

ABSTRACT
p16INK4A (hereafter called p16) is a faithful cellular ally in the fight against tumorigenesis. Although its canonical pathway through retinoblastoma (RB) is well delineated, RB-independent functions for p16 are beginning to emerge. Here we summarize non-canonical roles of p16, including our recent finding on its role in nucleotide metabolism.
KEYWORDS: Retinoblastoma, cell cycle, senescence, nucleotide metabolism, mTORC1, reactive oxygen species, NF-κB, JNK1/3, SP1, AP-1
Since its discovery in the early 1990s, p16INK4A (hereafter called p16) has been one of the most studied proteins, mostly due to its role in cancer and aging.1 As a cell cycle brake, p16 has proven to be a crucial regulator of tissue homeostasis by controlling cellular differentiation, growth, senescence, and apoptosis. Underscoring its physiological importance, overexpression of p16 has been found in many types of cellular senescence, including oncogene-induced senescence (OIS). OIS is considered a bona fide tumor suppressor mechanism in vivo. Although the stimuli that trigger OIS-mediated p16 upregulation are poorly understood, we and others have demonstrated that suppression of p16 bypasses OIS to allow for transformation and tumorigenesis.2,3 p16 is low or null in half of all human malignancies; however, there is currently no targeted therapy for these patients. It is, therefore, of the utmost importance to better delineate p16-related pathways to not only understand its tumor-suppressive functions, but also to identify novel ways to treat patients with p16-deficient cancers.
p16, encoded by the cyclin-dependent kinase inhibitor 2A (CDKN2A) gene, exerts its canonical function during the G1 to S transition. Upon different stresses, p16 is upregulated and bound to cyclin-dependent kinases (CDK4/6), which prevents the formation of the Cyclin D-CDK4/6 complex [reviewed in Ref1]. Predicted structural models as well as mutational analysis have determined that the third p16 ankyrin repeat is necessary for p16-mediated inhibition of CDK4. This maintains the Retinoblastoma (RB) protein in a hypophosphorylated state that allows the sequestration of E2F transcription factors, thereby preventing the expression of proliferation-associated genes (Figure 1). In addition to its canonical (RB-dependent) pathway, p16 can control cell homeostasis through other pathways. Recently, our laboratory has described a non-canonical, RB-independent role for p16 in the regulation of nucleotide biosynthesis.2 Maintaining healthy nucleotide and deoxyribonucleotide levels is critical for cellular homeostasis.4 Our previous work demonstrated that OIS cells restrain deoxyribonucleotide synthesis, and exogenous supplementation with nucleosides is sufficient to abrogate OIS.5 In our more recent work, we found that suppression of p16 increases both nucleotide and deoxyribonucleotide levels and bypasses OIS.2 Unexpectedly, this was not linked to p16-mediated control of the cell cycle or transcriptional activation of nucleotide metabolism genes. Further investigation revealed that loss of p16 promotes an increase in the mammalian target of rapamycin complex 1 (mTORC1)-mediated translation of the pentose phosphate pathway enzyme, Ribose 5-Phosphate Isomerase A (RPIA) in multiple senescence and tumor models. Indeed, knockdown of RPIA in p16-deficient, but not p16-proficient cells, induced senescence both in vitro and in vivo and may be a therapeutic target for p16-null cancers. This reinforces the idea that p16 has functions outside its canonical pathway. Consistent with our observation, other labs have described non-canonical p16 pathways, including inhibition of the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) complex, suppression of reactive oxygen species (ROS), impairing activation of activating protein-1 (AP1) via the mitogen-activated protein kinases (JNK1/3), inhibition of the eukaryotic translation elongation factor 1 alpha 2 (eEF1A2)-mediated translation, and promotion of tumor-suppressive miRNAs via SP1 transcription factor (Figure 1). We will briefly describe each publication below.
Figure 1.
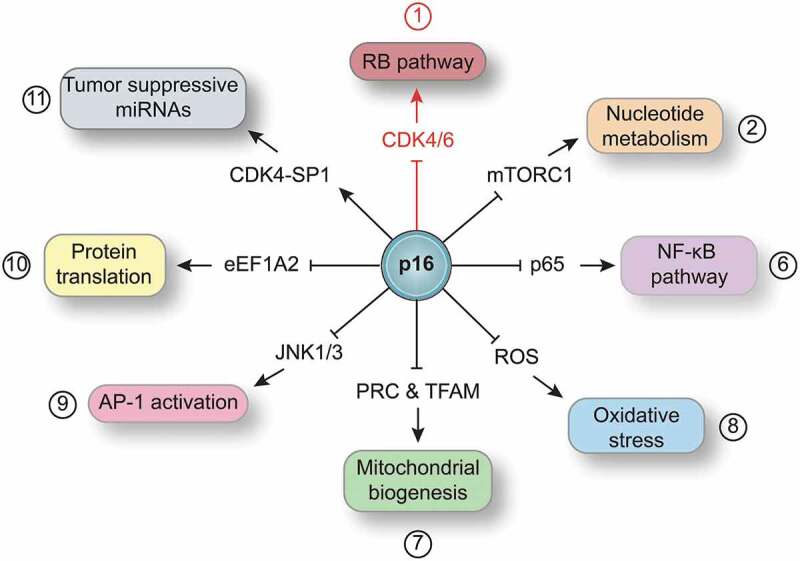
Canonical and non-canonical p16 signaling.
Schematic showing the p16 canonical (red) and non-canonical (black) pathways, including: p16–Retinoblastoma (RB) pathway; p16 regulation of nucleotide metabolism through the mammalian target of rapamycin complex 1 (mTORC1); p16 directly binds to p65 and inhibits the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) complex; p16 regulation of intracellular oxidative stress; p16 regulation of mitochondrial biogenesis (PRC: PGC-1-related coactivator; TFAM: Transcription Factor A, Mitochondrial); p16 directly binds to mitogen-activated protein kinases (JNK1/3) and regulates the activating protein-1 (AP1) transcription factor activity; p16 directly binds the eukaryotic translation elongation factor 1 alpha 2 (eEF1A2) and inhibits protein translation; and p16 forms a complex with the cyclin-dependent kinase 4 (CDK4) and SP1 transcription factor that promotes the transcription of tumor-suppressive miRNAs.
p16 directly interacts with the p65 subunit of the NF-κB complex.6 This interaction serves as brake of NF-κB-promoted tumorigenesis in the absence of its natural inhibitor IκBα (nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha). Additionally, p16-deficient cells have higher mitochondrial mass and decreased respiratory capacity, leading to elevated mitochondrial membrane potential and ROS production.7,8 Although the mechanism is not yet elucidated, suppression of RB or CDK4 activity did not phenocopy p16-deficient cells. Moreover, the N-terminal domain of p16 (first and second ankyrin repeats) directly interacts with and inhibits JNK1/3 as well as eEF1A2. In the case of JNK1/3, p16 directly binds to the Jun Proto-Oncogene (c-jun) binding site at the N-terminal domain of JNK1/3, impairing the activation of the AP-1 [c-jun/Fos Proto-Oncogene (c-fos)] transcription factor.9 In the absence of p16, UV-induced melanoma cells and RAS-induced Mouse Embryonic Fibroblasts (MEFs) activate the JNK-AP-1 pathway, promoting cellular transformation and tumorigenesis. In regards to eEF1A2, p16 directly binds to the eEF1A2 I/III domains to impair eEF1A2 translational activity, correlating to a decrease in colony-forming ability.10 Interestingly, p16 also modulates miRNA expression.11 Upon UV-induction, p16 forms a heterocomplex with CDK4 and the transcription factor SP1, facilitating the transcription of the tumor suppressors miR-141 and miR-146b-5p. The fourth p16 ankyrin repeat is necessary to bind SP1 to the p16-CDK4 complex. This finding suggests that p16 is not only capable of binding and regulating different proteins, but also different binding partners confer new protein conformations to p16 . This increases the catalog of possible p16 binding partners and its potential uncharacterized non-canonical functions. Together with our recent findings on how p16 loss increases nucleotide metabolism2, these studies suggest that non-canonical roles for p16 are important for the suppression of tumorigenesis.
The importance of p16 is clearly demonstrated by how its improper expression leads to unhealthy cellular phenotypes such as cancer. Both the canonical and non-canonical pathways of p16 prevent different aspects of oncogenic transformation, including suppression of nucleotide metabolism, which our recent study suggests is critical for the tumor-suppressive senescent phenotype2. Approximately 50% of cancer patients have a loss of functional p16 protein; however, there is no US Food and Drug Administration (FDA)-approved therapy that specifically targets p16-loss related pathways. While there are currently over 180 studies in the US targeting the canonical p16 pathway using CDK4/6 inhibitors (http://clinicaltrials.gov), our study and others indicate that p16-deficient tumors may require additional therapies as p16 has roles outside of CDK4/6 inhibition. For instance, our recent study2 suggests that p16-deficient tumors may be exquisitely sensitive to mTORC1 inhibitors or drugs that affect nucleotide metabolism. Therefore, it is critical to study the non-canonical p16 pathways in order to improve personalized cancer therapy for patients with p16-deficient cancer.
Funding Statement
This work was supported by an NIH/NCI grant (R00CA194309 to K.M.A.) and a Penn State Cancer Institute Postdoctoral Fellowship (R.B.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2(731–737). doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 2.Buj R, Chen CW, Dahl ES, Leon KE, Kuskovsky R, Maglakelidze N, Navaratnarajah M, Zhang G, Doan MT, Jiang H, et al. Suppression of p16 induces mTORC1-mediated nucleotide metabolic reprogramming. Cell Reports. 2019;28:1–3. doi: 10.1016/j.celrep.2019.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haferkamp S, Becker TM, Scurr LL, Kefford RF, Rizos H.. p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell. 2008;7(733–745). doi: 10.1111/j.1474-9726.2008.00422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buj R, Aird KM. Deoxyribonucleotide triphosphate metabolism in cancer and metabolic disease. Front Endocrinol (Lausanne). 2018;9(177). doi: 10.3389/fendo.2018.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aird KM, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Reports. 2013;3(1252–1265). doi: 10.1016/j.celrep.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolff B, Naumann M. INK4 cell cycle inhibitors direct transcriptional inactivation of NF-kappaB. Oncogene. 1999;18(2663–2666). doi: 10.1038/sj.onc.1202617. [DOI] [PubMed] [Google Scholar]
- 7.Tyagi E, Liu B, Li C, Liu T, Rutter J, Grossman D. Loss of p16INK4A stimulates aberrant mitochondrial biogenesis through a CDK4/Rb-independent pathway. Oncotarget. 2017;8(55848–55862). doi: 10.18632/oncotarget.19862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jenkins NC, Liu T, Cassidy P, Leachman SA, Boucher KM, Goodson AG, Samadashwily G, Grossman D. The p16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene. 2011;30(265–274). doi: 10.1038/onc.2010.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi BY, Choi HS, Ko K, Cho -Y-Y, Zhu F, Kang BS, Ermakova SP, Ma W-Y, Bode AM, Dong Z. The tumor suppressor p16(INK4a) prevents cell transformation through inhibition of c-Jun phosphorylation and AP-1 activity. Nature Structural & Molecular Biology. 2005;12(699–707). doi: 10.1038/nsmb960. [DOI] [PubMed] [Google Scholar]
- 10.Lee MH, Darehshouri A, Anderson KL, Page C, Lidke DS, Volkmann N, Hanein D, Wilson BS. Tumor suppressor p16(INK4a) inhibits cancer cell growth by downregulating eEF1A2 through a direct interaction. J Cell Sci. 2013;126(1744–1752). doi: 10.1242/jcs.113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Khalaf HH, Mohideen P, Nallar SC, Kalvakolanu DV, Aboussekhra A. The cyclin-dependent kinase inhibitor p16INK4a physically interacts with transcription factor Sp1 and cyclin-dependent kinase 4 to transactivate microRNA-141 and microRNA-146b-5p spontaneously and in response to ultraviolet light-induced DNA damage. J Biol Chem. 2013;288(35511–35525). doi: 10.1074/jbc.M113.512640. [DOI] [PMC free article] [PubMed] [Google Scholar]