

ABSTRACT
Unfolded protein response (UPRs) directs adaption or apoptosis depending on the severity of endoplasmic-reticulum (ER) stress. We found that apoptotic signaling by inositol requiring enzyme 1α (IRE1α), a transducer of UPRs, is suppressed by mitochondrial ubiquitin ligase MITOL/MARCH5 on ER-mitochondria contacts, suggesting that mitochondria regulate cell fate under ER stress.
KEYWORDS: Word, ER stress, UPR, IRE1α, MITOL/MARCH5, ER-mitochondria contact site
Introduction
The endoplasmic reticulum (ER) is the largest cellular organelle involved in the synthesis and folding of membrane/secretory proteins, lipid metabolism, and calcium storage. Therefore, adaptive ER response is a pivotal intracellular signaling for cell adaptation to intra – and extracellular environmental changes. Various physiological and pathological environmental changes, such as hypoxia, low-nutrition, oxidative stress, and increase in protein synthesis, promote adaptive ER reactions through the activation of ER stress responses, also known as the unfolded protein response (UPR) signaling. UPR signaling is initiated by three ER-sensor proteins, protein kinase R-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1α (IRE1α) (also known gene name EIF2AK3, ATF6, ERN1, respectively).1 When the ER perceives the imbalance in ER homeostasis, the UPR signaling mediates cell survival and adaptation to eliminate ER stress. However, when ER stress becomes severe and irreversible, UPR signaling triggers cell death.
The molecular understanding of the balance between cell adaptation and cell death in response to environmental changes may contribute to clarify the pathogenesis of various diseases caused by micro-environmental changes. Upregulation of ER functions has also been reported to allow tumor cells to adapt to cell-intrinsic and cell-extrinsic stresses. Several studies have indicated that tumor cells maintain their high proliferative capacity even in the severe stress conditions, such as hypoxia and acidic extracellular pH, depending on the adaptive UPR signaling.2 Importantly, although robust ER stress is observed in most tumor cells, they escape cell fate toward apoptosis triggered by apoptotic switch of UPR signaling.2
We have previously identified mitochondrial ubiquitin ligase (MITOL, also known gene name MARCH5), which is integrated into the mitochondrial outer membrane, and demonstrated that MITOL plays critical roles in mitochondrial homeostasis and signaling.3,4 We noticed that MITOL is abundantly localized in the proximal junction between the ER and mitochondria, suggesting a possibility that MITOL may regulate cellular signalings provoked not only from mitochondria but also from the ER.
Results
Recently, we have identified IRE1α, one of the three UPR sensor proteins, as a novel substrate for MITOL. IRE1α is a unique protein integrated in the ER membrane with dual catalytic activity, kinase and RNase.5 Under mild ER stress, IRE1α undergoes dimerization to induce cell adaptation by X-box-binding protein-1 (XBP1) mRNA splicing. In contrast, under severe or prolonged ER stress, IRE1α forms self-oligomers and cleaves various mRNA/miRNAs, including pro-survival mRNA and anti-apoptotic miRNA, thereby leading to cell death. We have demonstrated that MITOL directly ubiquitinates K481 of IRE1α at ER-mitochondria membrane contact sites. MITOL inhibits excessive oligomerization of IRE1α, thus, prevents IRE1α-dependent decay of mRNA/miRNAs by adding K63-linked polyubiquitin chain, which regulates the activity of substrate but not relates to proteasome degradation, to IRE1α. In MITOL deficient cells under ER stress, a drastic cell death was triggered by activation of IRE1α branch of the UPR signaling. Furthermore, overexpression of the IRE1α mutant K481R, unrelated to the ubiquitination by MITOL, phenocopied the enhanced alternative IRE1α signaling observed in MITOL deficient cells, such as excessive IRE1α oligomerization and remarkably decay of anti-apoptotic miRNA. Thus, our findings indicate that the ubiquitination of IRE1α K481 is one of the key regulatory mechanisms directing IRE1α signaling to apoptosis (Figure 1).
Figure 1.
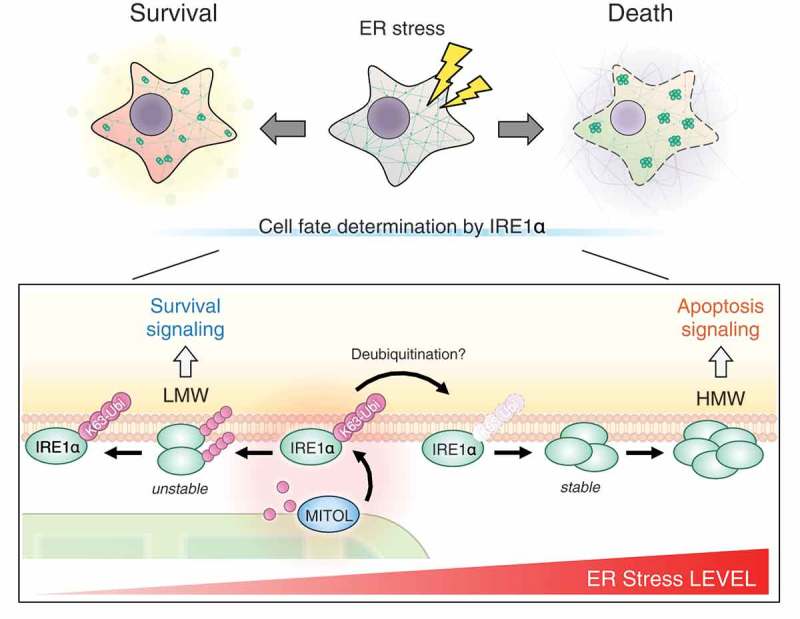
Mitochondrial ubiquitin ligase (MITOL)-dependent mitochondrial retrograde signaling in inositol requiring enzyme 1α (IRE1α) regulation. Under basal conditions or low level of endoplasmic-reticulum (ER) stress, MITOL interacts with and ubiquitinates IRE1α by the K63-linked polyubiquitin chain (K63-Ubi), preventing excessive oligomerization and continuous activation of IRE1α. This regulatory machinery mediated by MITOL contributes to cell survival under the permissible range of ER stress. Conversely, under irremediable ER stress, the IRE1α ubiquitination by MITOL is decreased by unclear mechanisms, thereby triggering IRE1α hyper-activation with mRNA/miRNA decay and apoptosis. LMW: low molecular weight, HMW: high molecular weight.
Discussion
Among the three UPR branches, the IRE1α-XBP1 pathway is strongly associated with tumor development.6,7 Cells inside solid tumors were exposed by stress conditions including hypoxia, glucose starvation, and an increase of protein synthesis for high proliferation. These stresses have reported to activate UPR signaling; thus, intrinsic/extrinsic environmental changes in tumor cells are considered to result in the activation of UPR signaling. Several studies have showed that XBP1 activation is pivotal for environmental adaptation and cell survival in tumor cells. In contrast, the IRE1α-dependent apoptosis is scarcely induced in the tumor cells in spite of its obvious activation of the IRE1α-XBP1 pathway. In a recent study, we found that a key mitochondrial regulator MITOL inhibits the apoptotic switch of ER-sensor IRE1α by K63-linked polyubiquitin chain at ER-mitochondria contact sites.5 Previous reports have indicated that MITOL is highly expressed in breast cancers, which caused efficient and excessive tumor proliferation.8 Therefore, there may be cases that hyperactivation of MITOL suppresses cell death by inhibiting excessive oligomerization of IRE1α in tumor cells. On the other hand, mutations of the MITOL/MARCH5 gene, which is identified in somatic endometrial cancers, lead to loss of catalytic-activity of MITOL.9 The dualistic role of MITOL in tumor development may be resulted from the multi-functional aspects of MITOL. Therefore, in some cases such as endometrial cancers, MITOL dysfunction and subsequent decrease in mitochondrial homeostasis may cause metabolic changes within the cancer and induce the Warburg effect, contributing to cancer development. Other proteins, such as BIM (also known gene name BCL2L11), have also been reported to regulate excessive IRE1α oligomerization by direct interaction, which is important for apoptotic switch of IRE1α signaling. Thus, in cases including endometrial cancers, other factors may inhibit apoptotic switch of IRE1α signaling independently of MITOL. Further understating of apoptotic switch of IRE1α signaling is required to develop a novel therapy targeting UPR signal switch.
Recently, ER stress has also reported to regulate anti-tumor immunity. The tumor microenvironmental conditions, such as secreted factors and acidosis, triggers ER stress in tumor-infiltrating immune cells. IRE1α contributes to secretion of inflammatory cytokines as well as proteostasis in the ER via XBP1 up-regulation, although excessive and sustained activation of IRE1α impairs cellular functions of the infiltrating immune cells, thereby leading to tumor immune evasion. These facts suggest that optimal ER stress response in tumor-infiltrating immune cells is required for anti-tumor immunity. Interestingly, we found that the K63-linked ubiquitination of IRE1α by MITOL is decreased following prolonged ER stress (Figure 1). Thus, under chronic ER stress, UPR signaling by IRE1α may shift to the alternative UPR signaling causing cell death by reduction of its inhibitory ubiquitination. Dissociation of K63-linked polyubiquitin chain from IRE1α is a faster than IRE1α protein turnover, suggesting that ubiquitin-specific proteases (USPs) or deubiquitinase-enzymes (DUBs) also determines the apoptotic switch of IRE1α via releasing of the polyubiquitin chain. Thus, the identification of unknown USP/DUBs involving in the IRE1α regulation may lead to more efficient drug development to control the apoptotic switch of IRE1α signaling, and the drug(s) may also be available for cancer treatment and immunotherapy.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Ron D, Walter P.. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:1–3. doi: 10.1038/nrm2199 PMID: 17565364. [DOI] [PubMed] [Google Scholar]
- 2.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692–706. doi: 10.1016/j.cell.2016.12.004 PMID: 28187289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S. Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem. 2014;155:273–279. doi: 10.1093/jb/mvu016 PMID: 24616159. [DOI] [PubMed] [Google Scholar]
- 4.Xu S, Cherok E, Das S, Li S, Roelofs BA, Ge SX, Polster BM, Boyman L, Lederer WJ, Wang C, et al. Mitochondrial E3 ubiquitin ligase MARCH5 controls mitochondrial fission and cell sensitivity to stress-induced apoptosis through regulation of MiD49 protein. Mol Biol Cell. 2016;27:349–359. doi: 10.1091/mbc.E15-09-0678 PMID: 26564796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Nagashima S, Shiiba I, Uda A, Tokuyama T, Ito N, Fukuda T, Matsushita N, Ishido S, Iwawaki T, et al. MITOL prevents ER stress-induced apoptosis by IRE1alpha ubiquitylation at ER-mitochondria contact sites. Embo J. 2019:e100999. doi: 10.15252/embj.2018100999 PMID: 31196886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119 PMID: 24670641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Logue SE, McGrath EP, Cleary P, Greene S, Mnich K, Almanza A, Chevet E, Dwyer RM, Oommen A, Legembre P, et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat Commun. 2018;9:3267. doi: 10.1038/s41467-018-05763-8 PMID: 30111846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang H, Peng S, Dong Y, Yang X, Yang P, Yang L, Yang BBao G. MARCH5 overexpression contributes to tumor growth and metastasis and associates with poor survival in breast cancer. Cancer Manag Res. 2019;11:201–215. doi: 10.2147/cmar.s190694 PMID: 30636894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SH, Park YY, Yoo YS, Cho H. Self-clearance mechanism of mitochondrial E3 ligase MARCH5 contributes to mitochondria quality control. Febs J. 2016;283:294–304. doi: 10.1111/febs.13568 PMID: 26476016. [DOI] [PubMed] [Google Scholar]