

Abstract
Hydroformylation utilizes dihydrogen, carbon monoxide, and a catalyst to transform alkenes into aldehydes. This work applies chiral bisdiazaphospholane (BDP) and bisphospholanoethane (BPE)-ligated rhodium complexes to the hydroformylation of a variety of alkenes to produce chiral tetrasubstituted aldehydes. 1,1’-Disubstituted acrylates bearing electron-withdrawing substituents undergo hydroformylation under mild conditions (1 mol% catalyst/BDP ligand, 150 psig gas, 60 °C) with high conversions and yields of tetrasubstituted aldehydes (e.g., 13:1 regioselectivity, 85% ee, and <1% hydrogenation for 1-fluoromethylacrylate). The scope also encompasses both acyclic 1,1’-disubstituted and trisubstituted, electron-poor alkenes, as well as di- and trisubstituted alkenes comprised of small rings with exocyclic and endocyclic unsaturation. For example, 1-methylene-β-lactam furnished the tetrasubstituted aldehyde with 98% selectivity and up to 83% ee. Notably, chiral trisubstituted bicyclic methyleneaziridines are transformed with >99% regioselectivity and >19:1 diastereoselectivity to tetrasubstituted aldehydes at rates >50 catalyst turnovers/hour. NMR studies of the non-catalytic reaction of HRh(BDP)(CO)2 with methyl 1-fluoroacrylate enable interception of tertiary alkyl-rhodium intermediates, demonstrating migratory insertion to acyl species is slower than formation of secondary and primary alkyl-rhodium intermediates. Overall, these investigations reveal how the interplay of sterics, electronics, and ring strain are harnessed to provide access to valuable α-tetrasubstituted aldehyde synthetic building blocks by promoting branched-selective hydroformylation.
Graphical Abstract

Introduction
Hydroformylation is a well-established commodity process for the synthesis of linear aldehydes from alkenes, carbon monoxide and dihydrogen.1 However, applications of the branch-selective asymmetric hydroformylation (AHF) to the syntheses of chiral aldehydes, which serve as useful synthons in the production of pharmaceuticals and fine chemicals, are underdeveloped. More recently, successful AHF of diverse monosubstituted alkenes2 have been reported using Rh complexes supported by chiral ligands (Figure 1) that include BisDiazaphos3 (L1), Ph-BPE (L2), Chiraphos (L3), DIOP (L4), BINAP (L5) and P-chirogenic ligands such as QuinoxP* (L6).4
Figure 1.

Common ligands for asymmetric hydroformylation.
In comparison to AHF reactions of monosubstituted alkenes, the analogous transformations of 1,1’- and 1,2-disubstituted alkenes have been significantly more challenging and display limited scope.5 In particular, 1,1’-alkene AHF reactions are scarcely reported, proceed more slowly than those of mono- or 1,2-disubstituted alkenes, and tend to favor linear selectivity (according to Keuleman’s rule).6 Despite the volume of literature describing enantioselective hydroformylation, only four examples of distinct 1,1’-disubstituted alkenes undergoing branch-selective AHF have been reported (Scheme 1).7 The groups of Gladiali,8 Alper,9 and Breit10 achieved successful AHF reactions of N-acylaminoacrylate, α-methylene-γ-butyrolactone and 1,3-dichloro-5-(3,3,3-trifluoroprop-1-en-2-yl)benzene, respectively. These reactions proceed with high branch-selectivity, but require high pressures and furnish only modest enantioselectivities. Alkene hydrogenation is a common side reaction, especially in the case of the acrylate derivatives. Buchwald11 found that Rh(CO)2acac supported by a (R,R)-QuinoxP* ligand promotes AHF of 1-(trifluoromethyl)ethenyl acetate to deliver the aldehyde in 91% ee; further oxidation furnished the 2-trifluoromethyl lactic acid in 99% ee. Finally, Zhang12 achieved the same transformation of 1-(trifluoromethyl)ethenyl acetate using a BIBOP ligand to give aldehyde in 80% ee (not shown).
Scheme 1.

Known examples of AHF of 1,1’-disubstituted alkenes.
The lack of branch-selective AHF reactions for the construction of tetrasubstituted, stereogenic centers is unfortunate, as this remains a formidable challenge in organic synthesis. The prevalence of quaternary methyl-bearing stereocenters in numerous drug candidates (Figure 2) and bioactive natural products inspired us to target α-tetrasubstituted aldehydes as particularly attractive intermediates, due to the synthetic versatility of the carbonyl group and the inability of the stereogenic center to racemize via enolization. Methods reported to generate enantioenriched α-tetrasubstituted aldehydes include organocatalysis13, chiral auxiliary-controlled14 or Pd-catalyzed asymmetric alkylations,15 reactions of prochiral enol derivatives,16 and stereocontrolled cycloadditions.17 However, these approaches generally suffer from limited substrate scope, lack of predictable regio- and stereoselectivity, and/or low cost-efficiency. Thus, branch-selective AHF of 1,1’-disubstituted or trisubstituted alkenes might offer a convenient strategy to produce enantioenriched α-tetrasubstituted aldehydes, provided the scope of the chemistry can be expanded.
Figure 2.

Representative examples of drugs and bioactive molecules with quaternary stereocenters.
Previous studies of AHF from the Landis group showed that 1,2-disubstituted alkenes bearing groups that withdraw electron density through the σ framework, including enol esters, enamides, vinyl arenes, vinyl fluorides or acrylates, did not follow the general steric preference for the linear aldehyde products and instead favored the branched regioisomer.2b,5 We hypothesized that strong electron-withdrawing groups might also promote the branched-selective AHF of a range of 1,1’-disubstituted alkenes to give tetrasubstituted aldehydes, a proposal that is supported by the results in Scheme 1. As a complement to electronic control, we were also curious to explore the potential of ring strain to improve the reactivity and branch-selectivity of Rh-catalyzed hydroformylations. Herein, we report highly selective and practical AHF reactions of diverse 1,1’-disubstituted and 1,1’,2-trisubstituted alkenes to furnish enantioenriched α-tetrasubstituted aldehydes.
Results and discussion
Electronic control of regioselectivity for the AHF of 1,1’-disubstituted alkenes to produce α-tetrasubstituted aldehydes.
Initial studies focused on methyl 2-fluoroacrylate 1 (Table 1), a commercially available compound expected to furnish the tetrasubstituted aldehyde 1b, due to the presence of the two electron-withdrawing substituents on the alkene. The α-fluoroaldehyde 1b has high synthetic value as a β-fluoroamine precursor, a motif occurring in several drug candidates.18 Current approaches to 1b rely on stereoselective, organocatalytic fluorination methods, but these strategies display limited scope and utilize expensive catalysts and non-atom-economic fluorinating agents.19 Successful branch-selective AHF would provide a selective and efficient route to 1b from commercially available starting materials.
Table 1.
Ligand screening in AHF of Me-2-fluoroacrylate 1.
| entry | ligand | % conversiona | % ee (1b)b | ||
|---|---|---|---|---|---|
| 1b | 1l | 1h | |||
| 1 | L1 | 96 | <1 | 3 | 85 |
| 2 | L2 | 55 | <1 | 5 | 84 |
| 3 | L3 | <1 | 0 | 0 | --- |
| 4 | L4 | 55 | <1 | 1 | 40 |
| 5 | L5 | <1 | 0 | 0 | --- |
| 6 | L6 | 18 | <1 | <1 | 71 |

Determined via 1H NMR of crude reaction mixture.
Determined via chiral GC of crude reaction mixture.
Ligands L1-L6 (Figure 1) were investigated for the AHF of 1 in the presence of a Rh(CO)2acac catalyst precursor at 400 psi CO/H2 (7:1) and 60 °C. Under these mild reaction conditions, L1 and L2 display high regio- and enantioselectivities (entries 1–2), with L1 delivering a conversion of 96% of 1 after 3 h. Ligands L4 and L6 (entries 4, 6) afford high branch-selectivity, but modest ee; L3 and L5 (entries 3, 5) show no appreciable conversion of 1 and only starting material was recovered. Given these results, L1 and L2 were chosen for further investigations.
The effects of CO/H2 ratios and temperature on the AHF of 1 using L1 are highlighted in Table 2. A general problem with AHF of 1,1’-disubstituted alkenes is competing hydrogenation; we hypothesized that a gas mixture enriched in CO, as compared to H2, would favor reaction of tetrasubstituted Rh-alkyl intermediate to give the aldehyde, rather than the hydrogenation, product. Higher CO partial pressures (entry 1 vs. entries 2–3) and lower temperatures (entry 3) do lead to higher hydroformylation:hydrogenation ratios, with a slight increase in ee noted at the lower temperature. Because the results at 150 psi (entry 1) revealed selectivities similar to 350 psi, all remaining studies were conducted at 150 psi in glass pressure bottles, a more practical and less expensive alternative to metal bomb reactors.
Table 2.
CO pressure and temperature effect in the AHF of 1.
| entry | Pco (psi) | PH2 (psi) | temp | 1b:1ha | % ee (1b)b |
|---|---|---|---|---|---|
| 1 | 100 | 50 | 60 °C | 13:1d | 85 |
| 2 | 300 | 50 | 60 °C | 32:1 | 85 |
| 3c | 300 | 50 | 40 °C | >40:1 | 89 |

Determined via 1H NMR of crude reaction mixture.
Determined via chiral GC of the crude reaction mixture.
77% conversion of 1.
ee of 1h is >90%.
Using the conditions established in Tables 1–2, we examined the scope of α-substituted acrylates, many of which are commercially available, in AHF (Table 3). This substrate class was chosen due to previous results showing that ester groups in 1,2-disubstituted alkenes could direct the regioselectivity in hydroformylation.5 Acrylates 1-10 were tested in the presence of both (S,S,S)-BisDiazaphos (L1) and (S,S)-Ph-BPE (L2). As previously mentioned, the methyl ester of 2-fluoropropenoic acid 1 (entry 1) gave excellent selectivity for the branched aldehyde 1b and a good ee of 85% using L1. In contrast, ligand L2 exhibits poor selectivity and conversion. With L1, replacement of the F substituent with the CF3 group of trifluoromethyl acrylate 2 (entry 2) affords modest selectivity for the branched aldehyde 2b; however, significant amounts of 2h were also observed. Comparison between 1 and 2 suggests that smaller electron-withdrawing groups are required for branch-selective hydroformylation; larger groups may slow the rate of desired CO migratory insertion in the alkyl-Rh intermediate in favor of competing hydrogenation.
Table 3.
AHF of α,β-unsaturated esters.
| entry | alkene | L | time (h) |
bb | % yielda l |
h | %
ee (b)c |
entry | alkene | L | time (h) |
bb | % yielda l |
h | 5
ee (b)c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |  |
L1 L2 |
2 2 |
90d (72) 12d |
<1 <1 |
3 9 |
85 85 |
6 |  |
L1 | 2 | 0d | 10 | 2 | nd |
| 2 |  |
L1 L2 |
2 2 |
43e 0 |
<1 0 |
57 >99 |
15 0 |
7 |  |
L1 L2 |
72 72 |
73d (61) 53d |
5 1 |
<1 <1 |
90 89 |
| 3 |  |
L1 L2 L1f |
2 2 14 |
>98 (86) 47d 96 |
<1 <1 0 |
<1 20 4 |
73 76 85 |
8 |  |
L1 L2 |
24 24 |
60d 87d (74) |
16 2 |
<1 3 |
na na |
| 4 |  |
L1 L2 |
24 24 |
28d 4 |
3 0 |
20 3 |
nd nd |
9 |  |
L1 L2 |
24 24 |
5g 26g |
22 7 |
<1 1 |
nd nd |
| 5 | 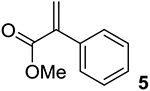 |
L1 L2 |
24 24 |
65d (51) 15d |
14 1 |
21 80 |
13 nd |
10 |  |
L1 L2 |
24 24 |
39g 93 (78) |
15 2 |
<1 5 |
25 95 |

Determined by crude 1H NMR vs. mesitylene.
Isolated yield In parenthesis after 2 steps (AHF, then Wittig olefination).
Determined via GC analysis of crude mixture or by 1H NMR after diastereomeric Imine formation with a chiral amine.
Remaining mass balance Is unreacted starting material.
350 psi CO/ 50 psi H2,
Reaction at 40 °C.
Remaining mass balance Is Isomerization of the starting material to a trisubstituted alkene.
The use of α-acetoxy acrylate 3 (entry 3) also results in excellent branched selectivity to deliver 3b in 98% yield over 2 h in the presence of Rh/L1; although Rh/L2 gives good branched selectivity, the yield of 3b is modest and significant hydrogenation to 3h is observed. Lowering the temperature from 60 to 40 °C improves the ee from 73% (entry 2) to 85% ee. Interestingly, the nature of the group on the enol ester has a significant impact on the ee; while the Me group in 3 gives 73% ee, replacing Me with Ph lowers the ee of the branched product to 16% ee with L1 (see the SI for details). Replacement of the enol ester with an amido group in 4 (entry 4) gives poor reactivity and selectivity, yielding significant hydrogenation product 4h in addition to the desired 4b. AHF of phenyl acrylate 5 (entry 5) proceeds predominantly with branched selectivity to give 5b in the presence of Rh/L1 (65% conversion after 24 h), but with poor enantiocontrol (13% ee). Rh/L2 largely gives hydrogenation to 5h under the same conditions. These results again point to the need for a small R group to achieve good selectivity for hydroformylation over hydrogenation.
The exclusive linear selectivity using 6 (Table 3, entry 6) gave us an opportunity to examine the reason behind the lack of branched product 6b (Scheme 2). Two general scenarios can be envisioned; in the first, only the linear Rh-alkyl intermediate is formed, which goes on to form the linear aldehyde 6l (Scheme 2A). A second scenario involves formation of both linear and branched alkyl intermediates, but only the linear one goes on to form 6l, while the branched one undergoes β-hydride elimination to regenerate 6. To probe if formation of the branched Rh-alkyl intermediate is reversible, deuterioformylation of 6 was conducted (Scheme 2B). This technique has been used to probe the reversibility of intermediate formation in similar reactions of 1,1’-diphenylethene20 and monosubstituted alkenes.21
Scheme 2.

Linear-selective hydroformylation (A) and deuterioformylation of 6 (B).
Incorporation of deuterium at the terminal vinyl position of 6 to yield 6-D would be indicative of reversible formation of the branched alkyl intermediate. Indeed, analysis of the crude deuterioformylation mixture at low conversion of 6 (3%) revealed two major products: the expected deuterated linear aldehyde 6l-D and the deuterated alkene 6-D, in an approximate ratio of 1.2:1. Scrambling of D into the substrate indicates that the tertiary alkyl-Rh intermediate forms, but reverts via β-hydride elimination more rapidly than the desired migratory insertion of CO that would produce the tetrasubstituted aldehyde product.
The scope of AHF to give tetrasubstituted aldehydes is not limited to alkenes bearing two electron-withdrawing groups. The methoxy-substituted acrylate 7 (entry 7) proceeds with excellent branched selectivity and 90% ee with both L1 and L2, displaying 73% and 53% conversion of 7 after 72 h, respectively. Hydroformylation of the less electronically activated methyl methacrylate22 8 (entry 8) provides excellent regioselectivity in the presence of Rh/L2. The AHF again proved sensitive to increased steric demand, as reaction of ethyl methacrylate 9 (entry 9) leads to poor branched selectivity, due to isomerization of the starting material to a trisubstituted alkene. Hydroformylation of 9 at 40 °C increased the extent of isomerization. Interestingly, hydroformylation of 8–10 with L2 results in greater reactivity and a higher branched-to-linear selectivity than with L1, although the reasons for this are not yet clear. In the case of 10 (entry 10), Rh/L2 afforded the highest reported ee to date for this alkene at 95% ee, contrasting with the 36% ee reported by Alper with the same substrate (see Scheme 1).9
Vinyl acetate derivatives also produce tetrasubstituted aldehydes under AHF conditions (Table 4). As shown in Table 3, entry 3 and Table 4, entry 1, AHF of methyl-2-acetoxyacrylate 3 with Rh/L1 proceeds with excellent selectivity for the tetrasubstituted aldehyde 2b and >70% ee. AHF of 1-(trifluoromethyl)vinyl acetate 11 (entry 2), previously investigated by Buchwald using ligand L6,11 affords high regioselectivity with both L1 and L2 (entry 2) and good ee of 85% and 92%, respectively. The AHF of 1-cyanovinyl acetate 12 (entry 3) gave 95% conversion to the tetrasubstituted aldehyde in 61% ee in the presence of Rh/L1, lower than the 85% ee observed for 11; however, lower reaction temperatures would be expected to increase the ee. Lastly, AHF of isopropenyl acetate 13 (entry 4) with Rh/L1 resulted in modest reactivity and selectivity for the tetrasubstituted aldehyde (29%), illustrating the importance of having at least one moderately electron-withdrawing group installed on the alkene precursor to achieve good branched selectivity and ee.
Table 4.
AHF of enol ester 1,1’-disubstituted alkenes.
| entry | alkene | L | time (h) |
bb | % yielda l |
h | % ee (b)c |
|---|---|---|---|---|---|---|---|
| 1 |  |
L1 L2 |
2 2 |
>98 (86) 47d |
<1 <1 |
<1 20 |
73 76 |
| 2 |  |
L1 L2 |
24 24 |
83d (71) 97d |
1 1 |
3 <1 |
85 92 |
| 3 |  |
L1 L2 |
2 2 |
95 (77) 5 |
0 0 |
5 5 |
61 nd |
| 4 |  |
L1 L2 |
24 24 |
29d 2d |
7 1 |
20 1 |
na na |

Determined by crude 1H NMR vs. mesitylene.
Isolated yield in parenthesis after 2 steps (AHF, Wittig olefination).
Determined via GC analysis of crude mixture or by 1H NMR after imine formation with a chiral amine.
Remaining mass balance is unreacted substrate.
The data in Tables 3 and 4 are helpful in determining which substituents efficiently yield α-tetrasubstituted aldehydes in AHF. General reactivity trends reveal that electron-deficient alkenes with small substituents result in the best reaction rates, regioselectivity and ee. The presence of the −CO2Me and –OAc groups on the alkene of 3 (Table 3, entry 3; Table 4, entry 1) give excellent b:l ratios with L1, along with promising ee. A direct comparison of the effect of –CO2Me vs. –OAc as a directing group was made between 2 (Table 3, entry 2) and 11 (Table 4, entry 2); both substrates exhibit good branched-to-linear selectivity, albeit with significant hydrogenation of 2 to 2h. The ee of 2b (15%) is also low, compared to the ee for 11b (85% with L1, 92% with L2). Another direct comparison could be made between 8 (Table 3, entry 8) and isopropenyl acetate 13 (Table 4, entry 4). In this case, the lower electronic activation provided by the Me substituent resulted in a better 8b:8l ratio (>40:1 with L2) as compared to 13b:13l (~2:1–4:1 at low conversion), again pointing to the benefit of having at least one strongly electron-withdrawing group present on the alkene.
Enol esters and vinyl fluorides conjugated with ester and cyano groups (Table 3, entries 1, 3; Table 4, entries 1, 3) show remarkably fast rates; full conversion of starting material is achieved within 2 h. In contrast, acrylates substituted with aryl, ether or alkyl groups require 24 h or more for satisfactory conversion at 1 mol % catalyst loading and 60 °C. Interestingly, rates obtained with 1,1’-alkenes (up to 50 turnovers/h, Table 3, entries 1, 3) can surpass those previously observed for 1,2-disubstituted enol esters and enamides (up to 17 turnovers/h) under the same conditions.5
In general, ligand L1 yields faster and more selective AHF than L2, although L2 shows better selectivity for alkyl-substituted acrylates, such as 8–9 (Table 3, entries 8–9) and α-methylene-ɣ-butyrolactone (entry 10). Thus, when screening new substrates, it is advisable to try both L1 and L2.
Attempted hydroformylation of 1,1’,2-trisubstituted alkenes.
A limitation of the AHF of 1,1’-disubstituted alkenes is that a methyl substituent is always present in the aldehyde product. Given the fast reaction rates in the AHF of 1,1’-disubstituted alkenes 3 and 12, we thought that electronically activated trisubstituted alkenes might perhaps be transformed to tetrasubstituted aldehydes under similar reaction conditions. Unfortunately, our initial attempts with a series of electronically activated alkenes were unsuccessful (Table 5). Entries 1 and 2 showed no appreciable conversion of either 14 or 15 to the desired tetrasubstituted aldehyde, while 16 formed only the hydrogenation product in excellent yield (entry 3).
Table 5.
Attempted AHFs of trisubstituted alkenes.
| entry | alkene | L | time (h) |
b | % yielda l |
h | % ee (b) |
|---|---|---|---|---|---|---|---|
| 1 |  |
L1 L2 |
24 24 |
7c <1d |
0 0 |
2 0 |
na na |
| 2 |  |
L1 | 24 | <1 | 0 | 0 | na |
| 3 |  |
L1 L2 |
24 24 |
0 0 |
0 0 |
>99 >99 |
na na |

Determined via crude 1H NMR vs. mesitylene.
400 psi 5:1 CO/H2.
The remaining mass balance is unreacted starting material.
The remaining mass balance Is a mixture of unreated starting material (82%) and isomerized starting material (18%).
Exploration of ring strain to produce α-tetrasubstituted aldehydes via regioselective AHF.
The disappointing results in Table 5 required us to either identify a more active catalyst or improve the reactivity of the alkene. We hypothesized that ring strain might both decrease the steric burden on the reaction and increase alkene reactivity. Exocyclic alkenes 17–18 (Table 6, entries 1–2) gave no branched selectivity and only linear products were observed. However, 19 gave small amounts (13%) of the tetrasubstituted aldehyde product (entry 3), suggesting that strain may play some role in controlling the regioselectivity of the hydroformylation reaction. The corresponding acyclic versions of these alkenes gave either poor conversion to the linear aldehyde product or significant isomerization (see compounds 39–40 in the Supporting Information for further details), due to the high preference for β-hydride elimination of the alkyl-Rh intermediate, as compared to productive CO insertion to form the branched aldehyde.
Table 6.
Exploring combinations of electronic effects and ring strain to control the regioselectivity of hydroformylation.
| entry | alkene | L | time (h) |
α | % yielda,b β |
h |
dr (or
ee (α)c |
entry | alkene | L | time (h) |
α aldehyde | α | % yielda,b β |
h |
dr (or
ee) (α)c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |  |
L1 | 24 | 0e | 82(5d) | 0(2f) | na | 11 |  |
L1 L2 |
3 15 |
 |
66 94 (87) |
34 6 |
0 0 |
>19:1 >19:1 |
| 2 |  |
L1 | 24 | 0 | 99(<1d) | 0 | na | 12 |  |
L1 L2 |
3 15 |
 |
45 84(72) |
55 164 |
0 0 |
na na |
| 3 |  |
L1 | 24 | 13 | 83(<1d) | 4 | na | 13 |
 |
L1 L2 |
3 15 |
 |
57 83(79) |
43 17 |
0 0 |
na na |
| 4 |  |
L1 L2 L2 |
2 2 2 |
98 98 26e |
1 <1 <1 |
1 1 0 |
40 71 83 |
14 |  |
L1 | 24 | 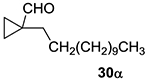 |
17f | 0 | 0 | na |
| 5 |  |
L1 L2 |
2 2 |
2e 12e |
7 4 |
7 2 |
na na |
15 |  |
L1 L2 |
2 2 |
 |
88 10e |
12 32 |
0 0 |
na na |
| 6 |  |
L1 | 24 | 85 | 15 | 0 | nd | 16 |  |
L1 L2 |
2 2 |
 |
99 (86) 25e,i,j |
<1 0 |
0 0 |
>19:1 >19:1 |
| 7 |  |
L1 L1 L2 |
24e 24g 24 |
6 <1 <1 |
0 0 0 |
89 99 99 |
nd nd nd |
17 |  |
L1 L2 |
2 2 |
 |
99 (84) <1e | <1 0 |
0 0 |
>19:1 nd |
| 8 |  |
L1 L2 |
24 24 |
0h 0e,h |
45(22) 17 |
0 0 |
nd nd |
18 |  |
L1 L2 |
2 2 |
 |
99 (84) 7j,k |
<1 0 |
0 0 |
>19:1 >19:1 |
| 9 |  |
L1 L2 |
3 15 |
61 93(86) |
39 15 |
0 0 |
>19:1 17.5:1 |
19 |  |
L1 L1 L2 |
2 4 - |
 |
14 99 - |
<1 <1 - |
0 0 |
>19:1 >19:1 - |
| 10 |  |
L1 L2 |
3 15 |
61 85(71) |
39 15 |
0 0 |
>19:1 18.7:1 |
20 |  |
L1 L2 |
 |
nd nd |
||||

Determined via crude 1H NMR vs. mesityiene.
Isolated yield in parenthesis.
Determined by GC analysis of crude mixture or by 1H NMR after diastereomeric imine formation with a chiral amine; dr determined by NMR.
Yield for isomerization to trisubstituted alkene.
Remaining mass balance is starting material.
Remaining mass balance is hydroformylation of isomerized substrate.
400 psi CO/IOO psi H2.
The remaining mass balance consists of enolized aldehyde.
The remaining mass balance is isomerization of alkene.
The remaining mass balance is a minor aldehyde.
Run with 5 mol % of catalyst.
The results in entries 1–3 demonstrated that ring strain alone is not sufficient to promote effective branch-selective hydroformylation. We therefore examined the addition of electron-withdrawing substituents to a series of strained 1,1’- disubstituted alkenes (Table 6, entries 4, 6–20). In many cases, these substrates exhibit enhanced reactivity in hydroformylation, typically reaching full conversion within 2 h using 1 mol % of catalyst. For example, hydroformylation of the β-lactam 20 gave excellent branched selectivity (98%) with Rh/L1 and Rh/L2 (entry 4); the ee of 20α using Rh/L2 increases from 71% to a promising 83% when the temperature is decreased from 60 °C to 40 °C. AHF of the analogous linear 2-methyl-N-phenylacrylamide 21 (entry 5) was conducted for the purposes of comparison, but resulted in both low reactivity (16–18% conversion) and poor branched selectivity, highlighting the influence of ring strain on the success of branch selective hydroformylation.
To further test the limits of this chemistry, a series of 1,1’,2-trisubstituted cyclobutenes (Table 6, entries 6–8) were tested to assess whether combining ring strain and electronic activation would enable a substrate class previously resistant to hydroformylation to undergo successful reaction (Table 5, vide supra). To our delight, hydroformylation of 2-ethylidene-cyclobutan-1-one 22 (Table 6, entry 6) exhibits complete conversion within 24 h and afforded the tetrasubstituted aldehyde in 85% yield. However, increasing the bulk of the alkene substituent from Me in 22 to cyclohexyl in 23 (entry 7) affords only 6% of the tetrasubstituted aldehyde, along with 89% of the hydrogenated product. Presumably, the CO migratory insertion step slows as a result of the increased steric bulk of the tertiary alkyl-Rh, thus favoring hydrogenolysis. Finally, the competing effects of strain and electronics in four-membered rings are highlighted using 24 (entry 8). The reaction produces only 24β in poor yields, suggesting that electronics may overrule ring strain in terms of dictating the regioselectivity of the hydroformylation event.
As cyclobutenes in general did not provide sufficient ring strain to strongly favor production of the α-tetrasubstituted aldehyde with broad scope, we turned our attention to methylenecyclopropanes. Interestingly, the aryl-substituted cyclopropenes 25–27 showed promise for dictating regioselectivity. Hydroformylation of 25 with the Rh/L1 catalyst affords 61% yield of the racemic tetrasubstituted aldehyde 25α and 39% of linear aldehyde 25β in excellent dr (>19:1), favoring the syn diastereomer via substrate control. While the regioselectivity was modest, greater selectivity (85% 26α) and dr (17.5:1 syn:anti) is observed using Rh/L2 (entry 9). Similar selectivity is observed for 26–27 (entries 10–11). The aryl group can be viewed as having an electronic, albeit remote, effect on the reaction; thus, we were surprised to discover AHF of the electronically unactivated alkenes 7-methylenebicyclo-[4.1.0]heptane 28 and 8-methylenebicyclo-[5.1.0]octane 29 (entries 12–13) yields 45% and 57% of the branched aldehydes 28α and 29α, respectively, using the Rh/L1 catalyst.
Furthermore, 84% and 83% of the same products are observed with the Rh/L2 catalyst system. This impressive level of branched selectivity unique for electronically unactivated alkenes and clearly emphasizes the important role that ring strain plays in influencing the regioselectivity in 1,1’-disubstituted alkenes. To further test the limits of this chemistry, two 1,1’,2-trisubstituted methylenecyclopropanes 30 and 31 (Table 6, entries 14–15) were tested. Increasing the steric bulk in 30 results in low conversion to the branched product (entry 14); isomerization of the alkene and subsequent hydroformylation of the resulting disubstituted alkene comprises the remaining mass balance. However, strained alkenes that cannot isomerize, such as 31 (entry 15), provide good b:l selectivity using Rh/L1, although the results were poor with Rh/L2 for reasons that are not yet clear.
Finally, a series of chiral bicyclic methyleneaziridines (MA), reported by the Schomaker23–25 group as key intermediates en route to a variety of complex amine stereotriads, were tested in the hydroformylation. The calculated ring strain of bicyclic MAs (~42 kcal/mol), coupled with the unusual enamide-type substituent on the alkene, led us to propose that MA hydroformylation would likely afford tetrasubstituted aldehydes with good rates. Indeed, hydroformylation of 32 (entry 16) affords the desired aldehyde 32α in 2 h using 1 mol % of Rh/L1. Hydroformylation can occur either syn or anti to the stereogenic carbon in 32 (Figure 3) to give two possible diastereomers. The reaction preferentially occurs anti to the more sterically demanding stereogenic carbon under the auspices of substrate control to give the anti diastereomer in >19:1 dr. Varying the R group in trisubstituted MAs (−Me vs. −C5H11 vs. −CH2CH2Ph vs −iPr, entries 16–19, 32–35) does not have a detrimental effect on the regioselectivity or dr of the reaction. However, slower rates are observed for the iPr-MA substrate 35. Tetrasubstituted aldehydes 32α−35α are obtained in excellent yields and >19:1 dr in favor of the anti stereoisomer. Effective transfer of axial-to-point chirality from the original allene precursor enables these compounds to be accessed in enantioenriched form. However, attempts to push the hydroformylation using a bicyclic methyleneaziridine containing a tetrasubstituted alkene 36 (entry 20) were not successful and only intractable mixtures of unidentifiable products were obtained.
Figure 3.

Stereoselective hydroformylation of 32.
Investigating catalyst speciation using low temperature NMR.
The presence of hydrogenated alkene by-products, particularly in the case of the trisubstituted alkene 23 (Table 6, entry 7), suggests a relatively slow CO migratory insertion step for sterically congested tertiary alkyl-Rh intermediates. This situation leads to competitive trapping of the Rh-alkyl intermediate III.5 (Figure 4) with H2 to give III.6 before migratory insertion can occur to ultimately furnish the desired III.8 through the intermediacy of III.7. We hypothesized that the alkyl intermediate may accumulate in high enough concentrations to be detectable by 1H NMR spectroscopy in the absence of H2. This hypothesis was tested by direct NMR observation of the reaction of pre- formed Rh(H)(CO)2(L7) catalyst 37 and 10 equiv of 1 as monitored by 31P{1H} NMR in the absence of H2 at 0 °C and under 1 atm CO (Figure 5). Under passive gas-liquid mixing conditions in a standard NMR tube, 37 (~83 ppm) was rapidly consumed and replaced by two major species, each with two distinct 31P resonances.26 One species comprises a doublet-of-doublet (dd) resonance at 71 ppm (1J P-Rh = 80 Hz, 2J P-P = 21 Hz) and a ddd resonance at 60 ppm (1JP-Rh = 167 Hz, 2JP-P = 21 Hz, 4JP-F = 12 Hz) whereas the second species exhibits a ddd resonance at 75 ppm (1J P-Rh = 81 Hz, 2J P-P = 11 Hz, 3JP-F = 36 Hz) and a ddd at 43 ppm (1JP-Rh = 143 Hz, 2JP-P = 11 Hz, 3JP-F = 29 Hz). The resonances at 71 and 60 ppm were assigned to the branched Rh-acyl species 38acyl and the resonances at 75 and 43 ppm to the branched Rh-alkyl species 38alkyl.
Figure 4.

Mechanistic model to rationalize the observed hydrogenation pathway during alkene hydroformylation.
Figure 5.

31P{1H} monitoring of reaction of [Rh(H)(CO)2(rac-L7)] 37 with 1 (10 equiv) at 0 °C under 1 atm CO in CD2Cl2. Spectra were taken every 3.5 min. Under actual AHF conditions, L7 is slower than L1, but both ligands display similar selectivity (1% Rh/L7, CO/H2 150 psi 1:1, 40 °C, 3 h, [1] = 1.4 M in CH2Cl2: 17% conversion of starting material, >50:1 branched to linear, 11:1 AHF to hydrogenation).
The larger differences in the 31P chemical shifts of 38alkyl vs. 38acyl are consistent with previous observations.21b,c The 1JP-Rh values of 80 and 143 Hz for 38alkyl and 81 and 167 Hz for 38acyl, respectively, are consistent with both intermediates existing as five-coordinate, trigonal bipyramidal complexes. Characteristics of 19F, 19F{1H} and 19F{31P} NMR spectra distinguish between the branched and linear isomers. The presence of quartets corresponding to 3JF-H = 21 Hz and 23 Hz for 38acyl and 38alkyl, respectively, are indicative of branched hydrocarbonyls; the linear complexes would display a 2JF-H coupling. In addition to 38acyl and 38alkyl, minor resonances are visible at 74 and 42 ppm (both ddd) that are slightly upfield from the 38alkyl resonances. The resemblance in chemical shift and coupling constants to 38alkyl suggest that this minor species is the diastereomer of 38alkyl.
Further details of the reaction progress are revealed by cooling a mixture of 38alkyl and 38acyl to −30 °C and following the changes that occur upon warming (Figure 6). At −30 °C, the resonances for the alkyl species 38alkyl sharpens to reveal two sets of resonances that we interpret as diastereomeric alkyls in a ratio of approximately 3:1. Upon slow warming, these peaks broaden somewhat and slowly transform to acyl resonances.26 Acyl formation is not complete unless more CO is added (established by separate experiments), because it is limited by the amount of CO dissolved in solution under passive gas-liquid conditions. Although the diastereomers of the 38acyl are not clearly resolved, it is clear that the major 38alkyl diastereomer is converted to 38acyl faster than the minor diastereomer. These studies establish a rare example of the interception of a trialkyl metal complex intermediate in a catalytic reaction and establish that, compared with secondary and primaryl alkyl-Rh intermediates, CO insertion into the tertiary alkyl-Rh bond is substantially slower. These studies suggest that more detailed kinetic and mechanistic analyses of hydroformylation processes to produce tetrasubstituted aldehydes are feasible.
Figure 6.

31P{1H} monitoring the reaction between [Rh(H)(CO)2(rac-L7)] 37 (83 ppm, 20 mM) with 1 (10 equiv) while temperature increases from −30°C to room temperature. Spectra were collected every 3.5 min.
Conclusions
Tetrasubstituted stereogenic centers occur in numerous drug candidates and natural products that are challenging to synthesize. AHF of 1,1’-disubstituted or 1,1’,2-trisubstituted alkenes is a convenient, atom-efficient strategy for making novel α-tetrasubstituted aldehydes. To date, branched-selective AHF of these substrate classes has been fraught with issues pertaining to control over the regio- and/or stereoselectivity of the reaction. In this work, we have shown that fast and selective hydroformylations of 1,1’-disubstituted alkenes bearing electron-withdrawing groups can be achieved using Rh catalysts supported by either BisDiazaphos or Ph-BPE ligands. Acrylates and acetates in particular displayed unprecedented rates (up to 50 turnovers/h) and selectivities (up to 95% ee and >50:1 branched selectivity) under mild conditions (150 psi CO/H2, 60 °C). NMR studies suggested that alkene insertion to form a branched Rh-alkyl intermediate is fast; however, the CO migratory insertion step is slow, leading to competitive trapping of this species by H2.
We have also reported the first examples to date of selective hydroformylations of 1,1’,2-trisubstituted alkenes, using a combination of ring strain and electronic effects and Rh catalysts supported by either BisDiazaphos or Ph-BPE ligands. The desired α-tetrasubstituted aldehyde products are furnished in good-to-excellent yields. Many of the 1,1’,2-trisubstituted alkenes show fast rates (up to 50 turnovers/h) and selectivities (>50:1 αβ, >19:1 dr) under mild conditions (150 psi CO/H2, 60 °C). NMR studies suggested that the hydroformylation proceeds through a tetrasubstituted Rh-alkyl intermediate; rapid migratory insertion of CO leads to the aldehyde in preference to competing hydrogenation products.
Branch-selective hydroformylation occurs in competition with simple hydrogenation. This work establishes correlations of substrate structure with hydrogenation vs. hydroformylation tendencies. Such correlations will inform future efforts directed towards expanding the scope of AHF of challenging substrates, exploring changes to the reaction conditions to further improve selectivity and decrease hydrogenation and investigations of further mechanistic details of these valuable transformations.
Experimental section
I. General information.
All glassware was either oven-dried overnight at 130 °C or flame-dried, cooled under vacuum and purged with nitrogen before use. Unless otherwise specified, reagents were used as obtained from the vendor without further purification. Mesitylene and (carbethoxymethylene)-triphenylphosphorane were purchased from Sigma. (S)-(+)-1-Methoxy-2-propylamine was obtained from Alfa Aesar. The (S,S)-Ph-BPE ligand (L2) was purchased from either Strem Chemicals or Sigma Aldrich. Synthesis gas (1:1 CO:H2) and CO gas were obtained from Airgas. The asymmetric hydroformylation (AHF) reactions were run in glass pressure bottles purchased from Ace Glass Inc. Reactors were assembled as previously reported.27 The bis-(S,S,S)-DiazaPhos ligand L1 and the Rh complex 37 were synthesized as previously reported.21b
Diethyl ether was distilled from purple Na/benzophenone ketyl. Tetrahydrofuran was distilled from purple Na/benzophenone ketyl or dried over alumni safety columns before use. Dichloromethane, acetonitrile and toluene were dried over CaH2 and distilled before use. All other solvents were purified using “Purification of Laboratory Chemicals”.28 All air- and moisture- sensitive reactions were performed using standard Schlenk techniques under an atmosphere of nitrogen. Analytical thin layer chromatography (TLC) was performed utilizing pre-coated silica gel 60 F254 plates containing a fluorescent indicator. Columns were conducted using a gradient method. KMnO4 was used as stains reagent. Rh(acac)(CO)2] was received from Dow Chemical and purified by passing it through cold toluene. Purified [Rh(acac)(CO)2] was stored in a N2-filled glovebox. Syngas as a 1:1 CO:H2 was obtained from Airgas. All pressures given are gauge pressures unless otherwise noted.
1H NMR, 13C NMR, and 19F NMR spectra were obtained using Bruker-300, Varian-300, Avance III 400, Varian Inova-500, or Varian Unity-500 spectrometers. 13C NMR spectra were obtained using 125 MHz or 75 MHz instruments. 1H NMR spectra chemical shifts were reported in accordance to residual protiated solvent peaks (δ 77.2, 39.5, 128.0 and 137.9 ppm for CDCl3, (CD3)2SO, C6D6, and CD3C6D5, respectively). Accurate mass measurements were acquired at the University of Wisconsin, Madison using a Micromass LCT (electrospray ionization, time-of-flight analyzer or electron impact methods). The NMR and Mass Spectrometry facilities are funded by the NSF CHE-1048642 and NIH 1S10OD020022–1. GC data was obtained on chiral BetaDex 225 column on a Shimadzu analytical GC-2010 Plus equipped with an FID detector. Optical rotation data were obtained on a Randolph digital polarimeter using a 1 mL cell with 0.5 dm path length at room temperature. The sign of the optical rotation is reported for products after Wittig olefination. Silica gel flash column chromatography was performed on Silicycle Siliaflash P60 230–400 mesh, 40–63 μm. Conversion and regioselectivity were determined by crude 1H NMR. Isolated yields are reported after 2 steps: AHF, followed by Wittig olefination of the crude AHF mixture with (carbethoxymethylene)triphenylphosphorane.
Methyl 2-fluoroprop-2-enoate (1).
The alkene was purchased from Alfa Aesar.
Methyl 2-(trifluoromethyl)-propenoate (2).
The alkene was purchased from Alfa Aesar.
Methyl 2-acetoxyacrylate (3).
The alkene was prepared according to a reported literature procedure.29
Methyl 2-acetamidoacrylate (4)
The alkene was purchased from sigma Aldrich.
2-Phenyl-acrylic acid methyl ester (5).
The alkene was prepared according to a reported literature procedure.30
Methyl 2-(2-methylphenyl)acrylate (6).
The alkene was prepared according to a reported literature procedure.30
Methyl 2-methoxylacrylate (7).
The alkene was prepared according to a reported literature procedure.31
Methacrylic acid methyl ester (8).
The alkene was purchased from sigma Aldrich.
Methyl 2-ethylacrylate (9).
The alkene was prepared according to a reported literature procedure.32 1H NMR (500 MHz, CDCl3) δ 6.14 – 6.12 (m, 1H), 5.55 – 5.52 (m, 1H), 3.76 (s, 3H), 2.33 (qt, J = 7.3, 1.3 Hz, 2H), 1.08 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.0, 142.3, 123.7, 51.9, 25.0, 12.8. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C6H11O2 115.0754, Found 115.0752.
α-Methylene-γ-butyrolactone (10).
The alkene was purchased from Sigma Aldrich.
1,1,1-Trifluoro-2-(acetyloxy)-2-propene (11).
The alkene was purchased from Alfa Aesar.
2-Acetoxyacrylonitrile (12).
The alkene was purchased from TCI.
Isopropenyl acetate (13).
The alkene was purchased from Sigma Aldrich.
Methyl (Z)-2-acetyloxybut-2-enoate (14).
The alkene was purchased from Sigma Aldrich.
(Z)-Methyl 3-(4-bromophenyl)-2-fluoroacrylate (15).
The alkene was prepared according to a reported literature procedure.33
3-Methylfuran-2,5-dione (16).
The alkene was purchased from Sigma Aldrich.
Methylenecyclohexane (17).
The alkene was purchased from Acros Organics.
Methylenecyclopentane (18).
The alkene was purchased from TCI.
Methylenecyclobutane (19).
The alkene was purchased from Sigma Aldrich.
3-Methylene-1-phenylazetidin-2-one (20).
The alkene was prepared according to a reported literature procedure.34
2-Methyl-N-phenylacrylamide (21).
The alkene was purchased from TCI.
2-Ethylidenecyclobutan-1-one (22).
The alkene was prepared according to a reported literature procedure.35
(E)-2-Cyclohexylcyclobutanone (23).
The alkene was prepared according to a reported literature procedure.35
(Z)-Ethyl 2-(2-oxocyclobutylidene)acetate (24).
The alkene was prepared according to a reported literature procedure.36 1H NMR (500 MHz, CDCl3) δ 5.58 (pentet, J = 2.3 Hz, 1H), 4.14 (q, J = 7.1 Hz, 2H), 3.13 (m, 2H), 2.87 – 2.78 (m, 2H), 2.09 (pentet, J = 8.0 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.0, 142.3, 123.7, 51.9, 25.0, 12.8. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C8H13O2 141.0910; Found 141.0908.
1-Methylene-2-phenylcyclopropane (25).
The alkene was prepared according to a reported literature procedure.37 1H NMR (500 MHz, CDCl3) δ 7.29 – 7.23 (m, 2H), 7.17 – 7.13 (m, 3H), 5.59 – 5.55 (m, 2H), 2.60 (ddt, J = 9.4, 4.8, 2.0 Hz, 1H), 1.74 (tt, J = 9.2, 2.3 Hz, 1H), 1.21 (ddt, J = 9.3, 4.9, 2.3 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 142.1, 135.6, 128.5, 126.5, 126.0, 104.6, 20.3, 14.7. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C10H11 131.0855; Found 131.0853.
2-(4-Chlorophenyl)-1-methylenecyclopropane (26).
The alkene was prepared according to a reported literature procedure.37 1H NMR (500 MHz, CDCl3) δ 7.24 – 7.18 (m, 2H), 7.12 – 7.03 (m, 2H), 5.60 – 5.55 (m, 2H), 2.56 (ddt, J = 9.2, 4.6, 2.0 Hz, 1H), 1.75 (tt, J = 9.2, 2.3 Hz, 1H), 1.17 (ddt, J = 9.2, 4.9, 2.3 Hz, 1H). 13C NMR (126 MHz, CDCl3) δ 140.6, 135.1, 131.6, 128.5, 127.9, 105.0, 19.7, 14.9. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C10H10Cl 165.0466; Found 165.0464.
1-(p-Methoxyphenyl)-2-methylenecyclopropane (27).
The alkene was prepared according to a reported literature procedure.37 1H NMR (400 MHz, CDCl3) δ 7.13 – 7.03 (m, 2H), 6.86 – 6.75 (m, 2H), 5.57 (q, J = 2.1 Hz, 2H), 3.77 (s, 3H), 2.56 (ddt, J = 9.1, 4.3, 1.9 Hz, 1H), 1.69 (tt, J = 9.2, 2.2 Hz, 1H), 1.20 – 1.06 (m, 1H). 13C NMR (126 MHz, CDCl3) δ 158.0, 135.7, 133.9, 127.0, 113.9, 104.5, 55.4, 19.6, 14.3. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C11H13O 161.0961; Found 161.0460.
7-Methylenebicyclo[4.1.0]heptane (28).
The alkene was prepared according to a reported literature procedure.37 1H NMR (400 MHz, CDCl3) δ 5.35 (t, J = 2.1 Hz, 2H), 1.86 – 1.71 (m, 2H), 1.71 – 1.59 (m, 2H), 1.57 – 1.46 (m, 2H), 1.31 – 1.16 (m, 4H). 13C NMR (126 MHz, CDCl3) δ 142.9, 101.4, 22.9, 21.4, 12.9. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C8H13 109.1012; Found 109.1011.
9-Methylidenebicyclo[6.1.0]nonane (29).
Prepared according to a reported literature procedure.37 1H NMR (500 MHz, CDCl3) δ 5.29 (t, J = 2.1 Hz, 2H), 1.97 (dq, J = 14.7, 3.4 Hz, 2H), 1.76 – 1.64 (m, 2H), 1.63 – 1.50 (m, 2H), 1.47 – 1.27 (m, 4H), 1.11 – 0.97 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 142.1, 101.1, 29.3, 26.4, 25.6, 19.4. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C10H17 137.1325; Found 137.1323.
Dodecylidenecyclopropane (30).
The alkene was prepared according to a reported literature procedure.38 1H NMR (500 MHz, CDCl3) δ 5.81 – 5.69 (m, 1H), 2.16 (m, 2H), 1.43 (m, 2H), 1.36 – 0.98 (m, 20H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 121.0, 118.6, 32.1, 32.0, 29.8, 29.8, 29.8, 29.7, 29.6, 29.5, 29.5, 22.9, 14.3, 2.3, 2.0.
Ethyl cyclopropenylideneacetate (31).
The alkene was prepared according to a reported literature procedure.36 1H NMR (500 MHz, CDCl3) δ 6.22 (p, J = 2.0 Hz, 1H), 4.21 (q, J = 7.2 Hz, 2H), 1.49 – 1.41 (m, 2H), 1.30 (t, J = 7.1 Hz, 3H), 1.27 – 1.18 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 166.4, 145.1, 111.1, 60.3, 14.5, 4.7, 2.2. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C7H11O2 127.0754; Found 127.0752.
(7E)-7-Ethylidene-3-oxa-1-azabicyclo[4.1.0]heptan-2-one (32).
The alkene was prepared according to a reported literature procedure.39
(7E)-7-Hexylidene-3-oxa-1-azabicyclo[4.1.0]heptan-2-one (33).
The alkene was prepared according to a reported literature procedure.39
(7E)-7-Benzylidene-3-oxa-1-azabicyclo[4.1.0]heptan-2-one-ethene (1/1) (34).
The alkene was prepared according to a reported literature procedure.39
II. General method for hydroformylation.
In a nitrogen-filled glovebox, a 15 mL Ace Glass glass pressure bottle equipped with a magnetic stir bar was charged with a THF stock solution of (0.1 M) Rh(acac)(CO)2 and either the bisdiazaphospholane ligand L1 or the (S,S)-Ph-BPE ligand L2 (0.04 or 0.05 M, respectively) using 1000 μL and 200 μL Eppendorf® pipets. The glass bottle was attached to a pressure reactor head and the apparatus was removed from the glovebox. The apparatus was placed inside a fume hood and was charged with 5 pressurization (150 psi)/depressurization (0 psi) cycles to completely replace the nitrogen atmosphere with syngas. The apparatus was filled with either 150 psi of a 1:1 CO:H2 syngas mixture or 90 psi of CO and an additional 60 psi of syngas. The apparatus was placed in an oil bath and the reaction mixture stirred vigorously for 30–60 min to ensure formation of the pre-activated catalyst. The reaction apparatus was then removed from the oil bath and cooled for 5 min. The pressure was reduced to 0 psi and a solution of the alkene in THF or a solution of alkene and mesitylene (1/3 eq) in THF was injected with a gas-tight syringe using a 12” needle. The reaction was then charged using 3 × 150 psi pressurization/0 psi depressurization cycles, taken to a final pressure of 150 psi and placed in the oil bath at the desired temperature. The reaction mixture was vigorously stirred for the specified period of time. The reactor was then removed from the oil bath, cooled to room temperature and depressurized. A crude NMR sample was taken in CDCl3 using 5 or 10 μL of mesitylene as an internal standard. The same crude NMR was used for GC analysis. Since CO is very toxic and H2 is flammable, a well-ventilated fume hood should be used at all times.
III. General procedure for asymmetric hydroformylations with pressures greater than 150 psi.
A CAT24 reactor is charged with either a 100 mM stock solution of Rh(acac)(CO)2 (1% catalyst loading, 48 μL) in toluene or THF, a 20 mM stock solution of ligand (1.2%, 264 μL) in either toluene or THF and the alkene substrate (0.48 mmol) inside a N2 glove box. The reactor is sealed and pressurized inside a fume hood, heated at the desired temperature with stirring at 700 rpm. At the end of the indicated reaction time, the reactor is allowed to cool down for 30 min and then depressurized. The crude reaction mixture is analyzed by 1H NMR and chiral GC analysis.
IV. General Procedure for Wittig olefination of the aldehydes.
The crude mixture of aldehydes from the hydroformylation reaction is transferred to a 50 mL oven-dried Schlenk flask. Under a flow of N2, (carbethoxymethylene)triphenylphosphorane (1.2 eq) is added as a solution in dichloromethane (7 mL) at room temperature. The mixture is allowed to stir at room temperature overnight under N2. The mixture is concentrated under reduced pressure and the residue purified via silica gel chromatography using 20% EtOAc/hexanes as the eluent.
V. General procedure for deuterioformylation.
An oven-dried, 15 mL glass pressure bottle equipped with a stir bar is charged with a 100 mM stock solution of Rh(acac)(CO)2 (1.0 mol% catalyst loading, 56.76 μL) in THF and a 30 mM stock solution of L1 (1.2 mol%, 189.2 μL) in THF inside a N2 glove box. The pressure bottle was then attached to a pressure gauge and placed inside a fume hood. The reactor was subjected to three pressurization (100 psig)/depressurization (20 psig) cycles with CO, taken to a final pressure of 120 psig CO/30 psig D2 and placed in an oil bath at 60 °C for 1 h. The reactor was allowed to cool for 5 min and depressurized to 10 psig. The alkene 2-(2-methylphenyl)acrylic acid methyl ester 6 (0.57 mmol, 100 mg) and the 1,4-bis-trimethylsilylbenzene internal standard (0.1 eq, 6.31 mg) in THF were injected into the reactor using a syringe (the amount of THF was adjusted to furnish a 1.6 M solution of the alkene). The reactor was then pressurized/depressurized 3 times with CO, taken to a final pressure of 120 psig CO/30 psig D2 and placed in the oil bath set at 60 °C for 19 h. The mixture was stirred vigorously, with the stir bar serving to break the gas-liquid interface for good gas-liquid mixing. After the specified reaction time, the reactor was allowed to cool for 30 min and then depressurized. An aliquot of the crude mixture was used for NMR analysis.
VI. General Procedure for Low Temperature Non-catalytic Experiments.27
An oven-dried, 15 mL glass pressure bottle equipped with a stir bar was charged with 37 (60 mg) and 1 mL CD2Cl2 inside a N2 glove box and then attached to a pressure gauge. Inside a fume hood, the reactor was subjected to three pressurization (150 psig)/depressurization (20 psig) cycles with syngas, then pressurized to 150 psig syngas and placed in an oil bath at 60 °C for 3 h. The reactor was allowed to cool for 5 min and depressurized to 10 psig. The reactor was pressurized-depressurized 5 times with CO to ensure complete removal of any remaining H2, then depressurized to 15 psig. The reactor was connected to an oven-dried and septum-capped NMR tube via a cannula and the system purged with CO for 2 min. The solution in the pressure bottle was then transferred to the NMR tube using the same cannula. The NMR tube was then sealed with parafilm and placed in an NMR spectrometer cooled to 0 °C. An initial NMR spectrum of the Rh(H)(L7)(CO)2 solution was obtained, the tube ejected from the spectrometer and methyl 2-fluoroacrylate 1 (18.26 μL) injected quickly through the septum using a gas-tight syringe. The NMR tube was inverted 3 times, quickly returned to the spectrometer and the 31P spectra collected immediately.
VII. Data for Hydroformylation of Alkenes.
Methyl (2R)-2-fluoro-2-methyl-3-oxopropanoate (1b).
1H NMR (500 MHz, CDCl3) δ 9.65 (d, J = 5.7 Hz, 1H), 3.85 (s, 3H), 1.70 (d, J = 21.6 Hz, 3H). 19F NMR (377 MHz, CDCl3) δ −167.30. The chiral separation was carried out via GC: 80 °C, hold 6 min, ramp 2 °C/min to 90 °C, hold 6 min, ramp 2 °C/min to 100 °C, hold 6 min, ramp 2 °C/min to 110 °C, hold 5 min, ramp 3 °C/min to 120 °C, hold 5 min, tR (S) 14.8 min, tR (R) 21.1 min. The crude aldehyde was oxidized to a carboxylic acid via a previously reported procedure,40 and the optical rotation [α]20D = +19.4 (c = 1.00, MeOH) compared to that of a previously reported41 (S)-(−)-α-fluoro-α-methylmalonic acid monoethyl ester, [α]20D = −22.5 (c = 2.20, MeOH).
1-Ethyl 5-methyl (2E)-4-fluoro-4-methylpent-2-enedioate (1c).
Hydroformylation was conducted on a 1.92 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 72% (0.28 g) isolation of 1c. 1H NMR (500 MHz, CDCl3) δ 7.01 (dd, J = 20.2, 15.7 Hz, 1H), 6.19 (d, J = 15.7 Hz, 1H), 4.22 (q, J = 7.5 Hz, 2H), 3.82 (s, 3H), 1.72 (d, J = 21.4 Hz, 3H), 1.31 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.6, 165.5, 143.4, 121.8, 93.1, 60.9, 53.2, 24.1, 14.2. 19F NMR (377 MHz, CDCl3) δ −158.20. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C9H13FO4Na 227.0690; Found 227.0690.
Methyl 3,3,3-trifluoro-2-formyl-2-methylpropanoate (2b).
1H NMR (400 MHz, CDCl3) δ 9.74 (s, 1H), 3.84 (s, 3H), 1.56 (s, 3H).
Methyl 2-(acetyloxy)-2-methyl-3-oxopropanoate (3b).
1H NMR (500 MHz, CDCl3) δ 9.87 (s, 1H), 3.83 (s, 3H), 2.17 (s, 3H), 1.60 (s, 3H). Chiral separation via GC: 50 °C, ramp 2 °C/min to 100 °C, hold 2 min, ramp 6 °C/min to 150 °C, hold 2 min tR (major) 12.7 min, tR (minor) 14.9 min.
1-Ethyl 5-methyl (2E)-4-(acetyloxy)-4-methylpent-2-enedioate (3c).
1H NMR (500 MHz, CDCl3) δ 7.16 (d, J = 15.9 Hz, 1H), 6.04 (d, J = 15.9 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 3.76 (s, 3H), 2.13 (s, 3H), 1.68 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 170.3, 169.5, 165.8, 144.9, 121.7, 79.2, 61.0, 53.2, 23.6, 21.1, 14.3. HRMS (ESI-TOF) m/z: [M+NH4]+ Calcd for C11H20O6N 262.1281; Found 262.1283.
Methyl 2-acetamido-2-methyl-3-oxopropanoate (4b).
1H NMR (500 MHz, CDCl3) δ 9.65 (s, 1H), 3.80 (s, 3H), 2.03 (s, 3H), 1.58 (s, 3H).
Methyl 2-methyl-3-oxo-2-phenylpropanoate (5b).
1H NMR (500 MHz, CDCl3) δ 9.87 (s, 1H), 7.43 – 7.38 (m, 2H), 7.37 – 7.31 (m, 1H), 7.26 – 7.15 (m, 2H), 3.81 (s, 3H), 1.69 (s, 3H). The ee was determined by 1H NMR after diastereomeric imine formation with (S)-(+)-1-Methoxy-2-propylamine as previously reported.41 One drop of acetic acid was added to the NMR tube to ensure fast imine formation. [α]20D = −22.6 (c = 1.00, CHCl3), literature: (R)-methyl 2-methyl-3-oxo-2-phenylpropanoate, [α]20D = +102.1 (c = 0.45, CHCl3; 77% ee).
1-Ethyl 5-methyl (2E)-4-methyl-4-phenylpent-2-enedioate (5c).
Hydroformylation was conducted on a 1.00 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 51% (0.40 g) isolation of 5c. 1H NMR (500 MHz, CDCl3) δ 7.48 (d, J = 16.0 Hz, 1H), 7.36 – 7.30 (m, 2H), 7.29 – 7.24 (m, 1H), 7.24 – 7.20 (m, 2H), 5.83 (d, J = 16.0 Hz, 1H), 4.21 (q, J = 7.2 Hz, 2H), 3.71 (s, 3H), 1.70 (s, 3H), 1.29 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.9, 166.3, 150.1, 141.6, 128.7, 127.4, 126.4, 121.5, 60.6, 53.3, 52.7, 23.4, 14.2. HRMS (ESI-TOF) m/z: [M+NH4]+ Calcd for C15H22O4N 280.1543; Found 280.1540. [α]20D = −2.2 (c = 1.00, CHCl3).
Methyl 2-methoxy-2-methyl-3-oxopropanoate (7b).
1H NMR (500 MHz, CDCl3) δ 9.62 (s, 1H), 3.81 (s, 3H), 3.42 (s, 3H), 1.53 (s, 3H). Chiral separation of the enantiomers was achieved via GC: 80 °C, hold 8 min, ramp 3 °C/min to 100 °C, hold 8 min, ramp 5 °C/min to 120 °C, hold 4 min, tR (minor) 13.9 min, tR (major) 15.0 min.
1-Ethyl 5-methyl (2E)-4-methoxy-4-methylpent-2-enedioate (7c).
Hydroformylation was conducted on a 1.00 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 61% (0.40 g) isolation of 7c. 1H NMR (500 MHz, CDCl3) δ 7.03 (d, J = 15.8 Hz, 1H), 6.13 (d, J = 15.8 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 3.33 (s, 3H), 1.55 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 171.9, 166.0, 146.4, 122.3, 80.1, 60.7, 52.9, 52.7, 22.9, 14.2. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C10H16O5Na 239.0890; Found 239.0891. [α]20D = +234 (c = 1.00, CHCl3).
Methyl 2,2-dimethyl-3-oxopropanoate (8b).
1H NMR (400 MHz, CDCl3) δ 9.66 (s, 1H), 2.27 (s, 3H), 1.36 (s, 6H).
1-Ethyl 5-methyl (2E)-4,4-dimethylpent-2-enedioate (8c).
Hydroformylation was conducted on a 2.66 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 74% (0.39 g) isolation of 8c. 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 16.0 Hz, 1H), 5.85 (d, J = 16.0 Hz, 1H), 4.20 (q, J = 7.2 Hz, 2H), 3.70 (s, 3H), 1.36 (s, 6H), 1.30 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 175.3, 166.5, 151.3, 119.7, 60.5, 52.4, 44.6, 24.5, 14.2. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C10H16O4Na 223.0941; Found 223.0939.
Methyl 2-methyl-3-oxo-2-(2-oxooxolan-3-yl)propanoate (10b).
1H NMR (400 MHz, CDCl3) δ 9.57 (s, 1H), 4.42 – 4.24 (m, 2H), 2.84 (ddd, J = 13.1, 7.8, 5.3 Hz, 1H), 2.07 (ddd, J = 13.2, 7.7 Hz, 1H), 1.53 (s, 3H). Analysis of the ee was carried out by 1H NMR following diastereomeric imine formation with (S)-(+)-1-Methoxy-2-propylamine as previously reported42 and by chiral GC after protection of the aldehyde with ethylene glycol as previously reported.43 Chiral separation of the enantiomers was carried out via GC: 100 °C, ramp 1 °C/min to 180 °C, hold 2 min, ramp 6 °C/min to 200 °C, hold 2 min, tR (minor) 41.1 min, tR (major) 41.4 min
1-Ethyl 5-methyl-(2E)-4-methyl-4-(2-oxooxolan-3-yl)pent-2-enedioate (10c).
Hydroformylation was conducted on a 3.00 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 78% (0.46 g) isolation of 10c. 1H NMR (500 MHz, CDCl3) δ 6.96 (d, J = 15.8 Hz, 1H), 5.96 (d, J = 15.9 Hz, 1H), 4.38 – 4.25 (m, 2H), 4.21 (q, J = 7.1 Hz, 2H), 2.47 – 2.37 (m, 1H), 2.29 – 2.20 (m, 1H), 1.44 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 178.0, 165.8, 146.9, 122.0, 65.1, 60.8, 45.1, 35.4, 22.5, 14.2. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C10H14O4Na 221.0784; Found 221.0784. [α]20D = +109.4 (c = 1.00, CHCl3).
1,1,1-Trifluoro-2-methyl-3-oxopropan-2-yl acetate (11b).
The characterization data agreed with previously reported data.11
Ethyl (2E)-4-(acetyloxy)-5,5,5-trifluoro-4-methylpent-2-enoate (11c).
Hydroformylation was conducted on a 3.00 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 71% (0.38 g) isolation of 11c. 1H NMR (500 MHz, CDCl3) δ 6.89 (d, J = 16.0 Hz, 1H), 6.08 (d, J = 15.9 Hz, 1H), 4.22 (q, J = 7.1 Hz, 2H), 2.12 (s, 3H), 1.83 (s, 3H), 1.31 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.3, 165.0, 140.7, 125.6, 124.7, 79.8, 61.1, 21.5, 17.3, 14.2. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C10H13F3O4 277.0658; Found 277.0657.
2-Cyano-1-oxopropan-2-yl acetate (12b).
1H NMR (400 MHz, CDCl3) δ 9.37 (s, 1H), 2.24 (s, 3H), 1.76 (s, 3H). Chiral separation via GC: 110 °C, hold 5 min, ramp 1.5 °C/min to 150 °C, hold 4 min, ramp 5 °C/min to 180 °C, hold 2 min, tR (major) 27.2 min, tR (minor) 27.9 min.

Ethyl (2E)-4-(acetyloxy)-4-cyanopent-2-enoate (12c).
Hydroformylation was conducted on a 3.00 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 77% (0.49 g) isolation of 12c. 1H NMR (400 MHz, CDCl3) δ 6.78 (d, J = 15.7 Hz, 1H), 6.33 (d, J = 15.7 Hz, 1H), 4.23 (q, J = 7.2 Hz, 2H), 2.14 (s, 3H), 1.85 (s, 3H), 1.31 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 168.3, 164.9, 141.6, 124.2, 116.5, 69.8, 61.2, 26.4, 20.8, 14.1. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C10H13NO4Na 234.0737; Found 234.0734. [α]20D = −18.6 (c = 1.00, CHCl3).
3-Methyl-2-oxo-1-phenylazetidine-3-carbaldehyde (20b).
1H NMR (400 MHz, CDCl3) δ 9.72 (s, 1H), 7.41 – 7.33 (m, 5H), 4.13 (d, J = 6.2 Hz, 1H), 3.49 (d, J = 6.2 Hz, 1H), 1.65 (s, 3H). Chiral separation via GC: 80 °C, hold 5 min, ramp 1 °C/min to 170 °C, ramp 4 °C/min to 190 °C, hold 3 min, tR (major) 94.7 min, tR (minor) 92.9 min.
1-Methyl-2-phenylcyclopropane-1-carbaldehyde (25b).
1H NMR (500 MHz, CDCl3) δ 8.96 (s, 1H), 7.28 (s, 2H), 7.11 (d, J = 8.4 Hz, 3H), 2.68 (dd, J = 9.3, 7.1 Hz, 1H), 1.69 (dd, J = 9.3, 5.5 Hz, 1H), 1.39 (dd, J = 7.1, 5.4 Hz, 1H), 0.97 (s, 3H).
Ethyl (2E)-3-(1-methyl-2-phenylcyclopropyl)prop-2-enoate (25c).
Hydroformylation was conducted on a 1.47 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 86% (0.30 g) isolation of 25c. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 2H), 7.24 – 7.13 (m, 3H), 6.70 (d, J = 15.6 Hz, 1H), 5.80 (d, J = 15.6 Hz, 1H), 4.21 (q, J = 7.1 Hz, 2H), 2.43 (t, J = 7.9 Hz, 1H), 1.40 – 1.20 (m, 5H), 0.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.3, 157.7, 137.4, 129.3, 128.3, 126.6, 116.5, 60.3, 32.6, 25.6, 20.5, 15.8, 14.5. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C15H19O2 231.1380; Found 231.1378.
2-(4-Chlorophenyl)-1-methylcyclopropane-1-carbaldehyde (26b).
1H NMR (500 MHz, CDCl3) δ 8.95 (s, 1H), 7.28 (s, 2H), 7.11 (d, J = 8.4 Hz, 2H), 2.68 (dd, J = 9.3, 7.1 Hz, 1H), 1.68 (dd, J = 9.4, 5.5 Hz, 1H), 1.39 (dd, J = 7.1, 5.5 Hz, 1H), 0.97 (s, 3H).
Ethyl (2E)-3-[2-(4-chlorophenyl)-1-methylcyclopropyl]prop-2-enoate (26c).
Hydroformylation was conducted on a 1.47 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 71% (0.27 g) isolation of 26c. 1H NMR (500 MHz, CDCl3) δ 7.31 – 7.21 (m, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.67 (d, J = 15.6 Hz, 1H), 5.80 (d, J = 15.5 Hz, 1H), 4.21 (q, J = 7.2 Hz, 2H), 2.41 – 2.31 (dd, 1H), 1.36 (dd, J = 8.9, 5.3 Hz, 1H), 1.30 (t, J = 7.2 Hz, 3H), 1.28 – 1.25 (m, 1H), 0.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.0, 157.0, 135.8, 132.3, 130.4, 128.3, 116.7, 60.2, 31.7, 25.4, 20.5, 15.7, 14.3. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C15H18ClO2 265.0990; Found 265.0986.
2-(4-Methoxyphenyl)-1-methylcyclopropane-1-carbaldehyde (27b).
1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 7.08 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 3.80 (s, 3H), 2.67 (dd, J = 9.3, 7.2 Hz, 1H), 1.66 (dd, J = 9.4, 5.3 Hz, 1H), 1.36 (dd, J = 7.1, 5.3 Hz, 1H), 0.96 (s, 3H).
Ethyl (2E)-3-[2-(4-methoxyphenyl)-1-methylcyclopropyl]prop-2-enoate (27c).
Hydroformylation was conducted on a 1.47 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 87% (0.33 g) isolation of 27c. 1H NMR (500 MHz, CDCl3) δ 7.08 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.7 Hz, 2H), 6.68 (d, J = 15.6 Hz, 1H), 5.79 (d, J = 15.6 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 3.79 (s, 3H), 2.37 (dd, J = 8.9, 6.9 Hz, 1H), 1.36 – 1.19 (m, 5H), 0.90 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.2, 158.3, 157.8, 130.1, 129.2, 116.2, 113.6, 60.1, 55.2, 31.8, 25.3, 20.6, 15.7, 14.3. HRMS (ESI-TOF) m/z: [M+Na]+ Calcd for C16H20O3Na 283.1305; Found 283.1301.
7-Methylbicyclo[4.1.0]heptane-7-carbaldehyde (28b).
1H NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 2.08 – 1.97 (m, 2H), 1.57 – 1.22 (m, 8H), 1.19 (s, 3H).
Ethyl (2E)-3-(7-methylbicyclo[4.1.0]heptan-7-yl)prop-2-enoate (28c).
Hydroformylation was conducted on a 1.47 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 72% (0.22 g) isolation of 28c. 1H NMR (500 MHz, CDCl3) δ 6.43 (d, J = 15.5 Hz, 1H), 5.67 (d, J = 15.5 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 2.00 – 1.85 (m, 2H), 1.49 – 1.39 (m, 2H), 1.39 – 1.21 (m, 7H), 1.19 (m, 2H), 1.13 (s, 3H). 13C NMR (126 MHz, CDCl3) δ 167.5, 161.0, 114.6, 60.0, 25.0, 23.3, 22.0, 18.9, 14.5, 10.9. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C13H21O2 209.1536; Found 209.1534.
9-Methylbicyclo[6.1.0]nonane-9-carbaldehyde (29b).
1H NMR (500 MHz, CDCl3) δ 8.66 (s, 1H), 2.41 (dd, J = 7.6, 2.1 Hz, 1H), 2.34 – 2.30 (m, 1H), 1.81 – 1.75 (m, 2H), 1.73 – 1.60 (m, 4H), 1.52 – 1.27 (m, 6H), 1.16 (s, 3H).
Ethyl (2E)-3-(9-methylbicyclo[6.1.0]nonan-9-yl)prop-2-enoate (29c).
Hydroformylation was conducted on a 1.47 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 79% (0.26 g) isolation of 29c. 1H NMR (500 MHz, CDCl3) δ 6.51 (d, J = 15.6 Hz, 1H), 5.69 (d, J = 15.6 Hz, 1H), 4.16 (q, 2H), 1.84 – 1.75 (m, 2H), 1.63 (dtd, J = 14.3, 7.0, 3.8 Hz, 4H), 1.45 – 1.32 (m, 4H), 1.27 (t, J = 7.1 Hz, 3H), 1.23 – 1.12 (m, 2H), 1.11 (s, 3H), 1.09 – 0.93 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 167.5, 161.2, 114.5, 60.0, 30.4, 29.4, 26.6, 24.6, 22.2, 18.0, 14.5, 10.5. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C15H25O2 237.1849; Found 237.1846.
7-Ethyl-2-oxo-3-oxa-1-azabicyclo[4.1.0]heptane-7-carbaldehyde (32b).
Hydroformylation was conducted on a 0.81 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 84% (0.13 g) isolation of 32c. 1H NMR (500 MHz, CDCl3) δ 8.91 (s, 1H), 4.48 – 4.31 (m, 2H), 3.02 (dd, J = 8.7, 6.9 Hz, 1H), 2.30 (ddt, J = 14.8, 6.9, 2.1 Hz, 1H), 2.10 – 1.94 (m, 1H), 1.65 – 1.51 (m, 1H), 1.49 – 1.36 (m, 1H), 1.09 (t, J = 7.5 Hz, 3H).13C NMR (126 MHz, CDCl3) δ 197.0, 156.3, 67.9, 53.4, 39.3, 19.3, 18.2, 9.4. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C8H12NO3 170.0812; Found 170.081.
7-Hexyl-2-oxo-3-oxa-1-azabicyclo[4.1.0]heptane-7-carbaldehyde (33b).
Hydroformylation was conducted on a 0.81 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 86% (0.17 g) isolation of 33c. 1H NMR (500 MHz, CDCl3) δ 8.98 (s, 1H), 4.55 – 4.40 (m, 2H), 3.10 (dd, J = 8.7, 6.9 Hz, 1H), 2.36 (dt, J = 14.8, 4.4 Hz, 1H), 2.04 (ddd, J = 14.8, 9.1, 3.6 Hz, 1H), 1.77 (ddd, J = 15.5, 9.5, 5.6 Hz, 1H), 1.72 – 1.59 (m, 1H), 1.44 – 1.19 (m, 8H), 0.88 (t, J = 6.6 Hz, 4H).13C NMR (126 MHz, CDCl3) δ 197.0, 156.3, 67.9, 52.7, 39.5, 31.4, 29.6, 24.9, 24.9, 22.5, 19.5, 13.99. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C12H20NO3 226.1438; Found 226.1443.
2-Oxo-7-(3-phenylpropyl)-3-oxa-1-azabicyclo[4.1.0]heptane-7-carbaldehyde(34b).
Hydroformylation was conducted on a 0.81 mmol scale. After completion of the reaction, the crude material was treated with (carbethoxymethylene)triphenylphosphorane to facilitate 84% (0.19 g) isolation of 34c. 1H NMR (500 MHz, CDCl3) δ 8.86 (s, 1H), 7.20 (d, J = 14.5 Hz, 2H), 7.14 – 7.06 (m, 3H), 4.40 – 4.22 (m, 2H), 2.98 (dd, J = 8.7, 6.9 Hz, 1H), 2.73 – 2.60 (m, 1H), 2.55 – 2.48 (m, 1H), 2.19 (ddt, J = 14.7, 6.9, 2.0 Hz, 1H), 2.10 (dddd, J = 16.6, 8.6, 6.1, 4.7 Hz, 1H), 2.03 – 1.90 (m, 1H), 1.61 – 1.52 (m, 1H), 1.42 – 1.31 (m, 2H).13C NMR (126 MHz, CDCl3) δ 197.0, 156.2, 141.0, 128.5, 128.4, 126.1, 67.9, 52.6, 39.4, 35.9, 26.5, 24.4, 19.3. HRMS (ESI-TOF) m/z: [M+H]+ Calcd for C15H18NO3 260.1281; Found 260.1276.
Supplementary Material
Acknowledgements
This work was partially funded through the NIH R01 GM11412 to JMS, the WiscAMP-BD project (NSF #HRD-1500138) to JE. The NMR facilities at UW-Madison are funded by the NSF (CHE-1048642, CHE-0342998) and NIH S10 OD012245. The National Magnetic Resonance Facility at Madison is supported by the NIH (P41GM103399, S10RR08438, S10RR029220) and the NSF (BIR-0214394). The purchase of the Thermo Q Exactive™ Plus in 2015 for mass spectrometry was funded by NIH Award 1S10 OD020022–1 to the Department of Chemistry.
Footnotes
Supporting Information
The supporting information contains NMR characterization data for all new and old compounds, reaction optimization conditions, unsuccessful substrates, GC spectra for the separation of enantiomers, and the relevant references.
References and Notes
- (1).(a) Franke R; Selent D; Börner A Applied hydroformylation. Chem. Rev 2012, 112, 5675–5732. [DOI] [PubMed] [Google Scholar]; (b) Whiteker G; Cobley C Applications of Rhodium-Catalyzed Hydroformylation in the Pharmaceutical, Agrochemical, and Fragrance Industries in Organometallics as Catalysts in the Fine Chemical Industry Beller M; Blaser H-U Eds.; Springer Berlin Heidelberg: 2012; Vol. 42, pp 35–46. [Google Scholar]
- (2).(a) Watkins AL; Hashiguchi BG; Landis CR Highly enantioselective hydroformylation of aryl alkenes with diazaphospholane ligands. Org. Lett 2008, 10, 4553–4556. [DOI] [PubMed] [Google Scholar]; (b) McDonald RI; Wong GW; Neupane RP; Stahl SS; Landis CR Enantioselective Hydroformylation of N-Vinyl Carboxamides, Allyl Carbamates, and Allyl Ethers Using Chiral Diazaphospholane Ligands. J. Am. Chem. Soc 2010, 132, 14027–14029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Risi RM; Burke SD Synthesis of the Prelog–Djerassi Lactone via an Asymmetric Hydroformylation/Crotylation Tandem Sequence. Org. Lett 2012, 14, 2572–2575. [DOI] [PubMed] [Google Scholar]
- (3).Clark TP; Landis CR; Freed SL; Klosin J; Abboud KA Highly active, regioselective, and enantioselective hydroformylation with Rh catalysts ligated by bis-3, 4-diazaphospholanes. J. Am. Chem. Soc 2005, 127, 5040–5042. [DOI] [PubMed] [Google Scholar]
- (4).(a) Yan Y; Zhang X A hybrid phosphorus ligand for highly enantioselective asymmetric hydroformylation. J. Am. Chem. Soc 2006, 128, 7198–7202. [DOI] [PubMed] [Google Scholar]; (b) Joe CL; Blaisdell TP; Geoghan AF; Tan KL Distal-Selective Hydroformylation using Scaffolding Catalysis. J. Am. Chem. Soc 2014, 136, 8556–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Klosin J; Landis CR Ligands for practical rhodium-catalyzed asymmetric hydroformylation. Acc. Chem. Res 2007, 40, 1251–1259. [DOI] [PubMed] [Google Scholar]; (d) Sakai N; Mano S; Nozaki K; Takaya H Highly enantioselective hydroformylation of olefins catalyzed by new phosphine phosphite-rhodium (I) complexes. J. Am. Chem. Soc 1993, 115, 7033–7034. [Google Scholar]
- (5).Abrams ML; Foarta F; Landis CR Asymmetric Hydroformylation of Z-Enamides and Enol Esters with Rhodium-Bisdiazaphos Catalysts. J. Am. Chem. Soc 2014, 136, 14583–14588. [DOI] [PubMed] [Google Scholar]
- (6).Keulemans AIM; Kwantes A; van Bavel T The structure of the formylation (OXO) products obtained from olefines and watergas. Recl. Trav. Chim. Pays-Bas 1948, 67, 298–308. [Google Scholar]
- (7).Deng YC; Wang H; Sun YH; Wang X Principles and Applications of Enantioselective Hydroformylation of Terminal Disubstituted Alkenes. ACS Catal. 2015, 5, 6828–6837. [Google Scholar]
- (8).Gladiali S; Pinna L Completely regioselective hydroformylation of methyl n-acetamidoacrylate by chiral rhodium phosphine catalysts. Tetrahedron-Asymm. 1990, 1, 693–696. [Google Scholar]
- (9).Lee CW; Alper H Influence of 1,4-bis(diphenylphosphino)butane on the hydroformylation of α,β-unsaturated esters catalyzed by zwitterionic, cationic, and neutral rhodium(I) complexes. The asymmetric hydroformylation of α-methylene-γ-butyrolactone. J. Org. Chem 1995, 60, 499–503. [Google Scholar]
- (10).Fanfoni L; Diab L; Smejkal T; Breit B Efficient Synthesis of New Fluorinated Building Blocks by means of Hydroformylation. Chimia 2014, 68, 371–377. [DOI] [PubMed] [Google Scholar]
- (11).Wang X; Buchwald SL Synthesis of Optically Pure 2-Trifluoromethyl Lactic Acid by Asymmetric Hydroformylation. J. Org. Chem 2013, 78, 3429–3433. [DOI] [PubMed] [Google Scholar]
- (12).Tan RC; Zheng X; Qu B; Sader CA; Fandrick KR; Senanayake CH; Zhang XM Tunable P-Chiral Bisdihydrobenzooxaphosphole Ligands for Enantioselective Hydroformylation. Org. Lett 2016, 18, 3346–3349. [DOI] [PubMed] [Google Scholar]
- (13).(a) Desmarchelier A; Coeffard V; Moreau X; Greck C Asymmetric organocatalytic functionalization of α, α-disubstituted aldehydes through enamine activation. Tetrahedron 2014, 70, 2491–2513. [Google Scholar]; (b) Lalonde MP; Chen YG; Jacobsen EN A Chiral Primary Amine Thiourea Catalyst for the Highly Enantioselective Direct Conjugate Addition of α,α-Disubstituted Aldehydes to Nitroalkenes. Angew. Chem. Int. Ed 2006, 45, 6366–6370. [DOI] [PubMed] [Google Scholar]; (c) Quintard A; Alexakis A Asymmetric addition of α-hetero-disubstituted aldehydes to vinyl sulfones: formation of highly functionalized tetrasubstituted carbon centres. Chem. Commun 2010, 46, 4085–4087. [DOI] [PubMed] [Google Scholar]
- (14).Vargas-Diaz ME; Joseph-Nathan P; Tamariz J; Zepeda LG Synthesis of a New (1R)-(−)-Myrtenal-Derived Dioxadithiadodecacycle and Its Use as an Efficient Chiral Auxiliary. Org. Lett 2007, 9, 13–16. [DOI] [PubMed] [Google Scholar]
- (15).Trost BM; Xu JY; Reichle M Enantioselective synthesis of α-tertiary hydroxyaldehydes by palladium-catalyzed asymmetric allylic alkylation of enolates. J. Am. Chem. Soc 2007, 129, 282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).DeBergh JR; Spivey KM; Ready JM Preparation of substituted enol derivatives from terminal alkynes and their synthetic utility. J. Am. Chem. Soc 2008, 130, 7828–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Snyder SA; Corey EJ Concise Total Syntheses of Palominol, Dolabellatrienone, β-Araneosene, and Isoedunol via an Enantioselective Diels−Alder Macrobicyclization. J. Am. Chem. Soc 2006, 128, 740–742. [DOI] [PubMed] [Google Scholar]
- (18).(a) Schulte ML; Lindsley CW Highly Diastereoselective and General Synthesis of Primary β-Fluoroamines. Org. Lett 2011, 13, 5684–5687. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liang T; Neumann CN; Ritter T Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]
- (19).Shibatomi K; Kitahara K; Okimi T; Abe Y; Iwasa S Enantioselective fluorination of α-branched aldehydes and subsequent conversion to α-hydroxyacetals via stereospecific C–F bond cleavage. Chem. Sci 2016, 7, 1388–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Lazzaroni R; Settambolo R; Rocchiccioli S; Paganelli S; Marchetti M Evidence for formation and different evolution of tertiary rhodium alkyl intermediates under rhodium-catalyzed deuterio-(hydro) formylation of 1-(n-pyridyl)-1-phenylethenes. J. Organomet. Chem 2005, 690, 1699–1704. [Google Scholar]; (b) Lazzaroni R; Uccello-Barretta G; Scamuzzi S; Settambolo R; Caiazzo A 2H NMR Investigation of the Rhodium-Catalyzed Deuterioformylation of 1, 1-Diphenylethene: Evidence for the Formation of a Tertiary Alkyl−Metal Intermediate. Organometallics 1996, 15, 4657–4659. [Google Scholar]
- (21).(a) Watkins AL; Landis CR Origin of Pressure Effects on Regioselectivity and Enantioselectivity in the Rhodium-Catalyzed Hydroformylation of Styrene with (S,S,S)-BisDiazaphos. J. Am. Chem. Soc 2010, 132, 10306–10317. [DOI] [PubMed] [Google Scholar]; (b) Nelsen ER; Landis CR Interception and characterization of alkyl and acyl complexes in rhodium-catalyzed hydroformylation of styrene. J. Am. Chem. Soc 2013, 135, 9636–9639. [DOI] [PubMed] [Google Scholar]; (c) Nelsen ER; Brezny AC; Landis CR Interception and Characterization of Catalyst Species in Rhodium Bis (diazaphospholane)-Catalyzed Hydroformylation of Octene, Vinyl Acetate, Allyl Cyanide, and 1-Phenyl-1,3-butadiene. J. Am. Chem. Soc 2015, 137, 14208–14219. [DOI] [PubMed] [Google Scholar]
- (22).Clarke ML; Roff GL Highly Regioselective Rhodium-Catalysed Hydroformylation of Unsaturated Esters: The First Practical Method for Quaternary Selective Carbonylation. Chem. Eur. J 2006, 12, 7978–7986. [DOI] [PubMed] [Google Scholar]
- (23).Boralsky LA; Marston D; Grigg RD; Hershberger JC; Schomaker JM Allene functionalization via bicyclic methylene aziridines. Org. Lett 2011, 13, 1924–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Rigoli JW; Guzei IA; Schomaker JM Aminodiols via Stereocontrolled Oxidation of Methyleneaziridines. Org. Lett 2014, 16, 1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) Rigoli JW; Weatherly CD; Alderson J; Vo BT; Schomaker JM Tunable, chemoselective amination via silver catalysis. J. Am. Chem. Soc 2013, 135, 17238–17241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schmid SC; Guzei IA; Schomaker JM A Stereoselective [3+ 1] Ring Expansion for the Synthesis of Highly Substituted Methylene Azetidines. Angew. Chem. Int. Ed 2017, 56, 12229–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Peak broadening is consistent with equilibration of a five-coordinate Rh-alkyl dicarbonyl species with a four-coordinate Rh-alkyl monocarbonyl species when the solution is starved in CO. The breadth of the peaks associated with 38alkyl at −30 °C is broadened when CO is sparged from solution and sharpened when CO is added.
- (27).Wong GW; Watkins AT; Clark TL; Landis CR Org. Synth 2012, 89, 243–254. [Google Scholar]
- (28).Armarego WLF; Chai C Purification of Laboratory Chemicals 6th ed. Elsevier: Burlington, MA, 2009 [Google Scholar]
- (29).Lee JJ; Kraus GA One-pot formal synthesis of biorenewable terephthalic acid from methyl coumalate and methyl pyruvate. Green Chem 2014. 16 2111–2116, DOI:10.1039/C3GC42487A [Google Scholar]
- (30).Zhang ZX; Ma BC; Zhu QQ; Ding Y; Wang CM; Song WF Sulfamic Acid as A Cost-Effective Catalyst for Synthesis of α-Acyloxyacrylate Esters as Candidate Monomers for Biobased Polymers by Acylation of Pyruvate Esters. Synth. Commun 2012, 42, 3053–3060. [Google Scholar]
- (31).Pietruszka J; Scholzel M Ene Reductase-Catalysed Synthesis of (R)-Profen Derivatives. Adv. Synth. Catal 2012, 354, 751–756. [Google Scholar]
- (32).Chen BF; Tasi MR; Yang CY; Chang JK; Chang NC A versatile approach to 6-substituted-5-methoxy-δ-lactam framework and application to the formal synthesis of (±)-homopumiliotoxin 223G. Tetrahedron 2004, 60, 10223–10231. [Google Scholar]
- (33).Farley AM; Sandford C; Dixon D Bifunctional Iminophosphorane Catalyzed Enantioselective Sulfa-Michael Addition to Unactivated α-Substituted Acrylate Esters. J. Am. Chem. Soc 2015, 137,15992–15995. [DOI] [PubMed] [Google Scholar]
- (34).Rousee K; Bouillon JP; Couve-Bonnaire S; Pannecoucke X Stereospecific Synthesis of Tri- and Tetrasubstituted α-Fluoroacrylates by Mizoroki–Heck Reaction. Org. Lett 2016, 18, 540–543. [DOI] [PubMed] [Google Scholar]
- (35).Wu L; Liu C; Zhang H; Ye K; Guanghui Z; Zhang W; Duan Z; You S; Lei A Palladium-Catalyzed Oxidative Carbonylation of N-Allylamines for the Synthesis of β-Lactams. Angew. Chem. Int. Ed 2014, 53, 2443–2446. [DOI] [PubMed] [Google Scholar]
- (36).Yu L; Wu Y; Cao H; Zhang X; Shi X; Luan X; Chen T; Pan Y; Xu Q Facile synthesis of 2-methylenecyclobutanones via Ca(OH)2-catalyzed direct condensation of cyclobutanone with aldehydes and (PhSe)2-catalyzed Baeyer–Villiger oxidation to 4-methylenebutanolides. Green Chem 2014, 16, 287–293. [Google Scholar]
- (37).Kim S; Chung Y Rhodium-Catalyzed Carbonylative [3+2+1] Cycloaddition of Alkyne-Tethered Alkylidenecyclopropanes to Phenols in the Presence of Carbon Monoxide. Org. Lett 2014, 16, 4352–4355. [DOI] [PubMed] [Google Scholar]
- (38).Kitatani K; Hiyama T; Nozaki H Stereoselective Monoalkylation of α-Halocyclopropyllithiums. A Versatile Method for the Synthesis of α-Alkylcyclopropyl Acetates and Alkylidenecyclopropanes. Bull. Chem. Soc. Jpn 1977, 50, 3288–3294. [Google Scholar]
- (39).Kippo T; Hamaoka K; Ryu I Bromine Radical-Mediated Sequential Radical Rearrangement and Addition Reaction of Alkylidenecyclopropanes. J. Am. Soc 2013, 135, 632–635. [DOI] [PubMed] [Google Scholar]
- (40).Foarta F; Landis CR Condensation oligomers with sequence control but without coupling reagents and protecting groups via asymmetric hydroformylation and hydroacyloxylation. J. Org. Chem 2016, 81, 11250–11255. [DOI] [PubMed] [Google Scholar]
- (41).Kitazume T; Murata K; Ikeya T Modification of Catalytic Activity Hydrolytic Enzymes with Fluorine-containing Molecules. J. Fluorine Chem 1986, 32, 233–238. [Google Scholar]
- (42).Chi Y; Peelen TJ; Gellman SH A Rapid 1H NMR Assay for Enantiomeric Excess of α-Substituted Aldehydes. Org. Lett 2005, 7, 3469–3472. [DOI] [PubMed] [Google Scholar]
- (43).Sterzycki R Pyridinium tosylate, a mild catalyst for formation and cleavage of dioxolane-type acetals. Synthesis-Stuttgart 1979, 9, 724–725. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.