeit, the murine osteons lack the highly organized Haversian structures, with which you are all familiar in the case of large mammals and humans.
Fig. 7.
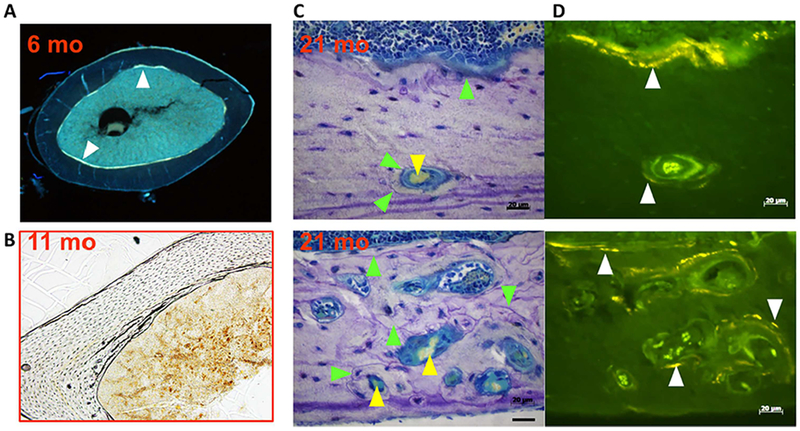
Cortical porosity results from de novo intracortical remodeling. (A, B) Cross-sections from the femoral diaphysis of 6-month-old and 11-month-old C57BL6/J mice. (C, D) Sequential sagittal sections of the femur of 21-month-old mice; the upper and lower panels are from two different sites. Toluidine blue–stained bright field images (C); and unstained adjacent section under fluorescence illumination (D), to visualize tetracycline given at 7 and 3 days before euthanasia. White arrowheads mark tetracycline labeling, green arrowheads mark cement lines, and yellow arrowhead mark red blood cells. In A, the endosteal surface is labeled by tetracycline. Please note lack of intracortical remodeling in B. Please note ample endocortical remodeling as indicated by scalloped cement lines in C and fluorochrome labeling in D. Cement lines and fluorochrome labeling form boundaries around both small and large pores containing a central capillary. Cement lines and tetracycline labels are similarly seen at the endosteal surface in C and D. Reproduced with permission from Piemontese and colleagues.(22)
Besides the loss of bone mass and architectural deterioration, aged mice develop spontaneous femur fractures. Indeed, in unpublished studies by Simon Melov and colleagues from the Buck Institute (personal communication), 200 C57BL6/J mice were surveyed every 3 months from 9 to 36 months of age, by serial μCT. Five to 10 percent of mice over the age of 25 months experienced spontaneous fractures of the femoral neck. Notably, these fractures were not associated with any other obvious phenotype. Because of the limited power of the cohort, Melov and coworkers have not been able to determine whether these fractures are more prevalent in one sex than the other.
Cellular and Molecular Mechanisms of the Effects of Estrogen Deficiency on Bone
Estrogens and androgens slow the rate of bone remodeling and estrogen or androgen deficiency accelerates it. Additionally, cell and biochemical studies revealed that the anti-remodeling effects of these two hormones result from their ability to restrain the birth rate of osteoclasts and shorten their lifespan.(17) Conditional deletion models of the estrogen receptor alpha or the androgen receptor have provided critical new insights into the cellular targets of sex steroid action in vivo.(27–34) This body of work, reviewed in detail elsewhere,(35,36) has produced two seminal but unexpected findings. First, the effects of estrogens and androgens on the cancellous bone compartment are mediated via different cell types. In females, estrogens protect against the loss of cancellous bone via direct actions on cells of the osteoclast lineage. In males, on the other hand, androgens protect against the loss of cancellous bone via actions on mature osteoblasts or osteocytes (Fig. 8). Second, and more striking, in both females and males, estrogens alone protect against the resorption of cortical bone, via actions on osteoprogenitors. In males, of course, the estrogens responsible for this effect derive from the aromatization of androgens.
Fig. 8.
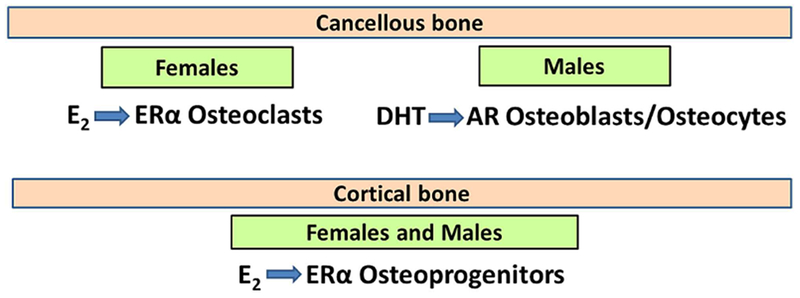
Target cells of estrogen and androgen action in bone.E2 = 17β-estradiol;DHT = dihydrotestosterone; ER = estrogen receptor; AR = androgen receptor.
More recently, work from the author’s laboratory has begun to identify the target genes of estrogen receptor α (ERα) signaling in the different bone envelopes, using ERα-deficient cells isolated from the mice with cell-targeted deletions. This approach establishes a priori that the putative target genes are: (i) expressed in cells that have been functionally validated in vivo and (ii) contextually relevant to bone mass regulation in the whole animal. The results suggest that in the estrogen-deficient state, increased expression of the calcium binding protein S100A8 in osteoclasts might contribute to the increased resorption of cancellous bone,(37–41) while an increase in the expression of the chemokine SDF1(34,38,39,42,43) and the protease MMP13(44–47) in osteoprogenitors might be involved in the increased resorption of cortical bone.
In addition to nuclear initiated actions, estrogens bind to subsets of their cognate receptors that are localized outside the nucleus, either on the cell membrane, the cytosol, or mitochondria. Binding of these ligands to receptors localized on the membrane initiates signal transduction cascades that activate cytoplasmic kinases, which in turn, phosphorylate substrate proteins and transcription factors that modulate gene transcription. During the last 20+ years, it has been shown that synthetic ligands that can selectively activate non-nuclear–initiated actions of the ERα, but have minimal or no effects on the nuclear-initiated actions of this receptor, at least partially replicate the beneficial effects of estrogens on skeletal maintenance without affecting reproductive organs, such as the uterus and the breast. To date, two types of agents have provided proof of principle for the selectivity of the nongenomic pathway.(48–50) The first is a cell membrane–impermeable estradiol conjugate, in which E2 is attached to a large, inert, positively charged nondegradable poly (amido)amine (PAMAM) dendrimer via a hydrolytically stable linkage. The second, and more recent agents, are termed pathway preferential estrogens (PaPes). These are novel estrogens resulting from a process of structural alteration of estrogenic ligands designed to preserve their essential chemical and physical features but greatly reduced their binding affinity for ERs. PaPEs are unable to stimulate reproductive and mammary tissues or breast cancer cells, but able to trigger beneficial responses both in metabolic tissues that reduced body weight gain and fat accumulation, and in the vasculature-accelerated repair of endothelial damage. The effect of the latter class on bone has not been determined yet.
At menopause, estrogen deficiency and aging inexorably overlap. This makes it very difficult to dissect their independent contribution to the cumulative bone anatomic deficit later in life. Moreover, it has remained unclear whether the molecular and cellular changes caused by aging and estrogen deficiency mechanistically interact. Different from humans, mice do not experience menopause, but are a faithful model of the adverse effects of estrogen deficiency (caused by gonadectomy) on both the cortical and cancellous bone compartments.(17) To dissect the contribution of estrogen deficiency to the involution of the mammalian skeleton, in a recent study from the author’s laboratory C57BL6/J mice were ovariectomized at 4 or 18 months of age.(51) Six weeks following the loss of ovarian function at either age, we observed the expected decrease in uterine weight and increase in body weight in both young and old mice. Hence, at least up to the age of 19.5 months these mice remained functionally estrogen-sufficient (Fig. 9). Nonetheless, femoral cortical thickness and vertebral cancellous bone volume declined and cortical porosity increased in the estrogen-intact mice between 5.5 and 19.5 months of age. This finding clearly demonstrates that the adverse effects of aging are independent of sex steroid status. Ovariectomy (OVX) in young mice caused a decline in both femoral cortical thickness and vertebral cancellous bone volume. Albeit, loss of ovarian function in the old mice had no effect on femoral cortical thickness, but it still caused a decline in vertebral cancellous bone volume. OVX at young or old age had no measurable effect on cortical porosity. These results demonstrate that the effects of aging on bone are independent of estrogen status in mice.
Fig. 9.
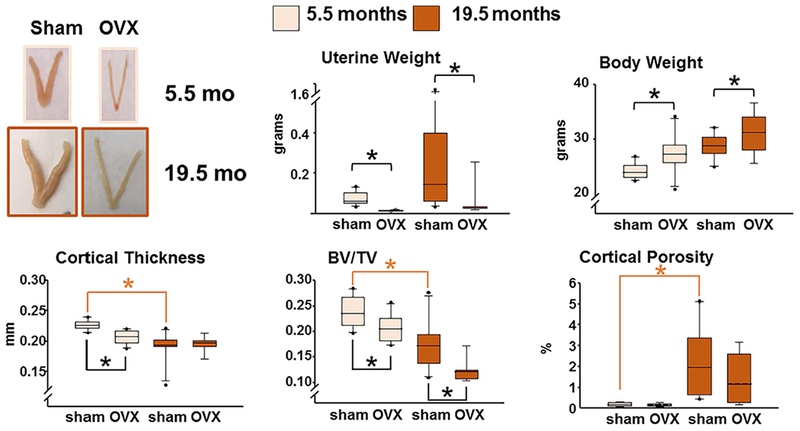
The effects of aging on bone are independent of estrogen status in mice. Mice were either sham-operated or ovariectomized (OVX) at 5.5 months or 19.5 months of age. Six weeks after the surgeries, the mice were euthanized. Femoral cortical thickness and cortical porosity were measured by μCT in the midshaft and the distal metaphysis, respectively. Cancellous bone (bone volume/tissue volume [BV/TV]) was measured in the 5th lumbar vertebra. Boxes depict values from the 25th to 75th quartiles, the middle line depicts the mean, and the vertical whiskers show the 10th and 90th percentiles; values outside this range are plotted as dots. n = 9 to 10; *p < 0.05 by two-way ANOVA. Reproduced with permission from Ucer and colleagues.(51)
Cellular and Molecular Mechanisms of the Effects of Aging on Bone
In a landmark article published in 2013, leading authorities in the field of aging biology proposed nine mechanisms as the hallmarks of aging in most, if not all, aged tissues(52): mitochondria dysfunction, cellular senescence, stem cell exhaustion, altered intercellular communication, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, and deregulated nutrient sensing. The criteria for defining these mechanisms as hallmarks of aging were that they are manifested during normal aging; their experimental aggravation accelerates aging; and experimental amelioration retards normal aging. Their common characteristics were an unequivocally negative effect on physiologic integrity (although may mediate beneficial effects at low levels but at high levels become deleterious) and co-occurrence during aging.
Parallel advances in the bone field, reviewed in detail elsewhere,(11,53) have strongly suggested that several of the hallmark mechanisms of aging are also manifested in aged bone and are indeed seminal culprits of skeletal aging. Michael Parfitt and I had reasoned 8 years ago that the bone cell types that are relevant to the effects of aging on bone must be long-lived.(54) In humans, osteoclasts live only for a few days or weeks and osteoblasts (while they are making bone) live only for a few months. The only long-lived cells in bone are the stem cells from which osteoclasts or osteoblasts are derived, cells of the periosteum about which still very little is known, and the lining cell-osteocyte syncytium consisting of terminally differentiated osteoblasts, which are the resident cells of bone. The notion that osteocytes have a lifespan that is relevant to pathophysiology was first conceived by Harold Frost over 50 years ago.(55)
Reactive Oxygen Species and Oxidative Stress
The levels of reactive oxygen species (ROS) in the bone marrow increase with age in both female and male mice; the same is true for the bone levels of phosphorylated p53,the active form of this tumor suppressor and seminal mediator of the responses of all mammalian cells to DNA damage.(21) Intriguingly, loss of sex steroids in females or males, caused by gonadectomy, have the same effects as aging. These changes are reversed in the gonadectomized mice by hormone replacement or administration of the antioxidant N-acetyl cysteine. Based on these observations, we had proposed that estrogen deficiency may accelerate the adverse effects of aging on bone. The FoxO family of transcription factors are an important defense mechanism against ROS.(56,57) ROS antagonize Wnt signaling in osteoblast progenitors, by diverting the limited pool of β-catenin from T-cell factor (TCF)-mediated to FoxO-mediated transcription. This indicates that, as is the case with several other defense responses against aging, FoxO activation eventually becomes a culprit of skeletal involution by suppressing Wnt signaling.(58–60)
Besides their cell-damaging effects in the setting of oxidative stress, ROS play an important role in physiological intracellular signaling by triggering proliferation and survival. During the last 30 years several studies, starting with one from the group of Greg Mundy, have documented that ROS accumulation promotes osteoclastogenesis and bone resorption, under both physiologic and pathologic conditions.(61–65) ROS are primarily generated in mitochondria during the respiratory cycle and H2O2 is the most abundant species of ROS. Catalase is an important antioxidant enzyme that eliminates H2O2. The transcription of the catalase gene is stimulated by FoxOs in osteoclasts; and gain or loss of FoxO function in osteoclasts increases and decreases bone mass, respectively, by altering osteoclast numbers. RANKL, on the other hand, inhibits the transcriptional activity of FoxOs by stimulating Akt.(66)
Importantly, restraining H2O2 accumulation by mitochondria-targeted catalase expression globally extends murine life span(67); prevents age-associated reductions in mitochondrial function and insulin resistance(68); partially rescues cardiac hypertrophy and dilatation, impairment of systolic and diastolic function, and increased cardiac fibrosis in mice with a homozygous mutation of mitochondrial DNA polymerase gamma(69); and reduces abnormal amyloid precursor protein processing and amyloid β production in a mouse model of Alzheimer’s disease.(70)
Based on the evidence that decreasing H2O2 production by targeted expression of catalase to mitochondria ameliorates several age-related pathologies, we generated transgenic mice expressing the mitochondria targeted catalase specifically in cells of the myeloid (using LysM-Cre) or the mesenchymal lineage (using Prx1-Cre). The OVX-induced or orchidectomy (ORX)-induced loss of cortical bone mass was prevented by restraining H2O2 accumulation in the cells of the myeloid lineage. This effect was associated with abrogation of the increase of osteoclast numbers in the endocortical surface.(51) However, restraining H2O2 accumulation in cells of the myeloid lineage did not prevent the age-dependent cortical or cancellous bone loss or cortical porosity.
Restraining H2O2 in cells of the mesenchymal lineage, on the other hand, did prevent the age-dependent decrease of cortical thickness, seen in the control mice. Specifically, at 22 months of age mice carrying the mitoCAT transgene had higher mineralizing surfaces as compared to their littermate controls. Albeit, in both genotypes, mineralizing surfaces decreased between 6 and 22 months of age, suggesting that other mechanisms, besides an increase in ROS, are responsible for this decline. The age-dependent increase in osteoclasts at the endocortical surface and cortical porosity, nonetheless, was indistinguishable between the two genotypes. Taken together, the evidence from the mitoCAT mice supports the notion that the adverse effects of estrogen deficiency and aging are mechanistically distinct. The effects of estrogen deficiency on cortical bone are primarily, if not exclusively, the result of increased resorption, whereas the adverse effects of aging are caused primarily by a decrease in bone formation combined with increased resorption.
Cellular Senescence
Cellular senescence is one of the hallmarks of aging. DNA damage and other signals that trigger senescence cause cells to stop dividing and undergo distinctive phenotypic alterations, including profound chromatin and secretome changes (Fig. 10). p53 and p16 act as a mediators of the DNA damage response by transmitting the signal from proteins that sense DNA damage to stop cell cycle progression. The transcription factor GATA4 functions as a key switch to activate the senescence associated secretory phenotype (SASP). Senescence-inducing signals block p62-dependent autophagic degradation of GATA4, resulting in NF-κB activation and SASP induction. Importantly, nonproliferating terminally differentiated cells also become senescent and exhibit the SASP.(71)
Fig. 10.
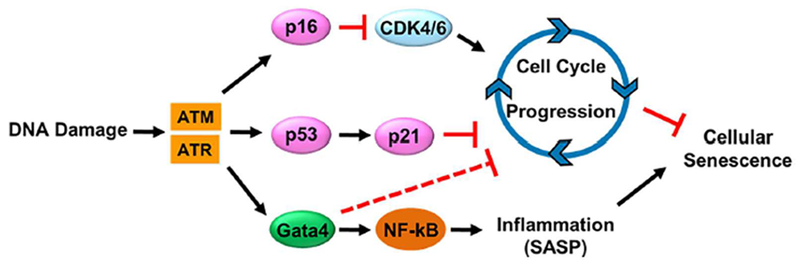
Cellular senescence and the key signaling cascades involved. ATM = ataxia-telangiectasia-mutated; ATR = ataxia telangiectasia and Rad3-related.
Maria Almeida in our group has spearheaded studies on osteoprogenitors expressing tomato red fluorescent protein under the control of Osx1-Cre. Using freshly isolated Osx1 cells from the bone marrow, we found that their number declines by more than 50% between 6 and 24 months of age in both female and male mice (Fig. 11A and B). This is caused by an arrest at the G1 phase of the cell cycle (Fig. 11C and D). Moreover the osteoprogenitors from the old mice exhibit markers of DNA damage and senescence, such as gamma H2AX foci, phosphorylation of p53, and increased p21 expression (Fig. 11E–H).(72)
Fig. 11.
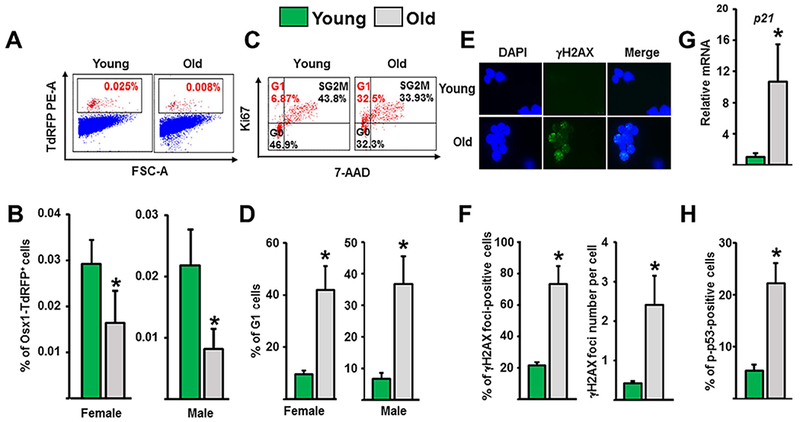
DNA damage, senescence, and decline off osteoprogenitors (Osx1) with age. (A, B) Osx1-TdRFP+ bone marrow cells were isolated from 4-month-old and 23-month-old Osx1-Cre;TdRFP mice (n = 4 to 6 mice/group). (A) Representative flow cytometric analysis of Osx1-TdRFP+ bone marrow cells. (B) Percentage of Osx1-TdRFP+ cells in the bone marrow. Osx1-TdRFP+ cells from old mice exhibit DNA damage and G1 arrest. (C) Representative FACS analysis of the cell cycle distribution of Osx1-TdRFP + cells from 3-month-old to 6-month-old and 23-month-old to 26-month-old Osx1-Cre;TdRFP mice (n = 5 mice/group). (D) Percentage of Osx1-TdRFP+cells at G1 in mice described in C. (E, F) Analysis of DNA double-strand breaks inTdRFP-Osx1+ cells from 6-month-old and 25-month-old Osx1-Cre;TdRFP male mice (n = 5 mice/group). (E) Representative photomicrographs of γH2AX immunofluorescence staining (green) and nucleic counterstaining with Hoechst-33342 (blue). (F) Percentage of γH2AX-positive cells (left) and number of γH2AX foci per cell (right). (G) P21 protein levels by Western blot analysis of Osx1-TdRFP+ cells from 6-month-old and 24-month-old Osx1-Cre;TdRFP female mice (n = 5 mice/group). (H) Percentage of p-p53-positive cells in Osx1-TdRFP+ cells from 3-month-old and 24-month-old Osx1-Cre;TdRFP male mice (n = 5 mice/group). Bars represent mean and vertical lines standard deviations; *p < 0.05. Reproduced with permission from Kim and colleagues.(72) γH2AX = phosphorylated histone H2AX.
Furthermore, osteoprogenitors from old mice exhibit elevated GATA4, activated NF-κB, and several SASP associated pro-osteoclastogenic cytokines, such as Tnf-α, IL-1α, MMP13, SDF1, and RANKL (Fig. 12A–C.
Fig. 12.
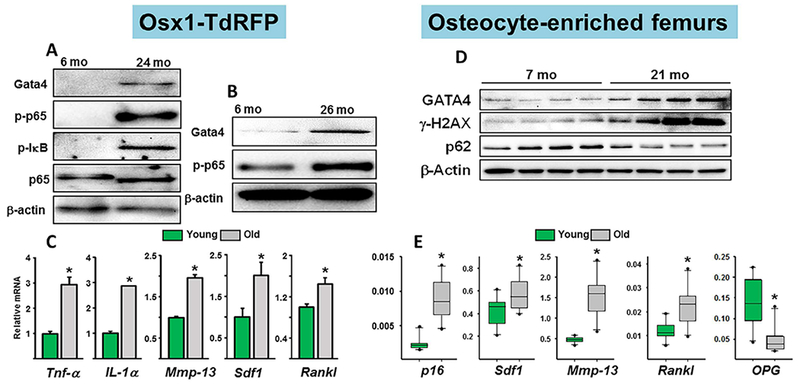
Increased GATA4, NF-κB, and SASP in osteoprogenitors and osteocytes from old mice. (A) Western blot analysis of lysates of Osx1-TdRFP+ cells freshly isolated from the bone marrow of 6-month-old and 24-month-old Osx1-Cre;TdRFP female mice (n = 5 mice/group). (B) Bone-marrow stromal cells from 6-month-old and 26-month-old Osx1-Cre;TdRFP female mice cultured with ascorbate and β-glycerophosphate for 7 days (triplicates). (C) mRNA levels by qRT-PCR in cells described in B. *p < 0.05 by Student’s t test. Bars represent mean and SD (error bars). Reproduced with permission from Kim and colleagues.(72) (D) Western blot analysis of femoral extracts from female C57BL6/J mice. (E) mRNA levels by qPCR in extracts from 7-month-old (n = 9) and 21-month-old (n = 10) female C57BL6/J mice. Box-plots represent the 25th to 75th percentile values and the line denotes the median value. Whiskers represent the 95% CI, and dots represent data falling outside this CI; *p < 0.05 versus 7-month-old animals. Reproduced with permission from Piemontese and colleagues.(22) γH2AX = phosphorylated histone H2AX; GATA4 = GATA binding protein 4.
The Contribution of Old Osteocytes to Skeletal Involution
Osteocytes from old mice exhibit markers of DNA damage and cellular senescence as well as the SASP, including some of the same cytokines found in the osteoprogenitors, such as SDF11 and MMP13 (Fig. 12D and E).(22) Osteocyte senescence has also been reported by Farr and colleagues.(73) As discussed earlier, MMP13 and SDF1 are likely candidates for the loss of cortical bone caused by estrogen deficiency. Therefore, the same cytokines may be responsible for the loss of cortical bone caused by both estrogen deficiency and old age. However, the culprit in old age is evidently cellular senescence and the SASP.
Deletion of Bak and Bax, two genes essential for apoptosis (Fig. 13A), in osteoblasts and osteocytes (Fig. 13B) greatly potentiates the effects of old age on cortical porosity (Fig. 13C).(74) Moreover, the age-dependent increase in cortical porosity in the Bak/Bax–deficient mice is accompanied by increased production of RANKL and vascular endothelial growth factor (VEGF) (Fig. 13D). Notably, attenuation of apoptosis stimulates cellular senescence.(75,76)
Fig. 13.
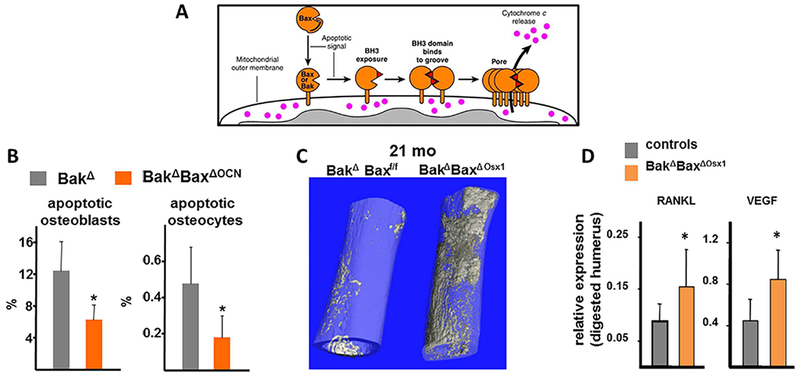
Prolongation of the lifespan of damaged osteoblasts/osteocytes by deletion of Bak/Bax intensifies the effects of aging on cortical porosity and is associated with increased RANKL and VEGF expression in osteocytes. (A) Apoptosis signaling promotes exposure of the BH3 domain of Bax and Bax leading to reciprocal BH3:groove interactions in the two proteins and formation of oligomeric complexes that form pores in the outer mitochondrial membrane, resulting in release of proapoptotic factors such as cytochrome c. Reproduced with permission from Dewson and Kluck.(102) (B) Cancellous osteoblast and cortical osteocyte apoptosis was assayed in bone sections by in situ end-labeling (ISEL).(C) Representative μCT images of femurs from 21-month-old female BakΔBaxΔOsx1 and BakΔBaxf/f littermates. (D) RANKL and VEGF mRNA levels were determined in collagenase-digested humeri, enriched in osteocytes, from 21-month-old female mice, n = 6 to 7/group; *p < 0.05 versus littermate controls. Reproduced with permission from Jilka and colleagues.(74)
Autophagy is a major adaptive response to cellular starvation and an essential protein/organelle quality control. Declining autophagy with advancing age is a big component of the loss of proteostasis, one of the hallmark mechanisms of aging.(52) Work from Charles O’Brien’s laboratory has demonstrated that attenuation of autophagy in osteocytes, by conditional deletion of the ATG7 gene in Dmp1-expressing cells, recapitulates most of the effects of old age in 6-month-old mice, including cortical porosity (Fig. 14).
Fig. 14.
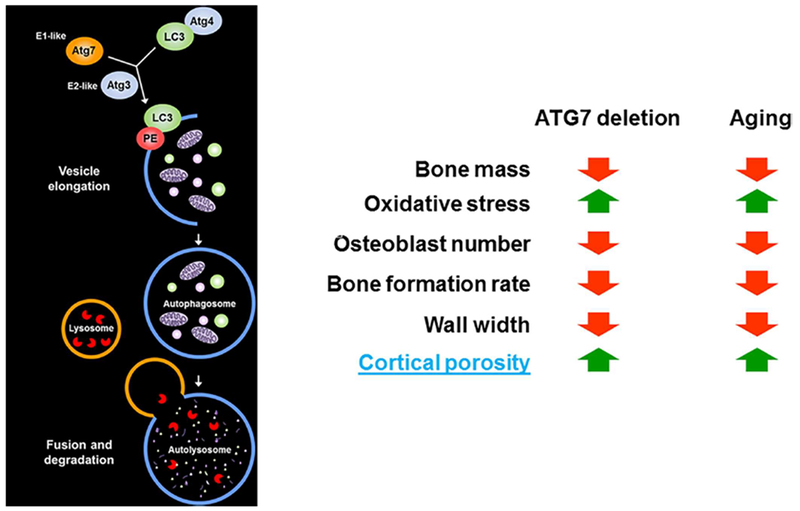
Suppression of autophagy in osteocytes mimics skeletal aging. The effects of ATG7 deletion are summary of results from Onal and colleagues.(103)
These findings, along with a series of studies from Mitch Schaffler’s group,(77–79) provide compelling evidence in support of the general idea, that in line with the seminal role of osteocytes in the choreography of bone remodeling, in conditions of overwhelming stress the physiological mechanisms of bone repair are exaggerated and become disease mechanisms.(80) Specifically, increased production of RANKL, MMP13, SDF1, and VEGF and decreased OPG by senescent, apoptotic, or dysfunctional osteocytes, and probably their affected neighbors (paracrine senescence), stimulate osteoclastogenesis, matrix degradation, focal bone resorption, and cortical porosity.
The Development of Drugs Targeting Mechanisms of Aging and Treating Several Degenerative Diseases Simultaneously
In the last part of this perspective, I discuss evidence that attenuating aging mechanisms represents a rational approach to treating several diseases of old age, including osteoporosis, simultaneously.
In Fig. 15, I have listed some of the new classes of drugs targeting mechanisms of aging that have been shown to be effective, in animal models, and in some cases in early stage clinical studies. The list includes inhibitors of the mTOR pathway, metformin and acarbose.(81,82) Cellular reprogramming to pluripotency by transient expression of the Yamanaka factors can also ameliorate age-associated symptoms, prolong lifespan in progeroid mice, and increase resistance to metabolic diseases and muscle injury homeostasis in older mice.(83) Very promising results have been also obtained with precursors of nicotinamide adenine dinucleotide (NAD) as well as activators of the histone deacetylases sirtuins.(84–86) Indeed, a large body of evidence from several organs and tissues has demonstrated that aging decreases NAD and the NAD-dependent SIRT1. Genetic evidence from our center and others, has shown that SIRT1 activation simultaneously stimulates osteoblastogenesis and suppresses osteoclastogenesis.(66,86–90) Last, targeted apoptosis of senescent cells with so-called senolytic agents restores tissue homeostasis in response to chemotoxicity and aging and attenuates atherosclerosis and posttraumatic osteoarthritis.(71,72,91–93)
Fig. 15.
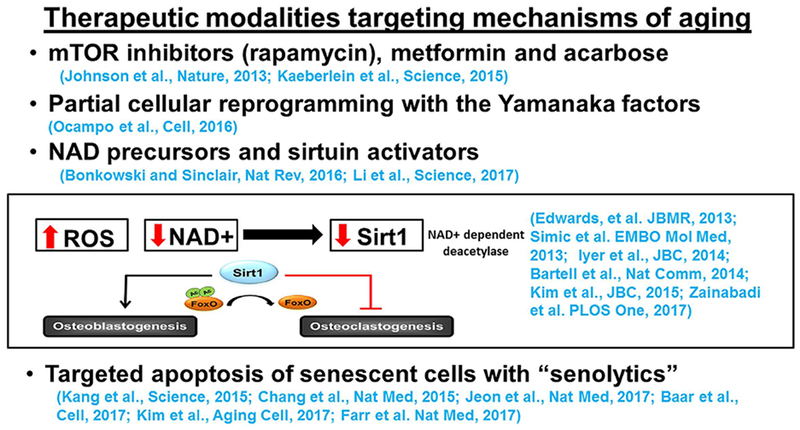
Therapeutic modalities targeting mechanisms of aging. (For details, see The Development of Drugs Targeting Mechanisms of Aging and Treating Several Degenerative Diseases Simultaneously.)
Ha-Neui Kim and Maria Almeida, in our center, have tested the effects of the senolytic drug ABT-263 on osteoprogenitors.(72) They found that it decreases GATA4 expression and improves the ability of osteoprogenitors to differentiate into bone-forming osteoblasts. Moreover, it attenuates the senescence-associated secretory phenotype and the ability of osteoprogenitors to support osteoclast formation. In line with our findings, Farr and colleagues(94) reported fairly recently that targeting cellular senescence increases bone mass in old mice. In their studies, they treated 20-month-old male mice with a combination of a tyrosine kinase inhibitor and a flavanol, once monthly for 4 months. The treatment caused an increase in both cancellous and cortical bone, a decrease in osteoclasts and an increase in osteoblasts numbers, and increased bone formation rate.
Oxidation-Specific Epitopes, innate Immunity, Atherosclerosis, and Osteoporosis
In the last section of this Perspective, I highlight some exciting recent work, suggesting yet another approach to the treatment of more than one disease of old age simultaneously, namely atherosclerosis and osteoporosis.
Atherosclerosis and osteoporosis are epidemiologically linked, and lipid peroxidation may be an important culprit in both.(95,96) Hyperlipidemia leads to atherogenesis by promoting the formation of oxidized forms of low-density lipoprotein (LDL) and phospholipids. These oxidation-modified moieties, in turn, induce potent inflammatory responses in the subendothelial matrix of the arteries, thereby triggering the pathogenetic changes that are ultimately responsible for the generation of the atherosclerotic lesion.(97) Consistent with this mechanism, genetic animal models of hyperlipidemia exhibit disruption of normal lipoproteins regulation and metabolism. Indeed, C57BL6/J mice maintained on an atherogenic-high fat diet (HFD), or genetically-modified C57BL6/J mice lacking the LDL receptor or its ligand (ApoE), develop hyperlipidemia and atherosclerosis as a result of reduced clearance of non–high-density lipoprotein (HDL) lipoproteins.(95)
BMD and LDL-cholesterol levels are inversely correlated in mice as well as rats,(95,98) and diet-induced hyperlipidemia adversely affects bone growth and BMD in these models. In line with the evidence for an adverse effect of lipids on bone, lipid accumulation has been demonstrated by histochemical staining in the subendothelial spaces of Haversian canals from patients with osteoporosis.(99) Based on this evidence, it has been hypothesized that, similar to the situation in the subendothelial space of the vasculature, lipid accumulation in the subendothelial spaces of the Haversian canals undergo non-enzymatic modifications, such as oxidation by ROS, rendering them capable of inducing inflammatory responses. Oxidized lipids may then induce bone loss by inhibiting the differentiation of bone-forming osteoblasts and promoting the differentiation of bone-resorbing osteoclasts.(98,100)
Lipid peroxidation results from ROS-induced oxidative damage and produces highly reactive degradation products, including oxidized phosphatidylcholine, malondialdehyde (MDA), and 4-hydroxynonenal (4-HNE).(101) These exposed neo-epitopes in turn interact with amino groups of proteins and other lipids to create oxidation-specific epitopes (OSEs). The same neo-epitopes that occur in oxidized LDL (OxLDL) occur on apoptotic cell bodies. OSEs are part of a larger group of proinflammatory molecules, collectively known as damage-associated molecular patterns (DAMPs), that are produced in response to cellular stressors, including oxidative stress. DAMPs share structural homology with another group of moieties present on microbes and collectively known as pathogen associated molecular patterns (PAMPs). Because of their shared molecular signatures, PAMPs and DAMPs bind to evolutionary conserved pattern recognition receptors (PRRs) of the innate immune system. In particular, the phosphorylcholine (PC) moiety of oxidized phosphatidylcholine is recognized by the IgM natural antibody (NAb) E06. Recent evidence indicates that when OSEs are not neutralized and/or their production is excessive, relative to the capacity of the innate immune system, the physiologic response is overwhelmed and becomes instead a disease-causing mechanism.(101)
Witztum and colleagues at UCSD have generated transgenic mice that overexpress a single-chain variable fragment of E06 (Witztum and colleagues, unpublished data). The fragment is secreted from liver and macrophages and achieves sufficient plasma levels to inhibit macrophage uptake of OxLDL. High fat diet (HFD)-fed LDL receptor null mice expressing E06 have 58% and 35% less atherosclerosis, after 4 and 12 months, respectively, compared to controls, despite the same high cholesterol levels of ~800 mg/dL.
Elena Ambrogini and Bob Jilka in our center have used the same mice to examine the influence of the E06 transgene on bone homeostasis (Ambrogini and colleagues, unpublished data). The bone loss that occurs in the HFD-fed LDL receptor (LDLR)-KO mouse was attenuated by the E06 transgene, as a result of increased osteoblast number and bone formation. Unexpectedly, the E06 transgene had an anabolic effect on the cancellous bone of C57BI/6J mice fed a normal diet. Last, sera from 26-month-old female C57BL/6J mice had dramatically decreased anti-PC IgM as compared to 6-month-old mice. This latter finding is consistent with evidence in humans that the cells that produce these IgMs decline with age; and low levels of anti-PC IgM antibodies are associated with increased incidence of cardiovascular diseases. These results indicate that OSEs chronically occurring with or without HFD in mice exert a restraining effect on bone formation; and that the age-related bone loss might be due in part to diminished innate immune system defense against OSEs. Anti-OSEs, therefore, may represent a novel therapeutic approach to the prevention and treatment of osteoporosis and atherosclerosis simultaneously.
Summary and Conclusions
The overarching cause of the involution of the mammalian skeleton and the degenerative diseases that accompany it, including the fracture syndrome of osteoporosis, is aging. Bone-intrinsic mechanisms, including mitochondria dysfunction, oxidative stress, declining autophagy, DNA damage, osteoprogenitor and osteocyte senescence, SASP, and lipid peroxidation may be primary culprits. Bone-extrinsic mechanisms, ie, age-related changes in other organs and tissues, such as the ovaries and the innate immune system are contributory. The effects of aging are independent of estrogen deficiency and mechanistically distinct. The extent to which these two mechanisms contribute to bone fragility in humans remains to be determined. Nonetheless, in view of the evidence that progressive bone loss in both sexes begins long before sex-steroid changes and evolutionary conserved aging mechanisms are inexorable, the diagnostic term “postmenopausal osteoporosis” is misleading and needs reappraisal. Several new classes of drugs targeting age-related mechanisms have the potential to treat more than one age-related disease, including osteoporosis, simultaneously. Skeletal involution, therefore, is not inexorable and may indeed be slowed or prevented by targeting aging mechanisms.
Acknowledgments
The author’s research is supported by the NIH (P01 AG013918, R01 AR056679), by the Department of Veterans Affairs (I01 BX001405), and by the University of Arkansas for Medical Sciences Tobacco Funds and Translational Research Institute (1UL1RR029884). The author thanks Katie H. Poe for help with the preparation of the manuscript; and his current and former co-workers (the late Michael A Parfitt, Robert Jilka, Rob Weinstein, Maria Almeida, Charles O’Brien, Haibo Zhao, Elena Ambrogini, Teresita Bellido, Stavroula Kousteni, Beata Lecka-Czernik, and Lilian Plotkin) for their invaluable contribution to the perspective and ideas discussed herein.
Footnotes
Presented as the Louis V. Avioli Lecture, ASBMR 2017 Annual Meeting, September 8–11, 2017, Denver, CO, USA.
Disclosures
SM is founder and equity holder of Radius Health, Inc.
References
- 1.Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374(9696): 1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Looker AC, Wahner HW, Dunn WL, et al. Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int. 1998;8(5): 468–89. [DOI] [PubMed] [Google Scholar]
- 3.Sambrook P, Cooper C. Osteoporosis. Lancet. 2006;367(9527): 2010–8. [DOI] [PubMed] [Google Scholar]
- 4.Popp AW, Zysset PK, Lippuner K. Rebound-associated vertebral fractures after discontinuation of denosumab-from clinic and biomechanics. Osteoporos Int. 2016;27(5):1917–21. [DOI] [PubMed] [Google Scholar]
- 5.Anastasilakis AD, Yavropoulou MP, Makras P, et al. Increased osteoclastogenesis in patients with vertebral fractures following discontinuation of denosumab treatment. Eur J Endocrinol. 2017;176(6):677–83. [DOI] [PubMed] [Google Scholar]
- 6.Parfitt AM. The two-stage concept of bone loss revisited. Triangle. 1992;31:99–110. [Google Scholar]
- 7.Parfitt AM. Bone effects of space flight: analysis by quantum concept of bone remodelling. Acta Astronaut. 1981; 8(9-10): 1083–90. [DOI] [PubMed] [Google Scholar]
- 8.Wakamatsu E, Sissons HA. The cancellous bone of the iliac crest. Calcif Tissue Res. 1969;4(2): 147–61. [DOI] [PubMed] [Google Scholar]
- 9.Wasserman SH, Barzel US. Osteoporosis: the state of the art in 1987: a review. Semin Nucl Med. 1987;17(4):283–92. [DOI] [PubMed] [Google Scholar]
- 10.Riggs BL, Khosla S, Melton U III. A unitary model for involutional osteoporosis: estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J Bone Miner Res. 1998;13:763–76. [DOI] [PubMed] [Google Scholar]
- 11.Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31 (3) :266–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rossouw JE, Anderson GL, Prentice RL, et al. Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–33. [DOI] [PubMed] [Google Scholar]
- 13.Droser ML, Gehling JG, Dzaugis ME, Kennedy MJ, Rice D, Allen MF. A new Ediacaran fossil with a novel sediment displacive life habit. J Paleontol. 2014;88(1):145–51. [Google Scholar]
- 14.Ricklefs RE. Insights from comparative analyses of aging in birds and mammals. Aging Cell. 2010;9(2):273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nussey DH, Froy H, Lemaitre JF, Gaillard JM, Austad SN. Senescence in natural populations of animals: widespread evidence and its implications for bio-gerontology. Ageing Res Rev. 2013;12(1): 214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21 (2):115–37. [DOI] [PubMed] [Google Scholar]
- 17.Almeida M, Laurent MR, Dubois V, et al. Estrogens and androgens in skeletal physiology and pathophysiology. Physiol Rev. 2017;97(1): 135–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wronski TJ, Pun S, Liang H. Effects of age, estrogen depletion, and parathyroid hormone treatment on vertebral cancellous wall width in female rats. Bone. 1999;25(4):465–8. [DOI] [PubMed] [Google Scholar]
- 19.Halloran BP, Ferguson VL, Simske SJ, Burghardt A, Venton LL, Majumdar S. Changes in bone structure and mass with advancing age in the male C57BL/6J mouse. J Bone Miner Res. 2002; 17(6):1044–50. [DOI] [PubMed] [Google Scholar]
- 20.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22(8):1197–207. [DOI] [PubMed] [Google Scholar]
- 21.Almeida M, Han L, Martin-Millan M, et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007;282(37):27285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piemontese M, Almeida M, Robling AG, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight. 2017;2( 17): e93771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parfitt AM, Villanueva AR, Foldes J, Rao DS. Relations between histologic indices of bone formation: implications for the pathogenesis of spinal osteoporosis. J Bone Miner Res. 1995; 10:466–73. [DOI] [PubMed] [Google Scholar]
- 24.Nirody JA, Cheng KP, Parrish RM, et al. Spatial distribution of intracortical porosity varies across age and sex. Bone. 2015; 75:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shanbhogue VV, Brixen K, Hansen S. Age- and sex-related changes in bone microarchitecture and estimated strength: a three-year prospective study using HRpQCT. J Bone Miner Res. 2016; 31 (8): 1541–9. [DOI] [PubMed] [Google Scholar]
- 26.Chesi A, Mitchell JA, Kalkwarf HJ, et al. A genomewide association study identifies two sex-specific loci, at SPTB and IZUMO3, influencing pediatric bone mineral density at multiple skeletal sites. J Bone Miner Res. 2017;32(6):1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura T, Imai Y, Matsumoto T, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130(5):811–23. [DOI] [PubMed] [Google Scholar]
- 28.Notini AJ, McManus JF, Moore A, et al. Osteoblast deletion of exon 3 of the androgen receptor gene results in trabecular bone loss in adult male mice. J Bone Miner Res. 2007;22(3):347–56. [DOI] [PubMed] [Google Scholar]
- 29.Chiang C, Chiu M, Moore AJ, et al. Mineralization and bone resorption are regulated by the androgen receptor in male mice. J Bone Miner Res. 2009;24(4):621–31. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Millan M, Almeida M, Ambrogini E, et al. The estrogen receptor-alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. 2010;24(2):323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinnesael M, Claessens F, Laurent M, et al. Androgen receptor (AR) in osteocytes is important for the maintenance of male skeletal integrity: evidence from targeted AR disruption in mouse osteocytes. J Bone Miner Res. 2012;27(12):2535–43. [DOI] [PubMed] [Google Scholar]
- 32.Määttä JA, Büki KG, Ivaska KK, et al. Inactivation of the androgen receptor in bone-forming cells leads to trabecular bone loss in adult female mice. Bonekey Rep. 2013;2:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Almeida M, Iyer S, Martin-Millan M, et al. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest. 2013;123(1 ):394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ucer S, Iyer S, Bartell SM, et al. The effects of androgens on murine cortical bone do not require AR or ERalpha signaling in osteoblasts and osteoclasts. J Bone Miner Res. 2015;30(7):1138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manolagas SC, O’Brien CA, Almeida M. The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol. 2013;9(12):699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanderschueren D, Laurent MR, Claessens F, et al. Sex steroid actions in male bone. Endocr Rev. 2014;35(6):906–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res. 2003;18(8):1404–18. [DOI] [PubMed] [Google Scholar]
- 38.Grassi F, Piacentini A, Cristino S, et al. Human osteoclasts express different CXC chemokines depending on cell culture substrate: molecular and immunocytochemical evidence of high levels of CXCL10 and CXCL12. Histochem Cell Biol. 2003;120(5): 391–400. [DOI] [PubMed] [Google Scholar]
- 39.Grassi F, Cristino S,Toneguzzi S, Piacentini A, Facchini A, Lisignoli G. CXCL12 chemokine up-regulates bone resorption and MMP-9 release by human osteoclasts: CXCL12 levels are increased in synovial and bone tissue of rheumatoid arthritis patients. J Cell Physiol. 2004;199(2):244–51. [DOI] [PubMed] [Google Scholar]
- 40.Wright LM, Maloney W, Yu X, Kindle L, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 binding to its chemokine receptor CXCR4 on precursor cells promotes the chemotactic recruitment, development and survival of human osteoclasts. Bone. 2005;36(5): 840–53. [DOI] [PubMed] [Google Scholar]
- 41.Kollet O, Dar A, Shivtiel S, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12(6):657–64. [DOI] [PubMed] [Google Scholar]
- 42.Zannettino AC, Farrugia AN, Kortesidis A, et al. Elevated serum levels of stromal-derived factor-1alpha are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res. 2005;65(5):1700–9. [DOI] [PubMed] [Google Scholar]
- 43.Gronthos S, Zannettino AC. The role of the chemokine CXCL12 in osteoclastogenesis. Trends Endocrinol Metab. 2007;18(3):108–13. [DOI] [PubMed] [Google Scholar]
- 44.Li J, Liao EY, Dai RC, Wei QY, Luo XH. Effects of 17 beta-estradiol on the expression of interstitial collagenases-8 and –13 (MMP-8 and MMP-13) and tissue inhibitor of metalloproteinase-1 (TIMP-1) in ovariectomized rat osteoblastic cells. J Mol Histol. 2004;35(8-9): 723–31. [DOI] [PubMed] [Google Scholar]
- 45.Schiltz C, Marty C, de Vernejoul MC, Geoffroy V. Inhibition of osteoblastic metalloproteinases in mice prevents bone loss induced by oestrogen deficiency. J Cell Biochem. 2008;104(5): 1803–17. [DOI] [PubMed] [Google Scholar]
- 46.Pivetta E, Scapolan M, Pecolo M, et al. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011;13(5):R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fu J, Li S, Feng R, et al. Multiple myeloma-derived MMP-13 mediates osteoclast fusogenesis and osteolytic disease. J Clin Invest. 2016;126(5):1759–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambliss KL, Wu Q, Oltmann S, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010; 120(7):2319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bartell SM, Han L, Kim HN, et al. Non-nuclear-initiated actions of the estrogen receptor protect cortical bone mass. Mol Endocrinol. 2013;27(4):649–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madak-Erdogan Z, Kim SH, Gong P, et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci Signal. 2016;9(429):ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ucer S, Iyer S, Kim HN, et al. The effects of aging and sex steroid deficiency on the murine skeleton are independent and mechanistically distinct. J Bone Miner Res. 2017;32(3):560–74. DOI: 10.1002/jbmr.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.López-Otiín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Almeida M, O’Brien CA. Basic biology of skeletal aging: role of stress response pathways. J Gerontol A Biol Sci Med Sci. 2013;68(10): 1197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. 2010;21 (6):369–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frost HM. In vivo osteocyte death. J Bone Joint Surg Am. 1960;42: 138–43. [PubMed] [Google Scholar]
- 56.Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends Biochem Sci. 2014;39(4):159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim H-N., Iyer S, Ring R, Almeida M. The role of FoxOs in bone health and disease. Curr Top Dev Biol. Forthcoming. Epub 2017 Dec 14 DOI: 10.1016/bs.ctdb.2017.10.004 [DOI] [PubMed] [Google Scholar]
- 58.Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21(11):2605–14. [DOI] [PubMed] [Google Scholar]
- 59.Iyer S, Ambrogini E, Bartell SM, et al. FOXOs attenuate bone formation by suppressing Wnt signaling. J Clin Invest. 2013;123(8): 3409–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manolagas SC. Wnt signaling and osteoporosis. Maturitas. 2014; 78(3):233–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990;85:632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ha H, Kwak HB, Lee SW, et al. Reactive oxygen species mediate RANK signaling in osteoclasts. Exp Cell Res. 2004;301(2):119–27. [DOI] [PubMed] [Google Scholar]
- 63.Lee NK, Choi YG, Baik JY, et al. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106(31:852–9. [DOI] [PubMed] [Google Scholar]
- 64.Bhatt NY, Kelley TW, Khramtsov VV, et al. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J Immunol. 2002;169(11):6427–34. [DOI] [PubMed] [Google Scholar]
- 65.Ishii KA, Fumoto T, Iwai K, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009;15(3):259–66. [DOI] [PubMed] [Google Scholar]
- 66.Bartell SM, Kim HN, Ambrogini E, et al. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nat Commun. 2014;5:3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schriner SE, Linford NJ, Martin GM, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–11. [DOI] [PubMed] [Google Scholar]
- 68.Lee HY, Choi CS, Birkenfeld AL, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 2010; 12(6):668–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dai DF, Chen T, Wanagat J, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9(4):536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mao P, Manczak M, Calkins MJ, et al. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet. 2012;21 (13):2973–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang C, Xu Q, Martin TD, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim HN, Chang J, Shao L, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 2017;16(4):693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farr JN, Fraser DG, Wang H, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res. 2016;31 (11):1920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jilka RL, O’Brien CA, Roberson PK, Bonewald LF, Weinstein RS, Manolagas SC. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates cortical porosity with age. J Bone Miner Res. 2014;29(1):103–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schmitt CA, Fridman JS, Yang M, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109(3):335–46. [DOI] [PubMed] [Google Scholar]
- 76.Crescenzi E, Palumbo G, Brady HJ. Bcl-2 activates a programme of premature senescence in human carcinoma cells. Biochem J. 2003;375(Pt 2):263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Emerton KB, Hu B, Woo AA, et al. Osteocyte apoptosis and control of bone resorption following ovariectomy in mice. Bone. 2010;46(3):577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kennedy OD, Herman BC, Laudier DM, Majeska RJ, Sun HB, Schaffler MB. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone. 2012;50(5):1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tomkinson A, Reeve J, Shaw RW, Noble BS. The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone. J Clin Endocrinol Metab. 1997;82(9):3128–35. [DOI] [PubMed] [Google Scholar]
- 80.Manolagas SC, Parfitt AM. For whom the bell tolls: distress signals from long-lived osteocytes and the pathogenesis of metabolic bone diseases. Bone. 2013;54(2):272–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013; 493(7432):338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaeberlein M, Rabinovitch PS, Martin GM. Healthy aging: the ultimate preventative medicine. Science. 2015;350(6265):1191–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ocampo A, Reddy P, Martinez-Redondo P, et al. In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell. 2016;167(7):1719–33.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016; 17(11):679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li J, Bonkowski MS, Moniot S, et al. A conserved NAD+ binding pocket that regulates protein-protein interactions during aging. Science. 2017;355(6331 ):1312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim HN, Han L, Iyer S, et al. Sirtuin 1 suppresses osteoclastogenesis by deacetylating FoxOs. Mol Endocrinol. 2015;29(10):1498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Edwards JR, Perrien DS, Heming N, et al. Silent information regulator (Sir)T1 inhibits NF-kappaB signaling to maintain normal skeletal remodeling. J Bone Miner Res. 2013;28(4):960–9. [DOI] [PubMed] [Google Scholar]
- 88.Simic P, Zainabadi K, Bell E, et al. SIRT1 regulates differentiation of mesenchymal stem cells by deacetylating beta-catenin. EMBO Mol Med. 2013;5(3):430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Iyer S, Han L, Bartell SM,et al. Sirtuin 1 (Sirtl) promotes cortical bone formation by preventing beta (b)-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J Biol Chem. 2014;289(35):24069–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zainabadi K, Liu CJ, Caldwell ALM, Guarente L. SIRT1 is a positive regulator of in vivo bone mass and a therapeutic target for osteoporosis. PLoS One. 2017;12(9):e0185236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeon OH, Kim C, Laberge RM, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med. 2017;23(6):775–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baar MP, Brandt RMC, Putavet DA, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169(1 ):132–47.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chang J, Wang Y, Shao L, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22(1):78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farr JN, Xu M, Weivoda MM, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. 2017;23(9): 1072–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tintut Y, Demer LL. Effects of bioactive lipids and lipoproteins on bone. Trends Endocrinol Metab. 2014;25(2):53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Makovey J, Chen JS, Hayward C, Williams FM, Sambrook PN. Association between serum cholesterol and bone mineral density. Bone. 2009;44(2):208–13. [DOI] [PubMed] [Google Scholar]
- 97.Steinberg D, Witztum JL. Oxidized low-density lipoprotein and atherosclerosis. Arterioscler Thromb Vase Biol. 2010;30(12):2311–6. [DOI] [PubMed] [Google Scholar]
- 98.Liu Y, Almeida M, Weinstein RS, O’Brien CA, Manolagas SC, Jilka RL. Skeletal inflammation and attenuation of Wnt signaling, Wnt ligand expression, and bone formation in atherosclerotic ApoE-null mice. Am J Physiol Endocrinol Metab. 2016;310(9): E762–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tintut Y, Morony S, Demer LL. Hyperlipidemia promotes osteoclastic potential of bone marrow cells ex vivo. Arterioscler Thromb Vase Biol. 2004;24(2):e6–10. [DOI] [PubMed] [Google Scholar]
- 100.Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. 2009; 284(40):27438–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Witztum JL, Lichtman AH. The influence of innate and adaptive immune responses on atherosclerosis. Annu Rev Pathol. 2014; 9:73–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009; 122(Pt 16):2801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Onal M, Piemontese M, Xiong J, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013;288(24): 17432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]