

ABSTRACT
Coxsackievirus A16 (CA16) has caused worldwide epidemics of hand, foot and mouth disease (HFMD), particularly in infants and pre-school children. Currently, there are no vaccines or antiviral drugs available for CA16-associated disease. In this study, a CA16-specific monoclonal antibody (MAb) NA11F12 was derived with an epidemic CA16 strain (GenBank no. JX127258). NA11F12 was found to have high cross-neutralization activity against different CA16 subgenotypes but not EV71 using RD cells. The neutralizing titers of NA11F12 ranged from 1:1024 to 1:12288 against A, B1, B2 and C subgenotypes of CA16 and was less than 8 against EV71 strain. In the neonatal mouse model, a single treatment of NA11F12 showed effective protection with a dose- and time-dependent relationship against lethal challenge by CA16 strain (GenBank no. JX481738). At day 1 post-infection, administering more than 0.1 μg/g of NA11F12 could protect 100% newborn mice from mobility and mortality challenged by CA16. With dose of 10 μg/g of NA11F12, a single administration fully protected mice against CA16-associated disease within 4 days post-infection. And there were 80% and 60% mice protected by administering NA11F12 at day 5 post-infection and day 6 post-infection when the control mice had shown clinical symptoms for 1- and 2-day, respectively. Immunohistochemical and histological analysis confirmed that NA11F12 significantly prohibited CA16 VP1 expression in various tissues and prevented CA16-induced necrosis. In conclusion, a CA16-specific MAb NA11F12 with high cross-neutralization activity was identified, which could effectively protect lethal CA16 challenge in mice. It could be a potential therapeutic MAb against CA16 in the future.
KEYWORDS: Hand, foot and mouth disease (HFMD), Coxsackievirus A16, monoclonal antibody, cross-neutralization, therapeutic effect
Introduction
Coxsackievirus A16 (CA16), belonging to the Enterovirus genus in the Picornaviridae family, is one of the major pathogens responsible for hand, foot and mouth disease (HFMD).1 Outbreaks of HFMD caused by CA16 have been reported worldwide since 1957, such as Vietnam,2 India,3 Singapore,4 and China.5-7 Although most CA16 infections are self-limited and cause mild symptoms in infants and children under five years old, severe complications such as myocarditis, aseptic meningitis, pneumonia and death have been increasingly describe.8 However, there is no effective treatment and prevention agent available against CA16-associated diseases.
MAbs are recognized as one of the effective ways to prevent and treat virus-inducing infectious diseases. Related reports have proved that MAbs against EV71, one of the main pathogen of HFMD, could provide effective protection.9-11 However, due to the negligence of CA16, only several MAbs against CA16 were reported, which were used to distinguish different types of CA16 viral particles.12 No therapeutic MAb against CA16 has been reported.
In this study, we characterized a CA16 monoclonal antibody (MAb) NA11F12 with high neutralization activity in vitro and efficacy in vivo, which could be a potential therapeutic MAb against CA16-associated diseases.
Result
In vitro cross-binding and cross-neutralizing abilities of MAb NA11F12
We produced a MAb NA11F12 by immunizing with the purified 190/CA16. Immunofluorescence Assay (IFA) showed specific binding ability of NA11F12 with CA16-infected cells, but not uninfected cells (Figure 1A). Enzyme-linked immunoreaction (ELISA) was used to further explore the potential binding ability of NA11F12, the results showed that NA11F12 could cross-bind different CA16 subgenotype strains but not EV71. (Figure 1B). A 32 kDa band indicated that NA11F12 could identify a linear epitope on VP1 protein by Western Blot (Figure 1C). NA11F12 could neutralize CA16 but not EV71 by neutralization assay. The neutralizing titers of NA11F12 against CA16 strains, covering the A, B, C subgenotypes, were higher than 1:1028. The titer against EV71 was significant difference with other groups (p < 0.05). The titer against CA16-C subgenotype was significant difference with other CA16 subgenotypes (p < 0.05) (Figure 1D). These results demonstrated that NA11F12 was a good candidate for testing in animals.
Figure 1.
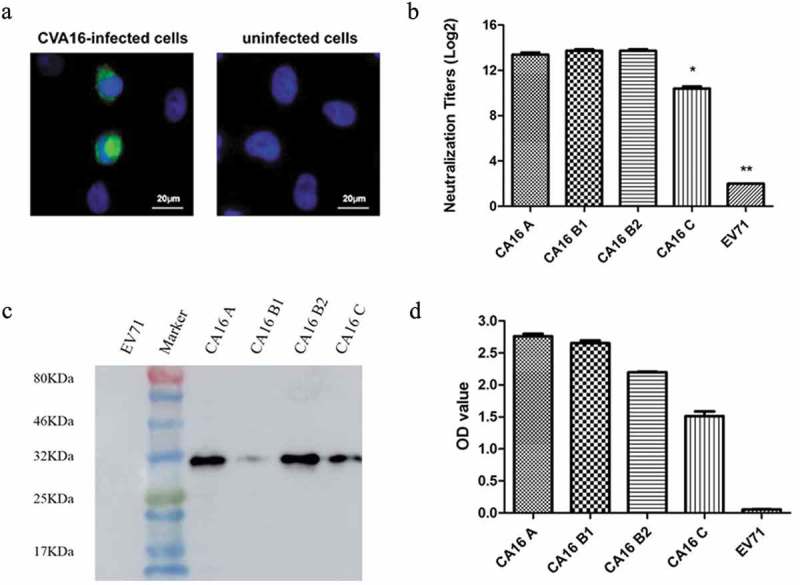
The cross-binding and cross-neutralizing abilities of MAb NA11F12. (A) Immunofluorescence assay of the MAb NA11F12 against the CA16-infected RD cells. Uninfected RD cells were used as the negative controls. The secondary antibody was FITC-conjugated anti-mouse (green). The nuclei were stained with DAPI (blue). (B) NA11F12 (6.25 μg) bound specifically with CA16 strains (5 × 104CCID50) covering A, B1, B2, C subgenotypes but not EV71 by ELISA. (C) The positive bands of MAb NA11F12 reacted with A, B1, B2, C subgenotypes CA16 strains were found at about 32 kDa. (D) Neutralizing titers of MAb NA11F12 against CA16 covering A, B1, B2, C subgenotypes by neutralization assay. The Bonferroni’s Multiple Comparison Test was used to compare the difference of the neutralizing titer. **The neutralizing titer against EV71 was significant difference with other groups, p < 0.05. *The titer against CA16 C subgenotype was significant difference with other CA16 subgenotypes, p < 0.05.
Dose-dependent effect of NA11F12 in vivo
To figure out the effect of different treatment dosage of NA11F12 at day 1 post-infection, groups of newborn BALB/c mice were injected i.p with serially diluted NA11F12 (10 μg/g–0.001 μg/g, respectively). Inactivity occurred in the controlled group treated with Minimal Essential Medium (MEM) at 5 days post-infection, and the symptom changed to forelimb weakness, followed with hind legs paralysis and quadriplegic, and died terminally at 7–11 days. In contrast, 10 μg/g, 1 μg/g and 0.1 μg/g dose of NA11F12 could fully protect mice against CA16 challenge with no visible clinical symptoms, whereas the 0.01 μg/g dose group could prevent 80% mice from paralysis and death compared with control group (all p < 0.01). Although all mice in the minimal dose (0.001 μg/g) group died, the time of presenting clinical symptom and death were delayed from day 5 to 7 and day 7 to 9 post-infection, respectively. The median effective dose (ED50) was calculated by Reed and Munch method to be 0.0042 μg/g (Figure 2).
Figure 2.
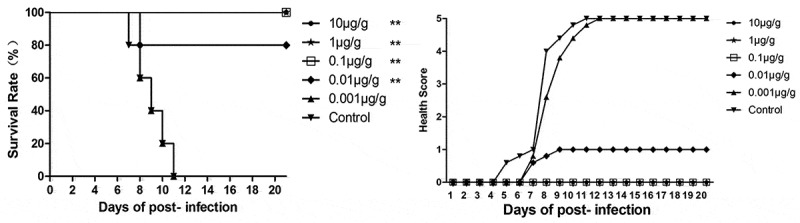
In vivo treatment of NA11F12 at different doses after CA16 infection. One-day-old BALB/c mice were i.p. challenged with 54CCID50/mouse BJCA08/CA16 and then i.p. treated with different doses of NA11F12 ranking from 10 μg/g to 0.001 μg/g (10-fold serial dilutions) at 1 day post-infection(DPI). The control mice were treated with Minimal Essential Medium (MEM, THERMO). Survival rates and health scores were monitored daily for 20 dpi. The Log-rank (Mantel-Cox) test was used to compare the survival of mice between each group and control group at 20 days of post-infection. **p < 0.01, *p < 0.05.
Time-dependent effect of NA11F12 in vivo
Different treatment times were studied to further evaluate the efficacy of NA11F12. After challenged by BJCA08/CA16, mice were treated i.p. with NA11F12 (10 μg/g) at day 1, 2, 3, 4, 5, 6 or 7 post-infection. The results showed that symptoms emerged in the control group as early as day 5 post-infection and aggravated to limb paralysis from day 6 post-infection. All the control mice died within 11 days post-infection, while the mice that received NA11F12 at day 1, 2, 3, 4 post-infection were totally healthy. However, treatment at day 5 or 6 post-infection could protect 80% or 60% mice from death respectively, indicating a significant treatment effect compared to the control group (all p < 0.05) (Figure 3). And 20% mice could be protected by treatment with NA11F12 at day 7 post-infection, when the mice began to die in the control group. These results show that NA11F12 was able to fully protect mice from CA16-induced paralysis and death within 4 days post-infection, which was very close to the time when the control mice began to present visible symptoms.
Figure 3.
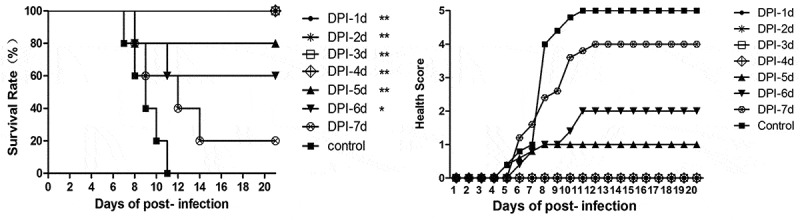
In vivo treatment of NA11F12 at different times after CA16 infection. Mice were challenged i.p. with 54CCID50/mouse BJCA08/CA16 and then treated with NA11F12 at different days post-infection from day 1 to day 7. The control mice were treated with Minimal Essential Medium (MEM, THERMO). Survival rates and health scores were monitored daily for 20 dpi. The Log-rank (Mantel-Cox) test was used to compare the survival of mice between each group and control group at 20 days of post-infection. **p < 0.01, *p < 0.05.
Histological and immunohistochemical staining
To confirm the treatment effect of NA11F12, hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC) examinations were performed. Compared to the normal morphology of NA11F12-treated mice, inflammatory cell infiltration was observed in the brain and severe necrosis was found in the heart and limb muscles in the MEM-treated group. Positive CA16 antigen was found in the heart, limb muscle and brain in MEM-treatment mice. In contrast, no pathological symptom change was observed in the NA11F12-treated group and health control group (Figure 4). NA11F12 was able to fully protect mice from CA16-induced morbidity and mortality within day 4 post-infection. These results show that the treatment of MAb NA11F12 was able to protect mice from CA16-induced paralysis and death.
Figure 4.
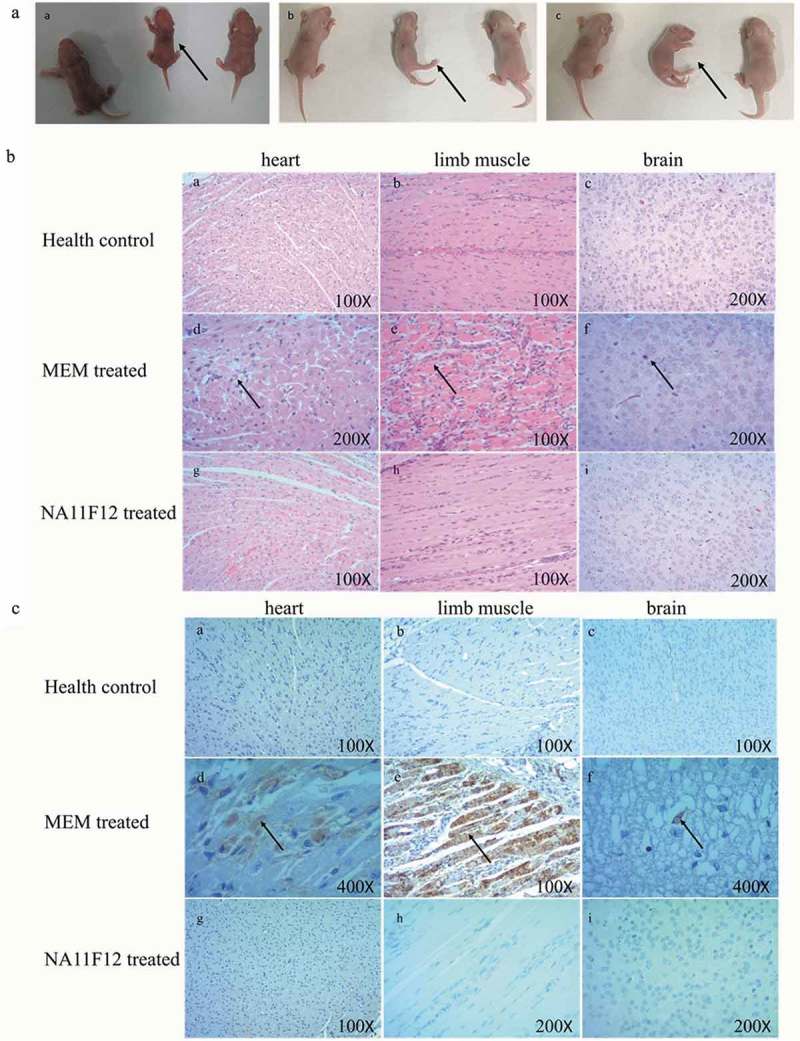
Histological examination and immunohistochemical results of in vivo study. One-day-old mice were i.p challenged with lethal doses of BJCA08/CA16 (54CCID50), then i.p. treated with 10 μg/g NA11F12 or MEM at day 1 post-infection. (A) Dynamic changes of clinical symptoms in newborn mice model. Uninfected CA16 mice (Health control) (left), MEM-treated mice (middle) and NA11F12-treated (right) in each picture. MEM-treated mice showed visible clinical symptoms: (a) waste, (b) hind legs paralysis, (c) quadriplegic (arrows). (B) HE staining of different tissues from BALB/c neonatal mice. Positive reactions were observed in heart, brain and limb muscle tissues by H&E staining. Infected mice (grades 4–5) exhibited severe inflammation in heart and limbs and cranial nerve necrosis (arrows). In contrast, no histological change was observed in the mice of health control group and NA11F12-treated group. Magnification×100 (a, b, e, g, h), Magnification×200 (c, d, f, i). (C) IHC staining of different tissues from BALB/c neonatal mice. Positive reactions were observed in heart, brain and limb muscle tissues by IHC straining. Numerous viral antigen-positive reactions were observed in the heart, limb muscle and brain (arrows) in the infected mice. In contrast, no viral antigen was observed in the mice of health control group and NA11F12-treated group. Magnification×100 (a, b, c, e, g), Magnification×200 (h, i), Magnification×400(d, f).
Discussion
CA16 and EV71 are the major pathogens of HFMD, which usually cause skin rash, herpes and fever in infants and children under 5 years old. Besides these common symptoms, central nervous system symptoms may appear in some patients, such us aseptic meningitis, brainstem encephalitis, neuronal pulmonary edema, and heart failure.8,13,14 It is universally accepted that severe HFMD is mainly caused by EV71 and mild cases are relevant to CA16 with wider epidemic.15,16 However, severe symptoms caused by CA16 have been reported in recent years,17 and mixed infection of CA16 and EV71 often causes more severe symptoms and elongates course of disease.18 Currently, there is no available effective method to treat and prevent CA16-related infections. Therefore, it is urgent to carry out researches in these aspects.
The first MAb OKT3 was approved by FDA in 1986 for therapy rejection reaction (www.fda.gov). Since then, characterized by low toxicity and high specificity, monoclonal antibodies have gained increasing attention and been approved for clinical use for diseases, such as cancer, autoimmune diseases, and inflammation, as well as in the infectious diseases.19 In the previous studies, passive transfer of specific neutralizing-MAbs could reduce corresponding virus-inducing severity, such as Venezuelan equine encephalitis,20 West Nile virus (WNA),21 influenza,9,10 Dengue Virus11 and EV71. Especially for EV71, another main member of Enterovirus genus, three neutralizing MAbs (nMAbs) could protect mice from lethal EV71 challenge at different time post-infection. Among them, an EV71-VP1 and EV71-VP2 epitope-targeted nMAb could protect mice from lethal EV71 challenge at day 1 post-infection.9,10 Another conformational nMAb CT11F9 was effective in preventing 100% mice from EV71-induced morbidity and mortality within three days post-infection, after which the mice began to show visible illness.11 These nMAbs provided potential treatment tool for HFMD caused by EV71.
There has been few reported studies on CA16 MAbs, Liu et al. discovered that the anti-CA16 MAb 8C4 (10 μg) was effective in partially preventing CA16-inducing death at 1 day post-infection.22 In this study, we produced an anti-CA16 MAb NA11F12 by immunizing an epidemic strain 190, which was separated in Taiwan in 2007. NA11F12 not only showed a high neutralization activity in vitro, but also could protect mice from lethal CA16 challenge with a good dose- and time-depended effect, respectively. NA11F12 at >0.1 μg and 0.01 μg could preventing 100% and 80% mice from mobility and mortality caused by CA16 challenge, respectively. 100% mice could be protected by therapy with NA11F12 within four days post-infection, after which the mice began to show visible illness. It was interesting that 80%, 60% and 20% mice could be protected by therapy with NA11F12 at 5, 6 and 7 days post-infection respectively. Compared with the significant damage and CA16-VP1 antigen in brain, heart and limb muscles in mice of the MEM-treatment group, no damage and CA16-VP1 antigen were found in the mice treated with NA11F12.
CA16 has been grouped into the genotype A, B and C according to the differences in VP4 nucleotide sequence.23 Since 1990s, the main epidemic strains have belonged to the B and C subgenotypes.24 It has been demonstrated that circulating CA16 virus is a group of complex recombinant viruses involving multiple type A HEVs, including coxsackievirus A4 (CA4), CA16 and EV71.25 There was >20% genetic difference between G10 prototype strain and the current epidemic strains.26 Besides the difference of nucleotide sequence, there were great differences in the antigenicity of different circulating strains.27 In the recent study,28 Yao et al. first discovered that anti-sera from ten circulating strains had different neutralizing abilities against G10 and these epidemic strains, in which these antisera could neutralize G10 with higher titer (GMT:69.3–210.7) but not ten epidemic strains(GMT < 8).These results indicated the epidemic strains were not easy to be neutralized, compared with the original strain G10. In this study, NA11F12 could not only neutralize G10 but also epidemic strains covering the B1, B2 and C subgenotypes, with high neutralization titers at 4096, 12288, 12288 and 1024 respectively. Therefore NA11F12 could be used as a potential therapeutic MAb for CA16 infection.
Vaccines are the most effective means of preventing infectious diseases. To prevent the outbreaks of HFMD in recent years, EV71 vaccines developed by three companies (Beijing Vigoo Biological, Sinovac Biotech Co. Ltd, and Institute of Medical Biology) have been approved on the market since 2015. Several companies and academic institutions launched projects to develop monovalent or bivalent HFMD vaccine against CA16.29 However, there are many challenges in CA16 and multivalent vaccine development, such as the low immunogenicity of CA16 and the difficulty of CA16 epidemic strains to be neutralized. In our study, NA11F12 could cross-bind and cross-neutralize different A, B1, B2 and C subgenotypes. NA11F12 had no activity in neutralizing and binding with EV71, which shared 80% sequence identity in capsid proteins with CA16,30 indicating its specificity to CA16. Further study on the neutralizing epitope of NA11F12 would be significant for the understanding of CA16 virus and the development of CA16 vaccine.
In conclusion, we produced a CA16-specific MAb NA11F12 with high cross-neutralizing titers against different CA16 subgentypes, which could protect 100% mice from lethal CA16 challenge. NA11F12 could be used as a potential therapeutic MAb against CA16 infection. Our findings played a positive role in the development of CA16 vaccine and better understanding of CA16. However, only CA16 C subgenotype (BJCA08/CA16) was used as the challenge strain to evaluate NA11F12 in newborn mice. More subgenotypes and epidemic strains should be used to evaluate the protective effect of NA11F12 in vivo. It would lay the foundation for further humanized antibody and clinical trial studies.
Materials and methods
Cells and viruses
Human muscular rhabdomyosarcoma (RD) cells and mouse myeloma cells (Sp2/0-Ag-14) were cultivated in MEM (THERMO) with 10% fetal bovine serum, 1% L-glutamine, 1% penicillin and 1% streptomycin at 37°C. Viruses including BJCA08/CA16, G10/CA16, 731/CA16 and 190/CA16 strain were grown in RD cells. As a control, 523/EV71 strain was also grown in RD cells (Table 1). All viruses were titrated for the 50% cell culture infectious dose (CCID50) in RD cells.
Table 1.
CA16 and EV71 strains.
Production of anti-CA16 monoclonal antibody
To obtain the anti-CA16 MAb NA11F12, BALB/c mice were immunized with the purified 190/CA16 (GenBank no. JX127258), a CA16 whole virus isolated from China in 2007, emulsified in complete Freund’s adjuvant and subcutaneously boosted twice at 2-week intervals with CA16 in incomplete Freund’s adjuvant. After final injection, splenocytes from the immunized mice were fused with Sp2/0-Ag-14. Antibodies against CA16 in hybridoma supernatant were screened by ELISA and neutralization assay. Positive wells were cloned at least twice. Ascetic fluid produced from a single clone of positive cells was purified by precipitating with 50% ammonium sulfate using protein chromatography (GE Healthcare)
Immunofluorescence assay (IFA)
RD cells were plated onto glass coverslips in 24-well plates and then infected with CA16. After incubating at 37°C for 12 h, cells were fixed with 4% paraformaldehyde for 30 minutes without light, followed by permeating with wash solution (PBS + 0.3% Triton X-100) for 10 minutes, and blocked with goat serum for 1 h. Pretreated cells were incubated with MAb NA11F12 (3 μg/mL) at 37°C for 1 h. After washing with wash solution, cells were incubated with fluorescein isothiocyanate-conjugated goat anti-mouse second antibody (GAM-FITC) at 37°C for 30 minutes and co-strained with 4ʹ,6-diamidino-2-phenylindole (DAPI) for 5 minutes. The results were obtained using a fluorescent microscopy (Axio Imager Z2, Zeiss).
Neutralization assay
An in vitro neutralization assay for CA16 was performed. Briefly, RD cells were seeded at 2 × 104 cells per well into 96-well plates (Corning). NA11F12 (1 mg/mL) was serially diluted in a 2-fold dilution from 1:8 to 1:16,384 and incubated with an equal volume of CA16 (100 CCID50) at 37°C for 2 h. The virus/MAb mixtures were added into starvation cells and then incubated at 37°C for 24 h. The neutralization titer was defined as the highest dilution in over >50% CPE. Each assay was processed independently three times.
Capture ELISA assays
ELISA was conducted to measure the immunoreactivity of NA11F12 with EV71 and CA16 inactivated virus. Briefly, 96-well microtiter plates (Corning) were coated with anti-EV71 or CA16 polyclonal antibody overnight at 4°C. After washing with wash solution (0.05% Tween 20 in PBS), the plates were blocked with casein at 37°C for 1h. 5 × 104CCID50 CA16 and EV71 viruses were added for 1h, and then 6.25 μg MAb NA11F12 (diluted with PBS) was added and incubated for 1h at 37°C. Goat anti-mouse antibody labeled with horseradish peroxidase (HRP) was added to the plates (1:10000 dilution) and incubated at 37°C for 1h. The wells were washed 5 times with PBST between each step. Visualization was achieved by adding 100 μl HRP substrate for 10 minutes at room temperature, followed by 50 μl 2M H2SO4. The OD value (A450/630) was measured by a microplate absorbance spectrophotometer (Bio-Rad). Each assay was processed independently three times.
Western blot analysis
Total proteins of CA16 and EV71 were used to analyze the immunoreactivity with NA11F12 and identity which capsid protein NA11F12 could bind. Proteins were resolved by SDS-PAGE and electro-blotted onto nitrocellulose membranes (Bio-Rad). Membrane were blocked in 5% blotting grade milk (diluted with PBS), and incubated in primary antibody solution for 1 h, followed by incubation in HRP-conjugated goat anti-mouse IgG at 1:10,000 dilution (Dako Cytomation) for another hour. The membranes were washed three times for 5 minutes in 0.1% Tween-20(diluted in TBS), and developed with ECL Western Blotting Substrate reagent (ThermoFisher), followed by color development with Amersham imager 600 (General Electric Company)
In vivo study in mice
All animal experiments were carried out in accordance with the guidelines of the National Institute for Food and Drug Control for the Care and Use of Laboratory Animals. In order to evaluate the antiviral efficiency of NA11F12, a clinical isolate (BJCA08/CA16) from a patient with HFMD was chosen as challenge strain, which had been used to establish a CA16 neonatal mouse model in the previous study.31 Newborn BALB/c mice were challenged intraperitoneally (i.p.) with BJCA08/CA16 (GenBank no. JXe81738) 54CCID50/mouse, and serially diluted NA11F12 (10 μg/g–0.001 μg/g) and MEM medium were inoculated at day 1. The mice in control group were treated with MEM. Each group (n = 5) contained two independent experiments. To further prove the protective effect of NA11F12, NA11F12 (10 μg/g per body weight) was given at day 1, 2, 3, 4, 5, 6, or 7 post-infection. In all experiments, the mice were monitored daily for body weight and clinical symptoms until day 20 post-infection. The grade of clinical disease was recorded as follows: 0, healthy; 1, wasting/inactivity; 2, forelimb weakness; 3, hind legs paralysis; 4, quadriplegic; 5, moribund and death (Table 2).
Table 2.
Grading score for clinical symptoms of CA16 infected mice.
| Clinical sign(s) | Grade |
|---|---|
| Healthy | 0 |
| Wasting/Inactivity | 1 |
| Forelimb weakness | 2 |
| Hind legs paralysis | 3 |
| Quadriplegic | 4 |
| Moribund and death | 5 |
Histology and immunohistochemistry analysis
Another two groups of newborn mice (n = 5) was challenged i.p. with CA16 BJCA08. At day 1 post-infection, the text group was given NA11F12 (10 μg/g per body weight), and another group was given MEM medium. The mice in control group were unchallenged by CA16. After 7 days, the animals were euthanized, and tissues including brain, kidney, spinal cord, heart, liver, lung, intestine and limb muscle were collected, and then fixed in 4% paraformaldehyde for at least 2 days. Hematoxylin and eosin (HE) and immunohistochemistry (IHC) staining were performed as in a previous study.31 Primary antibody against the VP1 region of CA16 was used at 1:32,000 dilution.
Statistical analysis
Data statistical analyses were performed using GraphPad Prism version 5, and p < 0.05 was considered as statistical significance. The health scores were shown as means.
Funding Statement
The study was sponsored by the Major Special Projects Funding Program (No. 2016ZX09101120, 2018ZX09711003-005-003 and 2017ZX10304402) from the Ministry of Science and Technology of the People’s Republic of China.
Abbreviations
- CA16
Coxsackievirus A16
- HFMD
Hand, foot and mouth disease
- MAb
monoclonal antibody
- EV71
Enterovirus 71
- nMAb
neutralizing Mab
- DPI
days of post-infection
- RD
Human muscular rhabdomyosarcoma
- MEM
Minimal Essential Medium
- CCID50
50% tissue culture infectious dose
- ELISA
enzyme-linked immunoreaction
- i.p
Intraperitoneally
- HE
hematoxylin and eosin
- IHC
immunohistochemistry
- ED50
the median effective dose
- IFA
Immunofluorescence Assay
Acknowledgments
We thank the Major Special Projects Funding Program (No. 2016ZX09101120) from the Ministry of Science and Technology of the People’s Republic of China for their support. We also thank the National Science and Technology Major Projects (2018ZX09711003-005-003 and 2017ZX10304402) for their support.
Consent for publication
All guardians of participants provided written informed consent.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Ethics approval and consent to participate
This study was done in accordance with the Declaration of Helsinki, Good Clinical Practice, and Chinese regulatory requirements.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Lim H, In HJ, Lee JA, Sik Yoo J, Lee SW, Chung GT, Choi YK, Chung JK, Cho SJ, Lee JW.. The immunogenicity and protection effect of an inactivated coxsackievirus A6, A10, and A16 vaccine against hand, foot, and mouth disease. Vaccine. 2018. June 7;36(24):3445–52. doi: 10.1016/j.vaccine.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Van Tu P, Thao NTT, Perera D, Truong KH, Tien NTK, Thuong TC, How OM, Cardosa MJ, McMinn PC. Epidemiologic and virologic investigation of hand, foot, and mouth disease, southern Vietnam, 2005. Emerg Infect Dis. 2007. November;13(11):1733–41. doi: 10.3201/eid1311.070632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kar BR, Dwibedi B, Kar SK. An outbreak of hand, foot and mouth disease in Bhubaneswar, Odisha. Indian Pediatr. 2013. January 8;50(1):139–42. [DOI] [PubMed] [Google Scholar]
- 4.Ang LW, Koh BK, Chan KP, Chua LT, James L, Goh KT. Epidemiology and control of hand, foot and mouth disease in Singapore, 2001-2007. Ann Acad Med Singapore. 2009. February;38(2):106–12. [PubMed] [Google Scholar]
- 5.Yin DQ, Wang CB, Ji SX. Epidemiology characteristics of human coxsackievirus A16 and enterovirus 71 circulating in Linyi, China. from 2009 to 2017. Jpn J Infect Dis. 2018. Nov 22;71(6):470–473. [DOI] [PubMed] [Google Scholar]
- 6.Xiao Y, Zhou J, Zhang H, Ding C, Shi P. Epidemiological and aetiological characteristics of hand, foot and mouth disease cases 2011–2017 in Yixing, China. Infect Dis (Lond). 2018. July;25:1–3. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Zhang H, Liu H, Zhang J, He L, Sun H, Huang X, Yang Z, Ma S. Molecular characteristics of hand, foot, and mouth disease for hospitalized pediatric patients in Yunnan, China. Medicine. 2018. August;97(31):e11610. doi: 10.1097/MD.0000000000011610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Legay F, Lévêque N, Gacouin A, Tattevin P, Bouet J, Thomas R, Chomelt JJ. Fatal coxsackievirus A-16 pneumonitis in adult. Emerg Infect Dis. 2007. July;13(7):1084–86. doi: 10.3201/eid1307.070295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He F, Kumar SR, Syed Khader SM, Tan Y, Prabakaran M, Kwang J. Effective intranasal therapeutics and prophylactics with monoclonal antibody against lethal infection of H7N7 influenza virus. Antiviral Res. 2013. October;100(1):207–14. doi: 10.1016/j.antiviral.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Zheng Q, Xia L, Wu WL, Zheng Z, Huo Y, Wu J, Liu Y, Yu H, Chen Y, Lau SY, et al. Properties and therapeutic efficacy of broadly reactive chimeric and humanized H5-specific monoclonal antibodies against H5N1 influenza viruses. Antimicrob Agents Chemother. 2011. April;55(4):1349–57. doi: 10.1128/AAC.01436-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li PC, Liao MY, Cheng PC, Liang JJ, Liu IJ, Chiu CY, Lin YL, Chang GJ, Wu HC. Development of a humanized antibody with high therapeutic potential against dengue virus type 2. PLoS Negl Trop Dis. 2012;6(5):e1636. doi: 10.1371/journal.pntd.0001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye X, Yang L, Jia J, Han J, Li S, Liu Y, Xu L, Zhao H, Chen Y, Li Y, et al. Development of sandwich ELISAs that can distinguish different types of coxsackievirus A16 viral particles. Appl Microbiol Biotechnol. 2016. March;100(6):2809–15. doi: 10.1007/s00253-016-7296-z. [DOI] [PubMed] [Google Scholar]
- 13.Goto K, Sanefuji M, Kusuhara K, Nishimura Y, Shimizu H, Kira R, Torisu H, Hara T. Rhombencephalitis and coxsackievirus A16. Emerg Infect Dis. 2009. October;15(10):1689–91. doi: 10.3201/eid1510.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang CY, Lu F L, Wu MH, Lee CY, Huang LM. Fatal coxsackievirus A16 infection.Pediatr. Infect Dis J. 2004. March;23(3):275–76. doi: 10.1097/01.inf.0000115950.63906.78. [DOI] [PubMed] [Google Scholar]
- 15.Chang LY, Lin TY, Huang YC, Tsao KC, Shih SR, Kuo ML, Ning HC, Chung PW, Kang CM. Comparison of enterovirus 71 and coxsackie-virus A16 clinical illnessesduring the Taiwan enterovirus epidemic, 1998. Pediatr Infect Dis J. 1999. December;18(12):1092–96. [DOI] [PubMed] [Google Scholar]
- 16.Perera D, Yusof MA, Podin Y, Ooi MH, Thao NT, Wong KK, Zaki A, Chua KB, Malik YA, Tu PV, et al. Molecular phylogeny of modern coxsackievirus A16. Arch Virol. 2007;152(6):1201–08. doi: 10.1007/s00705-006-0934-5. [DOI] [PubMed] [Google Scholar]
- 17.Goto K, Sanefuji M, Kusuhara K, Nishimura Y, Shimizu H, Kira R, Torisu H, Hara T. Rhombencephalitis and coxsackievirus A16. Emerg Infect Dis.. 2009. October;15(10):1689–91. doi: 10.3201/eid1510.090594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma S, Zhang Y, Du C, Yang T, Liu Q, Pan Y, Chen J, Shi H, Sun Q, Liu L, et al. Dynamic constitution of the pathogens inducing encephalitis in hand, foot and mouth disease in kunming, 2009–2011. Jpn J Infect Dis. 2015;68(6):504–10. doi: 10.7883/yoken.JJID.2014.428. [DOI] [PubMed] [Google Scholar]
- 19.Kuhn C, Weiner HL. Therapeutic anti-CD3 monoclonal antibodies: from bench to bedside. Immunotherapy. 2016. July;8(8):889–906. doi: 10.2217/imt-2016-0049. [DOI] [PubMed] [Google Scholar]
- 20.Hunt AR, Bowen RA, Frederickson S, Maruyama T, Roehrig JT, Blair CD. Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology. 2011. June 5;414(2):146–52. doi: 10.1016/j.virol.2011.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smeraski CA, Siddharthan V, Morrey JD. Treatment of spatial memory impairment in hamsters infected with West Nile virus using a humanized monoclonal antibody MGAWN1. Antiviral Res. 2011. July;91(1):43–49. doi: 10.1016/j.antiviral.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q, Shi J, Huang X, Liu F, Cai Y, Lan K, Huang Z. A murine model of coxsackievirus A16 infection for anti-viral evaluation. Antiviral Res. 2014. May;105:26–31. doi: 10.1016/j.antiviral.2014.02.015. [DOI] [PubMed] [Google Scholar]
- 23.Li L, He Y, Yang H, Zhu J, Xu X, Dong J, Zhu Y, Jin Q. Genetic characteristics of human enterovirus 71 and coxsackievirus A16 circulating from 1999 to 2004 in Shenzhen, People’s Republic of China. J Clin Microbiol. 2005;43(3):3835–39. doi: 10.1128/JCM.43.8.3835-3839.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iwai M, Masaki A, Hasegawa S, Obara M, Horimoto E, Nakamura K, Tanaka Y, Endo K, Tanaka K, Ueda J, et al. Genetic changes of coxsackievirus A16 and enterovirus 71 isolated from hand, foot, and mouth disease patients in Toyama, Japan between 1981 and 2007. Jpn J Infect Dis. 2009. July;62(4):254–59. [PubMed] [Google Scholar]
- 25.Zhao K, Han X, Wang G, Hu W, Zhang W, Yu XF. Circulating coxsackievirus A16 identified as recombinant type A human enterovirus, China. Emerg Infect Dis. 2011. August;17(8):1537–40. doi: 10.3201/eid1708.101719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Song JH, Park K, Shim A, Kwon BE, Ahn JH, Choi YJ, Kim JK, Yeo SG, Yoon K, Ko HJ. Complete sequence analysis and antiviral screening of medicinal plants for human coxsackievirus a16 isolated in Korea. Osong Public Health Res Perspect. 2015. February;6(1):52–58. doi: 10.1016/j.phrp.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang E, Zhao H, Zhang Y, Liu J, Liao Y, Wang L, Cui P, Yang L, Liu L, Dong C, et al. A comparative study of the characteristics of two Coxsackie A virus type 16 strains (genotype B). Sci China Life Sci 2012; 55(4): 336–342. 10.1007/s11427-012-4313-z [DOI] [PubMed] [Google Scholar]
- 28.Yao X, Mao Q, Li Y, Hao C, Bian L, Chen P, Gao F, Wu X, Lu W, Gao Q, et al. Poorly neutralizing polyclonal antibody in vitro against coxsackievirus A16 circulating strains can prevent a lethal challenge in vivo. Hum Vaccin Immunother. 2018. May 4;14(5):1275–82. doi: 10.1080/21645515.2018.1426420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Yan K, Feng Y, Huang X, Ku Z, Cai Y, Liu F, Shi J, Huang Z. A virus-like particle vaccine for coxsackievirus A16 potently elicits neutralizing antibodies that protect mice against lethal challenge. Vaccine. 2012. October 19;30(47):6642–48. doi: 10.1016/j.vaccine.2012.08.071. [DOI] [PubMed] [Google Scholar]
- 30.Ren J, Wang X, Zhu L, Hu Z, Gao Q, Yang P, Li X, Wang J, Shen X, Fry EE, et al. Structures of Coxsackievirus A16 capsids with native antigenicity: implications for particle expansion, receptor binding, and immunogenicity. J Virol. 2015. October;89(20):10500–11. doi: 10.1128/JVI.01102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mao Q, Wang Y, Gao R, Shao J, Yao X, Lang S, Wang C, Mao P, Liang Z, Wang J. A neonatal mouse model of coxsackievirus A16 for vaccine evaluation. J Virol. 2012. November;86(22):11967–76. doi: 10.1128/JVI.00902-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.