

Abstract
Although covalent inhibitors were used as therapeutics for more than a century, there was general resistance in the pharmaceutical industry for their further development attributing to safety concerns. This inclination has recently been reverted after the development of a large variety of covalent inhibitors for the intervention of human health conditions and the FDA approval of several covalent therapeutics for use in humans. Along with this exciting resurrection of an old drug discovery concept, this review surveys enzymes that can be targeted by covalent inhibitors for the treatment of human diseases. We focus on protein kinases, RAS proteins, and few other enzymes which have been studied extensively as targets for covalent inhibition, with the notion to address challenges in designing effective covalent drugs and provide suggestions in the area that are yet to be explored.
Keywords: Covalent Inhibitors, Protein kinases, RAS proteins, Cathepsin, Caspases, Acetyl Choline Esterase, Pancreatic Lipase, Methionine Amino Peptidase-2, Dipeptidyl Peptidase-4
Graphical Abstract

Introduction
Covalent Inhibitors are usually small molecules that bind to enzymes and inactivate them temporarily or permanently. In general, covalent inhibition is a two-step process.[1] First, an inhibitor reversibly associates with the target enzyme, by virtue of which the chemical warhead of the inhibitor comes within a close proximity of a targeted reactive amino acid residue of the enzyme. In the second step, reaction occurs between the two reactive entities in the inhibitor and the enzyme respectively to form a covalent bond. Reversible inhibitors differ from covalent inhibitors in that they do not involve the second step. A covalently conjugated inhibitor may undergo further chemical transformation(s) to get released from its target enzyme after a certain period of time. It may also permanently bind to the target leading to the enzyme to be locked in an inactive state. The use of small molecules as covalent inhibitors to target functionally critical enzymes in cells has found its implementation since late 19th century when Bayer started manufacturing aspirin as a painkiller and an anti-inflammatory drug.[2] Although it was in the drug market since the beginning of 20th century, its mechanism of action was not revealed until 1970s when Roth et al. showed that aspirin irreversibly inhibited cyclooxygenase-1 (COX-1), an enzyme that plays an instrumental role in the biosynthesis of prostaglandin.[3] When interacting with COX-1, aspirin acetylates an active site serine residue and thus inactivates COX-1 (Figures 1a, 1b, & 1c).[4] Besides aspirin, acetaminophen was also discovered in later 19th century and quickly introduced in the medical practice as a painkiller. Although its mechanism of action has not been clearly defined, the electron rich characteristics of acetaminophen makes it prone to oxidation, giving rise to quinone like structures. These quinone like molecules are susceptible to attack by nucleophilic protein/enzyme residues that may result in inhibition of proteins/enzymes.[5] Therefore, acetaminophen can also be considered as a covalent inhibitor. Another early covalent drug is penicillin. The serendipitous finding of penicillin as an antibiotic can be marked as one of the grandest discoveries in the history of drug discovery. Till date, a number of penicillin analogs have been approved for use in human patients. Together with penicillin, they all share a similar mechanism of action and contain a β-lactam as the chemical warhead. This β-lactam reacts with an active site serine residue in D-Ala-D-Ala transpeptidase that functions in the bacterial cell wall biosynthesis and therefore inactivates it, leading to the disruption of the bacterial cell wall biosynthesis and consequently the lysis of bacterial cells (Figures 1d, 1e, & 1f).[6] Other covalent antibiotics include some β-lactamase inhibitors such as avibactam[7], thienamycin[8], cephalosporin[9], etc. Although covalent inhibitors have long been found in applications of intervening human health conditions, the idea of covalent inhibition was not very popular until 1990s since many of the covalent drug metabolites were found to affect human health negatively, e.g. it has been found that cellular metabolites of acetaminophen are hepatotoxic.[10] During acetaminophen metabolism, it is oxidized by cytochrome P450 to highly reactive quinone intermediates (NAPQI and benzoquinone), which react with either glutathione (GSH) or the sulfhydryl group of cysteine residues present in proteins (Figure 1g) for covalent modifications.[5] Non-specific covalent drug-protein adducts potentially lead to unwanted immunogenic responses in patients. Against this drawback, several factors have revived interests of the pharmaceutical industry in the development of covalent drugs as therapeutic agents. First, there were successful covalent drugs like aspirin, penicillin etc. in the market. Second, not every covalent drug becomes toxic after undergoing metabolic activation. In addition, many natural products are covalent inhibitors. Covalent inhibitors also display advantages compared to reversible inhibitors such as having strong target affinity and prolonged acting lives in patients. As a result undesirable pharmacokinetic properties can often be tolerated as pharmacodynamics properties of these inhibitors outlast measurable inhibitor concentration in the plasma. Based on these observations, it has been concluded that if the reactivity of the warhead of a covalent inhibitor can be controlled, there should not be critical concerns of using it as a therapeutic. For these reasons, the development of covalent inhibitors has thrived in the past decade. A brief history about the covalent inhibitor discovery is summarized in Figure 2.[1, 2, 5]
Figure 1.

Early covalent drugs. a) The action mechanism of aspirin; b) COX-1 active site serine and its covalent adduct after its reaction with c) a bromo derivative of aspirin; d) The action mechanism of penicillin; e) Co-crystal structure of DD-transpeptidase bound covalently with ampicillin (PDB entry: 5HL9) f) The amphicillin-serine complex in the active site of DD-transpeptidase. g) The action mechanism of acetaminophen.
Figure 2.
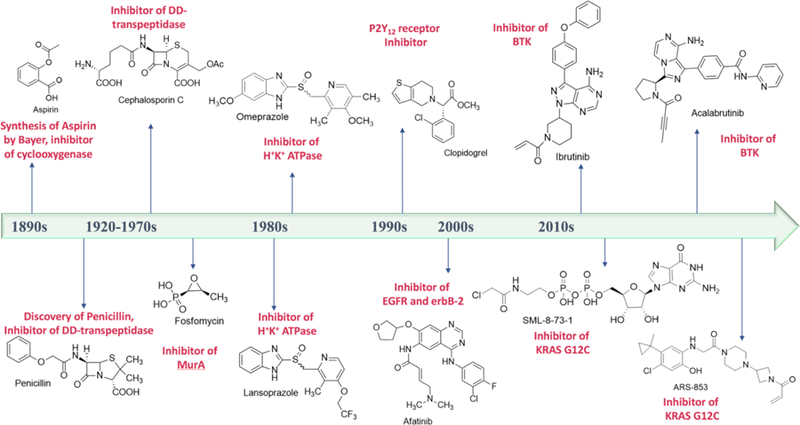
A brief timeline of covalent drug discovery. Structures of covalent inhibitors are provided along with the enzymes/proteins they inhibit.
In general, covalent inhibitors contain electrophiles that react with nucleophilic residues in enzymes. Till date, molecules with a variety of electrophilic warheads including epoxide, aziridine, ester, ketone, α, β-unsaturated carbonyl, nitrile, etc. have been identified as covalent inhibitors.[11] A new type of inhibitors known as “sulfur tethers” have also caught attention due to their ability to covalently conjugate cysteines in enzymes. In order to design a covalent inhibitor for a given enzyme target, three steps are typically involved. First, the structural analysis of the target provides crucial information regarding which nucleophile (e.g. cysteine, serine) is present at or around a potential binding pocket. The nucleophile needs to be unique in that protein family, otherwise there will be low selectivity. Second, a reversible inhibitor with some potency (μM to mM IC50) is found for which the binding mode and interactions are known. This inhibitor can come from archived inhibitors that were developed for related enzymes. Finally, an electrophilic ‘warhead’ is positioned in a selected reversible inhibitor to react specifically with the chosen nucleophile in the enzyme target. Isosteric replacement and analogue synthesis are typically used to obtain an active covalent inhibitor candidate. Although a co-crystal structure of an initial non-covalent scaffold bound to an enzyme target will provide more insights for designing a covalent inhibitor, it is not absolutely essential. In many cases, docking the initial non-covalent scaffold to a homology model has been proved to be sufficient. Structure-activity relationship studies can be further applied to optimize a lead covalent inhibitor for the identification of more potent compounds. Applications of computational chemistry in the designing of covalent inhibitors has been recently reviewed by Awoonor-Williams et al.[12] In the following sections, we will describe advances in targeted enzyme families including protein kinases and RAS proteins for the development of covalent inhibitors and briefly touch on other enzymes. Given the scope of this review, we apologize for not being able to cite other critical work on the covalent drug field.
Covalent Inhibitors of Protein Kinases
Although conceptually eye-catching, it is hard to design a covalent inhibitor because of the difficulty to maintain a right balance of selectivity, reactivity, and efficacy. This is even more critical when targeting protein kinases. Kinases are enzymes that transfer the γ-phosphate group from ATP to small molecules or amino acid residues in proteins. So far, 518 human kinases have been discovered, not to mention that over 900 genes in humans potentially encode kinases.[13] Although highly diversified in their amino acid sequences, the three-dimensional structures of human kinases are strikingly similar especially in the catalytically active ATP binding domain. Careful optimization of non-covalent binding affinity and reactivity of electrophiles are much needed for designing covalent drugs for protein kinases as there are very subtle changes in the active site environment across the kinome. On the basis of previously mentioned strategies of drug design, attempts have been made to design targeted covalent inhibitors (TCIs) for kinases. Some of these efforts are summarized below.
Irreversible Covalent Inhibitors of Epidermal growth factor receptor (EGFR)
EGFR belongs to the receptor tyrosine kinase family which catalyzes the protein tyrosine phosphorylation reaction to control signaling transduction. EGFR is a cell-surface protein which binds to its natural ligand, Epidermal growth factor (EGF) to induce tyrosine auto-phosphorylation and signals cell proliferation.[14] Specific mutations in this gene is linked to non-small cell lung cancer (NSCLC).[15] The most common are in-frame deletion of exon 19 (DelE746-A750) and single point mutation L858R in exon 21. They are called ‘activating mutations’ as they lead to ligand independent tyrosine kinase activity.[15] This elevated basal level kinase activity can be inhibited by ATP competitive, reversible drugs like gefitinib[16] and erlotinib[17] (Figure 3a). However, another active site mutation T790M reduces their efficacy by at least 50%.[18] It is believed that this mutation reduces the Km values for ATP and its analogues, therefore increasing the total amount of an inhibitor needed to inhibit EGFR T790M efficiently.
Figure 3.

a) Two reversible EGFR inhibitors, Gefitinib and Erlotinib; b) First generation irreversible covalent inhibitors of EGFR, PD168393, PF00299804 and EKB569. All contain an acrylamide moiety, highlighted in a box, as an electrophilic warhead. c) Neratinib and its complex with EGFR T790M (PDB entry: 2JIV). Gatekeeper residue (Met790) is shown in red. d) Afatinib and its complex with EGFR T790M (PDB entry: 4G5P).
The development of covalent inhibitors for the T790M mutant started in early 1990’s at Parke-Davis and Wyeth (now Pfizer).[19] Several irreversible inhibitors like PD168393,[20] PF00299804 (dacomitinib),[21] EKB569 (pelitinib)[22] etc. (Figure 3b) were reported but none succeeded to overcome drug efficacy problems related to the T790M mutation in long run. Unlike erlotinib and gefitinib, these covalent EGFR inhibitors contain the 4-anilinoquinazoline scaffold which is equipped with an electrophilic warhead, most of the time the acrylamide moiety that undergoes Michael addition with the conserved C797 in the EGFR active site. The covalent attachment increases the drug action time to inhibit EGFR T790M.[21] Neratinib (Figure 3c) is another covalent EGFR inhibitor that was thoroughly investigated for counteracting the T790M mutation. Albeit great promises, Neratinib exhibits low potency both in TCI-naïve patients and those who have taken TCIs before, probably because of insufficient bioavailability from diarrhea-imposed dose limit.[23] It is interesting to know that afatinib (Figure 3d), an ErbB family blocker, was the first FDA approved (2013) covalent EGFR inhibitor. Although afatinib alone elongated the progression free survival (PFS) time almost three times compared to those treated with placebo, a combination therapy with Cetuximab, a human–murine monoclonal antibody, produced far more convincing result, even in mice with L858R/T790M erlotinib-resistant tumors. Afatinib binds covalently to the ATP binding pocket of EGFR and hinders its tyrosine kinase activity partially. On the other hand, Cetuximab induces receptor degradation by blocking ligand binding but not effective enough to shut down ligand independent activity of receptors fully. Only in presence of both agents, the depletion level of EGFR is so high that it compels mutant EGFRs to reduce the amount of signaling below a certain threshold needed for cell survival.[24] Besides inhibiting the T790M mutant, traditional quinazoline-based inhibitors display similar inhibition on wild type EGFR, causing side effects like skin rash, diarrhea, etc.[23]
To alleviate side effects, Zhou et. al. developed a third-generation covalent inhibitor, WZ-4002 (Figure 4a), which shows high selectivity against EGFR T790M.[25] The crystal structure (PDB entry: 3IKA) showed that WZ-4002 approaches C797 from a unique angel. It is intriguing that the authors have described an interaction between chlorine on the pyrimidine ring and -SMe group of methionine that is annotated as ‘halogen bond’.[25, 26] It was claimed that this interaction makes the inhibitor more selective towards EGFR(T790M). In November 2015, US FDA approved TAGRISSO™ or osimertinib (formerly known as AZD9291 shown in Figure 4b) developed by AstraZeneca to target EGFR for advance NSCLC. The preclinical studies indicated IC50 value of 12 nM against the L858R mutant and 1 nM against the L858R/T790M mutant of EGFR. This drug showed approximately 200-fold greater potency against EGFR(L858R/T790M) than wild type EGFR. In vitro cellular phosphorylation along with phenotypic studies and biochemical profiling indicate that osimertinib is highly potent against EGFRm+ (EGFR containing ‘activating mutations’) and T790M positive EGFR mutants with a significantly broad margin of selectivity against wild type EGFR.[27] Although Cross et al. modelled osimertinib in the T790M mutant of EGFR (PDB entry: 4ZAU) and showed that C797 can form an irreversible adduct with it, an actual co-crystal structure of wild type EGFR with osimertinib does not exhibit a covalent linkage between C797 and the acrylamide moiety.[28] Yet, the C797 is positioned near enough to the electrophilic warhead so that slight movement of the loop containing C797 could induce the covalent linkage formation. Similar to previous generation drugs, secondary acquired resistance was reported for Osimertinib, usually observed after 8 or 9 months of treatment.[29]After analyzing the cell-free plasma DNA (cfDNA) collected from patients suffering from osimertinib-resistant advanced lung cancer with EGFRm+, a new C797S mutation was identified.[30] Although the C797S mutation is the most common mechanism of drug resistance,[31] there are other mechanisms as well, for example, ErbB2 receptor tyrosine kinase 2 gene (HER2) amplification,[32] MNNG HOS Transforming gene (MET) amplification,[33] an acquired BRAF V600E mutation,[34] EGFR G796D mutation,[35] etc.
Figure 4.

Third generation irreversible covalent EGFR inhibitors. a) WZ4002 and it complex with EGFR T790M (PDB entry: 3IKA); b) Osimertinib and its complex with wild type EGFR (PDB entry: 4ZAU); c) PF-06459988 and its complex with EGFR L858R/T790M (PDB entry: 5GH7); d) Other third generation irreversible covalent EGFR inhibitors.
There are several other third generation EGFR inhibitors under development in pharmaceutical companies including PF-06459988 (developed by Pfizer; Figure 4c),[36] Nazartinib (EFG 816; developed by Novartis),[37] Naquotinib (ASP8273; developed by Astellas),[38] Olmutinib (HM61713; developed by Hanmi),[39] Avitinib (AC 0010; developed by ACEA Bioscience),[40] etc. Most of these molecules are shown in Figure 4d. Highly potent (IC50 = 8 nM) inhibitor against the osimertinib resistant L858R/T790M/C797S mutant has also been developed.[41] It is interesting to note that rociletinib,[42] another 3rd generation EGFR irreversible covalent inhibitor for EGFRm+ developed by Clovis Oncology, exhibited good oral bioavailability (65%) as well as antitumor activity comparable to erlotinib and afatinib while having more than 20-fold more selectivity over WT EGFR. Unfortunately its development was terminated due to some inconsistency in the published data. Out of 130 patients who were enrolled for phase I/II trial, the response rate, according to RECIST 1.1 (Response Evaluation Criteria in Solid Tumors[43]) was 59% [95% confidence interval (CI) 45–73].[44] 20 of 92 patients, who were treated with Rociletinib, suffered from hyperglycemia due to undesirable inhibition of the type I insulin-like growth factor receptor (IGF-IR) and insulin receptor kinases by its metabolite, which could be suppressed by dose reduction. However, on November 2015, Clovis Oncology announced that the preliminary response rate of 59% was based on mainly unconfirmed responses and the confirmed response rate dropped down to 34% in 170 patients treated with an active dose of 625 mg b.i.d. (twice a day) and 28% among 79 patients with an active dose of 500 mg b.i.d.[45]
Irreversible Covalent Inhibitors of Bruton’s tyrosine kinase (BTK)
BTK is a non-receptor protein tyrosine kinase that belongs to the Tec family. It plays a key role in the maturation of B cells. Given this unique role, targeting BTK is an established treatment for B cell lymphoma and leukemia.[46] Mutations of BTK are also implicated in X-linked agammaglobulinemia (XLA), an inherited immunodeficiency disease.[47] XLA patients show defects strictly in their B cells but not in other immune cells, which makes BTK a potential target for treating XLA.[48] Ibrutinib (formerly known as PCI32765 shown in Figure 5a), a covalent BTK inhibitor, was first developed by Pharmacyclis LLC and approved by FDA as a second covalent kinase inhibitor drug. It was approved for the treatment of chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), and Waldenstrom’s macroglobulinemia.[49] Ibrutinib inhibits BTK by diminishing auto-phosphorylation at its Y223 residue. Its acrylamide moiety reacts with C481 to form a covalent bond in the BTK kinase domain, which locks BTK in an allosteric inhibitory state (Figure 5b). Although it succeeds in treating B cell lymphoma and leukemia, ibrutinib does lead to side effects such as bleeding, rash, diarrhea, and atrial fibrillation due to off-target binding to EGFR and other Tec family proteins[50] which also possess a similar cysteine residue at the active site.[51] Resistance to ibrutinib was also noticed.[52] To overcome side effects of ibrutinib, several second-generation BTK inhibitors have been developed.[53] One of these inhibitors, acalabrutinib (ACP-196, Figure 5c) exhibits high potency, rapid oral absorption, a short half-life as well as reduced binding to EGFR or other Tec family proteins.[54] Combination of acalabrutinib with other agents (for example, obinutuzumab, an anti-CD20 monoclonal antibody) looks promising for the treatment of B cell malignancies.[55] Hong Wu and co-workers developed CHMFL-BTK-11, another covalent BTK inhibitor, which suppress the activation of B-cells effectively and also blocks the secretion of different cytokines like IgG1, IgG2, IL-6 etc. It can be potentially used as a drug for rheumatoid arthritis.[56] Other second-generation inhibitors like ONO/GS-4059[57] and BGB-3111[53], PRN1008[58], CC-292[59] (Figure 5d) have also been developed. These inhibitors are in their early clinical testing stage.
Figure 5.

Covalent BTK inhibitors. a) Ibrutinib; b) A pyrrolopyrimidine precursor of ibrutinib and its complex with BTK (PDB entry: 3GEN); c) Acalabrutinib; d) Second generation irreversible covalent BTK inhibitors.
Irreversible Covalent Inhibitors of p90 ribosomal S6 kinases (RSKs)
The RSK family is a group of highly conserved Ser/Thr kinases that promote cell proliferation, growth, motility and survival. As they are almost exclusively activated downstream of extracellular signal-regulated kinases 1 and 2 (ERK1/2), therapeutic intervention by RSK inhibition is less likely to produce severe side effects such as those observed following inhibition of the upstream master regulators Raf (rapidly accelerated fibrosarcoma), MEK or MAPK (Mitogen activated protein kinase), and ERK1/2.[60] The RSK family constitutes of four closely related paralogs (RSK 1–4).[61] Structurally, RSKs consist of two kinase domains, a C-terminal kinase domain (CTD) that belongs to the Ca2+/calmodulin-dependent protein kinase family and a N-terminal kinase domain (NTD) that belongs to the AGC kinase group [mainly consists of protein kinase A (PKA), protein kinase G (PKG) and protein kinase C (PKC)]. The two domains are connected by a short linker with several regulatory phosphorylation cites. RSKs are well documented for phosphorylating proteins related to Na+/H+ exchanger NHE1,[62] the tumor suppressor kinase LKB1,[63] the translation initiation factor e1F4B,[64] etc. Abnormal RSK activity has been implicated in tumor cell invasion,[65] colonic epithelia,[66] and endothelial dysfunction.[42] A sequence alignment of the human kinome has led to the identification of a poorly conserved non-catalytic cysteine in the ATP binding site in RSK1–4 and seven other kinases.[67] It was also observed that RSK 1, 2, and 4 have a ‘gatekeeper’ threonine in the ATP binding pocket.[68] Inspired by the co-crystal structure of a pyrazolopyrimidine molecule (PP1 in Figure 6a) bound to hematopoietic cell kinase (HCK) (Figure 6c), a SRC-family kinase with a ‘gatekeeper’ threonine, a fluoromethylketone (FMK)-containing PP1 analog (Figure 6b) was designed to target RSKs.[69] Although FMK was supposed to inhibit CTD of RSK1, RSK2, and RSK4, both biochemical (IC50 = 15 nM) and cellular assays (EC50 = 300 nM) indicated that it only inhibits RSK2 significantly.[70] With the aid of computer simulation, it was expected that the poorly conserved C436 in RSK2 would take the similar position as V281 in HCK and react with FMK (Figure 6c). When the ‘gatekeeper’ threonine residue T493 in RSK2 is mutated to methionine or C436 is muted to valine, it confers resistance. Quite unexpectedly, FMK was extremely potent against a large panel of kinases.[71] In 2018, Gothelf and Nissen et. al. demonstrated that dimethyl fumarate acts as an allosteric covalent inhibitor of RSK2/mitogen- and stress-activated kinases (MSKs) and further studies are being pursued to improve the selectivity towards RSK2.[72] To the best of our knowledge, there is no other irreversible inhibitor of RSK1 and RSK4 that has been developed yet.
Figure 6.
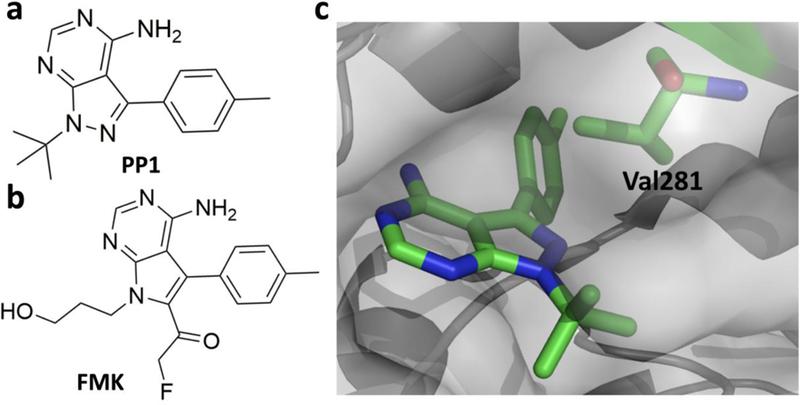
a) PP1, a Src family kinase inhibitor; b) FMK, an irreversible covalent inhibitor of RSK2; c) Co-crystal structure of the SRc family kinase HCK bound to PP1 (PDB entry: 1QCF) with N2 properly oriented to Val 281 (shown in green) which corresponds to C436 in RSK2.
Irreversible Covalent Inhibitors of other kinases
In accordance with the increased abundance of structural information of different kinase families, there has been an exponential growth of interest in developing irreversible covalent kinase inhibitors, although the scope of research has been confined mainly in receptor tyrosine kinases. A review by Zhao and Bourne[73] has summarized small molecule covalent inhibitors against kinases. Not surprisingly, EGFR inhibitors top the list. However, attempts have also been made to develop covalent inhibitors against other kinases. The fibroblast growth factor receptor (FGFR) is a kinase essential for cell proliferation and differentiation. It has been implicated in the development of colorectal, lung, and renal cell cancers and hepatocellular carcinoma.[74] Human FGFRs consist of four members (FGFR1–4) which can bind to a diverse set of 18 FGF ligands. Zhou et al. have developed a first generation covalent FGFR inhibitor FIIN-1 (Figure 7a) that inhibits FGFR1–4 selectively against a library of 402 different kinases, along with some off-target kinases like Flt1, Flt4, and VEGFR.[75] FIIN-1 features an acrylamide warhead which targets a conserved cysteine residue in all four FGFRs and confers EC50 values against FGFR1–3 in a range (~10 nM) comparable to BGJ398[76] and AZD4547[77], two potent reversible inhibitors of FGFRs. As mutations at the gatekeeper valine residue (V550 in FGFR4) in FGFRs induced strong resistance to AZD4547 and FIIN-1, Zhou et al. developed FIIN-2 and FIIN-3 to overcome these mutations (Figure 7a).[78] FIIN-3 shows potent inhibition against EGFR as well. Using structure-guided design, Brameld et al. developed another irreversible FGFR inhibitor PRN1371 (Figure 7b). This inhibitor is undergoing phase 1 clinical trial for the treatment of solid tumors.[79] Recently, Novartis developed FGF401, an aldehyde based covalent inhibitor of FGFR4 which can target a different non-conserved cysteine, Cys 552 of FGFR4.[80]
Figure 7.

a) The FIIN series inhibitors of FGFR and the co-crystal structure of FIIN3 complexed with FGFR V550L (PDB entry: 4R6V); b) PRN1371, a covalent FGFR inhibitor; c) Non-covalent CDK2 inhibitor NU6102, its corresponding covalent CDK2 inhibitor NU6300, and a co-crystal structure of CDK2 bound covalently with NU6300 (PDB entry: 5CYI) via K89; d) Covalent CDK7 inhibitor THZ1, THZ351, non-covalent CDK12 and CDK13 inhibitor THZ531, and a co-crystal structure of CDK12 bound covalently with THZ531 (PDB entry: 5ACB); e) PI3Kα inhibitor CNX-1351.
Covalent inhibitors that target cyclin-dependent kinases (CDK)[81] have also been reported recently. Anscombe et al. have developed NU6300 (Figure 7c), an irreversible covalent inhibitor of CDK2, from a known reversible inhibitor NU6102[82] by positioning a unique vinyl sulfone electrophile proximal to the residue K89 which is situated just outside the ATP binding cleft.[83] It achieves high selectivity as K89 is not conserved in closely related kinases. Kwiatkowski et al. have been able to target a remote cysteine residue located outside of the canonical kinase domain of CDK7 by THZ1 (Figure 7d) that bears a cysteine-reactive acrylamide moiety.[84] Sequence alignment, crystal structure and biochemical assays pointed out that CDK12 and CDK13 also possess C-terminal cysteines that can be modified by THZ1. Derived from THZ1, Zhang et. al. developed THZ531, a potent covalent inhibitor of CDK12 and CDK13 (Figure 7d) that has been demonstrated not only to reduce expression of genes associated with DNA damage response and super-enhancer-associated transcription factors, but also to cause substantial loss of elongating and hyperphosphorylated RNA polymerase II.[85] Another covalent kinase inhibitor worth mentioning is CNX-1351 (Figure 7e), which inhibits lipid phosphoinositide 3-kinase-α (PI3Kα) with EC50 < 100 nM. It targets the residue C862 that is unique to α isoform, thus providing specificity over PI3Kβ, -γ, and -δ isoforms.[86]
Reversible Covalent Inhibitors of Protein Kinases
Given long term effects of locking a kinase permanently in an inactive state are hard to predict, the development of inhibitors that covalently but reversibly bind to the kinase undoubtedly bears significant merits. It was previously known that the Michael addition reaction between a thiol and a 2-cyanoacrylate is reversible at neutral pH.[87] Based on this observation, Taunton and co-workers have successfully developed highly reactive 2-cyanoacrylate-based reversible covalent inhibitors of RSK2 (Figure 8a).[71] Based on the analysis of the binding mode of FMK, the pyrrolopyrimidine scaffold was modified so that C436 of the CTD of RSK2 can easily reach to the electrophile to form a reversible Michael addition adduct. Several inhibitors have been made. These include CN-NHiPr and CN-NHtBu that links covalently to C436 in its co-crystal form with RSK2. Both inhibitors target only C436 of RSK2 although there are many solvent exposed cysteine residues on the kinase. Furthermore, water soluble thiols like glutathione, even at millimolar concentrations, do not affect the inhibitory action of both reversible covalent inhibitors on RSK2. On the contrary, a mutation at C436 easily decimates the inhibitory effect from both inhibitors to the enzyme.[88] Reversible covalent inhibitors have also been developed for BTK. Bradshaw and co-workers explored the feasibility of designing reversible covalent inhibitors by introducing the cyanoacrylamide moiety in lieu of acrylamide and keeping the core structure of ibrutinib intact. They noticed that having a unique tert-butyl group as a β-substituent not only elongates the residual time of the inhibitor, but also makes it more durable under physiological conditions. The Cα proton, which has to be abstracted during the elimination of C481 thiol (Figure 8b) in the backward reaction, is not oriented properly inside the active site in terms of stereoelectronics to have enough kinetic and thermodynamic acidity. Also, both the capping tert-butyl group and the linker piperidine shields the Cα proton from any nearby base in the active site.[89] Therefore, a covalent complex form is strongly favored. Although a reversible covalent inhibitor with residual time comparable to irreversible inhibitors has several advantages over the later including less toxicity attributing to less permanent off-target adducts, this strategy cannot be generalized as the branched alkyl capping groups are on high steric demand which may not be suitable for relatively unhindered and solvent-exposed cysteines.
Figure 8.
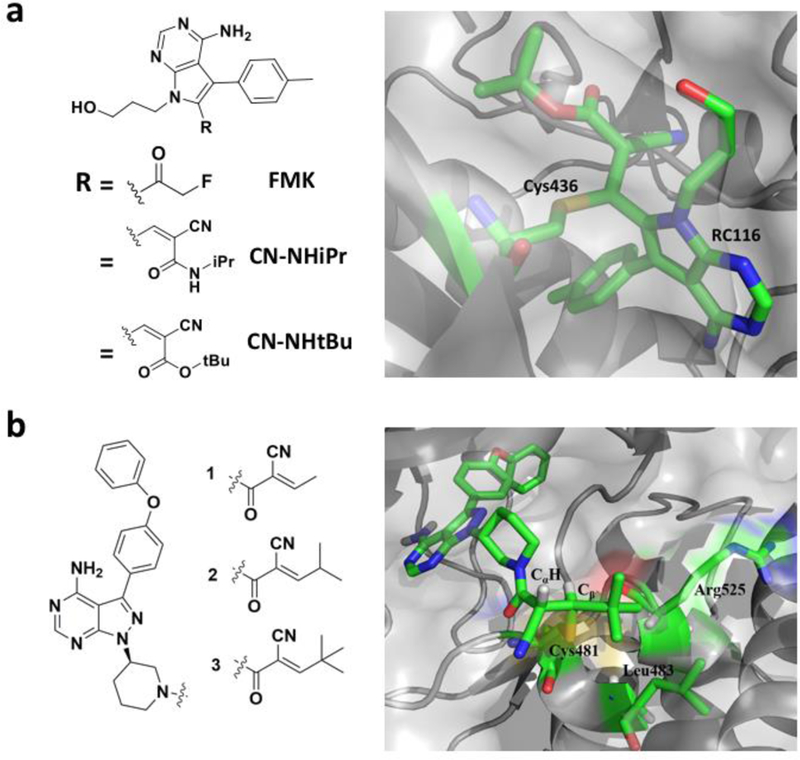
a) Reversible covalent inhibitors CN-NHiPr and CN-NHtBu of RSK2 that were derived from FMN and a co-crystal structure of RSK2 bound covalently with CN-NHtBu at C436 (PDB entry: 4D9U); b) Cyanoarylamide-based reversible covalent inhibitors of BTK and a co-crystal structure of BTK bound covalently with compound 3 at C481.
Covalent Inhibitors for Proteins of the RAS Family
For three decades since their discovery, RAS oncogenes are probably the most studied genes in cancer research. RAS proteins are also among the most mutated oncogenes ever known. Till date no efficient treatment is available for RAS oncogenic mutations. However, recent reports of the development of potent small covalent inhibitors of RAS proteins have revived the hope that these so called ‘undruggable’ proteins can be finally tuned to be druggable. RAS proteins are a family of related small GTPases, which are expressed in all animal cell lineages and organs. These proteins are involved in transmitting signals within cells. Mutations may permanently activate RAS superfamily proteins, leading to unintended and overactive signaling inside the cell, even if there are no incoming signals. As these signals result in cell growth and division, overactive RAS signaling can ultimately lead to cancer.[90] In spite of efficacious developments in the ATP dependent protein kinases family, very similar strategies with respect to RAS proteins have been unsuccessful. This is mainly due to the high picomolar binding interactions of RAS with GTP. In addition, the smooth spatial arrangements of RAS protein structures disappointed the search for small molecule covalent inhibitors and making RAS proteins as ‘undruggable’.[91] There are three RAS genes in humans including HRAS (Harvey rat sarcoma viral oncogene homolog), KRAS (Kristen rat sarcoma viral oncogene homolog), and NRAS (neuroblastoma RAS viral oncogene homolog). They are the most common oncogenes in human cancer.[92] In total, there are four RAS isoforms including HRAS, NRAS, KRAS4A and KRAS4B due to alternative splicing of KRAS during transcription.[93] The N-terminal domains of all four RAS proteins share almost 92–98% sequence similarity. The C-terminal regions that consist of 23–24 residues vary dramatically and therefore are called hypervariable regions (HVRs). HVRs contain a C-terminal CAAX box which for all four RAS proteins undergoes farnesyltransferase-catalyzed prenylation at the cysteine residue.[92] This is also the key post translational modification of RAS proteins for RAS activation.[94] Much effort in the previous anti-RAS drug discovery was the development of strategies that prevented RAS activation to associate with plasma membrane. This led to exhaustive effort in the 1990s to build libraries of farnesyltransferase inhibitors (FTIs).[95] Though there were some promising results in the preclinical studies, the results of clinical experiments with FTIs have not been encouraging. Some of these inhibitors including lonafarnib and tipifarnib work well for HRAS but not for KRAS and NRAS. This dissapointing outcome had dampened the interest of inhibiting RAS proteins based on the intervention of plasma membrane association, though lonafarnib is still under developement as a drug for the genetic disorder progeria.[94] Very recently Shokat et al. reported aboout a series of substrates for farnesyltransferase that can stop the alternative prenylation by geranylgeranyltransferase and mislocalize oncogenic KRAS in cells.[96] The development of other strategies for RAS inhibition is essential.
Oncogenic mutations in RAS proteins
The RAS proeins share a common mechanism of activation and downstream signalling which have been well charectarized. There are four main regions surrounding the nucleotide binding pocket of RAS proteins: the phosphate binding loop (p loop, residues 10–17), switch I (residues 30–38), switch II (reidues 60–67) and the base binding loops (residues 116–120 and 145–147). All these regions contribute to nucleotide dependent interactions. The arrangements of these regions play critical roles in RAS oncogenic mutations.[92] The most common oncogenic mutations occurring in RAS proteins are at the residues 12 and 13 (belonging to the p loop) and residue 61 (switch II). Key residues for mutations in KRAS and NRAS are residue 12 and residue 61 respectively whereas for HRAS it is almost equally distributed between residue 12 and 61. KRAS mutations are the most common overall and occur mostly in pancreatic, colorectal and lung cancer. HRAS mutations occur highly in bladder cancer and mutations of NRAS are mostly associated with melanoma and myeloid leukemia.[92] Most of the current drug discovery strategies are based on targeting these mutations in RAS subfamily proteins. One method of achieving mutant specificity is through covalent attachment of an inhibitor to the mutated residue itself.[92]
Covalent Inhibitors for KRAS mutants
Mutations of the two KRAS isoforms occur in 60% of pancreatic, 34% of colorectal, and 16% of lung cancers.[97] There are three most common sites for mutation in KRAS (residues 12, 13, and 61) which show minimized GTPase activating protein (GAP)-dependent GTPase activity. Except the mutation G12C, other mutations at residues 12,13, and 61 decrease affinity of RAS to its downstream kinase RAF (rapidly accelerated fibrosarcoma).[98] Some of these mutants are G12A, G12D, G13D, Q61L and G12V.[92] Being one of the most common KRAS mutations leading to cancer, the G12C mutation is particularly interesting in the sense that it has an active non-native cysteine residue that can be easily targeted for covalent inhibition without affecting wild-type KRAS. Recently, Shokat and colleagues reported a set of cysteine-reactive inhibitors that interact with the KRAS G12C mutant and subsequently form covalent adducts with G12C.[98] Using a disulfide tethering approach, they first screened a library of disulfide compounds to identify a series of compounds that selectively bind to KRAS G12C. In their experiments, fragments 2E07 and 6H05 showed greatest degrees of modification (Figure 9a). They also screened carbon based electrophiles such as acrylamides and vinyl sulfonamides that form irreversible covalent bonds with the G12C residue (Figures 9c & 9e). Crystal structures of KRAS G12C complexed with these identified molecules showed that these compounds bind to a newly exposed pocket on KRAS G12C, below the effector-binding switch-II region. Binding of these compounds to KRAS G12C leads to the nucleotide preference to GDP over GTP and thus results in blocked KRAS signaling. When the structures of KRAS G12C complexed with two kinds of covalent inhibitors are compared, it can be stated that disulfide compounds induce a small shift in the switch-II region leading to slight conformational change in switch-I. On the other hand, carbon based electrophiles showed more prominent effect by displacing the switch-II region which in turn disorder switch-I to a greater extent.[99] A series of such compound along with some of their co-crystal structures with KRAS G12C are shown in Figures 9b & 9d.[98]
Figure 9.

Tethering and electrophilic compounds that selectively bind to oncogenic KRAS G12C. a) Tethering compounds that binds covalently to KRAS G12C; b) Crystal structure of the KRAS G12C complex with compound 6 (PDB entry: 4LUC); c) Electrophiles with the vinyl sulphonamide moiety that bind covalently to KRAS G12C; d) Crystal structure of the KRAS G12C complex with compound 12; e) Electrophiles with the acrylamide moiety that bind covalently to KRAS G12C (PDB entry: 4LYF).
Gray et al. recently recognized that a GTP mimic with a reactive functional group with the aim to target the guanine nucleotide (GN) binding pocket because the natural content of this pocket dictate the signaling state of KRAS.[100] Using this structure based design, they synthesized SML-8–73-1 (Figure 10a), a GDP analogue with an electrophilic α-chloroacetamide moiety. With help from mass spectrometry analysis, they demonstrated SML-8–73-1 binds covalently at G12C. Hydrogen-exchange mass spectrometry analysis showed that this compound stabilizes the inactive form of KRAS G12C, leading to low affinity for effector proteins. Hunter et al. solved the crystal structure of KRAS G12C bound with this covalent inhibitor (PDB entry: 4NMM).[99] They showed that SML-8–73-1binds to KRAS G12C in a manner similar to that of GDP, forming a confirmation that is predicted to result in a non-productive state for activating downstream effectors (Figure 10b).[100] Due to the presence of two negatively charged phosphate groups, SML-8–73-1 cannot pass through the cell membrane. So, it was modified by capping the β-phosphate unit with an alanine ester phosphoramidate (SML-10–70-1) (Figure 10c) which penetrates through cell membranes and binds covalently with KRAS G12C.[32] Patrecelli et al. reported the compound ARS-853 which is a modified version of compound 12 in the previous Shokat’s study (Figure 10d).[101] ARS-853 shows robust cellular activity against KRAS G12C in the low micromolar range. Structural and iterative SAR studies based on compound 12 and some of its modified forms showed that the 5-chloro position on the benzene ring is very important for binding. Based on this information, multiple compound 12 derivatives were synthesized and screened against KRAS G12C to reveal ARS-853 as the most potent inhibitor. While compound 12 has an IC50 value around 100 µM, the IC50 value of the much-improved ARS-853 is about 1.6 µM. The structure of KRAS G12C complexed with ARS-853 is shown in Figure 10e.[101] Another very recent report by Patrecelli et al. demonstrated the structure based design and identification of ARS-1620 (Figure 10d) which is a covalent mechanism based inhibitor for KRAS G12C with high potency and selectivity. This compound can rapidly achieve sustained in vivo target occupancy to induce tumor regression. This is an important step toward proving that KRAS can be selectively targeted in vivo by ARS-1620 and also have scope for promising therapeutic potential.[102]
Figure 10.

Some other covalent inhibitors of KRAS G12C. a. SML-8–73-1; b. The structure of KRAS G12C bound with SML-8–73-1 (PDB entry: 4NMM); c. SML-10–70-1; d. ARS-853; e. The structure of KRAS G12C bound with ARS-853 (PDB entry: 5F2E).
New concepts are always emerging in discovering and designing covalent inhibitors for KRAS on a proteome wide scale. Current studies by Hansen et al. showed that ARS-853 and ARS-1620 gets activated by KRAS mediated catalysis of the chemical reaction in human KRAS G12C. This biochemical mechanism operates while the reversible binding affinity is weak. This mechanism of action is very similar to how enzymes activates their substrates.[103]
Covalent Inhibitors of HRAS and NRAS Mutants
In comparison to KRAS whose mutations occur in 21.6% of human cancer, mutations of NRAS and HRAS have relative low occurrence, with mutations in NRAS associated with 8.0% of human cancer and mutations in HRAS associated with 3.3% of human cancer.[104] Despite being most investigated historically, HRAS genes are least mutated in human cancer among all three RAS genes. As mentioned previously, HRAS mutations occur predominantly in bladder cancer with the key residues for mutation being G12 and Q61. Mutation of G12 in HRAS to any other amino acid except proline induces colony formation and anchorage-independent growth in rat fibroblasts. Although potent HRAS inhibitors have been developed, those that form covalent adducts with HRAS are yet to be identified.[105]
As of today, NRAS associated cancers are mostly found in melanoma and in some other cases in lung cancer and T-cell lymphoma. Major mutations associated with NRAS occurs at the residue 61 along with a few occurring at residues 12 and 13.[105] All these mutations are known to activate the RAS-RAF-MEK-ERK pathway. NRAS Mutants are also reported to activate the PI3K/mTOR signaling cascade.[105] Posch et al. reported that MEK inhibitors such as JTP-74057 and PD325901 showed good inhibition towards growth of NRAS mutant cells.[106] Again, in this case also the use of combined MEK inhibitors with PI3K or mTOR inhibitors showed greater efficiency towards reducing growth of NRAS mutant cells than using either of them individually. This combination of inhibitors reduces cell viability in vitro and decrease tumor size in vivo in a large panel of human melanoma NRAS cells.[106] Potent covlanet inhibitors for NRAS muants are also yet to be identified.
Covalent Inhibitors of Acetyl Choline Esterase (AChE)
Acetyl choline is a neurotransmitter that stimulates cholinergic receptors at chemical synapses in the central nervous system. Patients suffering from Alzheimer’s disease have decreased level of these receptors[107]. One strategy that has been used to combat Alzheimer’s disease is to increase the level of acetylcholine at neuronal synapses by inhibiting acetyl choline esterase (AChE) that regulates the level of endogenous acetylcholine. Two reversible drugs, tacrine and donepezil have been approved by FDA to treat Alzheimer’s disease[107]. However, they both inhibit AChE for a short time. A novel inhibitor, rivastigmine (Figure 11a) has been recently approved for the treatment of Alzheimer’s disease for its prolonged inhibition of AChE for about 10 hrs.[108] When it binds to the AChE active site, rivastigmine covalently modifies the protein.[107] From the crystal structure of AChE bound with choline that were reported by Bourne et al., we can tell that there are two key residues in the active site, S203 (esteratic site) and E202 (anionic site) (Figures 11b & 11c).[109] The anionic site plays an important role in stabilizing the positively charged nitrogen in the native substrate or in rivastigmine, which helps the binding of either the substrate or rivastigmine at the active site. Once rivastigmine binds to AChE, its carbamoyl group is located proximal to the active site serine that exchanges the carbamoyl group from rivastigmine to release a hydrolyzed phenolic derivative (Figure 11d)[109]. This results in a carbamoyl serine at the active site that slowly hydrolyzes to recover the activity of AChE. Another covalent inhibitor of AChE is metrifonate that shows even stronger inhibition of AChE.[110] Metrifonate is a prodrug which is hydrolyzed non-enzymatically to 2,2-dichlorovinyl dimethyl phosphate (DDVP)[110]. DDVP acts as the active agent, which binds at the esteratic site of the AChE to phosphorylate the active site serine. The hydrolysis of the O-P bond in the modified serine residue is extremely slow, leading to inactivation of AChE for a couple of weeks.
Figure 11.
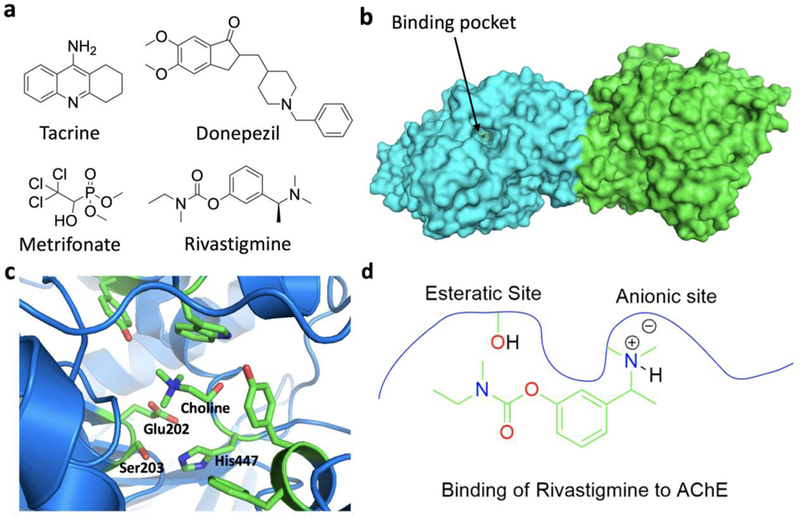
a. AchE inhibitors; b. Structure of mouse AChE; c. AChE with choline bound at the active site (PDB entry: 2HA3); d. Interactions of rivastigmine with AChE in the active site.
Covalent Inhibitors of Cathepsins
Cathepsins are a group of cysteine proteases that are involved in proteolysis in lysosome and control various signaling pathways in cells[111]. There are 11 cathepsins coded in humans (B, C, F, H, K, L, O, S, V, W and X). Hyperactivity of these enzyme is often related to disease development.[112] Among different cathepsins, CatK has been of high interest. It occurs abundant in osteoclasts and plays an important role in resorption and remodeling of bones. As such it has been a drug target for the treatment of osteoporosis in which bones decrease their density significantly and become fragile. Several covalent inhibitors were designed to target CatK for the treatment of osteoporosis but have been discontinued owing to their side effects. Among problems during the development of CatK inhibitors, one was that inhibitors identified to show good results on rodent CatK were not so efficient for human CatK.[113],[114] This is due to species variation of CatK. Another problem arose from attaching basic functional groups to inhibitors that increased specificity for CatK but also lysosomal accumulation. One such example is balicatib [115] (Figure 12a). Balicatib showed great selectivity for CatK with respect to other cathepsins but was discontinued after its Phase II clinical studies as it led to morphea-like skin lesions in some patients[116]. The lysosomal accumulation of balicatib gave rise to off-target activities in osteoclasts as well as in skin-fibroblasts[117]. Among many covalent inhibitors of CatK, odonacatib that is not lysosomotropic reached phase-III trials (Figure 12b).[118],[119] Odonacatib has an nitrile group that reacts with C25 of CatK to form an iminothioester adduct (Figure 12c; PDB entry: 5TDI).[120] Although odonacatib was established efficient in increasing bone mineral density and reducing hip or vertebrae fractures, its phase III trial was prematurely terminated. Evidences indicated that it increased the likelihood of cardiovascular complications like stroke in patients.[121] As of today, one CatK inhibitor that is undergoing clinical trials is MIV-711.[122] It is at the phase II clinical trial stage for osteoporosis and osteoartharitis.[116]
Figure 12.

Covalent cathepsin inhibitors. a. Balicatib; b. Odonacatib; c. The covalent adduct between odonacatib and Cys25 in the CatK active site (PDB entry: 5TDI); d. JPM-OEt; e. JPM-565.
Among other cathepsins, CatS has been found to play a unique role in mediating the immune response in dendritic and b cells. Hence inhibition of CatS can be an useful strategy to combat hyperactivation of immune systems against host antigens in several auto-immune diseases like rheumatoid arthritis, bronchial asthma etc.[123] Moreover, CatS was found overexpressed in psoriatic skin, where involvement of chronic antigens in dermal dendritic cells might influence psoriasis[116]. Efforts have been made to identify non-covalent CatS inhibitors for therapeutic intervention. In 2005, clinical trials of CatS inhibitor CRA-028129[124] was launched for the treatment of psoriasis but was discontinued after phase I. Another CatS inhibitor, RWJ-445380[116], which was launched for treating psoriasis and rheumatoid arthritis suffered the same fate due to its lack of efficacy. Another CatS inhibitor RO5461111 was used as an orally available drug successfully to suppress clinically advanced lupus nephritis in mouse model.[125] The development of SAR113137 as a CatS/CatK inhibitor for pain management was initially discontinued after carrying out safety studies but is now getting re-evaluated for treatment of Chagas disease as it probably cross react with cathepsin-like proteases in parasites.[126] Since upregulated activities of cathepsins are related to cancer development, there have also been efforts to use cathepsin inhibitors for treating cancer. Some of these inhibitors contain the epoxide functionality that potentially form covalent adducts with cathepsins. An epoxide based inhibitor JPM-OEt (Figure 12d) was tested successfully in treating pancreatic neuroendocrine cancer in mouse model but failed when tested against breast-to-bone metastasis and breast cancer in mammary gland.[116] The reason was explained to be the poor bioavailability of the drug. An improved version JPM-565[127] (Figure 12e) led to antitumor effects comparable to gene ablation studies in the same breast cancer model.
Covalent Inhibitors of Caspases
Caspases are a family of cysteine proteases that regulate protein cellular homeostasis.[128] They play essential roles in programmed cell death and inflammation. More than 10 caspases have been identified in humans. Malfunctions of these caspases have been implicated in the development a number of diseases and therefore targeting caspases have been an establishment for disease intervention[129]. Due to extensive studies of these enzymes, substrate preferences of different caspases have been known for some time, which has been used to design covalent inhibitors for caspases. These inhibitors generally contain a tetrapeptidyl moiety with an electrophilic warhead to react with the active site cysteine of a particular caspase. One covalent inhibitor of caspase 8 is presented in Figure 13a that also shows how it interacts with the active site C360 residue of caspase 8.[130] This inhibitor has a 2-oxoalkyl o,o-dichlorobenzoate moiety that reacts with C360 to form a covalent adduct. A similar strategy has also been applied to design covalent inhibitors for caspase 3 and caspase 6 (Figure 13b). These inhibitors have a 2-oxoalkyl tetrafluorophenyl ether moiety that reacts with a nucleophilic cysteine for releasing tetrafluorophenol, leading to a covalent 2-oxoalkyl cysteinyl ether adduct. The three molecules are considering for the treatment of Huntington Disease[131]. Various 2-acetic acid derivatives have also been synthesized and shown caspase-3 inhibition with micromolar IC50 values (Figure 13c). Many other caspase inhibitors are well known in the literature.[132] Interesting, a group of thiol-containing compounds have been recently identified to inhibit caspase activities.[133] Instead of targeting the active site cysteine residue, these compounds, known as “disulfide tethers” bind to an allosteric site. The binding of these “sulfur tethers” usually trap enzymes in their zymogen state and therefore prevents their activation. Compound 1 in Figure14a is one such “sulfur tether” which binds at the dimeric interface of caspase 7 to form a disulfide linkage with the C290 residue (Figures 14b & 14c), compound 2 inactivates caspase 3, compound 3 traps caspase 1, and compound 4 inhibit caspase 5.[134, 135]
Figure 13.
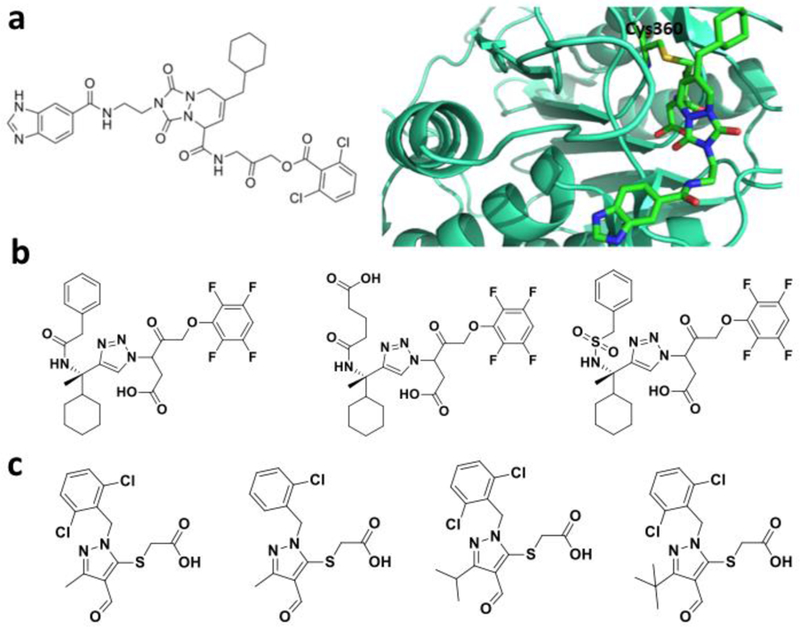
Covalent caspase inhibitors. a-b. A covalent caspase 8 inhibitor and its caspase 8 adduct structure (PDB entry: 3KJN); c. Three covalent caspase 3 and caspase 6 inhibitors that contain the tetrafluorophenyl ether moeity; d. Caspase 3 inhibitors that are 2-acetic acid derivatives.
Figure 14.
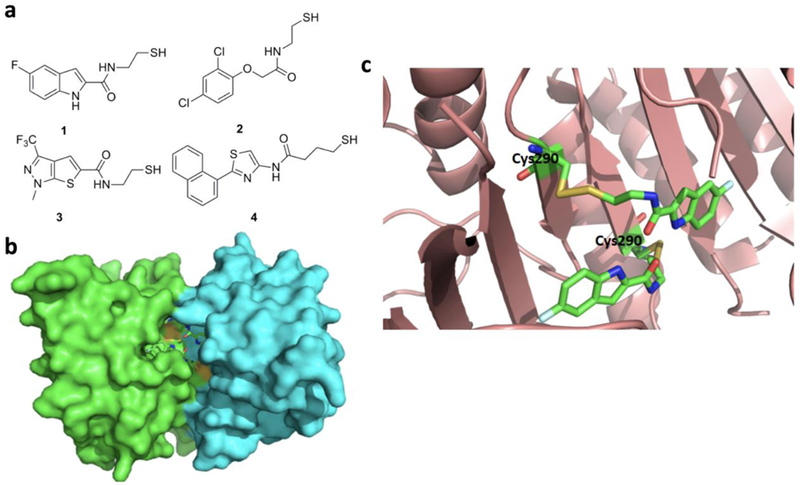
a. Thiol containing allosteric inhibitors of caspase enzymes; b. Crystal structure of caspase 7 bound with compound 1 (PDB entry:1SHL); c. an enlarged view at the active site for the structure shown in b.
Covalent Inhibitors of Several Other Enzymes for Treating Obesity and Diabetes
There are also many reports about covalent inhibitors for other enzymes. Inhibitors for three enzymes for treating obesity and diabetes are discussed here. Pancreatic lipase is an enzyme that hydrolyses triacylglycerol fatty acids. It has been implicated as one therapeutic target for treating obesity[136]. Pancreatic lipase has an active site serine that directly participates in the native hydrolysis reaction. One covalent inhibitor that targets this active site serine is orlistat that has a β-lactone moiety.[137] Orlistat (Figure 15) has been approved by FDA as an oral drug for obesity. Another well-known target to treat obesity is MetAP2. MetAP2 is responsible for cleaving the N-terminal methionine from the newly synthesized protein, which may further undergo posttranslational modifications. Although initially it was thought to be a target for cancer treatment, inhibition of MetAP2 was found to be useful to treat obesity. MetAP2 inhibition suppresses sterol regulatory element binding protein activity leading to lower level of lipid and cholesterol biosynthesis[138]. Beloranib (Figure 15) is a covalent inhibitor that is currently under clinical trials for treating obesity. Mechanistically the less hindered spiro-epoxide of beloranib reacts with residue H231 of MetAP2 at the active site, resulting in inhibition of the enzyme.[138] Another enzyme that has been targeted by covalent inhibition is DPP4. DPP4 is a serine protease that enhances human body’s own ability to control blood glucose by increasing incretin in human body and thus regulating insulin and glucagone secretion. As such, DPP4 has been researched as a potent target to treat Diabetes Mellitus[139]. Covalent inhibitors of DPP4 has been used to treat type 2 Diabetes Mellitus (T2DM) by enabling patients to produce their own insulin and control the glucose level in their blood.[140] Vidagliptin and Saxagliptin (Figure 15) are two DPP4 covalent inhibitors that are FDA-approved orally available drugs to treat T2DM and manage the glucose level in diabetic patients[140]. The warhead electrophilic center in both these molecules is a nitrile group which forms a covalent adduct with the active site residue S630 of DPP4.
Figure 15.
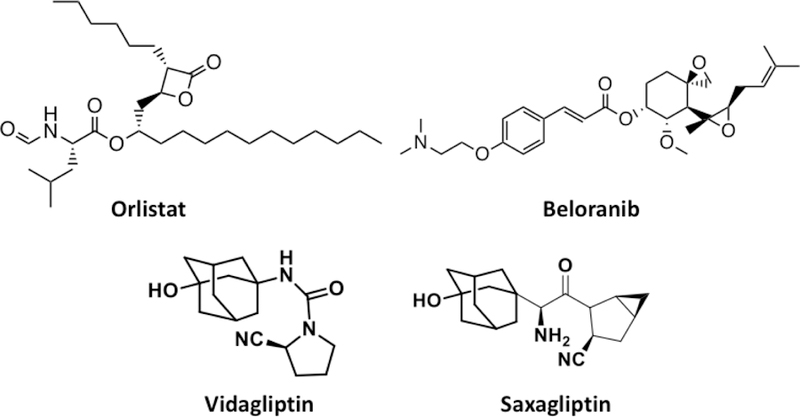
Structures of orlistat, beloranib, vildagliptin, and Saxagliptin.
Conclusion:
Despite a long history in the treatment of human health conditions, the direct use of covalent inhibitors for human enzymes was not popular in the pharmaceutical industry. Besides potential toxicity and off-target binding, another disadvantage of a covalent inhibitor is the over-dependence of its efficacy on a single residue in the targeted protein/receptor that can undergo mutation to acquire resistance. The targeted residues for both EGFR and BTK inhibitors have been shown to undergo mutation as a strategy to escape inhibition. For example, resistance was acquired against osimertinib generally within 9–13 months of treatment.[135] This acquired resistance raises serious concerns given huge investment during the development of a therapeutic drug. To relieve this potential problem, one strategy is to develop dual inhibitors that target two different enzymes that function in a same disease. For example, a patient suffering from NSCLC can be treated with inhibitors for both anaplastic lymphoma kinase and EGFR.[141] This strategy may be applied to other diseases. In order to improve selectivity and efficacy of a covalent drug, bivalent inhibitors that can target both the active site and an allosteric site might also be considered. It should be noted that allosteric inhibitors may also play a crucial role in fighting against covalent drug resistance. Several allosteric inhibitors for EGFR and BTK mutants have been developed.[142] Despite potential problems, the development of covalent inhibitors for human kinases has achieved an excellent start. Greater success is anticipated.
Although researched for more than three decades, targeting RAS proteins for drug development remained a huge challenge. RAS proteins have very high affinities for GTP, unique protein-protein binding modes, and high dynamic structures, which all contribute to the difficulty of developing efficient RAS inhibitors. However, recent progress in developing covalent RAS inhibitors has provided some hope to make these traditionally “undruggable” proteins druggable. One driving force for this change is the development of covalent inhibitors for KRAS G12C, although efficient covalent inhibitors for other RAS proteins are yet to be seen.
In summary, although some concerns related to covalent inhibitors remain, the past decade has witnessed the reversal of this trend with a number of covalent inhibitors developed for human enzymes. Several of these covalent inhibitors have already been approved by FDA for use in human patients. The development of covalent inhibitors for KRAS G12C is especially exciting. Improved potency is a clear advantage of using covalent inhibitors, which potentially improves the therapeutic window by providing a reduced dose. Using covalent inhibitors may also reduce the risk of idiosyncratic toxicity. Of course, cautions still need to be taken when applying covalent inhibitors as drugs. Clinical trials of several covalent inhibitors have resulted in death of patients.[1] These include beloranib. Teams working on covalent inhibitors must balance between the efficiency of the adduct formation of an inhibitor with its target enzyme and the dose with limited toxicity. Given the increased number of covalent therapeutics approved by FDA for the treatment of human diseases, we expect more covalent inhibitors will be developed and enter the clinical trials in the coming years.
Acknowledgement
Research support in Liu group is provided from National Institute of Health (grants R01GM121584 and R01GM127575), Cancer Prevention & Research Institute of Texas (grant RP170797), and Welch Foundation (grant A-1715). This review is the term paper of three leading student authors required for fulfilling the TAMU graduate level course Chem 630: Chemical Biology.
References
- [1].Baillie TA, Angew Chem Int Ed Engl 2016, 55, 13408–13421. [DOI] [PubMed] [Google Scholar]
- [2].Singh J, Petter RC, Baillie TA, Whitty A, Nat Rev Drug Discov 2011, 10, 307–317. [DOI] [PubMed] [Google Scholar]
- [3].Roth GJ, Stanford N, Majerus PW, Proc Natl Acad Sci U S A 1975, 72, 3073–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Picot D, Loll PJ, Garavito RM, Nature 1994, 367, 243–249; P. J. Loll, D. Picot, R. M. Garavito, Nat Struct Biol 1995, 2, 637–643. [DOI] [PubMed] [Google Scholar]
- [5].Baillie TA, Drug Metab Rev 2015, 47, 4–11. [DOI] [PubMed] [Google Scholar]
- [6].Yocum RR, Rasmussen JR, Strominger JL, J Biol Chem 1980, 255, 3977–3986. [PubMed] [Google Scholar]
- [7].Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL, Proc Natl Acad Sci U S A 2012, 109, 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kahan JS, Kahan FM, Goegelman R, Currie SA, Jackson M, Stapley EO, Miller TW, Miller AK, Hendlin D, Mochales S, Hernandez S, Woodruff HB, Birnbaum J, J Antibiot (Tokyo) 1979, 32, 1–12. [DOI] [PubMed] [Google Scholar]
- [9].Yotsuji A, Mitsuyama J, Hori R, Yasuda T, Saikawa I, Inoue M, Mitsuhashi S, Antimicrob Agents Chemother 1988, 32, 1848–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB, J Pharmacol Exp Ther 1973, 187, 195–202. [PubMed] [Google Scholar]
- [11].Bauer RA, Drug Discov Today 2015, 20, 1061–1073. [DOI] [PubMed] [Google Scholar]
- [12].Awoonor-Williams E, Walsh AG, Rowley CN, Biochim Biophys Acta Proteins Proteom 2017, 1865, 1664–1675. [DOI] [PubMed] [Google Scholar]
- [13].Zhang J, Yang PL, Gray NS, Nat Rev Cancer 2009, 9, 28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stamos J, Sliwkowski MX, Eigenbrot C, J Biol Chem 2002, 277, 46265–46272. [DOI] [PubMed] [Google Scholar]
- [15].Gazdar AF, Oncogene 2009, 28 Suppl 1, S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nakamura Y, Oka M, Soda H, Shiozawa K, Yoshikawa M, Itoh A, Ikegami Y, Tsurutani J, Nakatomi K, Kitazaki T, Doi S, Yoshida H, Kohno S, Cancer Res 2005, 65, 1541–1546. [DOI] [PubMed] [Google Scholar]
- [17].Schettino C, Bareschino MA, Ricci V, Ciardiello F, Expert Rev Respir Med 2008, 2, 167–178. [DOI] [PubMed] [Google Scholar]
- [18].Schwartz PA, Kuzmic P, Solowiej J, Bergqvist S, Bolanos B, Almaden C, Nagata A, Ryan K, Feng J, Dalvie D, Kath JC, Xu M, Wani R, Murray BW, Proc Natl Acad Sci U S A 2014, 111, 173–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Singh J, Dobrusin EM, Fry DW, Haske T, Whitty A, McNamara DJ, J Med Chem 1997, 40, 1130–1135. [DOI] [PubMed] [Google Scholar]
- [20].Fry DW, Bridges AJ, Denny WA, Doherty A, Greis KD, Hicks JL, Hook KE, Keller PR, Leopold WR, Loo JA, McNamara DJ, Nelson JM, Sherwood V, Smaill JB, Trumpp-Kallmeyer S, Dobrusin EM, Proc Natl Acad Sci U S A 1998, 95, 12022–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, Althaus IW, Gandhi L, Shapiro GI, Nelson JM, Heymach JV, Meyerson M, Wong KK, Janne PA, Cancer Res 2007, 67, 11924–11932. [DOI] [PubMed] [Google Scholar]
- [22].Yoshimura N, Kudoh S, Kimura T, Mitsuoka S, Matsuura K, Hirata K, Matsui K, Negoro S, Nakagawa K, Fukuoka M, Lung Cancer 2006, 51, 363–368. [DOI] [PubMed] [Google Scholar]
- [23].Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, Eaton K, Zacharchuk C, Freyman A, Powell C, Ananthakrishnan R, Quinn S, Soria JC, J Clin Oncol 2010, 28, 3076–3083. [DOI] [PubMed] [Google Scholar]
- [24].Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, Miller VA, Pao W, Cancer Discov 2014, 4, 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Janne PA, Nature 2009, 462, 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hernandes MZ, Cavalcanti SM, Moreira DR, de Azevedo Junior WF, Leite AC, Curr Drug Targets 2010, 11, 303–314. [DOI] [PubMed] [Google Scholar]
- [27].Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Brewer M. Red, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W, Cancer Discov 2014, 4, 1046–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yosaatmadja Y, Silva S, Dickson JM, Patterson AV, Smaill JB, Flanagan JU, McKeage MJ, Squire CJ, J Struct Biol 2015, 192, 539–544. [DOI] [PubMed] [Google Scholar]
- [29].Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, Mann H, Marotti M, Ghiorghiu S, Papadimitrakopoulou VA, Investigators A, N Engl J Med 2017, 376, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Thress KS, Paweletz CP, Felip E, Cho BC, Stetson D, Dougherty B, Lai Z, Markovets A, Vivancos A, Kuang Y, Ercan D, Matthews SE, Cantarini M, Barrett JC, Janne PA, Oxnard GR, Nat Med 2015, 21, 560–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, Kurtz DM, Stehr H, Scherer F, Karlovich CA, Harding TC, Durkin KA, Otterson GA, Purcell WT, Camidge DR, Goldman JW, Sequist LV, Piotrowska Z, Wakelee HA, Neal JW, Alizadeh AA, Diehn M, Nat Commun 2016, 7, 11815; H. N. Song, K. S. Jung, K. H. Yoo, J. Cho, J. Y. Lee, S. H. Lim, H. S. Kim, J. M. Sun, S. H. Lee, J. S. Ahn, K. Park, Y. L. Choi, W. Park, M. J. Ahn, J Thorac Oncol 2016, 11, e45–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Janne PA, Meyerson M, Marto JA, Engen JR, Gray NS, Angew Chem Int Ed Engl 2014, 53, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Planchard D, Loriot Y, Andre F, Gobert A, Auger N, Lacroix L, Soria JC, Ann Oncol 2015, 26, 2073–2078. [DOI] [PubMed] [Google Scholar]
- [34].Ho CC, Liao WY, Lin CA, Shih JY, Yu CJ, Chih-Hsin Yang J, J Thorac Oncol 2017, 12, 567–572. [DOI] [PubMed] [Google Scholar]
- [35].Zheng D, Hu M, Bai Y, Zhu X, Lu X, Wu C, Wang J, Liu L, Wang Z, Ni J, Yang Z, Xu J, Oncotarget 2017, 8, 49671–49679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cheng H, Nair SK, Murray BW, Almaden C, Bailey S, Baxi S, Behenna D, Cho-Schultz S, Dalvie D, Dinh DM, Edwards MP, Feng JL, Ferre RA, Gajiwala KS, Hemkens MD, Jackson-Fisher A, Jalaie M, Johnson TO, Kania RS, Kephart S, Lafontaine J, Lunney B, Liu KKC, Liu Z, Matthews J, Nagata A, Niessen S, Ornelas MA, Orr STM, Pairish M, Planken S, Ren S, Richter D, Ryan K, Sach N, Shen H, Smeal T, Solowiej J, Sutton S, Tran K, Tseng E, Vernier W, Walls M, Wang S, Weinrich SL, Xin S, Xu H, Yin M-J, Zientek M, Zhou R, Kath JC, J Med Chem 2016, 59, 2005–2024. [DOI] [PubMed] [Google Scholar]
- [37].Jia Y, Juarez J, Li J, Manuia M, Niederst MJ, Tompkins C, Timple N, Vaillancourt MT, Pferdekamper AC, Lockerman EL, Li C, Anderson J, Costa C, Liao D, Murphy E, DiDonato M, Bursulaya B, Lelais G, Barretina J, McNeill M, Epple R, Marsilje TH, Pathan N, Engelman JA, Michellys PY, McNamara P, Harris J, Bender S, Kasibhatla S, Cancer Res 2016, 76, 1591–1602. [DOI] [PubMed] [Google Scholar]
- [38].Wang S, Cang S, Liu D, J Hematol Oncol 2016, 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Liao BC, Lin CC, Yang JC, Curr Opin Oncol 2015, 27, 94–101. [DOI] [PubMed] [Google Scholar]
- [40].Zhang L, Zhao H, Hu B, Jiang J, Zheng X, Zhang Y, Ma Y, Ge J, Zou B, Fang X, Xu W, Xu X, Ann Oncol 2016, 27, 359O–359O; X. Xu, Chin J Cancer 2015, 34, 285–287.26658890 [Google Scholar]
- [41].Gunther M, Lategahn J, Juchum M, Doring E, Keul M, Engel J, Tumbrink HL, Rauh D, Laufer S, J Med Chem 2017, 60, 5613–5637. [DOI] [PubMed] [Google Scholar]
- [42].Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A, Cancer Discov 2013, 3, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J, Eur J Cancer 2009, 45, 228–247. [DOI] [PubMed] [Google Scholar]
- [44].Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, Garon EB, Heist RS, Logan J, Neal JW, Mendenhall MA, Nichols S, Piotrowska Z, Wozniak AJ, Raponi M, Karlovich CA, Jaw-Tsai S, Isaacson J, Despain D, Matheny SL, Rolfe L, Allen AR, Camidge DR, N Engl J Med 2015, 372, 1700–1709. [DOI] [PubMed] [Google Scholar]
- [45].Dhingra K, Ann Oncol 2016, 27, 1161–1164. [DOI] [PubMed] [Google Scholar]
- [46].Novero A, Ravella PM, Chen Y, Dous G, Liu D, Exp Hematol Oncol 2014, 3, 4; A. Aalipour, R. H. Advani, Br J Haematol 2013, 163, 436–443; R. H. Advani, J. J. Buggy, J. P. Sharman, S. M. Smith, T. E. Boyd, B. Grant, K. S. Kolibaba, R. R. Furman, S. Rodriguez, B. Y. Chang, J. Sukbuntherng, R. Izumi, A. Hamdy, E. Hedrick, N. H. Fowler, J Clin Oncol 2013, 31, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, Hammarstrom L, Kinnon C, Levinsky R, Bobrow M, et al. , Nature 1993, 361, 226–233; R. W. Hendriks, R. G. Bredius, K. Pike-Overzet, F. J. Staal, Expert Opin Ther Targets 2011, 15, 1003–1021. [DOI] [PubMed] [Google Scholar]
- [48].Uckun FM, Tibbles HE, Vassilev AO, Anticancer Agents Med Chem 2007, 7, 624–632. [DOI] [PubMed] [Google Scholar]
- [49].Broides A, Hadad N, Levy J, Levy R, J Clin Immunol 2014, 34, 555–560; J. J. Castillo, S. P. Treon, M. S. Davids, Cancer J 2016, 22, 34–39. [DOI] [PubMed] [Google Scholar]
- [50].Fabbro SK, Smith SM, Dubovsky JA, Gru AA, Jones JA, JAMA Oncol 2015, 1, 684–686; G. Cheng, Z. S. Ye, D. Baltimore, Proc Natl Acad Sci U S A 1994, 91, 8152–8155. [DOI] [PubMed] [Google Scholar]
- [51].Li X, Zuo Y, Tang G, Wang Y, Zhou Y, Wang X, Guo T, Xia M, Ding N, Pan Z, J Med Chem 2014, 57, 5112–5128. [DOI] [PubMed] [Google Scholar]
- [52].Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, Racchumi J, Xu G, Wu H, Ma J, Steggerda SM, Coleman M, Leslie C, Wang YL, N Engl J Med 2014, 370, 2352–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wu J, Liu C, Tsui ST, Liu D, J Hematol Oncol 2016, 9, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, Hillmen P, Stephens DM, Ghia P, Barrientos JC, Pagel JM, Woyach J, Johnson D, Huang J, Wang X, Kaptein A, Lannutti BJ, Covey T, Fardis M, McGreivy J, Hamdy A, Rothbaum W, Izumi R, Diacovo TG, Johnson AJ, Furman RR, N Engl J Med 2016, 374, 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rai KR, Barrientos JC, N Engl J Med 2014, 370, 1160–1162; C. Iragavarapu, M. Mustafa, A. Akinleye, M. Furqan, V. Mittal, S. Cang, D. Liu, J Hematol Oncol 2015, 8, 17. [DOI] [PubMed] [Google Scholar]
- [56].Wu H, Huang Q, Qi Z, Chen Y, Wang A, Chen C, Liang Q, Wang J, Chen W, Dong J, Yu K, Hu C, Wang W, Liu X, Deng Y, Wang L, Wang B, Li X, Gray NS, Liu J, Wei W, Liu Q, Sci Rep 2017, 7, 466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yasuhiro T, Sawada W, Klein C, Kozaki R, Hotta S, Yoshizawa T, Leuk Lymphoma 2017, 58, 699–707; J. Wu, M. Zhang, D. Liu, Oncotarget 2017, 8, 7201–7207. [DOI] [PubMed] [Google Scholar]
- [58].Smith PF, Krishnarajah J, Nunn PA, Hill RJ, Karr D, Tam D, Masjedizadeh M, Funk JO, Gourlay SG, Br J Clin Pharmacol 2017, 83, 2367–2376; J. K. Park, J. Y. Byun, J. A. Park, Y. Y. Kim, Y. J. Lee, J. I. Oh, S. Y. Jang, Y. H. Kim, Y. W. Song, J. Son, K. H. Suh, Y. M. Lee, E. B. Lee, Arthritis Res Ther 2016, 18, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, Zhu Z, Nacht M, Freed MI, Petter RC, Dubrovskiy A, Singh J, Westlin WF, J Pharmacol Exp Ther 2013, 346, 219–228. [DOI] [PubMed] [Google Scholar]
- [60].Neise D, Sohn D, Stefanski A, Goto H, Inagaki M, Wesselborg S, Budach W, Stuhler K, Janicke RU, Cell Death Dis 2013, 4, e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Hauge C, Frodin M, J Cell Sci 2006, 119, 3021–3023. [DOI] [PubMed] [Google Scholar]
- [62].Cuello F, Snabaitis AK, Cohen MS, Taunton J, Avkiran M, Mol Pharmacol 2007, 71, 799–806. [DOI] [PubMed] [Google Scholar]
- [63].Houde VP, Ritorto MS, Gourlay R, Varghese J, Davies P, Shpiro N, Sakamoto K, Alessi DR, Biochem J 2014, 458, 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N, EMBO J 2006, 25, 2781–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, Cohen MS, Johansen JV, Winther BR, Lund LR, Winther O, Taunton J, Hansen SH, Frodin M, Mol Cell 2009, 35, 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wang Y, Xiang GS, Kourouma F, Umar S, Br J Pharmacol 2006, 148, 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu Y, Bishop A, Witucki L, Kraybill B, Shimizu E, Tsien J, Ubersax J, Blethrow J, Morgan DO, Shokat KM, Chem Biol 1999, 6, 671–678; O. Buzko, K. M. Shokat, Bioinformatics 2002, 18, 1274–1275. [DOI] [PubMed] [Google Scholar]
- [68].Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J, Mol Cell 1999, 3, 639–648. [DOI] [PubMed] [Google Scholar]
- [69].Park BK, Boobis A, Clarke S, Goldring CE, Jones D, Kenna JG, Lambert C, Laverty HG, Naisbitt DJ, Nelson S, Nicoll-Griffith DA, Obach RS, Routledge P, Smith DA, Tweedie DJ, Vermeulen N, Williams DP, Wilson ID, Baillie TA, Nat Rev Drug Discov 2011, 10, 292–306. [DOI] [PubMed] [Google Scholar]
- [70].Cohen MS, Zhang C, Shokat KM, Taunton J, Science 2005, 308, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frodin M, Taunton J, Nat Chem Biol 2012, 8, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Andersen JL, Gesser B, Funder ED, Nielsen CJF, Gotfred-Rasmussen H, Rasmussen MK, Toth R, Gothelf KV, Arthur JSC, Iversen L, Nissen P, Nat Commun 2018, 9, 4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Zhao Z, Bourne PE, Drug Discov Today 2018, 23, 727–735. [DOI] [PubMed] [Google Scholar]
- [74].Tiong KH, Mah LY, Leong CO, Apoptosis 2013, 18, 1447–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS, Chem Biol 2010, 17, 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Guagnano V, Furet P, Spanka C, Bordas V, Le Douget M, Stamm C, Brueggen J, Jensen MR, Schnell C, Schmid H, Wartmann M, Berghausen J, Drueckes P, Zimmerlin A, Bussiere D, Murray J, Porta D. Graus, J Med Chem 2011, 54, 7066–7083. [DOI] [PubMed] [Google Scholar]
- [77].Gavine PR, Mooney L, Kilgour E, Thomas AP, Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, Brooks AN, Klinowska T, Cancer Res 2012, 72, 2045–2056. [DOI] [PubMed] [Google Scholar]
- [78].Schuttelkopf AW, van Aalten DM, Acta Crystallogr D Biol Crystallogr 2004, 60, 1355–1363; V. Chell, K. Balmanno, A. S. Little, M. Wilson, S. Andrews, L. Blockley, M. Hampson, P. R. Gavine, S. J. Cook, Oncogene 2013, 32, 3059–3070. [DOI] [PubMed] [Google Scholar]
- [79].Brameld KA, Owens TD, Verner E, Venetsanakos E, Bradshaw JM, Phan VT, Tam D, Leung K, Shu J, LaStant J, Loughhead DG, Ton T, Karr DE, Gerritsen ME, Goldstein DM, Funk JO, J Med Chem 2017, 60, 6516–6527. [DOI] [PubMed] [Google Scholar]
- [80].Knoepfel T, Furet P, Mah R, Buschmann N, Leblanc C, Ripoche S, Graus-Porta D, Wartmann M, Galuba I, Fairhurst RA, ACS Med Chem Lett 2018, 9, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lim S, Kaldis P, Development 2013, 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- [82].Davies TG, Bentley J, Arris CE, Boyle FT, Curtin NJ, Endicott JA, Gibson AE, Golding BT, Griffin RJ, Hardcastle IR, Jewsbury P, Johnson LN, Mesguiche V, Newell DR, Noble ME, Tucker JA, Wang L, Whitfield HJ, Nat Struct Biol 2002, 9, 745–749. [DOI] [PubMed] [Google Scholar]
- [83].Anscombe E, Meschini E, Mora-Vidal R, Martin MP, Staunton D, Geitmann M, Danielson UH, Stanley WA, Wang LZ, Reuillon T, Golding BT, Cano C, Newell DR, Noble ME, Wedge SR, Endicott JA, Griffin RJ, Chem Biol 2015, 22, 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, Jenkins CE, Hannett NM, McMillin D, Sanda T, Sim T, Kim ND, Look T, Mitsiades CS, Weng AP, Brown JR, Benes CH, Marto JA, Young RA, Gray NS, Nature 2014, 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, Manz T, Hao M, Bartkowiak B, Greenleaf AL, Marto JA, Geyer M, Bullock AN, Young RA, Gray NS, Nat Chem Biol 2016, 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Nacht M, Qiao L, Sheets MP, St Martin T, Labenski M, Mazdiyasni H, Karp R, Zhu Z, Chaturvedi P, Bhavsar D, Niu D, Westlin W, Petter RC, Medikonda AP, Singh J, J Med Chem 2013, 56, 712–721. [DOI] [PubMed] [Google Scholar]
- [87].Pritchard RB, Lough CE, Currie DJ, Holmes HL, Can J Chem 1968, 46, 775–+. [Google Scholar]
- [88].Lee CU, Grossmann TN, Angew Chem Int Ed Engl 2012, 51, 8699–8700. [DOI] [PubMed] [Google Scholar]
- [89].Bradshaw JM, McFarland JM, Paavilainen VO, Bisconte A, Tam D, Phan VT, Romanov S, Finkle D, Shu J, Patel V, Ton T, Li X, Loughhead DG, Nunn PA, Karr DE, Gerritsen ME, Funk JO, Owens TD, Verner E, Brameld KA, Hill RJ, Goldstein DM, Taunton J, Nat Chem Biol 2015, 11, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Goodsell DS, Stem Cells 1999, 17, 235–236. [DOI] [PubMed] [Google Scholar]
- [91].Ryan MB, Der CJ, Wang-Gillam A, Cox AD, Trends Cancer 2015, 1, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Downward J, Nat Rev Cancer 2003, 3, 11–22. [DOI] [PubMed] [Google Scholar]
- [93].Ostrem JM, Shokat KM, Nat Rev Drug Discov 2016, 15, 771–785. [DOI] [PubMed] [Google Scholar]
- [94].Ahearn IM, Haigis K, Bar-Sagi D, Philips MR, Nat Rev Mol Cell Bio 2012, 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Baker NM, Der CJ, Nature 2013, 497, 577–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Novotny CJ, Hamilton GL, McCormick F, Shokat KM, ACS Chem Biol 2017, 12, 1956–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Rudolph J, Stokoe D, Angew Chem Int Ed Engl 2014, 53, 3777–3779. [DOI] [PubMed] [Google Scholar]
- [98].Zebisch A, Troppmair J, Cell Mol Life Sci 2006, 63, 1314–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM, Nature 2013, 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD, Proc Natl Acad Sci U S A 2014, 111, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, Chen JH, Firdaus SJ, Babbar A, Ren P, Liu Y, Cancer Discov 2016, 6, 316–329. [DOI] [PubMed] [Google Scholar]
- [102].Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, Feng J, Chen JH, Li S, Li S, Long YO, Thach C, Liu Y, Zarieh A, Ely T, Kucharski JM, Kessler LV, Wu T, Yu K, Wang Y, Yao Y, Deng X, Zarrinkar PP, Brehmer D, Dhanak D, Lorenzi MV, Hu-Lowe D, Patricelli MP, Ren P, Liu Y, Cell 2018, 172, 578–589 e517. [DOI] [PubMed] [Google Scholar]
- [103].Hansen R, Peters U, Babbar A, Chen Y, Feng J, Janes MR, Li LS, Ren P, Liu Y, Zarrinkar PP, Nat Struct Mol Biol 2018, 25, 454–462; A. V. Statsyuk, Nat Struct Mol Biol 2018, 25, 435–437. [DOI] [PubMed] [Google Scholar]
- [104].Baines AT, Xu D, Der CJ, Future Med Chem 2011, 3, 1787–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M, Rogler G, Oncotarget 2015, 6, 42183–42196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, Daud AI, McCalmont TH, LeBoit PE, Ortiz-Urda S, Proc Natl Acad Sci U S A 2013, 110, 4015–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Jann MW, Pharmacotherapy 2000, 20, 1–12. [DOI] [PubMed] [Google Scholar]
- [108].Polinsky RJ, Clin Ther 1998, 20, 634–647. [DOI] [PubMed] [Google Scholar]
- [109].Bourne Y, Radic Z, Sulzenbacher G, Kim E, Taylor P, Marchot P, J Biol Chem 2006, 281, 29256–29267. [DOI] [PubMed] [Google Scholar]
- [110].Jann MW, Pharmacotherapy 1998, 18, 55–67; discussion 79–82. [PubMed] [Google Scholar]
- [111].Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, Turk D, Biochim Biophys Acta 2012, 1824, 68–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Palermo C, Joyce JA, Trends Pharmacol Sci 2008, 29, 22–28. [DOI] [PubMed] [Google Scholar]
- [113].Desmarais S, Masse F, Percival MD, Biol Chem 2009, 390, 941–948. [DOI] [PubMed] [Google Scholar]
- [114].Marquis RW, Ru Y, LoCastro SM, Zeng J, Yamashita DS, Oh HJ, Erhard KF, Davis LD, Tomaszek TA, Tew D, Salyers K, Proksch J, Ward K, Smith B, Levy M, Cummings MD, Haltiwanger RC, Trescher G, Wang B, Hemling ME, Quinn CJ, Cheng HY, Lin F, Smith WW, Janson CA, Zhao B, McQueney MS, D’Alessio K, Lee CP, Marzulli A, Dodds RA, Blake S, Hwang SM, James IE, Gress CJ, Bradley BR, Lark MW, Gowen M, Veber DF, J Med Chem 2001, 44, 1380–1395. [DOI] [PubMed] [Google Scholar]
- [115].Jerome C, Missbach M, Gamse R, Osteoporos Int 2011, 22, 3001–3011. [DOI] [PubMed] [Google Scholar]
- [116].Kramer L, Turk D, Turk B, Trends Pharmacol Sci 2017, 38, 873–898. [DOI] [PubMed] [Google Scholar]
- [117].Desmarais S, Black WC, Oballa R, Lamontagne S, Riendeau D, Tawa P, Duong LT, Pickarski M, Percival MD, Mol Pharmacol 2008, 73, 147–156; J. P. Falgueyret, S. Desmarais, R. Oballa, W. C. Black, W. Cromlish, K. Khougaz, S. Lamontagne, F. Masse, D. Riendeau, S. Toulmond, M. D. Percival, J Med Chem 2005, 48, 7535–7543. [DOI] [PubMed] [Google Scholar]
- [118].Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, DaSilva C, Santora AC, Ince BA, J Bone Miner Res 2010, 25, 937–947. [DOI] [PubMed] [Google Scholar]
- [119].Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong LT, Falgueyret JP, Kimmel DB, Lamontagne S, Leger S, LeRiche T, Li CS, Masse F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Therien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC, Bioorg Med Chem Lett 2008, 18, 923–928. [DOI] [PubMed] [Google Scholar]
- [120].Law S, Andrault PM, Aguda AH, Nguyen NT, Kruglyak N, Brayer GD, Bromme D, Biochem J 2017, 474, 851–864. [DOI] [PubMed] [Google Scholar]
- [121].Mullard A, Nat Rev Drug Discov 2016, 15, 669. [DOI] [PubMed] [Google Scholar]
- [122].Bromme D, Panwar P, Turan S, Expert Opin Drug Discov 2016, 11, 457–472. [DOI] [PubMed] [Google Scholar]
- [123].Baugh M, Black D, Westwood P, Kinghorn E, McGregor K, Bruin J, Hamilton W, Dempster M, Claxton C, Cai J, Bennett J, Long C, McKinnon H, Vink P, den Hoed L, Gorecka M, Vora K, Grant E, Percival MD, Boots AM, van Lierop MJ, J Autoimmun 2011, 36, 201–209; K. Deschamps, W. Cromlish, S. Weicker, S. Lamontagne, S. L. Huszar, J. Y. Gauthier, J. S. Mudgett, A. Guimond, R. Romand, N. Frossard, M. D. Percival, D. Slipetz, C. M. Tan, Am J Respir Cell Mol Biol 2011, 45, 81–87; H. Yang, M. Kala, B. G. Scott, E. Goluszko, H. A. Chapman, P. Christadoss, J Immunol 2005, 174, 1729–1737. [DOI] [PubMed] [Google Scholar]
- [124].Vasiljeva O, Reinheckel T, Peters C, Turk D, Turk V, Turk B, Curr Pharm Des 2007, 13, 387–403. [DOI] [PubMed] [Google Scholar]
- [125].Rupanagudi KV, Kulkarni OP, Lichtnekert J, Darisipudi MN, Mulay SR, Schott B, Gruner S, Haap W, Hartmann G, Anders HJ, Ann Rheum Dis 2015, 74, 452–463. [DOI] [PubMed] [Google Scholar]
- [126].Knezevic NN, Cicmil N, Knezevic I, Candido KD, Expert Opin Investig Drugs 2015, 24, 1631–1646. [DOI] [PubMed] [Google Scholar]
- [127].Mikhaylov G, Mikac U, Magaeva AA, Itin VI, Naiden EP, Psakhye I, Babes L, Reinheckel T, Peters C, Zeiser R, Bogyo M, Turk V, Psakhye SG, Turk B, Vasiljeva O, Nat Nanotechnol 2011, 6, 594–602. [DOI] [PubMed] [Google Scholar]
- [128].Wang Z, Watt W, Brooks NA, Harris MS, Urban J, Boatman D, McMillan M, Kahn M, Heinrikson RL, Finzel BC, Wittwer AJ, Blinn J, Kamtekar S, Tomasselli AG, Biochim Biophys Acta 2010, 1804, 1817–1831. [DOI] [PubMed] [Google Scholar]
- [129].MacKenzie SH, Schipper JL, Clark AC, Curr Opin Drug Discov Devel 2010, 13, 568–576. [PMC free article] [PubMed] [Google Scholar]
- [130].Shi Y, Protein Sci 2004, 13, 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Callus BA, Vaux DL, Cell Death Differ 2007, 14, 73–78. [DOI] [PubMed] [Google Scholar]
- [132].Hacker HG, Sisay MT, Gutschow M, Pharmacol Ther 2011, 132, 180–195; I. N. Lavrik, A. Golks, P. H. Krammer, J Clin Invest 2005, 115, 2665–2672. [DOI] [PubMed] [Google Scholar]
- [133].Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA, Proc Natl Acad Sci U S A 2004, 101, 12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gao J, Wells JA, Chem Biol Drug Des 2012, 79, 209–215; J. M. Scheer, M. J. Romanowski, J. A. Wells, Proc Natl Acad Sci U S A 2006, 103, 7595–7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gao X, Le X, Costa DB, Expert Rev Anticancer Ther 2016, 16, 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Drahl C, Cravatt BF, Sorensen EJ, Angew Chem Int Ed Engl 2005, 44, 5788–5809. [DOI] [PubMed] [Google Scholar]
- [137].Guerciolini R, Int J Obes Relat Metab Disord 1997, 21 Suppl 3, S12–23. [PubMed] [Google Scholar]
- [138].Joharapurkar AA, Dhanesha NA, Jain MR, Diabetes Metab Syndr Obes 2014, 7, 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Flatt PR, Bailey CJ, Green BD, Front Biosci 2008, 13, 3648–3660. [DOI] [PubMed] [Google Scholar]
- [140].Barnett A, Int J Clin Pract 2006, 60, 1454–1470. [DOI] [PubMed] [Google Scholar]
- [141].Jang J, Son JB, To C, Bahcall M, Kim SY, Kang SY, Mushajiang M, Lee Y, Janne PA, Choi HG, Gray NS, Eur J Med Chem 2017, 136, 497–510; A. Rossi, P. Maione, P. C. Sacco, A. Sgambato, F. Casaluce, M. L. Ferrara, G. Palazzolo, F. Ciardiello, C. Gridelli, Int J Oncol 2014, 45, 499–508; N. Karachaliou, R. Rosell, Cancer Biol Med 2014, 11, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, Janne PA, Eck MJ, Nature 2016, 534, 129–132; H. Patel, R. Pawara, A. Ansari, S. Surana, Eur J Med Chem 2017, 142, 32–47; A. R. Johnson, P. B. Kohli, A. Katewa, E. Gogol, L. D. Belmont, R. Choy, E. Penuel, L. Burton, C. Eigenbrot, C. Yu, D. F. Ortwine, K. Bowman, Y. Franke, C. Tam, A. Estevez, K. Mortara, J. Wu, H. Li, M. Lin, P. Bergeron, J. J. Crawford, W. B. Young, ACS Chem Biol 2016, 11, 2897–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]