

ABSTRACT
HCMV is a common pathogen for human with relatively high prevalence, which could be life-threatened in immunodeficient patients and lead to significant birth defects in newborns. In this study, we firstly report that HCMV infection significantly enhances the expression of microRNA-221 (miR-221) in Neural Precursor Cells (NPCs). We found that miR-221 directly targets at the 3ʹ-UTR of suppressor of cytokine signaling 1 (SOCS1) and suppresses SOCS1 expression at the both mRNA and protein levels. MiR-221 overexpression restrained HCMV replication by promoting type I interferon (IFN) and interferon stimulating genes (ISGs) production, whereas reintroduction of SOCS1 abrogated the miR-221-induced effects on HCMV replication. Importantly, miR-221 positively regulated the phosphorylation and activation of NF-κB by suppressing SOCS1. What’s more, miR-221 agomir alleviated MCMV-induced tissue injury by promoting type I IFN antiviral activities in vivo. Thus, miR-221 modulates the infection and replication of HCMV as an intrinsic antiviral factor, and could be developed as a treatment target for anti-HCMV treatment.
KEYWORDS: Mir-221, HCMV, SOCS1/NF-κB signaling, type I IFN
Introduction
Human cytomegalovirus (HCMV) is dsDNA virus, which belongs to herpesviridae, β-herpesvirus subfamily [1]. HCMV is a common pathogen for human with high infection rate [2]. Congenital HCMV infection is the leading cause of nonhereditary sensorineural hearing loss (SNHL) and can cause other long-term neurodevelopmental disabilities and health problems, including cerebral palsy, intellectual disability, vision impairment, and seizures [3]. It is of great importance to study the HCMV replication mechanisms in neuron cells and how it leads to developmental defeats in the nervous system.
MicroRNAs (miRNAs) are a family of small, endogenous non-coding RNAs, which can negatively regulate targeted gene expression by degrading mRNA [4]. MicroRNAs are widely involved in the regulation of host innate immune defense against viral infection [5]. Several miRNAs have been proved to involve in the regulation of HCMV replication and latency establishment and maintenance. In neural cells microRNA miR-21 was reported to attenuate HCMV replication through targeting Cdc25a [6]. MicroRNA-182 suppressed HCMV replication by promoting type I IFN response [7]. However, more researches are needed to demonstrate how microRNAs regulate HCMV replication and immune response.
Suppressor of Cytokine Signaling 1(SOCS1) is a critical negative regulator in TLR and cytokine-induced signaling [8,9]. By suppressing NF-κB signaling,SOCS1 plays a vital role in ameliorating inflammation [9]. SOCS1 is also involved in repressing the activation of type I IFN signaling upon viral infection [10]. Many cytokines, including IL-2, IL-6, IL-10, IFNα/β, IFNγ, TNFα could promote the expression of SOCS1. The activation of SOCS1 is also modulated by a panel of microRNAs. MiR-122 was reported to regulate the type I IFN expression through regulating the SOCS1 expression in HCV infected hepatic cells [11]. In macrophages miR-155 was markedly upregulated upon vesicular stomatitis virus infection. Through targeting SOCS1, miR155 positively regulated antiviral innate immune response [12].
Here, we firstly reported that HCMV infection upregulated the expression of miR-221 and miR-221 hindered the replication of HCMV by repressing the expression of SOCS1. Consistent with the roles of miR-221 in neural precursor cells, we found miR-221 agomir had antiviral function against HCMV in mice, suggesting it could be a potential target for anti-HCMV treatment.
Materials and methods
Cell culture
Neural Precursor Cells (NPCs) were purchased from the Cell Resource Center of the Chinese Academy of Medical Science. NPCs were grown in DMEM/F-12 medium supplemented with 10% fetal bovine serum, 2mM GlutaMAX, and 100 U/ml penicillin, and 100 mg/ml streptomycin at 37°C in humidified air atmosphere at 5% CO2. Cells were subcultured every 3–4 days depending on the confluence, and plated for experiments after cell counts.
Viral infection and plaque assay
The HCMV strain AD169 was purchased from the American Type Culture Collection (ATCC accession No. VR538,Vanassas, VA). To avoid NPC differentiation induced by fetal bovine serum, the virus for NPC infection was concentrated and resuspended in NPCs culture medium. 3 × 106 cells/10-cm dish NPCs were infected with HCMV at different MOIs.
Viral titers in the supernatants were determined by standard plaque assay. NPCs were seeded into 12-well plates (2 × 105 cells/well) and infected with HCMV in 1:10 dilutions, each dilution was triplicate. After 2 h, the virus-containing media were aspirated, and DMEM containing 0.5% carboxymethyl cellulose (CMC), 2% fetal bovine serum (FBS) were added into wells. After incubation at 37°C for 7 days the overlay media was removed and plaques were counted after crystal violet staining.
Immunofluorescence
NPCs were infected with HCMV for 24 h after miRNA mimic or inhibitor treatment and the infected cells were fixed for 10 min with fixation buffer (75% methanol, 25% glacial acetic acid) and subsequently washed with PBS. After permeabilization with 0.1% digitonin, the cells were probed with antibody against IE1 overnight at 4°C. The cells were examined under a fluorescence microscopy and images were analyzed using an Olympus FluoView FV10 confocal microscope (Tokyo, Japan).
Elisa
NPCs cells were transfected with miR-221 inhibitor, inhibitor NC or miR-221 mimics and mimics NC. Cells were infected with 1 MOI HCMV at 24 h after transfection. Supernatant was harvested at 12 h and 24 h post-infection for ELISA assay. IFN-α and IFN-β contents in supernatant were measured by ELISA kits (from PBL Biomedical Laboratories, Piscataway, NJ, USA) according to the manufacturers.
Quantitative RT-PCR
Total RNA was extracted with TRIzol(Invitrogen) according to the manufacturer’s instructions. NanoDrop 2000 (Thermo) was used for RNA quantification. Reverse transcription (RT) solution was made according to the instructions in MicroRNA Reverse Transcription Kit (TaKaRa). Real-time PCR was carried out on ABI 7900HT Fast Real-Time PCR System. U6 was chosen as internal controls for microRNA, the primer sequences were: the upstream, 5ʹ-CTCGCTTCGGCAGCACA-3ʹ, and the downstream, 5ʹ-AACGC-TTCACGAATTTGCGT-3ʹ. The upstream primers of miR-221 was 5ʹ-AGCUACAUUGUC UGCUGGGUUUC-3ʹ and the universal downstream primer provided in the kit. The fold changes in miRNA expression were calculated by the change-in-cycling-threshold (∆∆CT) method. The upstream primers SOCS1 were 5ʹ-CTGCGGCTTCTATTGGGGAC-3ʹ and the downstream were 5ʹ-AAAAGGCAGTCGAAGGTCTCG-3ʹ; the upstream primers IFN-α were 5ʹ-CTGAATGACTTGGAAGCCTG-3ʹ, and the downstream were 5ʹ-ATTTCTGCTCTGACAACCTC-3ʹ. The upstream primers IFN-β were 5ʹ-TAGCACTGGCTGGAATGAG-3ʹ, and the downstream were 5ʹ-GTTTCGGAGGTAACCTG TAAG-3ʹ. For IFN-stimulated gene 15 (ISG15), the primers were 5ʹ-GGTGTCCGTGACTAACTCCAT-3ʹ and 5ʹ-TGGAAAGGGTAA
GACCGTCCT-3ʹ.For MxA, the primers were 5ʹ-GCCAGGACCAGGTATACAG-3ʹ and 5ʹ-GCTCCTTCAGGAGCCAGA −3ʹ. For OSA, the primers were 5ʹ-TGTCCAAGGTGGTAAAGGGTG-3, and 5ʹ- CCGGCGATTTAACTGATCCT
G-3. The primer sequences were synthesized by Sangon Biotech (Shanghai).
HCMV UL83-CN F and R primers were used to quantitate HCMV DNA and GAPDH-CN F and R primers were used to quantitate cellular DNA. Tenfold serial dilutions of plasmids pcDNA3.0-UL83 and pcDNA3.0-GAPDH were used to generate standard curves. Reactions were denatured at 95°C for 3 min, followed by 40 two-step cycles at 95°C for 10 s and 60°C for 30 s. Viral genome copy numbers were normalized to GAPDH copies to produce viral genome copies/cell. All reactions were conducted in triplicate. The results reported are means the standard deviations (SD)
from three independent experiments.
RNA oligonucleotides, plasmids, and transfection
Scrambled miR, miR-221 mimics and miR-221 inhibitor were synthesized and purified by GenePharma (Shanghai, China). All miRNA oligonucleotides were transfected at a concentration of 50 μmol/mL with Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. The CDS sequence of SOCS1 was synthesized by Genewiz (Beijing, China) and subsequently cloned into the pcDNA3.1 vector (Promega) at the BamHI and HindIII sites. DNA sequencing(Sangon Biotech) was conducted to confirm the construct sequences. The cells were transfected until the cell confluence reached 70%. Transfections were carried out using Lipofectamine 2000 (Invitrogen).The mimic sequence was 5ʹ-AGCUACAUUGUCUG CUGGGUUUC-3ʹ, and the random sequence (scramble control) was 5ʹ-UUCUCCGAACGUGUCACGUT T-3ʹ. The Inhibitor sequence was
5ʹ-AGCUACAUUGUCUGCUGGGUUUC-3ʹ. The siRNA sequences are as follows: Control siRNA (si-Scramble), 5ʹ- TCGAACGCTGATAGTTGCCTACCCA −3ʹ; si-IFNAR1, 5ʹ- TCGCAAAGCTCAGATTGGTCCTCCA- 3ʹ; si-IFNAR2, 5ʹ- CAGCCTCGTGTTTGGTATTTCATAT- 3ʹ.
Western blot
Cells were lysed in lysis buffer (50 mM Tris, pH7.4, 150 mM NaCl, 1% Triton X-100 and 1 mM EDTA, pH 8.0) containing Complete mini-protease inhibitor cocktail(Roche) for 20 min on ice, and cell debris were removed by centrifugation at 13,000 rpm for 20 min. After adding loading buffer, incubate protein samples at 95°C for 5 min. Protein samples were resolved by 10% SDS-PAGE and transferred onto a PVDF membrane, which was then blocked with 5% milk and probed by indicated antibodies. The protein band was detected by chemiluminescence with Pierce ECL kits (Millipore). β-actin was used as an loading control. SOCS1 antibody (#3950), phospho-NF-κB p65 (#3033), phospho-IκBα (Ser32) (#2859), IκBα Antibody (#9242) and β-Actin (#3700) was from Cell Signaling Technology at 1:1000 dilution, IE2 antibody was from Abcam (ab53495) at 1:500 dilution.
Luciferase reporter assay
The wild type (wt) and mutant (mut) region at the 3ʹ-UTR of SOCS1 were synthesized and cloned into luciferase reporter vectors pGL3 (Promega) at the BamHI and NotI sites. The cells were cotransfected with vectors carrying the wt or mut 3ʹ-UTR together with miR-221 mimics or inhibitor using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instruction. The pDL3 control vector was transfected as a control. The Dual-Luciferase Reporter Assay (Promage) was used to detect the luciferase activity after 48 h of transfection.
Ethics statement
All animal experimentation was performed with the approval of the Animal Ethics and Experimentation Committee of the People’s Hospital of Zhengzhou University and according to the Guide for the Care and Use of Laboratory Animals published by the Chinese National Institutes of Health.
In vivo infection
Sisx to eight weeks old C57BL/6 mice were purchased from SLAC Laboratory Animal Co. Ltd. (Shanghai, China). The mice were maintained in the specific pathogen-free (SPF) environment, randomly divided into three groups of six animals each. For agomir injection, mice were injected with 20nmol agomir-221 intravenously or PBS as control intravenously. Two days after agomir injection, mice were injected intraperitoneally with 105 plaque-forming units (PFU) of MCMV strain K181, the control group was injected with phosphate-buffered saline. Seven days after a viral infection, mice were killed and organs collected and processed for RNA isolation and H&E staining. Relative viral titers were quantified by RT-PCR.
Statistical analysis
SPSS 19.0 software was employed to perform the statistical analysis. The data are presented as the mean ± SD of at least three independent experiments, and analyzed by Student’s t-test or one-way ANOVA with Tukey’s post hoc analysis. P < 0.05 was considered as statistical significance.
Results
MiR-221 is an inducible microRNA by different strains of HCMV infection
Through analyzing the miRNAs expression profile from GEO database (http://www.ncbi.nlm.nih.gov/geo) with the accession number of GSE33584 and GSE75305, we identified miR-221 which may be involved in HCMV infection (Figure 1(A)). The expression of miR-221 was significantly increased upon HCMV infection. It has been reported that miR-221 was also involved in HCV induced immune response [13,14]. We confirmed the array data by RT-qPCR in HCMV AD169 strain infected Neural Precursor Cells (NPCs) (Figure 1(B,C)). Different MOIs of HCMV AD169 strain infection dose-dependently increased the expression level of miR-221. Besides, the expression of miR-221 was induced 4-h post HCMV AD169 strain infection and reached the highest level in 24-h postinfection (Figure 1(B,C)). HCMV Towne strain infection also dose-dependently and time-dependently increased the expression level of miR-221 (Figure 1(D,E)). Taken together, miR-221 is an inducible microRNA by different strains of HCMV infection.
Figure 1.
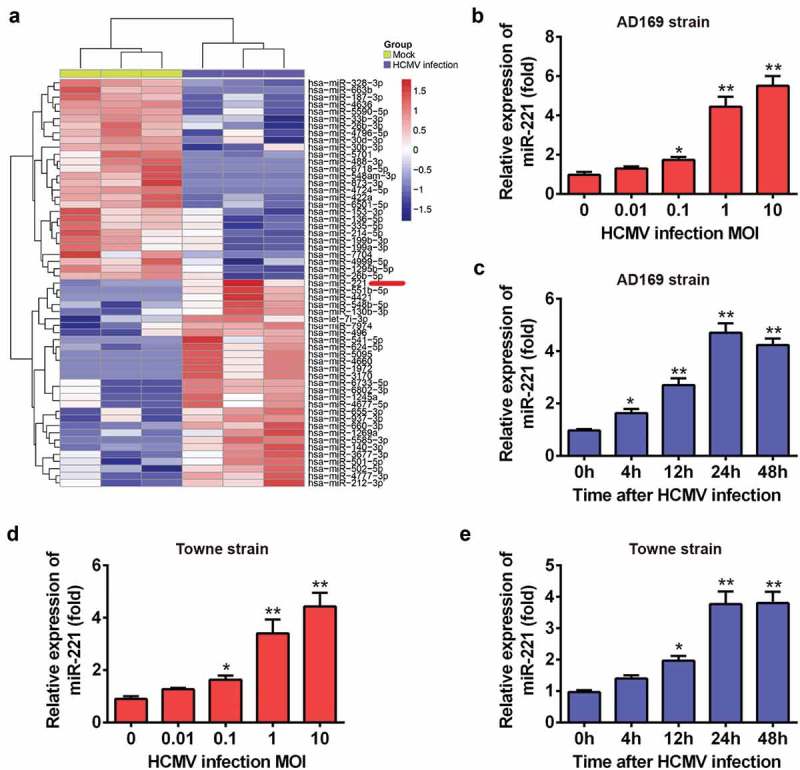
miR-221 is an inducible miRNA by different strains of HCMV infection. (A) Gene Expression Omnibus (GEO) data analyzed by heatmap. (B) NPCs (Neural Precursor Cells) was infected with indicated doses of HCMV AD169 strain, and the expression level of miR-221 was evaluated by qRT-PCR. (C) NPCs was infected with HCMV AD169 strain for indicated hours, and the expression level of miR-221 was checked by qRT-PCR. (D) NPCs was infected with indicated doses of HCMV Towne strain, and the expression level of miR-221 was evaluated by qRT-PCR. (E) NPCs was infected with HCMV Towne strain for indicated hours, and the expression level of miR-221 was checked by qRT-PCR. These experiments were conducted three times, and the representative data were shown mean ± SD(* p < 0.05, ** p < 0.01 vs 0 h or 0 MOI).
MiR-221 negatively regulated HCMV infection
To check whether miR-221 is involved in the regulation of HCMV infection, NPCs were transfected with mimics NC and miR-221 mimics (Figure 2(A)). The miR-221 expression level in miR-221 mimics transfected group was 10 times compared to the negative control (NC). HCMV genome copy number was compared in mimics NC and miR-221 mimics transfected groups at 4, 12 and 24 h posted infection by RT-qPCR (Figure 2(B)). MiR-221 mimics significantly reduced HCMV genome copy number at all these 3 time points by nearly 50% (Figure 2(B)). MiR-221 mimics also time-dependently repress the viral load (Figure 2(C)). On the contrary, miR-221 inhibitor transfection,which leads to 80% loss of miR-221 expression (Figure 2(D)), resulted in a significant increase of HCMV virus copy number and viral load at different time points postinfection (Figure 2(E,F)).
Figure 2.
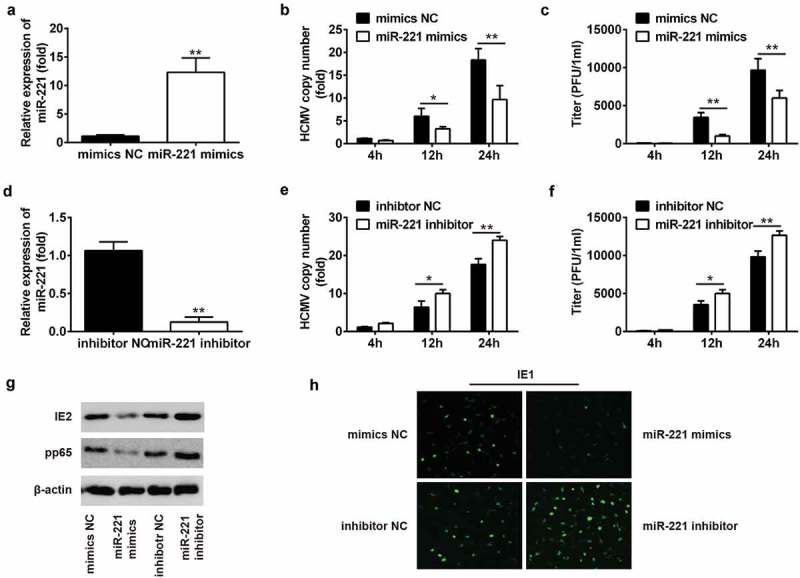
The replication of HCMV was suppressed by miR-221. (A) NPCs were transfected with mimics NC or miR-221 mimics for 24 h. The miR-221 expression level was checked by qRT-PCR. (B-C) NPCs were treated with mimics NC or miR-221 mimics for 24 h, followed by HCMV infection for 24 h. The HCMV copy numbers were checked by qRT-PCR assay at indicated times postinfection (B). HCMV titer was evaluated by plaque assay at indicated times post infection (C). (D) NPCs were treated with inhibitor NC or miR-221 inhibitor for 24 h, followed by HCMV infection for 24 h. The miR-221 expression level was checked by qRT-PCR. (E-F) NPCs were treated with inhibitor NC or miR-221 inhibitor for 24 h, followed by HCMV infection for 24 h. The HCMV copy numbers were checked by qRT-PCR assay at indicated times postinfection(E). HCMV titer was evaluated by plaque assay at indicated times post infection (F). (G-H) NPCs were treated with mimics NC, miR-221 mimics or inhibitor NC, miR-221 inhibitor for 24 h, followed by HCMV infection for 24 h. The expression levels of HCMV viral proteins IE2 and PP65 were checked by immunoblot (G). The expression levels of HCMV viral proteins IE1 was detected by immunofluorescence staining (H). All these experiments have been repeated three times, and the representative data were shown. (* p < 0.05, ** p < 0.01 vs mimics NC or inhibitor NC group).
By western blot and immunofluorescence, we also monitored the level of HCMV viral protein IE2, pp65 and IE1. MiR-221 mimics transfection decreased the expression of IE2, pp65 and IE1, while miR-221 inhibitor transfection led to increased expression of IE2, pp65 and IE1 (Figure 2(G,H)). Together, these data draw a conclusion that miR-221 could attenuate the replication of HCMV in NPCs.
MiR-221 positively regulated HCMV induced type I IFN response
MiR-221 was reported to regulate type I IFN response in HCV infection [14]. We made a hypothesis whether miR-221 attenuates HCMV replication by regulating HCMV triggered type I IFN response. Transfection of miR-221 mimics significantly increased of the production of IFN-alpha and IFN-beta in both mRNA expression level and protein level at 12 h and 24 h postinfection (Figure 3(A,B)), whereas miR-221 inhibitor inhibited the production of IFN-alpha and IFN-beta in both mRNA expression level and protein level in NPCs (Figure 3(C,D)). Through autocrine and paracrine, type I IFN could induce the expression of interferon-induced genes (ISG) by JAK-STAT pathway, and ISGs play an effective role in anti-viral defense [15]. We further check the expression of HCMV induced ISGs including ISG15, MxA and OSA. MiR-221 mimics significantly increased the expression of ISG15, MxA, and OSA; while miR-221 inhibitor significantly decreased the expression of ISG15, MxA and OSA (Figure 3(E,F)). These results suggested that miR-221 could positively regulate type I IFN response during HCMV infection.
Figure 3.
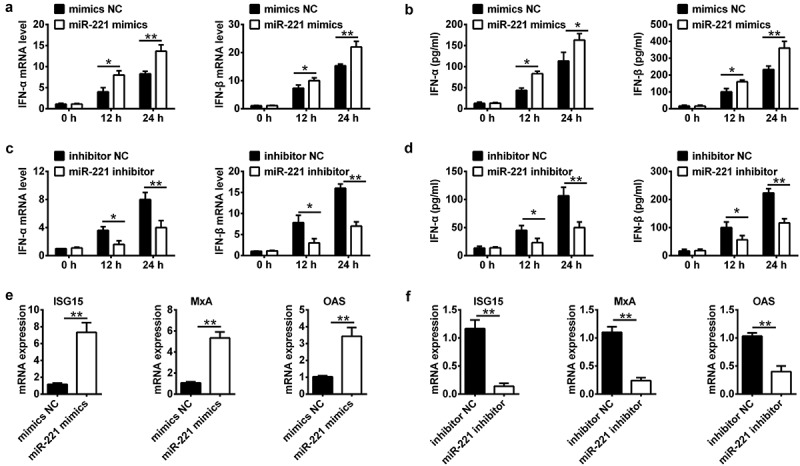
miR-221 is involved in HCMV induced type-I IFN production and ISG activation in NPCs. (A-B) NPCs were transfected with miR-221 mimic or mimic negative control, and infected with HCMV for indicated time. IFN-a and IFN-b mRNA expression was determined by qRT-PCR(A) and the levels of IFN-a and IFN-b cytokines in the cell culture supernatant was determined by enzyme-linked immunosorbent assay (ELISA)(B). (C-D) NPCs were transfected with miR-221 inhibitor or inhibitor negative control, and infected with HCMV for the indicated time. IFN-a and IFN-b mRNA expression was determined by qRT-PCR(C) and the levels of IFN-a and IFN-b cytokines in the cell culture supernatant was determined by enzyme-linked immunosorbent assay (ELISA)(D). (E-F) Experiments were performed as in A-D. Expression levels of ISG15, MxA and OAS were quantified by real-time PCR. All these experiments have been repeated three times, and the representative data were shown. (* p < 0.05, ** p < 0.01 vs mimics NC or inhibitor NC group).
MiR-221 attenuates HCMV replication by promoting type I IFN signaling
Both IFNα and IFNβ can bind to the cell membrane IFNα receptors, which consists of IFN-α receptor I (IFNAR1) and IFN-α receptor II (IFNAR2) and activate the downstream JAK-STAT pathway, thus interfere with a viral infection and modulate the host immune response by facilitating the transcription of hundreds of ISGs. To find out whether the effect of miR221 on the HCMV replication is through type I IFN signaling, we use siRNAs targeting IFNAR1 and IFNAR2 to block type I IFN signaling and check the effect of miR221 on HCMV induced ISGs expression and HCMV replication (Figure 4(A)). We found siRNAs targeting IFNAR1 and IFNAR2 successfully blocked the IFN induced ISG15, MX1, and OSA1 mRNA expression (Figure 4(B–D)). We also transfected NPCs cells with miR-221 mimics alone or co-transfected miR-221 mimics with si-IFNAR1 or si-IFNAR2, respectively (Figure 4(E,F)). The results shown that both the HCMV DNA copy number in the cell and the titers of infectious virus in the culture supernatants were significantly increased in co-transfection group (Figure 4(E,F)), which indicated that miR-221 attenuates HCMV replication through promoting type I IFN signaling.
Figure 4.
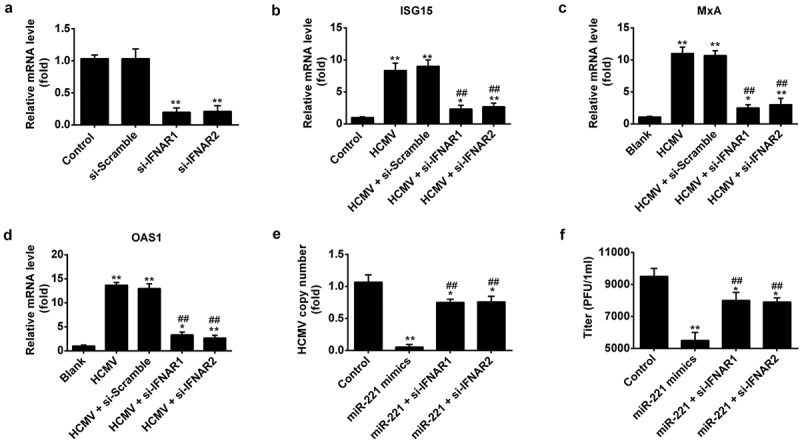
MiR-221 attenuates HCMV replication by promoting type I IFN signaling.(A-D) NPCs cells were transfected with siRNAs targeting IFNAR1 and IFNAR2 or si-scramble for 24h, followed by infected with HCMV at MOI of 1 for 24 h. Cells were harvested for mRNA and real-time PCR was conducted to determine the expression of IFNA receptor (A), ISGs expression like ISG15(B), MxA(C), OAS1(D). (E-F) NPCs cells were transfected with miR-221 mimics alone or co-transfected miR-221 mimics with si-IFNAR1 or si-IFNAR2 for 24h, followed by infected with HCMV at MOI of 1 for 24hr. (E) HCMV DNA copy number per cell was determined by qRT-PCR and standardized to cellular DNA (GAPDH) copy number. (F) Titers of infectious virus in the culture supernatants were determined by plaque assay. All these experiments have been repeated 3 times, and the representative data were shown. (* p < 0.05, ** p < 0.01 vs si-scramble group or HCMV Infection group).
SOCS1 is the direct target of miR-221
To figure out the mechanism of miR-221 in the regulation of HCMV replication, we used the TargetScan software for the prediction of miR-221 cellular targets. Suppressor of cytokine signaling-1 (SOCS1) was found to have a putative miR-221 binding site in its 3ʹUTR (Figure 5(A)). SOCS1 plays an important role in ameliorating inflammation response. Through inhibiting the activation NF-ĸB signaling pathway, SOCS1 works as a negative feedback inhibitor in cytokine signaling to avoid inflammation-induced tissue damage [8,16]. So we wanted to explore whether the expression of SOCS1 was modulated by miR-221 in HCMV infection condition. Our results suggested that SCOS1 expression could be significantly repressed by HCMV infection in a time-dependent and dose-dependent manners (Figure 5(B,C)). What’s more, miR-221 mimics markedly suppressed the protein level of SOCS1, whereas miR-221 inhibitor significantly increased SOCS1 expression (Figure 5(D,E)). To test whether SOCS1 expression was directly regulated by miR-221 binding, we clone the wild-type predicted target site in SOCS1 to a firefly luciferase reporter vector, together with a mutant vector as a negative control. Interestingly, the luciferase activity of wt-SOCS1 resulted in 60% reduction in the presence of miR-221 mimics compared to mimics NC, while blockage of cellular miR-221 by miR-221 inhibitor led to nearly 4 times of luciferase activity compared to inhibitor NC treatment (Figure 5(F)). However, neither a miR-221 inhibitor nor miR-221 mimics influenced the mutant SOCS1 3ʹUTR driven luciferase activity (Figure 5(F)). Collectively, these results suggested that SOCS1 is the direct target of miR-221.
Figure 5.
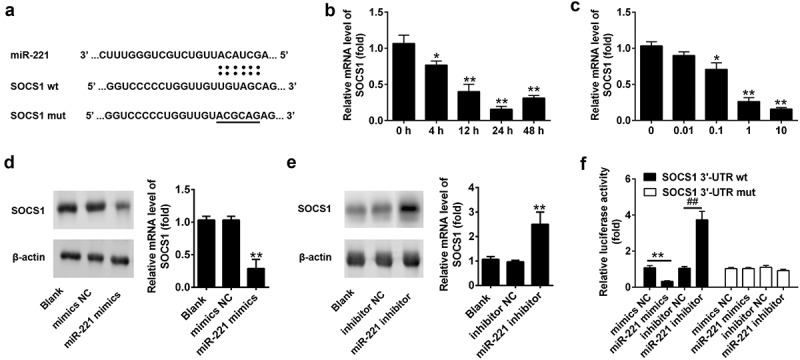
miR-221 targets 3ʹ-UTR of SOCS1 and suppresses the expression of SOCS1 during HCMV infection. (A) Potential base pairing is indicated by vertical lines between the seed sequence of miR-221 and its target sequences within the wild-type (wt) 3ʹ-UTR of SOCS1. Mutation (mut) of the miR-221 target sequence is predicted to eliminate miR-221-mediated repression. (B) NPCs were infected with HCMV for indicated hours, and the relative expression level of SOCS1 was determined by qRT-PCR. * p < 0.05, ** p < 0.01 vs 0 h group. (C) NPCs were infected with indicated doses of HCMV for 24 h, and the relative expression level of SOCS1 was determined by qRT-PCR. * p < 0.05, ** p < 0.01 vs 0 MOI group. (D) NPCs were untreated (Blank) or miR-221 mimic or mimic negative control for 24 h. The protein level of SOCS1 was detected by western blot, and quantified as indicated. * p < 0.05, ** p < 0.01 vs blank group. (E) NPCs were untreated (Blank) or miR-221 inhibitor or inhibitor negative control for 24 h. The protein level of SOCS1 was detected by western blot, and quantified as indicated. * p < 0.05, ** p < 0.01 vs blank group. (F) Luciferase report plasmids containing wild type or mutant SOCS1 3ʹ-UTR were transfected into the cells, followed by untreated or treated with miR-221 mimics or inhibitor. Relative luciferase activity was checked by luciferase activity assay.All these experiments have been repeated three times, and the representative data were shown. (* p < 0.05, ** p < 0.01 vs mimics NC or inhibitor NC group).
MiR-221 promotes type I IFN expression and inhibits HCMV replication by targeting SOCS1
To demonstrate the role of targeting SOCS1 in the antiviral response by miR-221, we firstly test whether miR-221 could target SOCS1 for degradation upon HCMV infection. Indeed, we found that upon HCMV infection the protein level of SOCS1 was down-regulated by miR-221 mimic, and could be rescued by SOCS1 overexpression (Figure 6(A)). Further, we found that HCMV copy number and viral titer in NPCs inhibited by miR-221 were significantly increased with SOCS1 overexpression (Figure 6(B)). Consistently, the mRNA and protein level of type I IFN is enhanced by transfected miR-221 mimics in HCMV infected NPCs, whereas co-transfection of pCDNA-SOCS1 dramatically attenuated the effect (Figure 6(C,D)). The ISGs expression levels gave the consistent results (Figure 6(E)). Hence, miR-221 promotes type I IFN expression and inhibits HCMV replication by targeting SOCS1.
Figure 6.
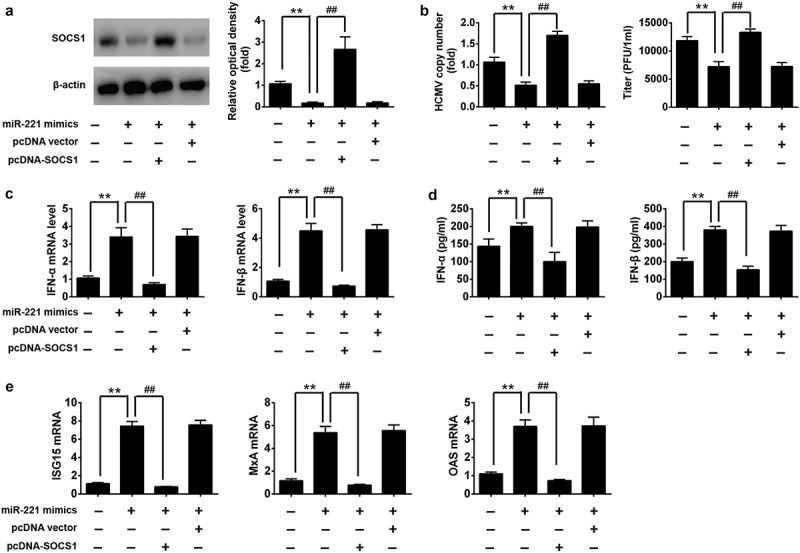
miR-221 exerts antiviral function by targeting SOCS1. NPCs were untreated or transfected with miR-221 mimics only, miR-221 mimics combined with SOCS1 overexpression plasmid, miR-221 mimics combined with control plasmid. Twenty-four hours after transfection, NPCs were infected by HCMV at MOI 1 for 24 h. (A) The protein level of SOCS1 was detected by western blot, and quantified as indicated. (B) The HCMV copy numbers was checked by qRT-PCR assay, and HCMV titer was evaluated by plaque assay. (C) IFN-a and IFN-b mRNA expression were determined by qRT-PCR. (D) The levels of IFN-a and IFN-b cytokines in the cell culture supernatant was determined by ELISA. (E) Expression levels of ISG15, MxA and OAS were quantified by real-time PCR. These experiments have been repeated three times, and the representative data were shown. (**p < 0.01 vs only HCMV infected group, ## p < 0.01 vs HCMV + miR-221 mimics).
MiR-221 promotes the activation of NF-κB by targeting SOCS1 upon HCMV infection
SOCS1 was reported to negatively regulate the activation of NF-κB [17]. We next explored whether miR-221 modulated the activation of NF-κB. IκB is the inhibitor of NF-κB, phosphorylation of IκB leads to the degradation of IκB and activation of NF-κB [18]. We found miR-221 mimics promoted the phosphorylation of IκB and activation of NF-κB subunit p65, while miR-221 inhibitor treatment diminished the phosphorylation of IκB and the level of phosphorylation of p65 in nuclear (Figure 7(A,B)). Phosphorylated NF-κB could enter the nuclear and play crucial roles in transcriptional induction of type I IFNs including IFNa and IFNb, which in turn signal through IFNa receptor to induce the expression of several IFN-stimulated genes (ISGs) for restraining virus replication [19–21]. These cytokines induced inflammation and lead to host defense to eliminate HCMV infection. To sum up, MiR-221 promotes the activation of NF-κB by targeting SOCS1 upon HCMV infection.
Figure 7.
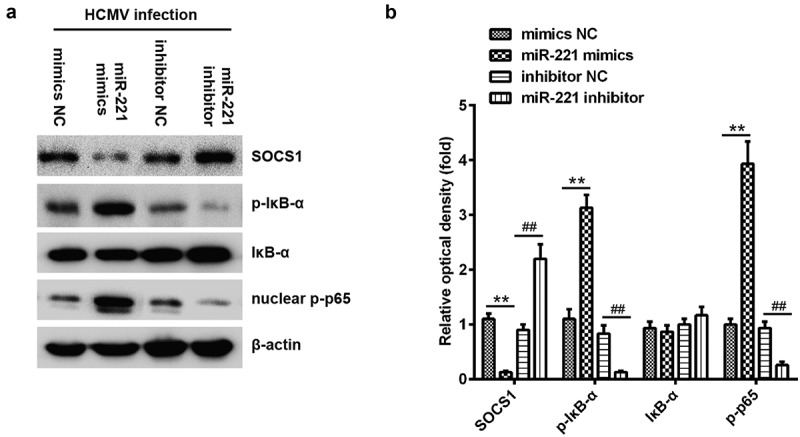
miR-221 promotes NF-κB activation by suppressing SOCS1. (A-B) NPCs were transfected with miR-221 mimic or mimic negative control and miR-221 inhibitor or inhibitor negative control as indicated. The protein levels of phosphorylated IkBa, total IkBa and nuclear-phosphorylated p65 in lysates were detected by immunoblot after HCMV infection for 24 h (A). The band intensity was calculated using ImageJ software (B). (** or ## p < 0.01 vs mimics NC or inhibitor NC, respectively).
MiR-221 attenuates cytomegalovirus replication in vivo
To investigate whether miR-221 can suppress cytomegalovirus infection in vivo, we inoculated BALB/C mice with 105 PFU MCMV K181 intravenously (i.v.) with or without the application of agomir-221. 7 days post-infection, the lungs, and livers were harvested for the evaluation of a miR-221 expression, MCMV load and histology analysis (Figure 8). Mice infected with MCMV displayed four fold expression level of miR-221 in both lung and liver, which was further increased by agomir-221 treatment (Figure 8(A,D)). It is noteworthy that agomir-221 treatment markedly decreased the MCMV copy number in both lung and liver (Figure 8(B,E)). In both tissues, MCMV-induced tissue damage was alleviated by agomir-221 treatment (Figure 8(C,F)). Collectively, these data indicated that cytomegalovirus infection promoted the expression of miR-221 in vivo, and miR-221 enhanced the anti-viral defense against cytomegalovirus.
Figure 8.
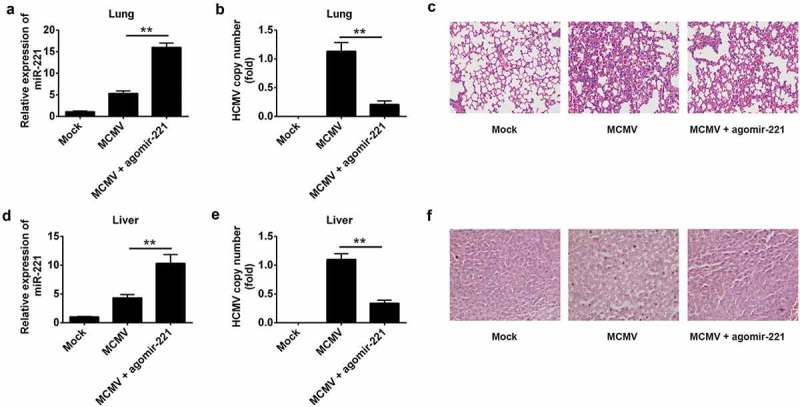
miR-221 agomir has an effective anti-MCMV function in vivo. (A) miR-221 expression level was checked by RT-qPCR in lung after MCMV K181strain infection or MCMV infection plus agomir-221 treatment. (B) MCMV genome copy was quantified by RT-qPCR in lung after MCMV infection or MCMV infection plus agomir-221 treatment. (C) The pathological analysis of lung by H&E staining after MCMV infection or MCMV infection plus agomir-221 treatment. (D) miR-221 expression level was checked by RT-qPCR in liver after MCMV infection or MCMV infection plus agomir-221 treatment. (E) MCMV genome copy was quantified by RT-qPCR in liver after MCMV infection or MCMV infection plus agomir-221 treatment. (F) The pathological analysis of liver by H&E staining after MCMV infection or MCMV infection plus agomir-221 treatment. These experiments have been repeated three times, and the representative data were shown. (** p < 0.01 vs MCMV group).
Discussion
HCMV is a ubiquitous pathogen which could establish a lifelong infection in human after primary infection [22]. HCMV infection during fetal development could lead to severe neural damage and neural developmental disorders [23]. Although the pathogenesis of HCMV in the neural system is not fully understood, viral replication in neuronal cells including NPCs is presumed to be the direct cause to neural damage [24].
Unlike other viruses in herpesviruses, human cytomegalovirus (HCMV) belongs to the betaherpesvirus family and have an ability to trigger a strong innate immune response in infected cells. Through activating the transcriptional factors IRF3 and NF-κB, HCMV induces the transcriptions of the Type I interferon and proinflammatory cytokines [25,26]. Type I IFNs include IFN-β (majorly secreted by epithelial cells and fibroblasts) and IFN-α subtypes 1 to 14 (majorly secreted by innate immune cell like macrophages and dendritic cells). IFN-α/β bind to and activate ubiquitously expressed IFNA receptor, leading to phosphorylation of signal transducer and activator of transcription 1 (STAT1) and STAT2. STAT1 and STAT2 activation lead to the transcription of ISGs which exert the anti-viral activities [25,27].
Micro-RNA have been discovered to play important roles in regulating cellular factors related to viral replication or host innate immune responses. Many miRNAs were reported to exert their regulatory functions in viral infection and replication. For instance, miR-215 can enhances HCV replication by targeting TRIM22 and inactivating NF-κB signaling [28]. miR-146a could facilitate the replication of dengue virus by dampening interferon induction through targeting TRAF6 [29]. MiRNA-221 was reported to be upregulated and involved in the carcinogenesis and pathogenesis of various kinds of human cancers [30]. Through the comparation of global miRNA expression profiles in HCMV infected NPCs and uninfected group, we found miR-221 could be induced by different strains of HCMV infection. We further demonstrated that miR-221 repressed HCMV replication by targeting negative regulator SOCS1 and promoting type I IFN signaling pathway(Figure 8). Additionally, the level of both mRNA and protein of IFNα and IFNβ were significantly increased by transfection of miR-221 mimics (Figure 3), implying that overexpression of miR-182 could be positively regulate the expression of type I IFN in NPCs cells challenged by HCMV. Through binding to the 3ʹUTR of SOCS1, miR-221 promotes the degradation of SOCS1 and facilitates the activation of NF-κB signaling and establishes an antiviral state against HCMV. Our findings promote a potential targets for anti-HCMV treatment.
SOCS1 belongs to the suppressor of cytokine signaling subfamily and takes part in a negative feedback loop to attenuate cytokine signaling. Several recent viral studies have shown that viral genes, such as Tax gene product (Tax), encoded by HTLV-1, could hijack SOCS1 to inhibit host antiviral pathways, as a strategy to evade host immunity [31]. In our study, SOCS1 is targeted by host miR-221 and downregulated SCOS1 promotes the activation of NF-κB signaling and establishes an antiviral state against HCMV. In conclusion, miR-221 modulates the infection and replication of cytomegalovirus via promoting type I IFN signaling in vitro and in vivo (Figure 9), suggesting it could be developed as a potential target for anti-HCMV treatment.
Figure 9.
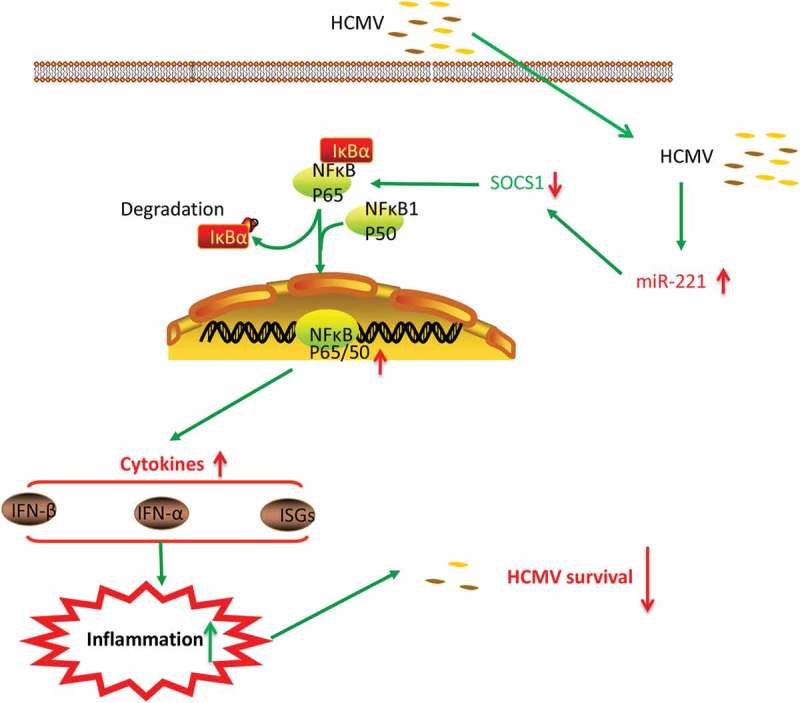
Model for the regulatory role of miR-221 in HCMV infection. HCMV infection induces the downregulation of SOCS1 protein level through promoting the expression of miR-221. As a negative regulator, SOCS1 downregulation leads to the enhanced activation of NF-κB pathway upon HCMV infection. Activated NF-κB enter the nuclear and facilitates the transcription of type I IFN (IFNα and IFNβ), leading to the activation of downstream ISGs effectors. The releases of type I IFN and ISGs setup an inflammation in the HCMV infected cell, and establishes an antiviral state against HCMV.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Jean Beltran PM, Cristea IM.. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev Proteomics. 2014;11:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Routon JR, Sherrill C.. Attitude toward physical education and self-concepts of asthmatic and nonasthmatic children taught by physical education specialists. Percept Mot Skills. 1989;68:1320–1322. [DOI] [PubMed] [Google Scholar]
- [3].Sia IG, Patel R. New strategies for prevention and therapy of cytomegalovirus infection and disease in solid-organ transplant recipients. Clin Microbiol Rev. 2000;13:83–121. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. [DOI] [PubMed] [Google Scholar]
- [5].Huang Y, Shen XJ, Zou Q, et al. Biological functions of microRNAs: a review. J Physiol Biochem. 2011;67:129–139. [DOI] [PubMed] [Google Scholar]
- [6].Fu YR, Liu XJ, Li XJ, et al. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J Virol. 2015;89:1070–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].He X, Teng J, Cui C, et al. MicroRNA-182 inhibits HCMV replication through activation of type I IFN response by targeting FOXO3 in neural cells. Exp Cell Res. 2018;369:197–207. [DOI] [PubMed] [Google Scholar]
- [8].Yoshimura A, Nishinakamura H, Matsumura Y, et al. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res Ther. 2005;7:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fujimoto M, Naka T. SOCS1, a negative regulator of cytokine Signals and TLR Responses, in Human Liver Diseases. Gastroenterol Res Pract. 2010;2010: 470468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Piganis RA, De Weerd NA, Gould JA, et al. Suppressor of cytokine signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon alpha receptor (IFNAR1)-associated tyrosine kinase Tyk2. J Biol Chem. 2011;286:33811–33818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li A, Song W, Qian J, et al. MiR-122 modulates type I interferon expression through blocking suppressor of cytokine signaling 1. Int J Biochem Cell Biol. 2013;45:858–865. [DOI] [PubMed] [Google Scholar]
- [12].Wang P, Hou J, Lin L, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185:6226–6233. [DOI] [PubMed] [Google Scholar]
- [13].Ding CL, Xu G, Ren H, et al. HCV infection induces the upregulation of miR-221 in NF-kappaB dependent manner. Virus Res. 2015;196:135–139. [DOI] [PubMed] [Google Scholar]
- [14].Xu G, Yang F, Ding CL, et al. MiR-221 accentuates IFNs anti-HCV effect by downregulating SOCS1 and SOCS3. Virology. 2014;462–463:343–350. [DOI] [PubMed] [Google Scholar]
- [15].Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yoshimura A, Ohishi HM, Aki D, et al. Regulation of TLR signaling and inflammation by SOCS family proteins. J Leukoc Biol. 2004;75:422–427. [DOI] [PubMed] [Google Scholar]
- [17].Yoshimura A, Ito M, Chikuma S, et al. Negative regulation of cytokine signaling in immunity. Cold Spring Harb Perspect Biol. 2018;10:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. 2017;168:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Czerkies M, Korwek Z, Prus W, et al. Cell fate in antiviral response arises in the crosstalk of IRF, NF-kappaB and JAK/STAT pathways. Nat Commun. 2018;9:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015;36:150–160. [DOI] [PubMed] [Google Scholar]
- [21].McNab F, Mayer-Barber K, Sher A, et al. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Britt WJ. Congenital human cytomegalovirus infection and the enigma of maternal immunity. J Virol. 2017;91:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Revello MG, Zavattoni M, Furione M, et al. Preconceptional primary human cytomegalovirus infection and risk of congenital infection. J Infect Dis. 2006;193:783–787. [DOI] [PubMed] [Google Scholar]
- [24].Gabrielli L, Bonasoni MP, Santini D, et al. Congenital cytomegalovirus infection: patterns of fetal brain damage. Clin Microbiol Infect. 2012;18:E419–27. [DOI] [PubMed] [Google Scholar]
- [25].DeFilippis VR, Alvarado D, Sali T, et al. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J Virol. 2010;84:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Marques M, Ferreira AR, Ribeiro D. The interplay between human cytomegalovirus and pathogen recognition receptor signaling. Viruses. 2018;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jackson SE, Mason GM, Wills MR. Human cytomegalovirus immunity and immune evasion. Virus Res. 2011;157:151–160. [DOI] [PubMed] [Google Scholar]
- [28].Tian H, He Z. miR-215 enhances HCV replication by targeting TRIM22 and inactivating NF-kappaB signaling. Yonsei Med J. 2018;59:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu S, He L, Li Y, et al. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J Infect. 2013;67:329–341. [DOI] [PubMed] [Google Scholar]
- [30].Garofalo M, Quintavalle C, Romano G, et al. miR221/222 in cancer: their role in tumor progression and response to therapy. Curr Mol Med. 2012;12:27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Charoenthongtrakul S, Zhou Q, Shembade N, et al. Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF-kappaB-dependent induction of SOCS1. J Virol. 2011;85:6955–6962. [DOI] [PMC free article] [PubMed] [Google Scholar]