

ABSTRACT
Heavy chain (Hc) heterodimers represent a majority of bispecific antibodies (bsAbs) under clinical development. Although recent technologies achieve high levels of Hc heterodimerization (HD), traces of homodimer contaminants are often present, and as a consequence robust purification techniques for generating highly pure heterodimers in a single step are needed. Here, we describe two different purification methods that exploit differences in Protein A (PA) or Protein G (PG) avidity between homo- and heterodimers. Differential elution between species was enabled by removing PA or PG binding in one of the Hcs of the bsAb. The PA method allowed the avidity purification of heterodimers based on the VH3 subclass, which naturally binds PA and interferes with separation, by using a combination of IgG3 Fc and a single amino acid change in VH3, N82aS. The PG method relied on a combination of three mutations that completely disrupts PG binding, M428G/N434A in IgG1 Fc and K213V in IgG1 CH1. Both methods achieved a high level of heterodimer purity as single-step techniques without Hc HD (93–98%). Since PA and PG have overlapping binding sites with the neonatal Fc receptor (FcRn), we investigated the effects of our engineering both in vitro and in vivo. Mild to moderate differences in FcRn binding and Fc thermal stability were observed, but these did not significantly change the serum half-lives of engineered control antibodies and heterodimers. The methods are conceptually compatible with various Hc HD platforms such as BEAT® (Bispecific Engagement by Antibodies based on the T cell receptor), in which the PA method has already been successfully implemented.
KEYWORDS: Antibody, BEAT, bispecific, engineering, heterodimer, Protein A, Protein G, purification
Introduction
Bispecific antibodies (BsAbs) combine specificities of two antibodies in a single antibody construct that is able to bind two different epitopes on the same or on different antigens. In cancer treatment, bsAbs are considered one of the most promising novel therapeutic modalities.1–3 Aside from enhancing efficacy through co-targeting, bsAbs offer the design of new targeting strategies such as recruitment of immune cells to targeted cancer cells or cross-linking of two different receptors.4 The heavy chain (Hc) heterodimer format represents a major class of bispecific drug candidates as this Fc-containing architecture intrinsically benefits from a long serum half-life and the ability to mediate effector functions.5,6 To date, it is the only bispecific format to have received two market approvals. Catumaxomab (Removab®, Trion Pharma, Germany),7 the first marketed bsAb, is an anti-EpCAM/anti-CD3ϵ Hc heterodimer produced by a mouse-rat quadroma cell line.8 Production was only possible because of the difference in Protein A (PA) avidity between homo- and heterodimers, as rat homodimers did not bind PA whereas mouse homodimers bound more strongly than the mouse-rat heterodimers, thereby allowing separation.9 The current clinical-stage Hc heterodimers are human or humanized bsAbs produced in Chinese hamster ovary cells where the majority of the candidates have been enhanced for Hc heterodimerization (HD) through a pair of engineered CH3 domains. An example is emicizumab, an anti-factor (F)IXa/X bsAb, that was recently approved for prevention and reduction of bleeding in patients with hemophilia A with factor VIII inhibitors.10
Regardless of the HD level achieved, Hc heterodimer preparations contain at least traces of homodimer contaminants following scale up.11–13 This contamination affects antibody discovery and development because homo- and heterodimers have similar composition, and one or more chromatographic steps are usually required to reach homogeneity. Mimicking the inherent difference in PA avidity found in the mouse-rat heterodimer, recent purification strategies have focused on a heterodimer design wherein one Fc chain originates from human immunoglobulin (Ig) G3, a natural IgG subclass that does not bind PA (at least for most allotypes).14 Hc heterodimers based on a mixed human IgG1/IgG3 subclass have been developed either by using a complete IgG3 CH3 domain15 or a pair of IgG3 CH3 derived substitutions16 in one of the two Hcs. Recently, Tustian et al. reported that using VH3 domains in these Hc heterodimers poses challenges because the VH3 subclass binds PA and reduces the avidity difference between homo- and heterodimers, overall interfering with the differential purification process and requiring additional purification steps.17 This bottleneck was also identified in our laboratory, as many of our bsAb candidates utilize the VH3 subclass, often in both Hcs.
The discovery of bispecific Hc heterodimers benefits from purification techniques that will provide homogenous heterodimer preparations from a single affinity purification step, similar to the PA purification of conventional monoclonal antibodies. To this end, we set out to further optimize the current PA avidity strategies by engineering VH3 domains that do not bind PA, as well as to explore a new strategy based on a difference in Protein G (PG) avidity.
PA and PG have a strong affinity for the interface of the CH2 and CH3 domains of human IgG1, a structural region that also includes the FcRn binding site (Figure 1).14,18 FcRn regulates antibody serum half-life by mediating transcytosis across epithelial barriers.19 As a consequence, removing PG binding while preserving normal antibody pharmacokinetic (PK) properties and PA binding is not trivial. PG also binds the antigen-binding fragment (Fab) of IgG1 with low affinity in the CH1 region.14 Although this interaction may be classified as weak, removal of this second binding site is needed to enable efficient avidity separation, similarly to what is observed for the PA techniques with regard to VH3 domains.
Figure 1.
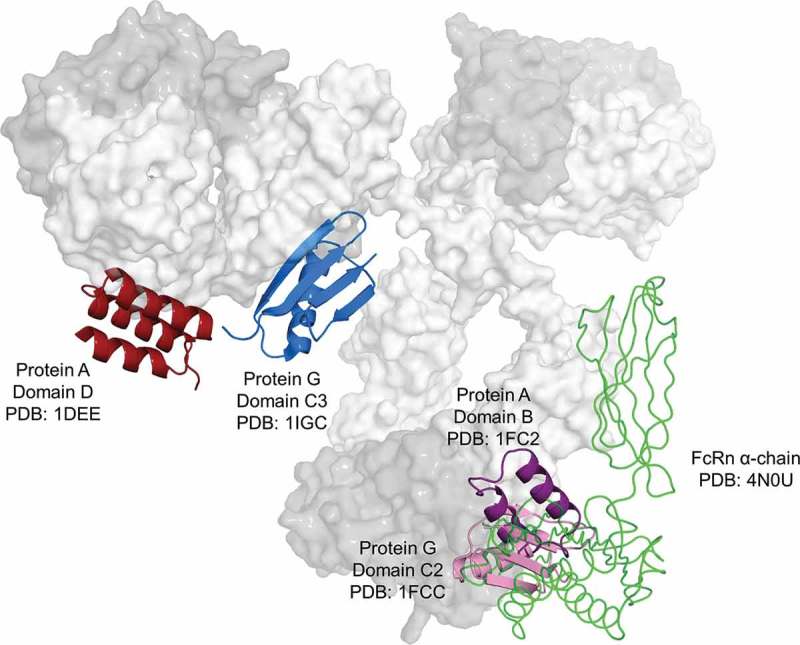
Compiled 3D representation of PA, PG and FcRn interactions with human IgG1. For illustration purposes, different regions from different isotypes and species were superimposed. PA and PG interacting with the VH3-Fab portion are colored in dark red and blue, respectively. PA, PG, and FcRn (only the extracellular domain of the α-chain is shown) in complex with the Fc region are colored purple, pink, and green, respectively. PDB codes as indicated. Lcs and CH3 domains are shaded in dark gray.
Here, we describe two purification methods for bsAbs based on a difference in PA or PG avidity, which have been engineered via structure-guided design. Modifications were made in both the Fab and the Fc regions to enable efficient separation without affecting heterodimer properties and in vitro stability. Moreover, only small to moderate changes in FcRn affinities were found, and rat PK profiles were close to the profile observed for human IgG1.
Both methods can be implemented as single-step purification techniques to achieve homogeneous Hc heterodimer preparations for discovery and scale up. Furthermore, the methods are compatible with CH3 HD technologies such as knobs-into-holes20 or BEAT® (Bispecific Engagement by Antibodies based on the T cell receptor).15 Lastly, the PA method was successfully used to manufacture clinical-grade material for a bispecific T-cell engager antibody, currently in a Phase 1 study.21
Results
Engineered Hc without PA binding
PA from Staphylococcus aureus includes five highly similar domains (from N-terminus: E, D, A, B, and C), and each domain can bind Fc.14 Additionally, all PA domains bind the VH3 subclass with an affinity in the µM range.22 Most of the site interacting with the Fab portion is structurally separate from the domain surface that mediates Fc binding. Next-generation PA resins such as MabSelect™ SuRe™ are based on alkaline-resistant recombinant versions of the protein that have been optimized for antibody manufacturing.23 MabSelect™ SuRe™ is a tetramer of an engineered version of the B domain, the so-called Z domain. Although MabSelect™ SuRe™ has been reported to lack VH3-Fab binding,22,24 the resin still binds VH3-F(ab’)2 fragments,25 which is the likely root cause for the lack of separation between hetero- and homodimers in PA avidity-based methods.
We first designed an IgG that included a VH3 variable domain and the following Hc constant domains: IgG1 CH1, IgG1 hinge, IgG3 CH2, and IgG3 CH3 (abbreviated IgG 1133, wherein the numerals in the name correspond to the IgG isotype subclass of each domain in the order of: CH1/hinge/CH2/CH3), and found that the IgG was still able to bind PA in spite of having the CH2 and CH3 domains of human IgG3 (Figure 2(a)). We deduced that the avidity created by the two VH3-Fab portions was sufficient to restore PA binding and set out to mutate PA binding in VH3 domains. Although substitutions at Kabat position 57 in complementarity-determining region (CDR)-H2 have been reported to abrogate MabSelect™ SuRe™ binding of VH3-F(ab’)2 fragments,25 this result prompted us to further engineer the framework region of the VH3 subfamily in order to find a more systematic, framework-embedded solution.
Figure 2.
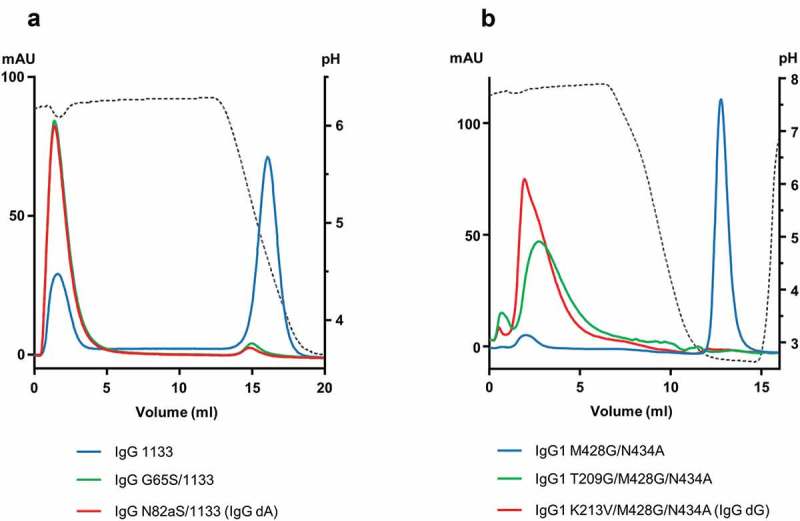
PA and PG binding assessment of engineered antibodies by linear-gradient chromatography. (a) Overlay of HiTrap® MabSelect™ SuRe™ PA chromatograms (RT). An IgG3-like antibody (IgG 1133) based on the VH3 subclass (blue) still bound the MabSelect™ SuRe™ resin even though elution occurred at a mild pH (~pH 4.2). Adding the Fab substitution G65S (green) or N82aS (red) completely abolished binding (Kabat numbering). (b) Overlay of HiTrap® PG HP chromatograms (RT). An IgG1 antibody carrying the M428G/N434A substitutions in its Fc region (blue) still bound PG, while the same antibody with the added Fab substitution T209G (green) or K213V (red) was found in the flow through (Eu numbering).
As a starting point for engineering, we used the crystal structure of a Fab from the VH3 subfamily bound to the D domain of PA (Figure S1).26 In the complex, the Fab interacts with the α-helices II and III of the D domain via a surface composed of four VH3 framework β-strands, mainly hydrophilic, involving polar interactions and salt bridges. Based on amino acid sequence differences between the VH3 subclass and all other subclasses (Figure S2) and known PA interacting residues, various single substitutions were designed and tested. In total, five substitutions located outside of CDRs were selected: G65S, R66Q, T68V, Q81E, and N82aS (Kabat numbering). Mutated Fabs were prepared in the background of the variable domains of trastuzumab, an anti-human HER2 antibody utilizing the VH3 subclass (brand name Herceptin®, Roche-Genentech),27 and purified by Protein L before testing by gradient chromatography on a non-modified PA resin, i.e., known to bind Fabs, the MabSelect™ resin.25 G65S, Q81E and N82aS were found to completely abrogate PA binding (Figure S3). We omitted Q81E from further characterization to focus on charge neutral substitutions. Even though these framework residues are not directly involved in target engagement, conformational changes impacting antibody binding cannot be excluded. Thus, we introduced the G65S or N82aS substitution in several VH3-Fabs and subsequently measured the affinity for their target by surface plasmon resonance (SPR) (Table S1). All tested Fabs retained their original affinities, supporting our original observations that the selected framework substitutions did not change CDR conformation.
Individual substitutions, G65S or N82aS, were further tested in the context of an IgG 1133 antibody. Variants were purified by PG and subsequently injected on the MabSelect™ SuRe™ resin (Figure 2(a)). Both mutations successfully abolished PA binding and antibodies were found in the unbound fraction. The variants were then further characterized by differential scanning calorimetry (DSC) and size exclusion high performance liquid chromatography (SE-HPLC). Thermal stability of the Fc portion of IgG 1133 (termed Fc 133, wherein the numerals in the name correspond to the IgG isotype subclass of each domain in the order of: hinge/CH2/CH3) was lower than that of IgG1, but very similar to IgG4. As CH3 domains are known to have a higher thermal unfolding midpoint compared to CH2 domains, the 68 °C transition likely corresponded to the melting point of the CH2 domains and the 72 °C transition to the midpoint of the CH3 domains (Figure S4 and Table S2).28 Adding the N82aS mutation did not affect Fab stability or induce aggregation; the same was observed for G65S (data not shown). A low risk of immunogenicity was determined for the N82aS substitution using in the Lonza Epibase™ in silico immunogenicity prediction platform (detailed in the supplemental results section and Table S3; note that G65S was not tested).29 This mutation in combination with Fc 133 was selected to exemplify our method in proof-of-concept heterodimers. Antibodies and single-chain variable fragments (scFv) or Fab arms of heterodimers having no binding to PA are denoted dA below.
Engineered Hc without PG binding
Engineering of the Fc site – PG, a cell-surface protein found in Gram-positive bacteria such as Streptococcus, contains multiple copies of two different small protein domains that can independently bind albumin and IgGs.14 The IgG-binding region consists of three domains: C1, C2 and C3, exhibiting high sequence homology with only a few residue differences. Each domain is able to bind independently, thereby creating avidity. Unlike PA, PG was never used for the avidity purification of Hc heterodimers. PG, PA, and FcRn have overlapping binding sites within the Fc region of IgG1, all of which have been precisely mapped by X-ray crystallography (Figure 1).30–33 We aimed to design an Fc chain with no PG binding while retaining affinities for PA and FcRn, and thus primarily selected Fc residues for mutagenesis that are known to interact with PG, but not with PA or FcRn. Five Fc residues were selected as a first set for engineering: M252, E380, E382, S426, and M428 (Eu numbering). Aside from M252, which interacts with FcRn, these residues are known to interact with PG, but have no interaction with PA or FcRn. Selecting M252 was motivated by previous mutagenesis studies wherein various substitutions at position 252 were shown to improve FcRn binding.34 In addition, it is known that mutating M252 to alanine has no effect on FcRn binding.35
The PG C2 domain comprises one α-helix positioned diagonally across a four-stranded β-sheet (Figure S5), and the protein-protein interface between PG and Fc can be divided into three different regions on PG, previously reported as I (K28 and Q32), II (E27 and K31), and III (N35, V39, D40, E42, and W43);33 PG residue numbering according to Protein Data Bank (PDB) code 1FCC. Our first selection of residues was therefore clustered at one end of the α-helix.
E380, E382, S426 and M428 are CH3 residues interacting with region I via an extensive network of interactions centered on PG K28, which includes two side chain-side chain hydrogen bonds with E380 and E382, and a hydrophobic interaction with M428 side chain. S426 plays a supporting role by forming a side chain-side chain hydrogen bond with E382. Finally, M252, a CH2 residue, interacts with PG K28 and E27 residues via hydrophobic interactions.
Our choice of substituting amino acids was based on the residues found in IgA (Figure S6), a non-PG binding Ig isotype. Three-dimensional (3D) equivalent positions between the CH2 and CH3 domains of IgG1 and IgA1 were identified via the IMGT database and numbering system (http://www.imgt.org/IMGTrepertoire/Proteins/; section: C-DOMAIN).36,37 All variants were produced as IgG1 Fcs and purified by gravity-flow PA purification before being assessed by gradient PG chromatography. The variants were then further characterized by DSC, SE-HPLC, and FcRn binding by SPR. Results are summarized in Table 1 and S4.
Table 1.
Biochemical characterization of the different Fc variants engineered for reduced PG binding.
| No | Fc proteins | PG binding | CH2 Tm (°C) | CH3 Tm (°C) |
Monomer (%) | FcRn nor. aff.* |
Expression (mg/l) |
|---|---|---|---|---|---|---|---|
| IgG1 | + | 72.6 | 85.5 | 98.7 | 1.00 | 52 | |
| 1 | M252L/E380R/E382L/S426M/M428G | - | 55.9 | 84.7 | 96.2 | NB | 30 |
| 2 | E380R/M428G/N434A/Q438K | - | 57.1 | 80.6 | 96.7 | 0.20 | 11 |
| 3 | E380A/E382A/N434A | - | 62.5 | 81.6 | 96.5 | 2.97 | 32 |
| 4 | E380R/N434A/Q438K | - | 60.5 | 83.9 | 97.2 | 2.24 | 31 |
| 5 | E380R/N434A | - | 60.8 | 82.8 | 94.3 | 2.87 | 46 |
| 6 | N434A/Q438K | + | 69.4 | 85.4 | 99.1 | ND | 63 |
| 7 | E380R/Q438K | + | 60.9 | 85.9 | 94.3 | ND | 50 |
| 8 | M428G/N434A | - | 65.3 | 79.5 | 98 | 0.95 | 41 |
| 9 | E380R/M428G | + | 58.0 | 81.1 | 92.8 | ND | 33 |
| 10 | M428G/Q438K | + | 65.1 | 85.1 | 97.9 | ND | 42 |
| 11 | E380A/N434A | -/+ | 64.4 | 82.8 | 97.9 | 4.59 | 50 |
| 12 | M252G/N434A | - | 65.1 | 82.6 | 94.5 | NB | 16 |
| 13 | M428G | + | 66.0 | 82.3 | 96.3 | 0.23 | 36 |
| 14 | N434A | + | 70.7 | 82.4 | 98.5 | 4.61 | 46 |
SPR binding affinities to FcRn were normalized against human IgG1 Fc. (ND) not done. (NB) no binding. (*) for each measurement, the relative KD value of the test antibody was calculated vs. IgG1 control wherein the control was measured on the same day with the same experimental setup (KD value of the test antibody/average KD value of control), the normalized affinity (nor. aff.) shown here corresponds to the 1/average of the different relative KD values (n ≥ 3).
The first Fc variant based on these IgA1-derived substitutions (variant 1) successfully lost PG binding, but also lost its binding to FcRn as well as some thermal stability in the CH2 domains. We then designed a series of Fc variants having fewer IgA1-derived substitutions, but which included the N434A substitution with the goal to restore FcRn binding, as previous mutagenesis studies have shown that this change can enhance FcRn binding by 3.5 fold without affecting PA purification.35 N434 is an important Fc residue that mediates a strong network of interactions with PG, including two side chain-side chain hydrogen bonds in region III of the protein-protein interface: one with PG N35, a residue situated on the opposite end of PG’s α-helix with regard to PG K28, and another bond with PG W43, a residue located in the β-strand closest to the α-helix near PG N35 (Figure S5). At this stage, we also selected Q438 because this position makes a side chain-side chain hydrogen bond with PG Q32 (region I) and does not interact with FcRn (Figures S5–6). In addition to Q438A, we tested Q438K to remove the interaction with PG Q32, since a lysine residue is found at the 3D equivalent position in human IgM (position IMGT 118), another non-PG-binding Ig isotype.
Among the different combinations tested, the combination of the two IgA1-derived substitutions E380R and M428G with N434A and Q438K was the most successful, with a recovery of 20% of the original FcRn binding (variant 2). Next, substitutions at positions E380, E382 and N434 were tested as triple-mutant Fc molecules, and found not only to recover but also to strongly enhance FcRn binding (variants 3 and 4). Regrettably, these variants showed an overall loss of stability in their CH2 domains and were deemed unsuitable for further engineering.
Instead, to reduce the number of mutations and improve biophysical properties, eight double mutants were generated on the basis of the previous results and assessed in a similar manner (variants 5–12). Three of these double mutants had lost PG binding, but had similar or enhanced FcRn binding compared to human IgG1: E380R/N434A, M428G/N434A and E380A/N434A (variants 5, 8, and 11, respectively). As E380A/N434A (variant 11) showed signs of residual PG binding (data not shown) and E380R/N434A (variant 5) still exhibited a significant loss of thermal stability in its CH2 domains, we selected M428G/N434A (variant 8) as our final candidate.
This pair of substitutions was found to completely abrogate PG binding with little impact on FcRn binding, which was only decreased by 5% compared to human IgG1 Fc. In terms of thermal stability, this variant showed a loss of ~7 and 6 °C for its CH2 and CH3 domain melting transitions, respectively. Aside from this moderate loss in thermal stability, HEK293E transient expression levels were comparable to human IgG1 Fc without any sign of aggregation (98% monomeric by SE-HPLC). The risk of immunogenicity for the M428G/N434A substitutions was determined as low using the above mentioned in silico prediction tool (supplemental results section and Table S3).
Both M428G and N434A were individually tested to assess the contribution of each substitution to PG binding (Figure S7). Neither M428G nor N434A were able to abrogate PG binding on their own, although each substitution was able to decrease retention time. Interestingly, the pair of substitutions appeared to complement each other, as M428G reduced FcRn binding by five-fold (variant 13) whereas N434A enhanced FcRn binding by about 4.5-fold (variant 14), and therefore resulted in little to no loss of binding (Table 1).
Engineering of the Fab site – PG also interacts with the Fab portion of IgGs through a secondary binding site located in their CH1 domain, a domain which is highly conserved across IgG subclasses and species.18 Similarly to PA, a separate region from the Fc binding site is involved in binding to the Fab. It has been shown that the affinity for the Fab is much lower compared to that of the Fc (µM vs nM, respectively).14 Unlike PA, the low affinity of the PG-Fab interaction limits PG usage as a Fab affinity reagent, yet F(ab’)2 fragments are efficiently bound due to avidity.38 As shown in Figure 2(b), introducing the M428G/N434A substitutions was not sufficient to abrogate PG binding in IgG1 because enough interactions remained through the Fab arms. Hence, efficient PG avidity separation chromatography requires the abrogation of PG binding in CH1. The PG-Fab complex has been studied in detail by X-ray crystallography in the context of a mouse IgG1.39 The C3 domain of PG binds mainly to CH1 via an antiparallel β-sheet interaction mediated by its second β-strand and the β-strand G of the CH1 domain (IMGT nomenclature). The interaction is mainly driven through hydrogen bonds that involve main chain atoms, as well as two additional hydrogen bonds coming from side chain contacts (Figure S8). β-strands G are highly conserved, and thus this interaction can be translated to human IgGs. Despite being structurally close, other human Ig isotypes, such as IgA and IgM, do not bind PG. Based on a structural alignment, we designed IgA1- or IgM-derived substitutions in the β-strand G of IgG1 CH1 at positions 209, 210, 213, and 214 (Eu numbering) that interact directly with the PG C3 domain (Figure S9(a)). Mutations were introduced in an anti-HER3 IgG1 antibody40 carrying the M428G/N434A substitutions and PG binding was assessed by gradient chromatography.
Among the four selected positions, only the single substitutions targeting the two residues involved in the side chain contacts were successful at preventing binding: T209G, T209P, and K213V (Figure S9(b)). This result is in line with the importance of these two positions in the CH1-C3 interaction because T209 and K213 make hydrogen bonds with PG C3 domain residues T21 and T16 (numbering according to PDB code 1IGC), respectively (Figure S8). Calorimetric measurements showed that T209G and K213V had no impact on CH1 domain thermal stability, and Fab Tms were comparable to control, at approximatively 79 °C (Table S5). Conversely, substituting a proline at position 209, structurally corresponding to the start of the β-strand G, was destabilizing and Fab thermal stability was decreased by nearly 8 °C. Affinity to HER3 was assessed by SPR, showing that all three mutants retained full binding to recombinant HER3 compared to the non-substituted control antibody. Both, K213V and T209G, completely abolished PG binding when individually introduced in the previously substituted M428G/N434A antibody (Figure 2(b)). The above mentioned in silico prediction tool was used to investigate the immunogenicity potential of the three substitutions, with K213V exhibiting the lowest profile of the group (supplemental results section and Table S6). Accordingly, the K213V substitution combined with the M428G/N434A mutations was selected to exemplify our method in proof-of-concept heterodimers. Antibodies and scFv or Fab arms of heterodimers having no binding to PG are denoted dG below.
Fc receptor interactions of engineered Hcs
Since our engineering focused on the CH2-CH3 domain interface, we further investigated the affinity of our engineered Hcs for human FcRn. Additionally, we measured the affinities for the different Fcγ receptors to probe for other alterations. Affinities were assessed in the antibody format, i.e., homodimeric IgG constructs using SPR (Table 2, S7). Human IgGs have two independent FcRn binding sites with identical affinities,41 and FcRn molecules have been shown to bivalently engage IgGs, thereby creating an avidity effect that positively impacts half-life.19 In the case of our engineered heterodimers, the affinities of the two FcRn binding sites will differ and may create some averaging when measuring affinities by SPR. Importantly, we chose to measure FcRn interactions with mutated antibodies by immobilizing IgGs rather than FcRn to avoid avidity effects. Engineered antibodies were designed with the variable domains of trastuzumab.
Table 2.
SPR binding affinities of the trastuzumab variants to human Fc receptors.
| Antibody | FcγR1a | FcγR2a | FcγR2b | FcγR3a | FcRn | |
|---|---|---|---|---|---|---|
| IgG1 | KD ± SD* | 106 ± 13 | 2761 ± 976 | 6335 ± 1629 | 271 ± 112 | 501 ± 214 |
| Nor. aff. # | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | |
| IgG dG | Nor. aff. # | 0.74 | 1.04 | 0.97 | 1.02 | 0.92 |
| p value | <.0001 | .250 | .399 | .160 | .0028 | |
| Significant | Yes | No | No | No | Yes | |
| IgG dA | Nor. aff. # | 1.16 | 0.91 | 1.06 | 0.79 | 0.67 |
| p value | .0001 | .0001 | .17 | <.0001 | <.0001 | |
| Significant | Yes | Yes | No | Yes | Yes | |
| IgG1 RF | Nor. aff. # | 0.98 | 1.29 | 0.90 | 1.40 | 0.76 |
| p value | .66 | <.0001 | .14 | <.0001 | <.0001 | |
| Significant | No | Yes | No | Yes | Yes |
Average KD values were normalized (nor. aff.) and statistically compared against trastuzumab average KD values. (*) average KD based on all replicates (nM) ± standard deviation (SD) between individual replicates, independently of the day of measurement. (#) for each measurement, the relative KD value of the test antibody was calculated vs. control wherein the IgG1 control was measured on the same day with the same experimental setup (KD value of the test antibody/average KD value of control), the normalized affinity (nor. aff.) shown here corresponds to the 1/average of the different relative KD values (n ≥ 3).
For the PA method, we set out to compare the two main strategies to abrogate PA binding, and measured affinities of the IgG dA antibody (N82aS substitution in combination with Fc 133), as well as those of an IgG1 antibody carrying the H435R/Y436F substitutions (denoted IgG1 RF, Eu numbering), thereby comparing usage of an IgG3 Fc portion vs. the known IgG3-derived substitutions that abrogate PA binding.15,16 KD values for both approaches were consistent with a moderate loss in affinity for FcRn. Affinity was reduced by 33% for the IgG dA and by 24% for the IgG1 H435R/Y436F, although the difference between the two methods was not statistically significant (p value was 0.2).
Similarly, we used the variable domains of trastuzumab in combination with the K213V, M428G, and N434A substitutions to build an IgG1 antibody without PG binding (denoted IgG dG). Measurements on the IgG dG antibody showed an 8% loss in FcRn affinity, an additional 3% decrease compared to our initial measurements on the engineered Fc M428G/N434A fragment. Taken together, SPR data confirmed a moderate to small loss in FcRn affinity for the PA and PG methods, respectively. Further measurements on Fcγ receptors showed a small decrease in FcγR3a and FcγR2a binding for IgG dA (~20% and ~10%, respectively) whereas IgG dG only exhibited a moderate loss in FcγR1a binding (~25%). Conceivably, heterodimers based on these methods will typically have a single engineered Fc chain while the second chain will remain unmodified, therefore the small impact on FcRn binding observed with these antibodies is expected to be further minimized in the context of a heterodimeric IgG.
Avidity purification of heterodimeric IgGs via a single PA or PG affinity step
Reducing the number of purification steps represents substantial savings in cost of goods during manufacturing. Having successfully engineered Hcs without PA or PG binding, we set out to design Hc heterodimers that can be efficiently purified from their homodimeric species via a single chromatography step. We used the variable domains of trastuzumab and the Fab-scFv-Fc format to circumvent the light chain pairing problem. Reverse phase high performance liquid chromatography coupled to mass spectrometry (RP-HPLC-MS) was used to determine heterodimer purity. To exemplify our PA method, we co-transfected the Hc and the light chain (Lc) of the IgG dA antibody described above together with an scFv-Fc chain, thus producing a mix of two homodimeric species and one heterodimer with a different avidity for PA (Figure 3(a)). The main challenge was to define a pH window to separate the elution of the heterodimer, which only binds PA through one Fc chain, from the scFv-Fc homodimer, which binds PA through both Fc chains. The non-PA binding homodimer (IgG dA) should be in the unbound fraction. During development, we determined that, when run at room temperature (RT), the best elution conditions used an eluent at pH 4.1 followed by a strip buffer at pH 3.0, with the pH of the heterodimer main peak fraction measured at 4.1 and the pH of the PA-binding homodimer main peak fraction measured at 3.5. When run at 4 °C as exemplified here, the elution of the heterodimer could be performed at pH 4.2 instead of 4.1; thereby adding 0.1 units to the pH window between bound species. These conditions, at 4 °C and at RT (data for RT not shown), allowed baseline-resolution between the elution peaks of the two bound species (Figure 3(b)). Heterodimer purity was at 93.5% as determined by RP-HPLC-MS (Figure 3(c)).
Figure 3.
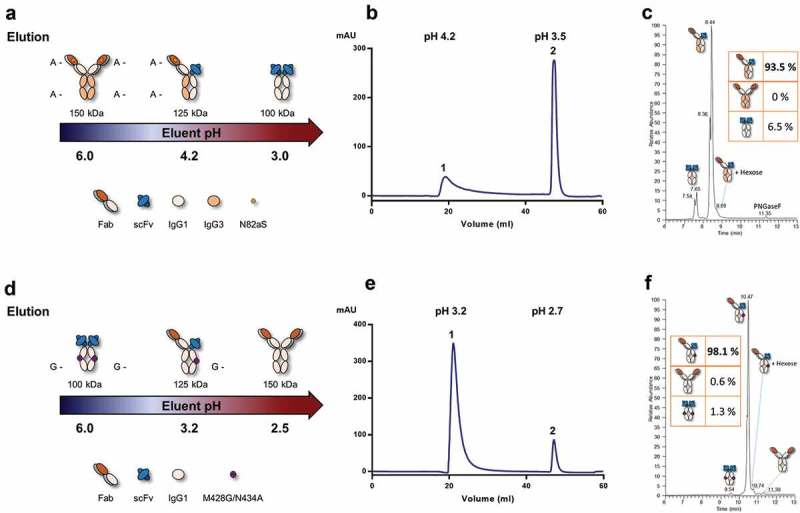
Single-step avidity purification of heterodimeric IgGs. (a) Schematic of single-step PA avidity purification. The IgG dA homodimer did not bind PA, the heterodimeric IgG was eluted with NaAc pH 4.2 (4 °C) or 4.1 (RT), and the scFv-Fc homodimer was stripped from the column with glycine buffer pH 3 (RT or 4 °C). (b) HiTrap® MabSelect SuRe™ chromatogram at 4 °C, pH measurements of the main peak fractions are indicated. (c) Total ion chromatogram obtained by RP-HPLC-MS of the main peak (1) fraction with baseline-separation between homo- and heterodimers. Fractional abundances were obtained by integrating the extracted ion chromatograms of homo- and heterodimer species (inset). (d) Schematic of single-step PG avidity purification. The scFv dG homodimer did not significantly bind PG, the heterodimeric IgG was eluted with glycine buffer pH 3.2 (RT or 4 °C), and the IgG1 antibody was stripped from the column with glycine buffer pH 2.5 (RT or 4 °C). (e) HiTrap® PG HP chromatogram at 4 °C, pH measurements of the main peak fractions are indicated. (f) RP-HPLC-MS analysis of the main peak (1) fraction as in (c). (A-) denotes a nonfunctional PA binding site. (G-) denotes a nonfunctional PG binding site.
Similarly, for our PG method, we co-transfected the Hc and Lc of an IgG1 antibody along with an scFv-Fc chain carrying the M428G/N434A substitutions to generate a mix of two homodimeric species and one heterodimer with a different avidity for PG (Figure 3(d)). To achieve baseline-separation of the PG binding species, heterodimer eluent pH had to be lowered to pH 3.2 and the strip buffer to pH 2.5, with the pH of the heterodimer main peak fraction measured at 3.2 and the pH of the PG-binding homodimer main peak fraction measured at 2.7 (Figure 3(e)). Heterodimer purity was at 98% as determined by RP-HPLC-MS (Figure 3(f)). Besides running the method at 4 °C, RT was also tested and elution pHs were the same (data not shown). The pH window for separation was therefore similar to the one observed with the PA method, i.e. 0.5 vs. 0.6–0.7 pH units, even though PG has a higher affinity toward human Fc and usually requires harsher elution conditions as observed here.42 While both differential affinity purification techniques, PA and PG based, performed well, it is worth noting that the two methods can be combined and run sequentially without chromatography system wherein strong acidic pH conditions can be used at each step to elute the bound material. Using this two-step approach, the heterodimer was readily prepared with over 95% purity as judged by SDS-PAGE (Figure S10).
Retrospectively, with the availability of in-house common light chain (cLc) antibody phage libraries, we set out to further exemplify the PG method in the context of a full-length IgG bsAb, thereby demonstrating the utility of abrogating PG binding in the Fab region (Figure 4(a)). To this end, we selected two Fab portions (termed Fab #1 and Fab #2) sharing a cLc, but each having specificity for a different oncology target. We co-transfected the cLc with Fab #1 Hc carrying the K213V mutation in its CH1 domain, as well as the M428G/N434A substitutions in the CH3 domain and Fab #2 Hc consisting of non-mutated IgG1 domains. Baseline-separation between the heterodimer and the PG binding homodimer could be achieved using the same chromatographic method used for the Fab-scFv-Fc described above (Figure 4(b)). Despite minimal differences between homo- and heterodimers in the cLc format, i.e., having the same architecture and utilizing the same VH and VK frameworks, baseline-separation of the different species could be observed by RP-HPLC, allowing the quantification of heterodimer purity (Figure 4(c)). Similarly to the Fab-scFv-Fc construct that did not require the PG abrogation in CH1, the cLc bsAb exhibited a high heterodimer purity at 97%.
Figure 4.
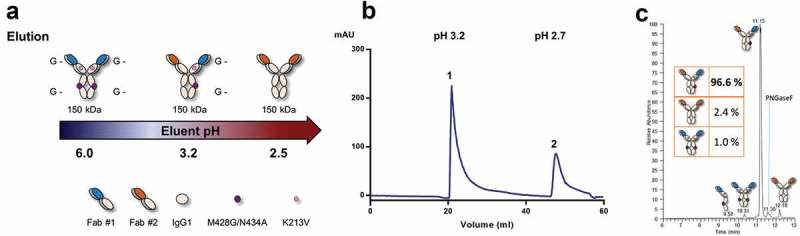
Single-step avidity purification of a full-length heterodimeric common light chain IgG. (a) Schematic of single-step PG avidity purification. The Fab #1 dG homodimer did not significantly bind PG, the heterodimeric IgG was eluted with glycine buffer pH 3.2, and Fab #2 IgG1 antibody was stripped from the column with glycine buffer pH 2.5. (b) HiTrap® PG HP chromatogram at 4 °C, pH measurements of the main peak fractions are indicated. (c) Total ion chromatogram obtained by RP-HPLC-MS of the main peak (1) fraction with baseline-separation between homo- and heterodimers. Fractional abundances were obtained by integrating the extracted ion chromatograms of homo- and heterodimer species (inset). (G-) denotes a nonfunctional PG binding site.
PK properties of antibodies and heterodimers in rats vs. Fc thermal stability
Different antibodies and Fab-scFv-Fc heterodimers were produced using the PA and PG methods or combinations thereof. To avoid PK differences due to non-specific binding events mediated by the variable regions, the variable domains of trastuzumab were used in all constructs. This was also to provide consistency in the detection assay and the subsequent PK analysis. Thus, PK profiles were expected to be driven by stability and FcRn binding properties. Both antibodies and heterodimers were cleared from the circulation in a bi-phasic manner, showing a rapid distribution phase followed by a prolonged elimination phase characteristic of antibodies (Figure 5). Although antibodies and heterodimers used the same variable domains, non-specific binding was not assessed in this study, and it is possible that heterodimers had increased non-specific interactions originating from the scFv arm. Additionally, scFv moieties are generally less stable compared to Fabs.43 Preliminary studies also using trastuzumab-based constructs showed that a change of format from antibody to scFv-Fc fusion protein, led to a three-day reduction in half-life, from 10 to 7 days, respectively (data not shown).
Figure 5.
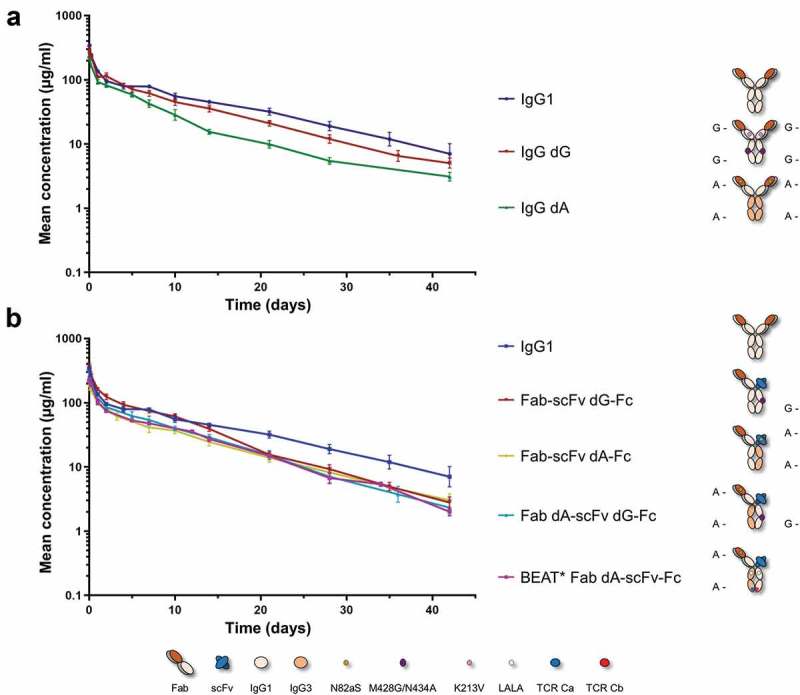
PK profiles of engineered antibodies and heterodimeric IgGs in Sprague-Dawley rats. Blood concentrations over time are shown. Antibodies and heterodimers were administrated at 10 mg/kg in a single intravenous bolus injection. Data points represent the mean concentration ± SD. (a) PK profile comparison of engineered antibodies: trastuzumab dA (IgG dA) and trastuzumab dG (IgG dG) vs. trastuzumab analog (IgG1). (b) PK profile comparison of the different heterodimeric IgGs. The trastuzumab analog profile (IgG1) generated from the same in vivo experiments is shown, heterodimers as indicated. (A-) denotes a nonfunctional PA binding site. (G-) denotes a nonfunctional PG binding site. (*) denotes the presence of the L234A and L235A substitutions in both BEAT Fc chains (Eu numbering).
Overall, no significant differences were observed between the different antibodies tested with half-lives close to the trastuzumab analog (Figure 5(a) and Table 3). Other PK parameters, such as clearance and volume of distribution were in the range of previously reported therapeutic antibodies.44–46 The IgG dA antibody exhibited the highest clearance rate and a lower area under the curve despite having a half-life of 11.8 days, yet clearance was similar to that of pertuzumab, which was also measured in rats at the same dose regimen.44 A lower Cmax is also to be taken into account. The IgG dG antibody gave very similar results to those of the trastuzumab analog.
Table 3.
PK parameters of engineered antibodies following a single IV bolus injection in female Sprague-Dawley rats.
| PK parameter | IgG1 | IgG dG | IgG dA |
|---|---|---|---|
| Cmax (µg/ml) | 351 ± 8 | 281 ± 51 | 238 ± 21 |
| AUC0-inf (hr*µg/ml) | 45318 ± 4025 | 36176 ± 4022 | 23564 ± 1328 |
| CL (ml/day/kg) | 5.3 ± 0.4 | 7.2 ± 1.4 | 10.1 ± 0.6 |
| Vss (ml/kg) | 77 ± 7 | 83 ± 8 | 117 ± 4 |
| t1/2 (day) | 10.4 ± 2.4 | 9.5 ± 0.5 | 11.8 ± 0.6 |
(AUC0-inf) area under the serum concentration-time curve from time zero to infinity. (CL) clearance. (Cmax) maximum serum concentration. (t½) elimination half-life. (Vss) apparent volume of distribution at steady state. Standard deviation for each parameter is indicated.
PK properties of heterodimeric IgGs encompassing dA and/or dG Hcs in the Fab-scFv-Fc format were also investigated (Figure 5(b) and Table 4). Heterodimers containing a dG Hc combined with either an IgG1 Hc or a dA Hc (Fab-scFv dG-Fc and Fab dA-scFv dG-Fc) showed a small reduction in half-life compared to the trastuzumab analog (~8 days). The Fab-scFv dA-Fc heterodimer exhibited the longest half-life (9.5 days). Combining our PA avidity method with CH3 engineering (BEAT Fab dA-scFv-Fc; note that the BEAT heterodimer also included the L234A and L235A substitutions known to abrogate Fcγ receptor binding47) resulted in a heterodimeric IgG with a very similar PK profile and half-life value compared to the non-CH3 engineered dG-based heterodimers.
Table 4.
PK parameters of heterodimeric IgGs following a single IV bolus injection in female Sprague-Dawley rats.
| PK parameter | Fab-scFv dG-Fc | Fab-scFv dA-Fc | Fab dA-scFv dG-Fc | BEAT* Fab dA-scFv-Fc |
|---|---|---|---|---|
| Cmax (µg/ml) | 383 ± 42 | 237 ± 14 | 308 ± 18 | 230 ± 28 |
| AUC0-inf (hr*µg/ml) | 39507 ± 1755 | 26099 ± 3013 | 29314 ± 2179 | 26371 ± 116 |
| CL (ml/day/kg) | 6.1 ± 0.3 | 9.3 ± 1 | 8.2 ± 0.6 | 9.1 ± 0.05 |
| Vss (ml/kg) | 58 ± 5 | 108 ± 8 | 84 ± 7 | 98 ± 3 |
| t1/2 (day) | 8.1 ± 0.6 | 9.5 ± 0.4 | 8.2 ± 0.4 | 7.9 ± 0.2 |
(*) denotes the presence of the L234A and L235A substitutions in both BEAT Fc chains (Eu numbering). (AUC0-inf) area under the serum concentration-time curve from time zero to infinity. (CL) clearance. (Cmax) maximum serum concentration. (t½) elimination half-life. (Vss) apparent volume of distribution at steady state. Standard deviation for each parameter is indicated.
Both antibodies and heterodimers exhibited good thermal stability and a low aggregation profile (Figures S4, S11-12 and Tables S2, S8–9). CH2 and CH3 thermal stabilities varied across methods and formats, with each value being almost identical to an average stability calculated from the two different Fc arms of the heterodimer when considering the previously observed CH2 or CH3 stabilities in Fc 133, Fc M428G/N434A, and IgG1 Fc.
The scFv moiety was previously shown to transition at 68 °C in the scFv-Fc format (data not shown), and this was unchanged in all heterodimers. Thus, scFv melting overlapped with CH2 melting. As a result, CH2 stabilities in heterodimers could only be inferred from the observation that melting transitions were sharp and no double peak could be observed with mid-points measured at 67–68 oC, suggesting stabilities in line with average values derived from the different homodimeric Fc formats. Fab melting mid-point was also unchanged across heterodimers (82.2–83.8 °C) and matched IgG1 control (83.5 °C). A partial overlap between Fab and CH3 melting transitions was also noticeable across heterodimers; in our IgG1 control, this overlap was complete, as we previously found CH3 to melt with a mid-point at 85.5 °C in the IgG1 Fc format (Table 1). Nonetheless, individual transitions were successfully deconvoluted upon data processing. Like for the CH2 stabilities, CH3 mid-point values in heterodimers were almost identical to the average of the CH3 melting values observed in the homodimeric Fc constructs, with a discrepancy of less than one degree (Table S9). A strong increase of ~7 °C in CH3 stability was observed for the Fab-scFv dA-Fc heterodimer vs. Fc 133 with a mid-point equivalent to the average of the CH3 melting values observed in Fc 133 and IgG1 Fc (79. 2 oC vs. 78.9 oC for the average value). CH3 melting transitions of the Fab-scFv dG-Fc and Fab dA-scFv dG-Fc heterodimers were measured at 81.6 °C and 75.4 ° C, respectively.
Nevertheless, as observed here, these differences in CH3 thermal stabilities did not translate into meaningful differences in vivo; the same remark can be made for the BEAT heterodimer whose CH3 stability was previously measured at 70 oC.15 Taken together, all PK profiles overlaid well and half-lives were very similar to one another, suggesting that heterodimer PK properties were mostly influenced by the Fab-scFv-Fc format rather than the nature of the Fc region.
Discussion
Herein we report two independent methodologies to prepare Hc heterodimers, both of which allow for homogenous preparations with minimal homodimer contamination in a single purification step. Current methods based on a difference in PA avidity between Fc chains are suboptimal since PA binding can be rescued via Fab arms using the VH3 subclass,17 a key VH family in terms of antibody discovery and engineering.48,49 Thus, an efficient PA avidity technology not only needs to create a PA binding asymmetry between the two Fc chains, but also needs to address PA binding mediated by VH3 domains. Our structure-guided investigation identified two possible framework substitutions that individually can abrogate PA binding in VH3-Fabs without introducing charged residues. N82aS or G65S (Kabat numbering) abolished PA binding without affecting affinity (Figure S3 and Table S1). These mutations translated into VH3-IgGs having a mixed human IgG1/IgG3 subclass without loss of stability or inducing aggregation (Figure 2(a), S4 and Table S2), and further allowed for the preparation of highly pure and stable Hc heterodimers in the Fab-scFv-Fc format (>93%) from a single PA purification step (Figure 3(a–c), S12 and Table S9).
At the same time that we were improving PA methods, we set out to explore a difference in PG avidity to prepare Hc heterodimers, a method which, to the best of our knowledge, has never been reported. Using our concept of 3D equivalent positions between Ig domains,15 structure-guided design, and Fc residues known to modulate FcRn binding, we successfully abolished PG binding in the Fc region of IgG1. Importantly, our engineering did not affect PA binding, and most of the FcRn interaction was preserved (95% of the native binding, Table 1). Only a moderate loss of thermal stability for the CH2 and CH3 domains was observed (~7 and 6 °C, respectively). Two substitutions in the CH3 domains, M428G and N434A, were sufficient to abrogate binding. M428G was directly identified by 3D equivalence between the CH3 domains of IgG1 and IgA1, while N434A was implemented to restore FcRn binding. Surprisingly, substitutions were not able to abrogate binding on their own, and only the combination of the two mutations was successful (Figure S7). Subsequent design of Hc heterodimers based on a difference in PG avidity between the two Hcs was hampered by the additional PG binding sites found in the Fab arms. This observation was reminiscent of the known issues of the previously reported PA methods. PG binding to the CH1 domains was therefore the subject of a second engineering campaign. Using the same concept of 3D equivalence between IgG1 and IgA1 or IgM, and the 3D structure of the complex between Fab and PG, we found two possible mutations that could abrogate PG binding in CH1, T209G or K213V, without affecting affinity and Fab stability (Figure 2(b), S11 and Tables S5, S8). The combination of the three substitutions: M428G, N434A, and K213V, successfully abolished PG binding in human IgG1 without much impact on antigen, PA, and FcRn affinity (Table 2). PG abrogation in the Fc portion enabled the design and rapid purification of highly pure and stable Hc heterodimers in the Fab-scFv-Fc format (98%) using a single PG purification step (Figure 3(d–f), S12 and Table S9). Retrospectively, with the availability of in-house cLc antibody phage libraries, we tested the utility of abrogating PG binding in the Fab region (K213V) and the Fc region (M428G and N434A) in the context of a full-length IgG bsAb (Figure 4(a–b)). Similarly to the Fab-scFv-Fc construct that did not require the PG abrogation in CH1, we found that the cLc bsAb exhibited a high level of purity (97%, Figure 4(c)).
A low immunogenicity profile was predicted for the N82aS, M428G and N434A substitutions using the Epibase in silico modeling tool,29 in line with the non-modified antibody control used in our study, trastuzumab, for which the reported immunogenicity in humans has been shown to be low50 (supplemental results section and Table S3). Regarding our PG binding mutants in the CH1 region, K213V was our preferred choice since this substitution had the lowest predicted immunogenicity profile of the group (Table S6).
Notably, both technologies achieved baseline-resolution between the homo- and heterodimeric species wherein the resin can be extensively washed at the pH of elution of the heterodimer of interest with minimal contamination by the bound homodimer species. Thus, our results contrast with Zwolak et al.51 who recently described a reduction rather than a complete removal of PA binding in one of the Fc chains to avidity purify bispecific Hc heterodimers. As stated by the authors, the three different elution steps of the three different species required careful pH calibration with one homodimeric species eluting at pH 4.6 and the heterodimer of interest starting to elute at pH 4.5. Moreover, abrogating rather than modulating binding in one of the two Hcs results in one of the homodimer species not binding the affinity resin, thereby simplifying separation because only two binding species instead of three need to be separated.
For large-scale manufacturing chromatography, small product pool volumes with a high product concentration are preferred.52 Both methods allowed stable baseline separation between the two retained species with a pH difference ≥0.5, and are therefore compatible with large scale manufacturing. The methods are run in bind-elute mode with a single elution step for the heterodimer, whereas the bound homodimer species is eluted in the column cleaning step. Indeed, the PA method presented here was successfully combined with the BEAT® HD technology15 and used on a large scale (250 l) to manufacture an anti-HER2 T-cell engager BEAT® antibody currently in Phase 1 clinical trial for treatment of HER2-positive cancers (GBR 1302, ClinicalTrials.gov Identifier: NCT02829372).21 Both binding arms of the anti-HER2 T-cell engager BEAT® antibody used a VH3 domain, and consequently one domain had to be abrogated for PA binding to enable avidity purification. Purity post PA was 95.7% (data not shown), and therefore very similar to the purity observed in this study without HD technology. Naturally, the supernatant that was applied to the PA resin had significantly higher heterodimer content than the supernatants for constructs that were engineered without HD technology, i.e., a greater heterodimer yield can be obtained when applying the HD technology. While PA resins for manufacturing are standard to the industry, PG resins are only available for analytical, i.e., non-GMP purposes. Alkaline-resistant versions of the PG C2 domain were reported by two research groups for manufacturing, although these are not commercially available.53,54 Still GMP-grade PG resins could be easily custom made (GE Healthcare, personal communication); alternatively, biomimetic PA or PG resins could be used, but have yet to be tested with our methods.
Since both technologies showed a small to moderate reduction in FcRn binding (Table 2), we investigated the impact of our engineering in vivo by exploring the PK profiles of various antibodies and heterodimers. First, we looked at antibodies (i.e., Hc homodimers) wherein PA or PG binding was completely abolished (Figure 5(a) and Table 3). For the PA abrogated IgG1, clearance was twofold compared to the trastuzumab analog, but not outside of the reported range observed for other therapeutic antibodies such as pertuzumab,44 whereas half-life was similar to that of the trastuzumab analog. Importantly, the PK parameters were found to be in the normal range, whereas FcRn binding was found to be reduced by 33% in our SPR measurements (Table 2). Overall, no significant difference in half-life or clearance was observed between the PG abrogated IgG1 and the trastuzumab analog. This showed that our engineering was successful at abrogating PG binding without any impact on in vivo stability.
Next, we tested various Hc heterodimers built with our PA and/or PG methods and the Fab-scFv-Fc format. In addition, we built an Hc heterodimer based on our PA method in combination with our CH3 engineering method, BEAT®. PK profiles were very similar for all heterodimers irrespective of the technology used, with half-lives between ~8-9.5 days (Figure 5(b) and Table 4), suggesting that PK behavior was mostly influenced by the Fab-scFv-Fc format rather than the engineering of the Fc region. Importantly, differences in CH3 thermal stabilities did not translate into meaningful differences in vivo.
These results are in agreement with the small to moderate impact the different technologies had in vitro. As such, these Hc heterodimers are very close to native IgG1 in terms of FcRn binding since one Fc chain is native IgG1 and the other Fc chain is engineered with either ~70% (PA) or ~90% (PG) of the native IgG1 binding. Our results are also in agreement with those published by two other research groups, both reporting normal serum half-lives for H435R/Y436F variants as a bispecific heterodimer utilizing a cLc or in the antibody format.16,51
In conclusion, we have shown that PA or PG binding can be abolished in IgG1 without much impact on affinity and in vitro/vivo stability. The two different methods reported herein allow for the highly homogenous preparation of Hc heterodimers in a single affinity-purification step upon which bsAbs can be designed and developed at large scale.
Materials and methods
Cell culture
HEK293E cells (LGC Standards, catalog no. ATCC-CRL-10852) were maintained in suspension at 37 °C under 5–8% CO2 conditions using EX-CELL® 293 serum-free medium (Sigma-Aldrich, catalog no. 14571C) supplemented with 10 mM L-glutamine and 10% of Pluronic F-68.
Cloning and expression of recombinant antibodies, Fcs, and heterodimeric IgGs
Variable domains were obtained from previously described antibodies: U3-159 (anti-human HER3),40 trastuzumab (anti-human HER2),27 pertuzumab (anti-human HER2),55 and G6-31 (anti-human VEGF).56 For the full-length cLc heterodimeric IgG, Fabs sharing the same light chain sequence (VK3-15) were selected from the same cLc antibody phage display library, each Fab having specificity for a different oncology target. Both Fabs utilized the same VH subfamily (VH1-69) and only varied by their CDRs. cDNA coding sequences of IgG1 Hc, kappa Lc, and the mixed IgG1/IgG3 Fc were gene synthetized by Geneart AG (ThermoFisher Scientific). Hcs, Fcs, Fabs, and scFv-Fcs and variants thereof were generated by site-directed mutagenesis using standard overlap-PCR techniques. Fcs engineered for reduced PG binding had their C-terminal lysine residue replaced by a short amino acid linker and a C-tag57 sequence (GGGGSEPEA) to support SPR analysis. PCR products were digested with BamHI (5´end) and NotI (3´end) restriction enzymes (New England Biolabs) and ligated in a modified pcDNA™3.1 plasmid (ThermoFisher Scientific, catalog no. V79020) carrying a CMV promoter, the bovine growth hormone poly-adenylation signal, and oriP, which is the origin of plasmid replication of Epstein-Barr virus. In all expression-vectors, secretion was driven by the murine VJ2C leader peptide. Each chain was cloned independently and plasmids were transiently transfected in HEK293E cells using branched polyethyleneimine (Sigma-Aldrich, catalog no. 408727). For all constructs, equal quantities of each chain-vector were co-transfected. Supernatants were collected and filtered 4 to 5 days after transfection.
Purification of recombinant antibodies and Fcs
Antibodies and Fcs were purified in batch mode using affinity resins before being assessed by gradient PA or PG chromatography at RT. Antibodies engineered for reduced binding to PA were purified using PG sepharose 4 Fast Flow (GE Healthcare Europe GmbH Europe GmbH, catalog no. 17–0618-01). Conversely, antibodies engineered for reduced binding to PG were purified with PA sepharose (Repligen, catalog no. CA-PRI-0100). VH3-Fabs engineered for reduced PA binding were purified via Protein L (Genescript, catalog no. L00239) before gradient PA chromatography.
PA and PG binding assessment by affinity chromatography
PA and PG interactions were monitored by affinity chromatography at RT. Antibodies, Fc or Fabs were injected on affinity resins pre-equilibrated in phosphate-citrate buffer (McIlvaine’s buffer) pH 8.0. PG binding mutants were loaded on a 1 ml HiTrap® PG HP column (GE Healthcare Europe GmbH Europe GmbH, catalog no. 17–0404-01), whereas IgG 1133 antibody variants were loaded on a 1 ml HiTrap® MabSelect™ SuRe™ column (GE Healthcare Europe GmbH, catalog no. 11–0034-93). VH3-Fabs engineered for reduced PA binding were assessed on a 1 ml HiTrap® MabSelect™ column (GE Healthcare Europe GmbH, catalog no. 28–4082-55). Elutions were performed with a linear pH-gradient of phosphate-citrate buffer ranging from pH 8.0 to pH 3.0, and followed by UV reading at 280 nm using an ÄKTA™ purifier chromatography system (GE Healthcare Europe GmbH).
One-step affinity purification of heterodimeric IgGs
Affinity chromatography was performed at RT and 4 °C. Supernatants were conditioned to reach pH 6.0 and were loaded on a 1 ml column of the appropriate affinity resins, i.e., HiTrap® MabSelect™ SuRe™ or HiTrap® PG HP. After loading, bound proteins were washed extensively with phosphate-citrate buffer pH 6.0 (15 col. vol.). Elution (30 col. vol.) was performed using 50 mM sodium acetate pH 4.1 (RT), pH 4.2 (4 °C) or 0.1 M glycine pH 3.2 (RT or 4 °C) for dA and dG heterodimers, respectively. Affinity resins were stripped (10 col. vol.) with either 0.1 M glycine pH 3.0 or 2.5 for MabSelect™ SuRe™ and HiTrap® PG HP, respectively. Flow rate was 1 ml/min.
Protein characterization methods
The presence and purity of proteins were confirmed by RP-HPLC-MS, SDS-PAGE (4–12% acrylamide) and SE-HPLC. Protein concentrations were determined using absorbance readings at 280 nm. Differential scanning calorimetry – Calorimetric measurements were carried out on a VP-capillary differential scanning microcalorimeter (Malvern Instruments Ltd., UK). The cell volume was 0.128 ml, the heating rate was 200 °C/h, and the excess pressure was kept at 65 p.s.i. All proteins were used at a concentration of 0.9–1.6 mg/ml in phosphate-buffered saline (PBS, pH 7.4) with the exception of IgG dG, which was measured at 3.6 mg/ml. The molar heat capacity was estimated by comparison with duplicate samples containing identical buffer from which the protein had been omitted. The partial molar heat capacities and melting curves were analyzed using standard procedures. Thermograms were baseline corrected and concentration normalized before being further analyzed using a Non-Two State model in the software Origin v7.0 (supplied by Malvern Instruments Ltd). Size-Exclusion chromatography – Analytical SE-HPLC was performed using a Tosoh Bioscience TSKgel G3000SWxl column (Tosoh Bioscience AG, catalog no. 08541) at room temperature with 0.1 M sodium phosphate buffer, 0.15 M sodium chloride, pH 6.8 as eluent at 1 ml/min flow rate, on a Waters Alliance 2695 HPLC system with a Waters 2998 PDA detector (Waters AG), monitoring at 214 nm and 280 nm. RP-HPLC-MS – Samples were first treated for one hour with carboxypeptidase B to remove the C-terminal lysine (when present) and then deglycosylated overnight with PNGase F. RP-HPLC-MS was performed on a Vanquish chromatography system (ThermoFisher Scientific) coupled to an extended mass range hybrid electrospray-quadrupole_Orbitrap mass spectrometer (Q-Exactive Plus BioPharma, ThermoFisher Scientific). The purified proteins were injected on a polyphenyl reverse phase column (Bio BioResolve RP mAb 2.1 × 150 mm 450 Å, 2.7 µm column, Waters UK). Column temperature was set to 70 °C. The mobile phase consisted of 0.08% formic acid and 0.02% trifluoroacetic acid in water (solvent A) and 0.08% formic acid and 0.02% trifluoroacetic acid in acetonitrile (solvent B). The proteins were loaded at 5% solvent B with a 0.4 ml/min flow rate. Elution was performed with a linear 30% to 38% gradient of solvent B. The mass spectrometer was operated in the positive ion mode scanning the m/z 1500–5000 range with a resolution of 17500 at m/z 200. In-source collision energy was set to 60 eV to avoid TFA adducts. The acquired data was processed using the BioPharma Finder software (ThermoFisher Scientific). Peak detection was performed using the sliding window with deconvolution performed using the ReSpect® algorithm. Identifications were performed by comparing the measured masses with those calculated for the different expected species. Purity was assessed by integrating the areas of the extracted ion chromatograms for the detected protein species.
SPR analyses
Experiments were performed on a Biacore 2000 or Biacore T200 instrument (GE Healthcare Europe GmbH) at room temperature. Data fitting was performed using the BIAevaluation software version 4.1 or the Biacore T200 Evaluation software version 3.0 (GE Healthcare Europe GmbH). Binding affinities for human FcRn (Fc-C-tagged constructs) – Human FcRn α-chain and β2 microglobulin extracellular domains were cloned, expressed, and purified in-house. KD values for FcRn (UniProt no. P55899 and P61769) were measured by capture of the Fc-C-tagged constructs via the CaptureSelect™ Biotin Anti-C-tag conjugate (ThermoFisher Scientific, catalog no. 7103252100) previously coated on a streptavidin sensor chip (GE Healthcare Europe GmbH, catalog no. BR100531). Dissociation was monitored for two min. Regeneration was performed by injecting 10 µl of 10 mM glycine buffer, pH 2.0 (GE Healthcare Europe GmbH). All data were processed using the steady-state fitting model. HBS-EP+ buffer adjusted to pH 6.0 was used as running buffer. Affinity measurements of engineered Fabs for their target antigens – Antigens were cloned, expressed, and purified in-house as human Fc tagged proteins. Fabs were used as analytes, whereas Fc-tagged antigens were captured using a monoclonal mouse anti-human Ig (Fc) antibody coupled CM5 sensor chip (Human Antibody Capture Kit, GE Healthcare Europe GmbH, catalog no. BR100839). Capture levels were approximatively of 200 response units (RUs). HBS-EP (GE Healthcare Europe GmbH, catalog no. BR100188) was used as running buffer and flow path one used for referencing. Dilution series of the Fab mutants were injected for four min at a flow rate of 30 µl/min. Dissociation time was seven min. Data were fitted with a 1:1 Langmuir binding model. After each binding event, the surface was regenerated with 3 M MgCl2. To account for the experimental variations in antibody capture, the Rmax value was set to local in all fits. Measurements were performed in duplicate, and included zero-concentration samples for referencing. Both Chi2 and residual values were used to evaluate the quality of a fit between the experimental data and individual binding models. Binding affinities for human Fc receptors – Human Fc receptor extracellular domains were cloned, expressed, and purified in-house. KD values for FcγR2a (Uni-Prot no. P12318, variant H167), FcγR2b (UniProt no. P31994), FcγR3a (UniProt no. P08637, variant V176), and FcRn (UniProt no. P55899 and P61769) were measured via direct covalent coupling of the antibody onto a CM5 sensor chip (GE Healthcare Europe GmbH). Dissociation was monitored for 2 min (FcγR2a, FcγR2b, and FcRn) or 10 min (FcγR3a). No regeneration step was needed for FcγR2a, FcγR2b, and FcγR3a as complete dissociation was observed; for these Fc receptors, HBS-EP+ buffer was used as running buffer. For FcRn measurements, HBS-EP+ adjusted to pH 6.0 was used as running buffer and regeneration was performed by injecting 2 × 10 µl of HBS-EP+, pH 7.4 (GE Healthcare Europe GmbH). All data were processed using the steady-state fitting model. Affinities for FcγR1a (UniProt no. P12314) were measured via capture of the antibody on a CM5 sensor chip previously coated with human HER2 (Acrobiosystems, catalog no. HE2-H5225) via amine coupling. Experimental data for FcγR1a were processed using a 1:1 Langmuir model. After each binding event, the surface was regenerated with 10 mM glycine buffer, pH 1.5. Data processing, normalization and statistical analysis – Measurements were normalized as significant variability in affinity measurements from day to day could not be excluded (e. g., variability in capture efficiency). To normalize data, a reference standard was analyzed with the test samples (trastuzumab IgG1 or IgG1 Fc-C-tag protein). Normalization was performed as follows: for each test sample, measurements were performed at least in triplicates, and the KD value of each measurement was divided by the average KD value for the reference standard calculated from replicate measurements recorded using the same experimental setup, on the same day as all samples were part of the same multi-cycle process. Average relative KD and standard deviations were calculated on these normalized KD values. Normalized affinity was calculated as follow: 1/average relative KD. Average relative KD values were further used as inputs for statistical comparison. Each test sample was compared with the reference standard using the unpaired T-test with two-tailed p value (GraphPad Prism v7.02, GraphPad Software Inc.). A p value below 0.05 (p < .05) was considered significant (95% confidence level).
PK studies
Analyses were conducted in female Sprague-Dawley rats. Each group contained four rats. Rats received 10 mg/kg of antibody or heterodimeric IgG by intravenous bolus injection. Blood samples were collected at multiple time points between 0.25 and 1008 h after drug administration. Serum levels of antibodies were determined by a sandwich ELISA. HER2-Fc antigen (extracellular portion of HER2 fused to a human Fc and produced in-house) was coated onto 96-well ELISA plates at a concentration of 2 µg/ml and incubated overnight at 4 °C. After the plates were blocked with PBS 3% BSA, calibration standards, quality control samples, and study samples were added to the plate and incubated for one hour at room temperature. After washing to remove unbound antibody, peroxidase-conjugated goat anti-human IgG_F(ab’)2 fragment specific detection antibody (Jackson ImmunoResearch Laboratories, catalog no.109–035-006) was added and developed by standard colorimetric tetramethylbenzidine substrate (Pierce-ThermoFisher Scientific) according to the manufacturer’s recommendations. Absorbance values at 450 nm were recorded on a plate reader and the concentrations of antibody in serum samples were interpolated using the calibration standard curve generated in the sample plate utilizing four parametric regression models. PK parameters were evaluated by non-compartment analysis using WinNonlin™ version 5.3 (Pharsight Corporation, Mountain View, CA, USA). PK parameters are shown as mean ± standard deviation.
Abbreviations
- BEAT®
Bispecific Engagement by Antibodies based on the T cell receptor
- bsAbs
bispecific antibodies
- cLc
common light chain
- DSC
differential scanning calorimetry
- FcRn
neonatal Fc receptor
- GMP
good manufacturing practice
- Hc
heavy chain
- HD
heterodimerization
- HER2
human epidermal growth factor receptor 2
- HER3
human epidermal growth factor receptor 3
- Ig
immunoglobulin
- Lc
light chain
- PA
Protein A
- PG
Protein G
- PK
pharmacokinetic
- SE-HPLC
size exclusion high performance liquid chromatography
- RP-HPLC-MS
reverse phase high performance liquid chromatography coupled to mass spectrometry
- SPR
surface plasmon resonance
- VEGF
vascular endothelial growth factor
Acknowledgments
We thank Dr. Darko Skegro and Dr. Krzysztof Bielikowicz for helpful discussions; we thank Dr. Jeremy Loyau for providing the common light chain Fabs.
Author contributions
R.O., P.W., T.M., C.S. and S.B. designed research; R.O., P.W., T.M., C.R.F., S.G., V.C.A. and D.A. performed research; R.O., P.W., T.M., C.R.F., S.G., V.C.A., D.A., C.S., G.S.G and S.B. analyzed data; and R.O., C.S. and S.B. wrote the paper.
Disclosure of potential conflicts of interest
All authors were employees of Glenmark Pharmaceuticals SA at the time the work presented here was conducted.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Garber K. Bispecific antibodies rise again. Nat Rev Drug Discov. 2014;13:799–801. doi: 10.1038/nrd4478. [DOI] [PubMed] [Google Scholar]
- 2.Sheridan C. Amgen’s bispecific antibody puffs across finish line. Nat Biotechnol. 2015;33:219–21. doi: 10.1038/nbt0315-219. [DOI] [PubMed] [Google Scholar]
- 3.Sheridan C. Despite slow progress, bispecifics generate buzz. Nat Biotechnol. 2016;34:1215–17. doi: 10.1038/nbt1216-1215. [DOI] [PubMed] [Google Scholar]
- 4.Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–47. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Brinkmann U, Kontermann RE. The making of bispecific antibodies. mAbs. 2017;9:182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9:86–120. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seimetz D, Lindhofer H, Bokemeyer C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev. 2010;36:458–67. doi: 10.1016/j.ctrv.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 8.Chelius D, Ruf P, Gruber P, Ploscher M, Liedtke R, Gansberger E, Hess J, Wasiliu M, Lindhofer H. Structural and functional characterization of the trifunctional antibody catumaxomab. mAbs. 2010;2:309–19. doi: 10.4161/mabs.2.3.11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a Single-step Purification of Bispecific Antibodies. J Immunol. 1995;155:219–25. [PubMed] [Google Scholar]
- 10.Scott LJ, Kim ES. Emicizumab-kxwh: first global approval. Drugs. 2018;78:269–74. doi: 10.1007/s40265-018-0861-2. [DOI] [PubMed] [Google Scholar]
- 11.Jackman J, Chen Y, Huang A, Moffat B, Scheer JM, Leong SR, Lee WP, Zhang J, Sharma N, Lu Y, et al. Development of a two-part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J Biol Chem. 2010;285:20850–59. doi: 10.1074/jbc.M110.113910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein C, Sustmann C, Thomas M, Stubenrauch K, Croasdale R, Schanzer J, Brinkmann U, Kettenberger H, Regula JT, Schaefer W. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs. 2012;4:653–63. doi: 10.4161/mabs.21379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sampei Z, Igawa T, Soeda T, Okuyama-Nishida Y, Moriyama C, Wakabayashi T, Tanaka E, Muto A, Kojima T, Kitazawa T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8:e57479. doi: 10.1371/journal.pone.0057479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boström T, Nilvebrant J, Hober S. Purification systems based on bacterial surface proteins In: Ahmad R, editor. Protein purification. InTech; 2012. p. 89–136. Available from: http://www.intechopen.com/books/protein-purification/purification-systems-based-on-bacterial-surface-proteins. [Google Scholar]
- 15.Skegro D, Stutz C, Ollier R, Svensson E, Wassmann P, Bourquin F, Monney T, Gn S, Blein S. Immunoglobulin domain interface exchange as a platform technology for the generation of Fc heterodimers and bispecific antibodies. J Biol Chem. 2017;292:9745–59. doi: 10.1074/jbc.M117.782433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshner JR, Canova L, Menon J, et al. A novel, native-format bispecific antibody triggering T-cell killing of B-cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2015;5:17943. doi: 10.1038/srep17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tustian AD, Endicott C, Adams B, Mattila J, Bak H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs. 2016;8:828–38. doi: 10.1080/19420862.2016.1160192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nezlin R, Ghetie V. Interactions of immunoglobulins outside the antigen-combining site. Adv Immunol. 2004;82:155–215. doi: 10.1016/S0065-2776(04)82004-2. [DOI] [PubMed] [Google Scholar]
- 19.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 20.Atwell S, Ridgway JB, Wells JA, Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J Mol Biol. 1997;270:26–35. doi: 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- 21.Wermke M, Schmidt H, Nolte H, Ochsenreither S, Back J, Salhi Y, Bayever E. A phase I study of the bispecific antibody T-cell engager GBR 1302 in subjects with HER2-positive cancers. J Clin Oncol. 2017;35:TPS3091–TPS. doi: 10.1200/JCO.2017.35.15_suppl.TPS3091. [DOI] [Google Scholar]
- 22.Jansson B, Uhlen M, Nygren PA. All individual domains of staphylococcal protein A show Fab binding. FEMS Immunol Med Microbiol. 1998;20:69–78. doi: 10.1111/j.1574-695X.1998.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 23.Healthcare G. MabSelect SuRe–studies on ligand toxicity, leakage, removal of leached ligand, and sanitization. GE application note 2004; 11-0011-64 AA.
- 24.Seldon TA, Hughes KE, Munster DJ, Chin DY, Jones ML. Improved Protein-A separation of V(H)3 Fab from Fc after papain digestion of antibodies. J Biomol Tech. 2011;22:50–52. [PMC free article] [PubMed] [Google Scholar]
- 25.Bach J, Lewis N, Maggiora K, Gillespie AJ, Connell-Crowley L. Differential binding of heavy chain variable domain 3 antigen binding fragments to protein A chromatography resins. J Chromatogr A. 2015;1409:60–69. doi: 10.1016/j.chroma.2015.06.064. [DOI] [PubMed] [Google Scholar]
- 26.Graille M, Stura EA, Corper AL, Sutton BJ, Taussig MJ, Charbonnier JB, Silverman GJ. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A. 2000;97:5399–404. doi: 10.1073/pnas.97.10.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, Shepard HM. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A. 1992;89:4285–89. doi: 10.1073/pnas.89.10.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355:751–57. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 29.Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther. 2007;7:405–18. doi: 10.1517/14712598.7.3.405. [DOI] [PubMed] [Google Scholar]
- 30.Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–70. [PubMed] [Google Scholar]
- 31.Martin WL, West AP Jr., Gan L, Bjorkman PJ. Crystal structure at 2.8 A of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol Cell. 2001;7:867–77. [DOI] [PubMed] [Google Scholar]
- 32.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall’Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. 2014;289:7812–24. doi: 10.1074/jbc.M113.537563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauer-Eriksson AE, Kleywegt GJ, Uhlen M, Jones TA. Crystal structure of the C2 fragment of streptococcal protein G in complex with the Fc domain of human IgG. Structure. 1995;3:265–78. [DOI] [PubMed] [Google Scholar]
- 34.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169:5171–80. doi: 10.4049/jimmunol.169.9.5171. [DOI] [PubMed] [Google Scholar]
- 35.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 36.Lefranc MP. Immunoglobulin and T cell receptor genes: IMGT((R)) and the birth and rise of immunoinformatics. Front Immunol. 2014;5:22. doi: 10.3389/fimmu.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lefranc MP, Giudicelli V, Duroux P, Jabado-Michaloud J, Folch G, Aouinti S, Carillon E, Duvergey H, Houles A, Paysan-Lafosse T, et al. IMGT(R), the international ImMunoGeneTics information system(R) 25 years on. Nucleic Acids Res. 2015;43:D413–22. doi: 10.1093/nar/gku1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Proudfoot KA, Torrance C, Lawson AD, King DJ. Purification of recombinant chimeric B72.3 Fab’ and F(ab’)2 using streptococcal protein G. Protein Expr Purif. 1992;3:368–73. [DOI] [PubMed] [Google Scholar]
- 39.Derrick JP, Wigley DB. The third IgG-binding domain from streptococcal protein G. An analysis by X-ray crystallography of the structure alone and in a complex with Fab. J Mol Biol. 1994;243:906–18. doi: 10.1006/jmbi.1994.1691. [DOI] [PubMed] [Google Scholar]
- 40.Rothe M, Treder M, Hartmann S, Freeman D, Radinsky B, Borges E. Antibodies directed to HER-3 and uses thereof, PCT publication No WO2007/077028. 2007. doi: 10.1094/PDIS-91-4-0467B. [DOI]
- 41.Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. mAbs. 2015;7:331–43. doi: 10.1080/19420862.2015.1008353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe H, Matsumaru H, Ooishi A, Feng Y, Odahara T, Suto K, Honda S. Optimizing pH response of affinity between protein G and IgG Fc: how electrostatic modulations affect protein-protein interactions. J Biol Chem. 2009;284:12373–83. doi: 10.1074/jbc.M809236200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gil D, Schrum AG. Strategies to stabilize compact folding and minimize aggregation of antibody-based fragments. Adv Biosci Biotechnol. 2013;4:73–84. doi: 10.4236/abb.2013.44A011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng R, Iyer S, Theil FP, Mortensen DL, Fielder PJ, Prabhu S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? mAbs. 2011;3:61–66. doi: 10.4161/mabs.3.1.13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kelley SK, Gelzleichter T, Xie D, Lee WP, Darbonne WC, Qureshi F, Kissler K, Oflazoglu E, Grewal IS. Preclinical pharmacokinetics, pharmacodynamics, and activity of a humanized anti-CD40 antibody (SGN-40) in rodents and non-human primates. Br J Pharmacol. 2006;148:1116–23. doi: 10.1038/sj.bjp.0706828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nnane IP, Xu Z, Zhou H, Davis HM. Non-clinical pharmacokinetics, prediction of human pharmacokinetics and first-in-human dose selection for CNTO 5825, an anti-interleukin-13 monoclonal antibody. Basic Clin Pharmacol Toxicol. 2015;117:219–25. doi: 10.1111/bcpt.12391. [DOI] [PubMed] [Google Scholar]
- 47.Hezareh M, Hessell AJ, Jensen RC, van de Winkel JG, Parren PW. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J Virol. 2001;75:12161–68. doi: 10.1128/JVI.75.24.12161-12168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol. 2003;325:531–53. doi: 10.1016/s0022-2836(02)01237-8. [DOI] [PubMed] [Google Scholar]
- 49.Rouet R, Lowe D, Christ D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014;588:269–77. doi: 10.1016/j.febslet.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 50.Strohl WR, Strohl LM Development issues: antibody stability, developability, immunogenicity, and comparability In: Strohl WR, Strohl LM, editors. Therapeutic antibody engineering. Philadelphia (PA): Woodhead Publishing; 2012. p. 377–595. Available from: https://www.sciencedirect.com/book/9781907568374/therapeutic-antibody-engineering [Google Scholar]
- 51.Zwolak A, Leettola CN, Tam SH, Goulet DR, Derebe MG, Pardinas JR, Zheng S, Decker R, Emmell E, Chiu ML. Rapid purification of human bispecific antibodies via selective modulation of protein a binding. Sci Rep. 2017;7:15521. doi: 10.1038/s41598-017-15748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu HF, Ma J, Winter C, Bayer R. Recovery and purification process development for monoclonal antibody production. mAbs. 2010;2:480–99. doi: 10.4161/mabs.2.5.12645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gulich S, Linhult M, Stahl S, Hober S. Engineering streptococcal protein G for increased alkaline stability. Protein Eng. 2002;15:835–42. [DOI] [PubMed] [Google Scholar]
- 54.Palmer B, Angus K, Taylor L, Warwicker J, Derrick JP. Design of stability at extreme alkaline pH in streptococcal protein G. J Biotechnol. 2008;134:222–30. doi: 10.1016/j.jbiotec.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 55.Franklin MC, Carey KD, Vajdos FF, Leahy DJ, de Vos AM, Sliwkowski MX. Insights into ErbB signaling from the structure of the ErbB2-pertuzumab complex. Cancer Cell. 2004;5:317–28. [DOI] [PubMed] [Google Scholar]
- 56.Liang WC, Wu X, Peale FV, Lee CV, Meng YG, Gutierrez J, Fu L, Malik AK, Gerber H-P, Ferrara N, et al. Cross-species vascular endothelial growth factor (VEGF)-blocking antibodies completely inhibit the growth of human tumor xenografts and measure the contribution of stromal VEGF. J Biol Chem. 2006;281:951–61. doi: 10.1074/jbc.M508199200. [DOI] [PubMed] [Google Scholar]
- 57.Djender S, Beugnet A, Schneider A, de Marco A. The biotechnological applications of recombinant single-domain antibodies are optimized by the C-terminal fusion to the EPEA sequence (C Tag). Antibodies. 2014;3:182. doi: 10.3390/antib3020182. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.