

ABSTRACT
As reported here, we developed and optimized a purification matrix based on a Protein A-derived domain, ZCa, displaying calcium-dependent antibody binding. It provides an alternative to the acidic elution conditions of conventional Protein A affinity chromatography for purification of sensitive antibodies and other Fc-based molecules. We describe the multimerization of ZCa to generate a chromatography resin with higher binding capacity. The highest order multimeric variant, ZCaTetraCys, demonstrated a considerably high dynamic binding capacity (35 mg IgG/ml resin) while preserving the specificity for IgG. High recovery was obtained and host cell protein and DNA content in purified fractions proved to be comparable to commercial MabSelect SuRe and MabSelect PrismA. Various elution conditions for use of this domain in antibody purification were investigated. The purification data presented here revealed variations in the interaction of different subclasses of human IgG with ZCaTetraCys. This resulted in diverse elution properties for the different IgGs, where complete elution of all captured antibody for IgG2 and IgG4 was possible at neutral pH. This optimized protein ligand and the proposed purification method offer a unique strategy for effective and mild purification of antibodies and Fc-fusion proteins that cannot be purified under conventional acidic elution conditions due to aggregation formation or loss of function.
KEYWORDS: Protein A, purification, calcium-dependent, antibody, multimerization, elution
Introduction
Antibodies are highly valuable, commonly used tools within biomolecular research and analysis, as well as medical diagnosis and therapy. Monoclonal antibodies (mAbs) constitute the largest class of approved biotherapeutics, and the number of antibody drugs that are available for clinical use is increasing continuously.1 For this reason, there is an increasing demand for efficient antibody purification strategies. The most widely used method for purification of antibodies is affinity chromatography based on Protein A.2 Protein A chromatography is a simple and highly selective method relying on the strong and specific interaction between Protein A and the crystallizable fragment (Fc) of the antibody.3,4 Due to this, high yields and exceptional purity can be achieved. Alternatives to Protein A have been explored, such as cation exchange,5–8 and non-chromatographic methods like precipitation,9–11 but they have not been able to compete with the well-established Protein A platform when applied to industrial-scale bioprocessing.12
However, the elution step poses a weakness for Protein A purification because lowering the pH is required to break the interaction between the antibody and Protein A. These acidic conditions can be harmful for some antibodies and Fc-fused proteins, increasing the risk of aggregation and loss of function of the purified molecules.13–17 To address this drawback of Protein A, we previously developed the molecule ZCa, which displays calcium-dependent binding to IgG.18 ZCa is an engineered protein domain, based on Z derived from the B domain of Protein A.19 The Z domain is a small and stable protein of 58 amino acids that folds into a three-helix bundle with high affinity for the Fc region of IgG. By introducing a randomized calcium-binding loop between helix two and three of the Z domain, phage display was used to select for binders with a calcium-dependent IgG-binding. ZCa was shown to selectively capture IgG from a complex cell culture supernatant, after which calcium-dependent elution of the IgG could be performed using ethylenediaminetetraacetic acid (EDTA) at pH 5.5.
As the market for biotherapeutics expands and advances in upstream processing lead to accelerating cell culture titers, the pressure on downstream processing to develop more efficient processes is increasing. Since Protein A capture commonly is the initial purification step and a vital part of the downstream mAb process, it risks becoming a bottleneck for the whole manufacturing process. Thus, it is of great importance to increase the binding capacity of Protein A resins. Currently, various new Protein A resins offering higher dynamic binding capacities (DBCs) and improved process productivity are being launched to handle the high feed concentrations and large throughput. Many of these commercially available resins are made up from several repeated binding units, which in many cases have been shown to significantly increase the DBC of the ligand.20
Here, we examined whether multimerization of the ZCa domain can lead to a higher binding capacity of the ZCa resin in an effort to optimize the domain for use as an affinity ligand in current biomanufacturing processes. The variants that were tested included a monomer, dimer, trimer and tetramer. The effect of the multimerization on the binding performance of the proteins was evaluated both in an affinity chromatography setup as well as using surface plasmon resonance (SPR). We have also investigated the optimal method for purification of IgG using ZCa, both in regard to the ligand and purification conditions, and we present a method for more efficient purification of acid-sensitive antibodies or Fc-fused proteins with mild elution conditions.
Results
Multimerization of ZCa
In order to optimize ZCa for development of a chromatography resin with high binding capacity, three multimeric versions (di-, tri- and tetrameric) of the domain were constructed and successfully produced, all containing a C-terminal cysteine for directed coupling. As reference, the monomeric version, also equipped with a C-terminal cysteine, was used. For comparison of the ZCa variants as affinity ligands in IgG purification, the proteins were covalently and irreversibly immobilized onto a purification matrix via the C-terminal cysteine. Similar coupling degrees of ~ 50% were obtained for all proteins. Columns were packed and saturated with pure human polyclonal IgG and the captured protein was eluted with 100 mM EDTA at pH 5.5, which has previously been shown to efficiently elute all captured IgG from a column coupled with a ZCa monomer.18 Triplicates were performed and the eluted protein amounts were measured (Figure 1(a)). To verify that all protein was successfully eluted with EDTA, elution with acetic acid (HAc) at pH 3.3 was performed after the EDTA elution. This resulted in negligible amounts of protein for all ligands. In Figure 1(a), significant differences in the amount of eluted antibody between the multimeric variants and the monomer can be observed. All multimeric variants were able to capture more antibody than the monomer.
Figure 1.
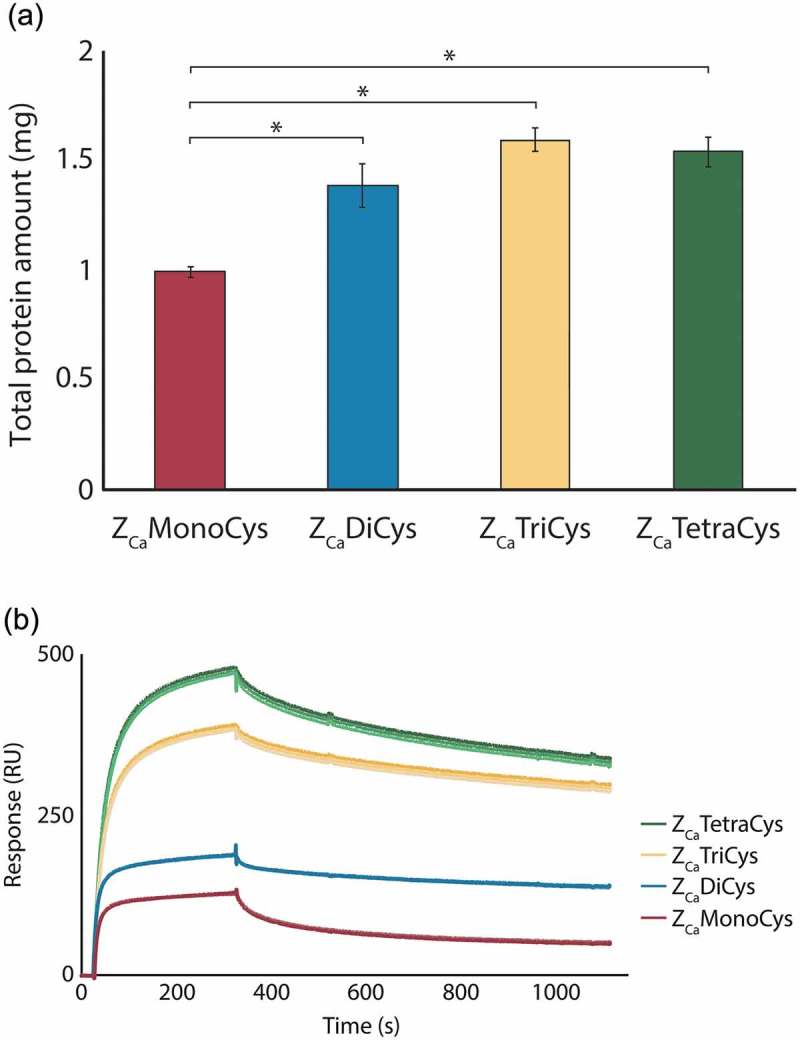
Monomeric and multimeric ZCa variants with C-terminal cysteines analyzed as ligands in a column purification setup and by SPR (a) The average amount (±SD) of human polyclonal IgG eluted with EDTA at pH 5.5 from columns coupled with the mono- (red), di- (blue), tri- (yellow) and tetrameric (green) ZCa, based on triplicate experiments. The multimeric variants of ZCa capture significantly more protein as compared to the monomer (*P < .001). (b) Sensorgrams from triplicate injections of 500 nM IgG flown over surfaces with equimolar amounts of each ZCa variant immobilized show that larger multimers exhibit improved binding performance.
The multimeric variants were further evaluated by SPR. Using directed thiol coupling, the proteins were immobilized to equimolar amounts on separate surfaces and 500 nM human polyclonal IgG was flown over. Sensorgrams in triplicates are shown in Figure 1(b), which, similar to the test purification, indicate that the multimeric proteins have a higher binding performance than the monomeric protein. They also demonstrate a trend among the multimeric variants toward improved binding performance with the increasing number of binding domains. ZCaTriCys and ZCaTetraCys indisputably generate the highest signals and have thus the potential to function as the most efficient affinity ligands.
For a more thorough comparison of the most interesting variants, ZCaTriCys and ZCaTetraCys were coupled to a purification matrix using directed coupling through their unique cysteine residue, and packed into 1 ml HiTrap™ columns. In a purification setup, human polyclonal IgG was added until the columns were saturated and eluted with EDTA. The eluted amounts of IgG can be found in Table 1 together with the coupling degrees of the two ligands and the number of binding sites. ZCaTriCys and ZCaTetraCys generated similar elution profiles when using pH 5.5 and 100 mM EDTA (Fig. S1). However, ZCaTetraCys displayed a better binding performance, as it bound 1.7 times more IgG per molecule and 1.3 times more IgG per binding site compared to ZCaTriCys.
Table 1.
Comparison of the amount of bound antibody per molecule and binding site for ZCaTriCys and ZCaTetraCys on 1 ml columns. Coupling degrees in mg amino acid per ml matrix and M (Mw ZCaTriCys: 23 036 Da, ZCaTetCys: 30 675 Da), number of binding sites and the average amounts of IgG eluted with EDTA at pH 5.5 (n = 3) for ZCaTriCys and ZCaTetraCys. This data suggests that the tetrameric variant of ZCa has the highest binding performance.
| Coupling degree (mg amino acid/ml matrix) |
Coupling degree (M)1 | Molecules per column (mol)2 | Binding sites per molecule3 | Amount eluted IgG (mg) | Eluted IgG per molecule (mg/mol)4 | Eluted IgG per binding site (mg/mol)5 | ||
|---|---|---|---|---|---|---|---|---|
| ZCaTriCys | 4.5 | 2.0 * 10−7 | 3 | 3.6 | 1.8 * 107 | 6.0 * 106 | ||
| ZCaTetraCys | 3.2 | 1.0 * 10−7 | 4 | 3.1 | 3.1 * 107 | 7.8 *106 | ||
1 Coupling degree (mg/ml)/Molecular weight.2 Coupling degree (M)*Column volume.3 Number of binding domains per multimer.4 Amount eluted IgG/Molecules per column.5 Amount eluted IgG/(Molecules per column*Binding sites per molecule).
To further investigate the difference in binding capacity between ZCaTriCys and ZCaTetraCys, breakthrough curves were generated at a residence time of 5 min using human IgG1 (trastuzumab) at 1 mg/ml (Figure 2). From these experiments, a greater distinction between the two ligands could be made. The DBC at 10% breakthrough could be estimated as 15 mg antibody/ml resin for ZCaTriCys while the ZCaTetraCys resin captured as much as 35 mg antibody/ml resin, making the tetrameric ZCa the most promising ligand.
Figure 2.
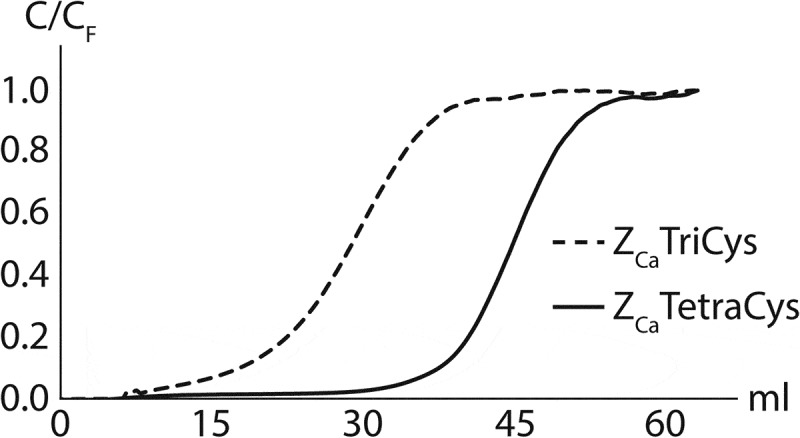
Breakthrough curves on columns coupled with ZCaTriCys (dashed) and ZCaTetraCys (solid) at a residence time of 5 min. Trastuzumab (IgG1) was loaded at a concentration of 1 mg/ml. ZCaTetraCys displays significantly higher dynamic binding capacity compared to ZCaTriCys.
Purifications using tetrameric ZCa
Since ZCaTetraCys could capture the most IgG, both per molecule and per binding site, and exhibited a remarkable DBC for a non-optimized resin, the conditions for eluting human polyclonal antibodies from the ZCaTetraCys column were further examined. Calcium-dependent elution with 100 mM EDTA at pH 5.5 resulted in an almost identical elution curve to acidic elution at pH 3.3 (Figure 3(a)). Elution at pH 5.5 in the presence of calcium gave a minor peak with barely any eluted antibody.
Figure 3.
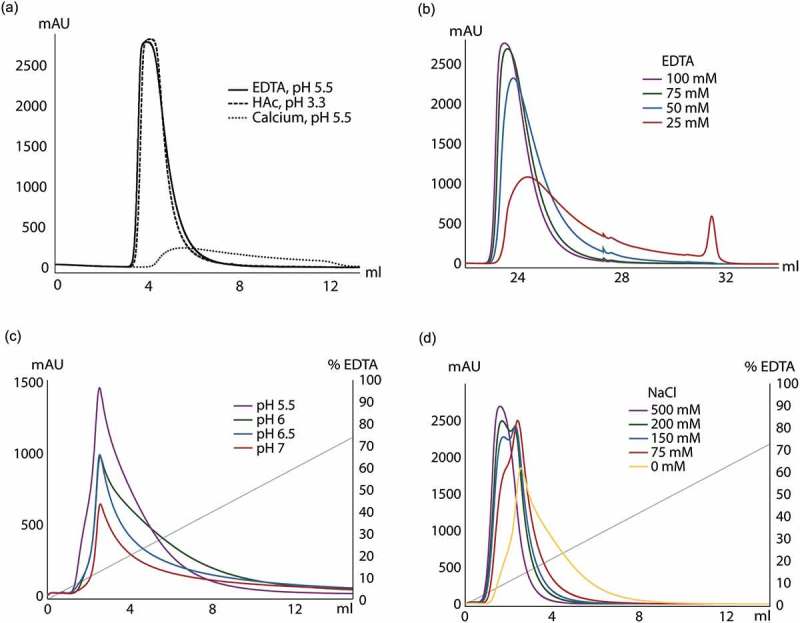
Investigation of elution buffer composition for elution of human polyclonal IgG on a ZCaTetraCys column. (a) IgG was eluted using three different elution buffers: 0.3 M HAc at pH 3.3, 100 mM EDTA at pH 5.5 and 1 mM CaCl2 at pH 5.5. Elution with EDTA at pH 5.5 (solid) gives a very similar elution peak as acidic elution with HAc (dashed), whereas almost no antibody could be eluted at pH 5.5 with calcium present (dotted). (b) Elution using different concentrations of EDTA at pH 5.5 are overlaid: 25 mM (red), 50 mM (blue), 75 mM (green) and 100 mM (purple). Lowering the EDTA concentration to 50 mM or less significantly altered the elution profile. (c) Overlay of gradient elutions between 0–200 mM EDTA at different pH (5.5 in purple, 6 in green, 6.5 in blue and 7 in red) with the gradient profile in gray. This data clearly shows that pH 5.5 renders a more favorable elution profile while also giving the largest amount of product. (d) Gradient elution using 0–200 mM EDTA (gray) at different sodium chloride concentrations: 75 mM (red), 150 mM (blue), 200 mM (green) and 500 mM (purple). The addition of sodium chloride to the elution buffer leads to elution at lower EDTA concentrations as well as more concentrated elution peaks compared to elution with only EDTA (yellow).
Different concentrations of EDTA in the elution buffer were tested to investigate the minimum EDTA concentration that could be used for complete IgG elution, from 25 mM to 100 mM (Figure 3(b)). When eluting human polyclonal IgG with 25 mM EDTA, a very broad, tailing peak was observed with a small, additional peak following. Significant amounts of antibody were retained on the column and could subsequently be eluted at low pH. Also, at 50 mM, the elution profile differed compared to at 100 mM and part of the antibody was retained on the column. A concentration of 75 mM EDTA was needed for elution of all bound antibody, which differs considerably from purifications on a column with monomeric ZCa where a concentration as low as 10 mM was enough for complete elution (Fig. S2a, b).
ZCa has repeatedly been shown to release all bound antibody with EDTA at pH 5.5, but higher pH values of the elution buffer are also desirable. Here, pH values closer to neutral pH (6, 6.5 and 7) were explored by utilizing a gradient elution of EDTA from 0 mM to 200 mM to elute human polyclonal IgG from a ZCaTetraCys column (Figure 3(c)). Elution at increasing pH above pH 5.5 resulted in much lower and broader peaks with increasing amounts of antibody retained on the columns after elution. Still, the percentage of antibody that was eluted from the column compared to that eluted at pH 5.5 was 90%, 75% and 50% at pH 6, 6.5 and 7, respectively. Elution at higher pH values on a ZCa monomer column, on the other hand, generated elution profiles that were fairly similar to pH 5.5, although at a higher EDTA concentration, with 90%, 80% and 75% eluted antibody at pH 6, 6.5 and 7, respectively, compared to pH 5.5 (Fig. S2c). Alternative elution buffer reagents were explored by utilizing a ZCa monomer column. Elution at both pH 5.5 and higher pH was investigated, but no significant improvement over current elution conditions were detected. EDTA was exchanged for ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) (Fig. S3a), a metal chelator that is similar to EDTA, and citrate, which is also known for chelating calcium ions, was added to the EDTA elution buffer at pH 6.5 (Fig. S3b), both performing equal to EDTA alone.
In another attempt to improve the elution conditions, the role of conductivity was investigated by the addition of sodium chloride to the elution buffer in a gradient elution of 0–200 mM EDTA (Figure 3(d)). The elution profile was significantly improved by addition of 75 mM sodium chloride, and further increase in concentration of sodium chloride led to sharper elution peaks, yielding a more concentrated product. Interestingly, a slight separation between the different antibodies in the polyclonal pool, with two overlapping peaks, could be observed at a sodium chloride concentration of ~ 150–200 mM.
To explore possible variations in elution behavior between different subclasses of IgG, purifications of mAbs were conducted on the ZCaTetraCys column. Monoclonal IgG1, IgG2 and IgG4 were added to the column and eluted using a gradient of 0–200 mM EDTA at pH 5.5. A comparison of the elution curves of the mAbs is shown in Figure 4(a). The elution profiles of IgG2 and IgG4 at pH 5.5 are nearly identical, while IgG1 is eluted at a higher concentration of EDTA, exhibiting a somewhat broader peak. This diverging behavior of IgG1 is reflected in Figure 4(b), where a gradient of 0–200 mM EDTA at pH 7 was used for elution of the mAbs. Here, IgG1 shows no defined peak with practically no antibody eluted, whereas all captured IgG2 and IgG4 could be successfully eluted at pH 7, with no residual antibody on the column when lowering the pH to 3.3 after EDTA elution (data not shown).
Figure 4.
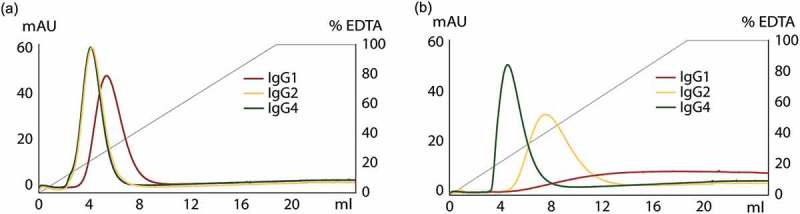
Chromatograms from purifications of mAbs on the ZCaTetraCys column using different pH. (a) Elution of human IgG1 (red), IgG2 (yellow) and IgG4 (green) using a gradient of 0–200 mM EDTA (gray) at pH 5.5. IgG1 required a higher EDTA concentration than IgG2 and IgG4 to be eluted. (b) Elution of IgG1 (red), IgG2 (yellow) and IgG4 (green) using a gradient of 0–200 mM EDTA (gray) at pH 7. All bound IgG2 and IgG4 could be eluted at a neutral pH whereas most IgG1 was retained on the column.
We also tested ZCaTetraCys’ ability to selectively capture IgG from a Chinese hamster ovary (CHO) cell supernatant containing recombinantly produced human IgG1 (HPC4). As seen in Figure 5(a), an elution peak with bound and subsequently eluted protein could be observed. The eluate was shown to contain pure IgG by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western Blot analysis, and the flow through was completely devoid of IgG, indicating high antibody recovery (Figure 5(b)). For analysis of the host cell protein (HCP) and DNA impurity levels in eluted fractions containing purified antibody, trastuzumab was captured from a CHO cell culture supernatant by injection of equal amounts on ZCaTetraCys as well as MabSelect SuRe and MabSelect PrismA. The elution pools were analyzed for HCP and DNA levels, which were normalized to antibody content, and log reduction values (LRVs) were calculated (Table 2). ZCaTetraCys demonstrated a similar LRV (2.3) to MabSelect SuRe (2.5) and MabSelect PrismA (2.4) for HCP content. Regarding DNA levels, the measured LRVs were also satisfactory, although a slightly higher DNA level could be detected in the IgG fraction that was eluted from ZCaTetraCys (LRV 3.2) compared to MabSelect SuRe and MabSelect PrismA (LRVs 4.4 and 5.7, respectively).
Figure 5.
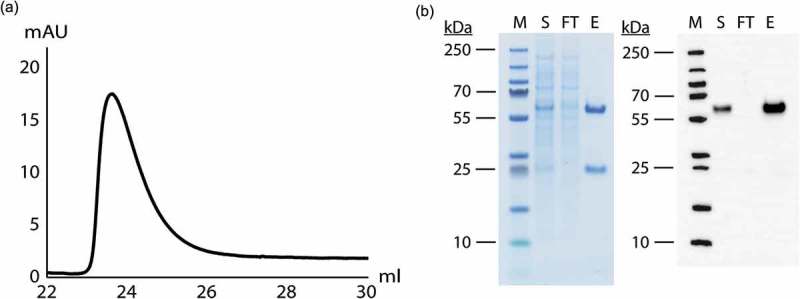
CHO cell culture supernatant containing recombinantly produced IgG1 (HPC4) was successfully purified on the ZCaTetraCys column. (a) A chromatogram displaying elution of the captured IgG with 100 mM EDTA at pH 5.5. (b) SDS-PAGE and Western Blot analysis of the supernatant (S), the flow through (FT) and the eluate (E) next to a molecular weight marker (M) with bands at 10, 15, 25, 35, 55, 70, 100, 130 and 250 kDa, showing that recombinantly produced IgG can be successfully captured on the ZCaTetraCys column and no antibody can be detected in the flow through by Western Blot analysis.
Table 2.
HCP and DNA content in a CHO cell culture supernatant and eluted fractions after purification of trastuzumab on columns with ZCaTetraCys, MabSelect SuRe and MabSelect PrismA, including the log reduction values (LRVs). ZCaTetraCys demonstrates comparable HCP LRVs to MabSelect SuRe and MabSelect PrismA and a sufficient DNA LRV.
| Sample HCP | Elution HCP | LRV | Sample DNA | Elution DNA | LRV | |
|---|---|---|---|---|---|---|
| (ng/mg mAb) | (ng/mg mAb) | HCP | (pg/mg mAb) | (pg/mg mAb) | DNA | |
| ZCaTetraCys | 7.2 * 102 | 2.3 | 1.2 * 103 | 3.2 | ||
| MabSelect SuRe | 1.6 * 105 | 5.2 * 102 | 2.5 | 1.9 * 106 | 81 | 4.4 |
| MabSelect PrismA | 5.9 * 102 | 2.4 | 4.0 | 5.7 |
Discussion
In this work, we designed a calcium-dependent purification matrix and developed a corresponding protocol for purification of IgG. The purification matrix is based on the previously developed protein domain ZCa, with calcium-dependent binding to IgG.18 To achieve a high binding capacity of the matrix, multimeric versions of ZCa (ZCaDiCys, ZCaTriCys and ZCaTetraCys), and a control monomeric version (ZCaMonoCys), were constructed with unique C-terminal cysteines to ensure site-directed coupling of the ligands, and coupled to individual purification columns. While ZCaMonoCys and the multimeric variants were all able to release the captured antibodies using EDTA (pH 5.5), the IgG purifications revealed considerable differences in binding performance between the monomer and the multimeric constructs (Figure 1(a)). With higher-order multimers of ZCa, more antibody could be captured, which was confirmed by SPR analysis (Figure 1(b)). Interestingly, the SPR data showed only a small difference in binding performance between the mono- and dimer, as well as between the tri- and tetramer. This might occur because one additional domain cannot provide an extra IgG binding site due to intra-ligand steric hindrance, but merely increases the distance from the solid phase, which leads to increased accessibility of the ligands for the IgG molecules. The second additional domain of ZCaTriCys may possibly accommodate another IgG molecule, in addition to being even more accessible due to the increased length, whereas the third additional domain of ZCaTetraCys again increases the accessibility rather than adding another binding site compared to the trimer. In comparing ZCaTriCys and ZCaTetraCys on HiTrap™ columns, we found that ZCaTetraCys is able to capture more IgG both per full length molecule and per binding site (Table 1). Furthermore, ZCaTetraCys displayed an astonishing DBC at 10% breakthrough, comparable to commercially available Protein A resins,21 and highly superior to ZCaTriCys despite the higher ligand density of the ZCaTriCys column (Figure 2). The higher binding capacity of ZCaTetraCys is likely a consequence of more functional domains that are accessible for capture of IgG molecules and more space to allow simultaneous binding of additional molecules.
Based on these promising results where multimerization has had a great effect on binding capacity, even higher order multimers would possibly increase the binding capacity further. Other alternatives that might increase the accessibility include insertion of a spacer arm between the matrix and the ligand or addition of longer linker sequences between the domains of the multimeric proteins to ensure that each domain is able to bind to one IgG molecule. However, linkers could pose a threat to the multimers by being sensitive to degradation. Introduction of a longer linker region would therefore require careful evaluation of linker length and composition.22
In order to further examine ZCaTetraCys and identify the optimal conditions for antibody purification using ZCa, purifications of IgG were conducted on a column with ZCaTetraCys immobilized via thiol coupling. Elution from the ZCaTetraCys column at pH 3.3 or with EDTA at pH 5.5 gives strikingly similar elution curves (Figure 3(a)), which differs from previous experiments with a column with a monomeric ZCa immobilized via amine coupling, where EDTA elution resulted in a lower and broader peak.18 The difference in coupling chemistries used to immobilize the monomeric and tetrameric variant, which were randomly and site-specifically immobilized, respectively, could potentially explain these results.
To optimize the purification protocol, lower concentrations of EDTA in the elution buffer were examined. For elution of all captured antibody from the tetrameric column, a much higher concentration of EDTA was needed, 75 mM (Figure 3(b)) compared to 10 mM for the monomeric version (Fig. S2b). Increasing the binding performance of ZCaTetraCys thus required more EDTA in the purification process. This might be explained by the avidity of the multimerized ligand, which in this case requires higher EDTA concentrations to break the interactions.
We examined whether elution of captured human polyclonal antibodies could be done at neutral pH or slightly below to pose less risk to antibodies or Fc-fusion proteins that are sensitive even to pH 5.5, but this did not result in complete elution, neither from ZCaTetraCys (Figure 3(c)) nor the monomeric ZCa (Fig. S2c). At pH 6, 90% of all captured antibody could be eluted from both columns with rapidly decreasing fractions closer to pH 7. The elution profiles from the two columns differed at higher pH, with ZCaTetraCys showing lower peaks and substantial peak tailing. This is probably also a consequence of the avidity effect of the multimeric ligand, which requires the disruption of several possible binding interactions simultaneously to allow elution of the antibodies. In an attempt to facilitate elution at higher pH values, the metal chelator citrate23 was added to the EDTA elution buffer at pH 6.5 for purification on a ZCa monomer column. However, the combination of the two metal chelators did not increase the amount of eluted antibody compared to only EDTA (Fig. S3b). EGTA was also evaluated as an alternative elution agent due to its reportedly higher selectivity for calcium ions and lower affinity for magnesium than EDTA,24,25 but no improvement was observed and the elution peak was not as sharp as for EDTA (Fig. S3a). These results, in combination with the substantially higher costs of using EGTA over EDTA, renders EGTA an unfavorable alternative for elution.
The effect of sodium chloride on the elution properties at pH 5.5 was studied for ZCaTetraCys (Figure 3(d)). Considerably sharper elution peaks at low EDTA concentrations were found for concentrations of 75 mM NaCl and higher, using an EDTA gradient, with a reduction of elution volume by half for 500 mM NaCl compared to an elution buffer without sodium chloride. However, precipitation of the EDTA buffer with 500 mM NaCl could be observed with time, and lower sodium chloride concentrations should preferably be used. Due to the weakening effect of sodium chloride on the affinity interaction of the IgG molecules and ZCaTetraCys, further studies of the use of sodium chloride together with EDTA could be of interest to improve elution at higher pH values than 5.5. Elution of IgG at concentrations of 150 and 200 mM NaCl resulted in an elution profile with two overlapping peaks, providing separation of the IgG subclasses at this particular ionic strength, which could be useful for the preparation of individual IgG subclasses if further optimized.
Human monoclonal antibodies of subclasses IgG1, IgG2 and IgG4, which are commonly used for therapeutics, were purified using ZCaTetraCys. For elution of IgG1 at pH 5.5, a higher concentration of EDTA was required than for IgG2 and IgG4, which displayed very similar elution behaviors (Figure 4(a)). When increasing the pH of the elution buffer, almost no IgG1 was released from the ZCaTetraCys column (Figure 4(b)). Remarkably, both IgG2 and IgG4 could be fully eluted at pH 7. Taken together, these results indicate that a large part of the polyclonal antibody sample that could not be eluted with EDTA at pH 7 (Figure 3(c)) could be IgG1. This reasoning is supported by the fact that Protein A has been shown to have the highest affinity for IgG1 of the three subclasses.26
Before industrial use of an optimized ZCa matrix, ligand stability in terms of leaching of the Protein A ligand must be investigated. The addition of EDTA, which functions as a metalloprotease inhibitor, to the elution buffer has previously been shown to reduce the co-elution of Protein A fragments with the antibody for several commercial matrices.27 Measurement of leached Protein A ligand was not included in this study. This needs to be investigated in further studies of the matrix. For use of ZCaTetraCys in large-scale production processes, the virus inactivation also has to be taken into account since this is frequently accomplished by a low pH hold directly after Protein A elution. This method could, for instance, seamlessly be replaced by solvent/detergent treatment.28–30
The ZCaTetraCys matrix has been shown to perform equal to or better than commercial Protein A matrices in important aspects which include highly selective capture of IgG from complex feed solutions (Figure 5(b)). With regards to residual HCP levels after purification of trastuzumab from a CHO cell supernatant, ZCaTetraCys demonstrated a similar LRV to MabSelect SuRe and MabSelect PrismA. The reduction of DNA levels was well within the acceptable range for commercially used Protein A matrices (Table 2).31
This study provides an extensive evaluation of the optimal purification parameters for use of the novel calcium-dependent domain ZCa to purify antibodies. In particular, ZCaTetraCys provided the most efficient antibody purification with the highest DBC of the chromatography resins evaluated here, as well as good antibody recovery. We found that the most successful way of eluting IgG from ZCaTetraCys is by a combination of EDTA and sodium chloride, which yields a very concentrated product. Elution should occur at pH 5.5 to ensure dissociation of all bound polyclonal IgG. Interestingly, the optimal pH for elution was shown to depend on the subclass of the IgG. Antibodies of subclasses IgG2 and IgG4 can successfully be released from ZCaTetraCys at neutral pH while antibodies of the IgG1 subclass require a lower pH of 5.5 for complete elution. Thus, for the purification of Fc-fusion proteins, the origin of the Fc portion used will affect the pH at which that particular protein can be efficiently eluted. Our results indicate that this protein purification matrix is a novel and valuable tool for mild antibody and Fc-fusion protein purification.
Materials and methods
Enzymes were purchased from New England Biolabs (NEB), unless stated otherwise. PCR reactions were performed using Phusion DNA polymerase (ThermoFisher, M0530L) and PCR screens using DreamTaq DNA polymerase (ThermoFisher, EP0702). PCR reactions were purified using the QIAquick PCR Purification Kit (Qiagen, 28106) and gel extractions were performed using the QIAquick Gel Extraction Kit (Qiagen, 28706). Enzymes and purification kits were used according to the instructions of the manufacturer.
Cloning
Genes encoding monomer and multimeric variants (ZCaMonoCys, ZCaDiCys, ZCaTriCys and ZCaTetraCys) of ZCa constructed head-to-tail and equipped with a C-terminal cysteine were purchased from Invitrogen. The 3ʹ and 5ʹ ends were designed to be unique sequences to allow for amplification of each entire gene. A standard PCR protocol was followed using a forward and reverse primer (TAG Copenhagen A/S) matching the unique sequences of the 3ʹ end and the 5ʹ end of the genes and with overhangs containing a NcoI and XhoI restriction site, respectively. The purified PCR products of the multimeric variants were run on and extracted from a 2% GTG agarose gel. The ZCa constructs and the vector pET-45b(+) (Novagen, 71327), containing a T7 promoter and ampicillin (Amp) resistance, were cleaved with NcoI (R3193S) and XhoI (R0146S) restriction enzymes and gel extracted. Cleaved, purified constructs and cleaved, gel extracted vector were ligated with a 3x excess of construct using T4 DNA ligase (M0202L). The ligation mixtures were chemically transformed to Escherichia coli strain Top10. To verify the correct sequences of the inserted constructs, Sanger Cycle Sequencing was conducted at Microsynth Seqlab (Göttingen, Germany) after a PCR-screen using standard T7 sequencing primers.
Protein production and purification
Individual colonies carrying the sequence-verified constructs were grown in tryptic soy broth (30 g/l (Merck, 1054590500), TSB) with Amp (100 µg/ml) and incubated at 150 rpm, 37°C overnight (ON). Plasmid DNA was isolated using the QIAprep Spin Miniprep Kit (Qiagen, 27106). The purified pET-45b(+) containing the ZCa constructs were transformed to BL21 (DE3) cells (Novagen®, Merck Biosciences, 69450) by heat shock and plated on Amp-plates (200 µg/ml), incubated at 37°C ON. Single colonies were inoculated in TSB+Y (TSB supplemented with 5 g/l yeast extract (Merck, 1037530500)) with Amp (100 µg/ml) and incubated at 150 rpm, 37°C ON.
Cultivations were initiated by the addition of 1 ml ON culture to 100 ml TSB+Y with Amp (100 µg/ml) and CaCl2 (1 mM) and incubated at 150 rpm, 37°C until OD600 reached 0.5–1. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to induce protein expression. The cultures were grown at 150 rpm, 25°C ON and harvested by centrifugation at 2400 × g, 4°C for 8 min. The pellets were resuspended in 20 ml 1xTBST-C (1xTBS (50 mM Tris, 150 mM NaCl), 0.05% (v/v) Tween20, 1 mM CaCl2, pH 7.5) followed by sonication using a Vibra-cell (Sonics, CT, USA) at 40% amplitude with 1.0/1.0 s pulsing for 1 min and 30 s. Centrifugation at 10 000 × g, 4°C for 20 min was performed to remove cell debris.
The proteins were purified from the cell lysates by affinity chromatography using columns packed with 5 ml IgG-sepharose 6 Fast Flow (GE healthcare, 17096902). The columns were pulsed with 20 ml sterile H2O, 20 ml 1xTBST-C and 20 ml 0.3 M HAc (pH 3.3) followed by equilibration with 50 ml 1xTBST-C. The cell lysates were added and the columns were washed with 75 ml 1xTBST-C and 50 ml 5 mM NH4Ac with 1 mM CaCl2 (pH 5.5–6). The bound product was eluted with 0.3 M HAc (pH 3.3) in 15 × 1 ml fractions followed by regeneration with 20 ml 0.3 M HAc and washing with 20 ml 1xTBST-C and 20 ml 1xTBST (20% EtOH). The fractions containing protein, determined by absorbance at 280 nm, were freeze dried using an Automatic Environmental SpeedVac System (Savant, MA, USA) or a CoolSafe freeze dryer (ScanVac, Sweden) and dissolved in 1xTBS with 1 mM CaCl2. A bicinchoninic acid assay (BCA), using bovine serum albumin as a standard, was used to determine the protein concentrations (Thermo Scientific, 23225).
Protein analysis
Mass spectrometry
The molecular weights of the proteins were determined using matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS). Since the proteins contain cysteines, they were alkalized before mass spectrometry analysis in order to examine the monomeric forms of the proteins. A 1:1 mixture of alkalized protein sample and α-cyano-4-hydroxycinnamic acid (5 mg/ml, Bruker, 8255344) or sinapinic acid (5 mg/ml, Sigma-Aldrich, 85429-5G), depending on the expected size of the protein, was spotted on a MALDI-target and analyzed with a 4800 MALDI time-of-flight (TOF)/TOF Analyzer (Applied Biosystems, CA, USA).
Electrospray ionization MS was also used to determine the molecular weights, after desalination of the alkalized protein samples on a C4 column (μ-Precolumn Cartridge Acclaim PepMap™ 300 C4, 5 µm, 300Å, 300 μm i.d. 5 mm, Dionex, 163591). Analysis of the samples was conducted with an Agilent 6520 Accurate-Mass Q-TOF LC/MS system (Agilent Technologies, CA, USA).
SDS-PAGE
Protein purity was determined by SDS-PAGE. The samples were mixed with reducing buffer to a final concentration of 20 mM Tris-HCl, 1 mM EDTA, 88 mM SDS, 720 mM β-mercaptoethanol and 17% glycerol. Samples were heated to 95°C for 5 min and loaded on a NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen, NP0323BOX) together with the marker LMW-SDS marker kit (GE Healthcare, 17-0446-01) after which it was run in 1xMES (50 mM MES, 50 mM TRIS, 1 mM EDTA, 0.1% SDS, pH 7.3) running buffer at 200 V, 4°C for 30 min. Afterward, washing of the gel in deionized water was repeated 3 × 5 min, GelCode® Blue (Thermo Scientific, 24596) was added to stain the gel for 1 hour followed by destaining in deionized water ON.
Western Blot
The gel from the SDS-PAGE was transferred to a 0.45 μm Invitrolon™ PVDF/Filter Paper Sandwich (Novex, LC2007) at 30 V, for 90 min in 1xTransfer buffer (25 mM Bicine, 25 mM BisTris, 1 mM EDTA, 10% ethanol, pH 7.2). PageRuler™ Plus Prestained Protein Ladder (Thermo Scientific, 26620) was used as the molecular weight marker. Blocking buffer (5% w/v dry milk) was added to the blot and incubated at 4°C ON after which it was washed, once quickly, followed by 5 × 5 min in 20 mL 1xTBST. The blot was incubated with horseradish peroxidase (HRP)-coupled goat anti-human antibody (ThermoFisher, A18805) at a 1:5000 dilution in 5 ml blocking buffer at room temperature for 1 h and washed as previously described with 1xTBST. The blot was developed using Immobilon™ Western Chemiluminescent HRP Substrate (Merck, WBKLS0500).
Surface plasmon resonance
The ZCa variants were analyzed on a Biacore 3000 instrument (GE Healthcare). The proteins were coupled to a CM5 chip by thiol‐based chemistry via their C‐terminal cysteines, using a Thiol Coupling Kit (GE Healthcare, BR100557) according to the provided immobilization protocol for ligand thiol coupling. The different ZCa variants were diluted in 0.1 M sodium acetate pH 4 and 1 mM CaCl2 and injected onto the chip with the aim to reach an immobilization level of 25 RU for ZCaMonoCys, 50 RU for ZCaDiCys, 75 RU for ZCaTriCys and 100 RU for ZCaTetraCys in order to couple a comparable number of molecules of each protein. ZCaDiCys was, due to dimerization, reduced before immobilization with dithiothreitol to a final concentration of 20 mM and buffer exchanged to 0.1 M sodium acetate, 1 M NaCl pH 4 and 1 mM CaCl2. 1xHBST (1xHBS (20 mM HEPES, 150 mM NaCl, pH 7), 0.05% Tween20) with 1 mM CaCl2 was used as running buffer. Human polyclonal IgG was diluted to 500 nM in 1xHBST with 1 mM CaCl2 and triplicates were injected at 50 µl/min with 300 s association and 600 s dissociation. Regeneration of the surfaces was conducted using 10 mM HCl.
Purification using ZCa ligands
Resin coupling
In house coupling of resin for bench purification
All ZCa variants were covalently and irreversibly immobilized to columns via the C-terminal cysteines using the SulfoLink® Coupling Resin (Thermo Scientific, 20401), which is activated with iodoacetyl groups. Approximately 70 nmole of each protein was coupled to individual 1 ml SulfoLink® Coupling Resin beds according to the manufacturer’s instructions. The coupling reaction took place in the dark and the resins were packed in columns. To make the sulfhydryl groups accessible for coupling, tris(2-carboxyethyl)phosphine (pH 7, Sigma, C4706-10G) was added to the protein dilutions prior to coupling, according to instructions, and incubated at room temperature for 30 min. 1xTBST-C (20% ethanol) was used as storage buffer. Coupling degrees were determined by comparing the absorbance at 280 nm of the protein added to the coupling resin in the purification columns with the absorbance of the noncoupled flow through collected from the columns.
Coupling and pre-packing of resin for ÄKTA purification
ZCaTriCys and ZCaTetraCys were buffer exchanged to 1xHBS with 1 mM CaCl2 and coupled to a solid support and packed in 1 ml HiTrap™ columns (GE Healthcare) by GE healthcare. Their coupling degrees were determined through amino acid analysis.
Antibody purification
Bench purification
Purifications on the packed SulfoLink® columns were performed in triplicates, using a large excess of human polyclonal IgG (25 mg/ml) in 2 ml 1xTBST-C. Pulsing of the columns with 4 ml 0.3 M HAc (pH 3.3) and 4 ml 1xTBST-C twice was followed by addition of the polyclonal IgG. The columns were washed with 10 ml 1xTBST-C and elution buffer (100 mM NH4Ac, 100 mM EDTA, pH 5.5) was added in fractions of 10 × 300 µl. Washing was done with 3 ml 5 mM NH4Ac, 1 mM CaCl2 (pH 5–6) before a second elution with 0.3 M HAc in 10 × 300 µl fractions. The columns were regenerated with 4 ml 0.3 M HAc, 4 ml 1xTBST-C and 4 ml 1xTBST (20% EtOH). Eluted protein amounts were determined based on absorbance measurements at 280 nm. A two-tailed t test was used to determine the statistical significance.
ÄKTA purification
The ZCa variants coupled to a solid support and packed in HiTrap™ columns, as well as the monomeric ZCa, without a C-terminal cysteine, as developed in Kanje et al.,18 and coupled to HiTrap™ NHS-Activated HP columns (GE Healthcare, 17071601), were analyzed here. Purifications were also run on 1 ml HiTrap™ columns with MabSelect SuRe and MabSelect PrismA resins for comparison. The columns were connected to an ÄKTA Pure chromatography system (GE Healthcare) for purification of human polyclonal IgG, human monoclonal IgG1 (rituximab, Roche), IgG2 (Department of Biotechnology, University of Natural Resources and Life Sciences, Vienna) and IgG4 (BioInvent), as well as two different CHO cell supernatants from cells producing human IgG1 (HPC4 antibody, (produced in-house) or trastuzumab (GE Healthcare)), at a flow rate of 1 ml/min. The ZCa-based columns were pulsed with 6 column volumes (c.v.) 1xTBST-C and 6 c.v. of the elution buffers found in Table S1. Gradient elutions were performed for 20 c.v. (Table S1). Next, the columns were equilibrated with 13 c.v. 1xTBST-C and sample injected through a 2-ml sample loop. Washing was conducted with 13 c.v. 1xTBST-C and 5 c.v. 5 mM NH4Ac, 1 mM CaCl2, (pH 5–6) followed by elution with 6 c.v. of elution buffer. For purification at different NaCl concentrations, the same NaCl concentrations that were used in the elution buffers (Table S1) were used in the purification buffers above. The eluate was collected in fractions of 500 µl. The columns were regenerated using 3 c.v. elution buffer, 6 c.v. 1xTBST-C and 2 c.v. 1xTBST-C (20% ethanol). Purifications for HCP and DNA analysis of ZCaTetraCys, MabSelect SuRe and MabSelect PrismA eluates were conducted with equal volumes of CHO cell supernatant containing trastuzumab. Elution was performed with 100 mM EDTA (pH 5.5, Table S1) from ZCaTetraCys and according to the corresponding application note for MabSelect SuRe and MabSelect PrismA.
Dynamic binding capacity experiments
ZCaTriCys and ZCaTetraCys columns were connected to an ÄKTA pcc system (GE Healthcare). Pure trastuzumab was loaded at a concentration of 1 mg/ml in equilibration buffer with a flow rate of 0.2 ml/min. The column equilibration, wash and regeneration were performed as previously described but using 20 c.v. elution buffer. The protein concentration based on the obtained absorbance at 280 nm was plotted as a fraction of the inlet protein concentration against the volume of loaded sample, and DBC at 10% breakthrough was determined using the UNICORN DBC calculation extension.
HCP and DNA analysis
Detection of CHO HCP and DNA in cell culture supernatant and eluates was conducted by Sobi. The HCP and DNA content in the eluates was normalized to the antibody content. Log reduction values (LRV) were calculated by the common logarithm ratio of HCP or DNA content in the unpurified sample to the HCP or DNA content in the eluates.
Funding Statement
This work was supported by VINNOVA [2016-05181];
Abbreviations
- Amp
Ampicillin
- CHO
Chinese hamster ovary
- DBC
Dynamic binding capacity
- EDTA
Ethylenediaminetetraacetic acid
- EGTA
Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- Fc
Fragment crystallizable
- HAc
Acetic acid
- HCP
Host cell protein
- IgG
Immunoglobulin G
- LRV
Log reduction value
- mAb
Monoclonal antibody
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SPR
Surface Plasmon Resonance
Acknowledgments
This work was funded by Vinnova (the AdBIOPRO excellence center). The authors would like to extend their gratitude to Björn Norén at GE Healthcare for the directed coupling of the purification molecules on a matrix. GE Healthcare has also provided the CHO cell supernatant containing trastuzumab and supplied the ÄKTA pcc system that was used for the breakthrough curves. Ulrika Regnéll and Sajit Thottathil Oommen at Sobi kindly performed the HCP and DNA analysis. The authors also want to thank Alois Jungbauer’s lab at the Department of Biotechnology, University of Natural Resources and Life Sciences, Vienna and BioInvent for generously sharing monoclonal IgG2 and IgG4, respectively.
Disclosure of Potential Conflicts of interest
SK and SH have filed a patent application regarding the novel ZCa domain.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kaplon H, Reichert JM.. Antibodies to watch in 2019. MAbs. 2019;11(2):219–38. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gagnon P. Technology trends in antibody purification. J Chromatogr A. 2012;1221:57–70. doi: 10.1016/j.chroma.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 3.Hober S, Nord K, Linhult M. Protein A chromatography for antibody purification. J Chromatogr B Anal Technol Biomed Life Sci. 2007;848:40–47. doi: 10.1016/j.jchromb.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 4.Boström T, Nilvebrant J, Hober S. Purification systems based on bacterial surface proteins In: Ahmad R, editor. Protein purification. INTECH Open Access Publisher; 2012. p. 89–136. [Google Scholar]
- 5.Urmann M, Graalfs H, Joehnck M, Jacob LR, Frech C. Cation-exchange chromatography of monoclonal antibodies: characterization of a novel stationary phase designed for production-scale purification. MAbs. 2010;2:395–404. doi: 10.4161/mabs.2.5.13089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao Y, Ibraheem A, Conley L, Cecchini D, Ghose S. Evaluation of high-capacity cation exchange chromatography for direct capture of monoclonal antibodies from high-titer cell culture processes. Biotechnol Bioeng. 2014;111:1354–64. doi: 10.1002/bit.25192. [DOI] [PubMed] [Google Scholar]
- 7.Joucla G, Le Sénéchal C, Bégorre M, Garbay B, Santarelli X, Cabanne C. Cation exchange versus multimodal cation exchange resins for antibody capture from CHO supernatants: identification of contaminating host cell proteins by mass spectrometry. J Chromatogr B Anal Technol Biomed Life Sci. 2013;942–943:126–33. doi: 10.1016/j.jchromb.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 8.DiLeo M, Ley A, Nixon AE, Chen J. Choices of capture chromatography technology in antibody manufacturing processes. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1068–1069:136–48. doi: 10.1016/j.jchromb.2017.09.050. [DOI] [PubMed] [Google Scholar]
- 9.McDonald P, Victa C, Carter-Franklin JN, Fahrner R. Selective antibody precipitation using polyelectrolytes: A novel approach to the purification of monoclonal antibodies. Biotechnol Bioeng. 2009;102:1141–51. doi: 10.1002/bit.v102:4. [DOI] [PubMed] [Google Scholar]
- 10.Brodsky Y, Zhang C, Yigzaw Y, Vedantham G. Caprylic acid precipitation method for impurity reduction: an alternative to conventional chromatography for monoclonal antibody purification. Biotechnol Bioeng. 2012;109:2589–98. doi: 10.1002/bit.v109.10. [DOI] [PubMed] [Google Scholar]
- 11.Tscheliessnig A, Satzer P, Hammerschmidt N, Schulz H, Helk B, Jungbauer A. Ethanol precipitation for purification of recombinant antibodies. J Biotechnol. 2014;188:17–28. doi: 10.1016/j.jbiotec.2014.07.436. [DOI] [PubMed] [Google Scholar]
- 12.Kelley B, Blank G, Lee A. Downstream processing of monoclonal antibodies: current practices and future opportunities In: Gottschalk U, editor. Process scale purif antibodies. Hoboken, NJ: Wiley; 2017. p. 1–21. [Google Scholar]
- 13.Paul AJ, Schwab K, Hesse F. Direct analysis of mAb aggregates in mammalian cell culture supernatant. BMC Biotechnol. 2014;14:99. doi: 10.1186/s12896-014-0099-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vázquez-Rey M, Lang DA. Aggregates in monoclonal antibody manufacturing processes. Biotechnol Bioeng. 2011;108:1494–508. doi: 10.1002/bit.23155. [DOI] [PubMed] [Google Scholar]
- 15.Shukla AA, Gupta P, Han X. Protein aggregation kinetics during Protein A chromatography. Case study for an Fc fusion protein. J Chromatogr A. 2007;1171:22–28. doi: 10.1016/j.chroma.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 16.Liu B, Guo H, Xu J, Qin T, Xu L, Zhang J, Guo Q, Zhang D, Qian W, Li B, et al. Acid-induced aggregation propensity of nivolumab is dependent on the Fc. MAbs. 2016;8:1107–17. doi: 10.1080/19420862.2016.1196521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazzer AR, Perraud X, Halley J, O’Hara J, Bracewell DG. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. J Chromatogr A. 2015;1415:83–90. doi: 10.1016/j.chroma.2015.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanje S, Venskutonytė R, Scheffel J, Nilvebrant J, Lindkvist-Petersson K, Hober S. Protein engineering allows for mild affinity-based elution of therapeutic antibodies. J Mol Biol. 2018;430:3427–38. doi: 10.1016/j.jmb.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 19.Nilsson B, Moks T, Jansson B, Abrahmsén L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlén M. A synthetic IgG-binding domain based on staphylococcal protein a. Protein Eng Des Sel. 1987;1:107–13. doi: 10.1093/protein/1.2.107. [DOI] [PubMed] [Google Scholar]
- 20.Müller E, Vajda J. Routes to improve binding capacities of affinity resins demonstrated for Protein A chromatography. J Chromatogr B Anal Technol Biomed Life Sci. 2016;1021:159–68. doi: 10.1016/j.jchromb.2016.01.036. [DOI] [PubMed] [Google Scholar]
- 21.Pabst TM, Thai J, Hunter AK. Evaluation of recent protein. A stationary phase innovations for capture of biotherapeutics. J Chromatogr A. 2018;1554:45–60. doi: 10.1016/j.chroma.2018.03.060. [DOI] [PubMed] [Google Scholar]
- 22.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65:1357–69. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glusker JP. Citrate conformation and chelation: enzymatic Implications. Acc Chem Res. 1980;13:345–52. doi: 10.1021/ar50154a002. [DOI] [Google Scholar]
- 24.Arena G, Musumeci S, Purrello R, Sammartano S. Calcium- and magnesium-EDTA complexes. Stability constants and their dependence on temperature and ionic strength. Thermochim Acta. 1983;61:129–38. doi: 10.1016/0040-6031(83)80309-8. [DOI] [Google Scholar]
- 25.Smith GL, Miller DJ. Potentiometric measurements of stoichiometric and apparent affinity constants of EGTA for protons and divalent ions including calcium. Biochim Biophys Acta. 1985;839:287–99. doi: 10.1016/0304-4165(85)90011-X. [DOI] [PubMed] [Google Scholar]
- 26.Reis KJ, Ayoub EM, Boyle MD. Streptococcal Fc receptors. I. Isolation and partial characterization of the receptor from a group C streptococcus. J Immunol. 1984;132:3091–97. [PubMed] [Google Scholar]
- 27.Carter-Franklin JN, Victa C, McDonald P, Fahrner R. Fragments of protein A eluted during protein A affinity chromatography. J Chromatogr A. 2007;1163:105–11. doi: 10.1016/j.chroma.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 28.Nur I, Bar L. Method of the inactivation of viruses by a solvent-detergent combination and nanofiltration, (US6468733B2). 2002.
- 29.Roberts PL. Virus inactivation by solvent/detergent treatment using Triton X-100 in a high purity factor VIII. Biologicals. 2008;36:330–35. doi: 10.1016/j.biologicals.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Roberts PL, Lloyd D, Marshall PJ. Virus inactivation in a factor VIII/VWF concentrate treated using a solvent/detergent procedure based on polysorbate 20. Biologicals. 2009;37:26–31. doi: 10.1016/j.biologicals.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Butler MD, Kluck B, Bentley T. DNA spike studies for demonstrating improved clearance on chromatographic media. J Chromatogr A. 2009;1216:6938–45. doi: 10.1016/j.chroma.2008.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.