

ABSTRACT
The analysis of monoclonal antibodies (mAbs) by a middle-down mass spectrometry (MS) approach is a growing field that attracts the attention of many researchers and biopharmaceutical companies. Usually, liquid fractionation techniques are used to separate mAbs polypeptides chains before MS analysis. Gas-phase fractionation techniques such as high-field asymmetric waveform ion mobility spectrometry (FAIMS) can replace liquid-based separations and reduce both analysis time and cost. Here, we present a rapid FAIMS tandem MS method capable of characterizing the polypeptide sequence of mAbs light and heavy chains in an unprecedented, easy, and fast fashion. This new method uses commercially available instruments and takes ~24 min, which is 40-60% faster than regular liquid chromatography-MS/MS analysis, to acquire fragmentation data using different dissociation methods.
KEYWORDS: FAIMS, gas-phase fractionation, light and heavy chains, mAb, middle-down analysis
Monoclonal antibodies (mAbs) based on immunoglobulins G (IgGs) are multichain glycoproteins with an approximate molecular weight of 150 kDa. They consist of four polypeptide chains: two light chains (Lc) of ∼25 kDa each and two heavy chains (Hc) of ∼50 kDa each, all linked together by disulfide bonds.1 MAbs are the fastest growing class of human therapeutics; the U.S. Food and Drug Administration has already approved over 80 therapeutic mAbs since 2018,2 and they are being used in the treatment of diseases related to cardiovascular, respiratory, hematology, kidney, immunology, and oncology systems.3,4 As with any other therapeutic, molecular structure of mAbs must be well characterized to ensure drug safety, efficiency, batch-to-batch consistency, and stability over time.5
Bottom-up, middle-down, and top-down mass spectrometry (MS) strategies are often used to fulfill and streamline molecular characterization requirements.1 Bottom-up approaches digest the mAb into peptides before analysis,6 top-down MS methods analyze intact molecules,7-10 and middle-down procedures are performed by measuring the mass and subsequent fragmenting of large pieces or subunits from mAbs (typically 25–50 kDa) that are more suitable for state-of-the-art liquid chromatography tandem MS (LC–MS/MS) methods and techniques.11-16 The subunits or parts of the polypeptide chain can be obtained through the chemical reduction of disulfide bonds, yielding free Lc and Hc,17 or by using a specific enzymatic proteolysis (i.e., digestion with IdeS or IdeZ) that usually generates F(ab’)2 (∼100 kDa) and Fc (∼50 kDa) pieces.18 The S-S bonds in these pieces can be further reduced, resulting in three ∼25 kDa subunits: one Lc and two portions of Hc named Fc/2 and Fd.19
Even the simplest mixture of two unique polypeptide chains obtainable via disulfide bond reduction (without proteolysis), Lc and Hc, cannot be analyzed effectively by MS without some type of front-end fractionation that can isolate or partially separate the chains. Both the overlap of their charge state envelopes and ionization suppression effects can lower their spectral signal-to-noise ratio (S/N), particularly for the larger Hc, and could result in co-isolation during fragmentation experiments (tandem MS or MS/MS). Fractionation methods are typically based on liquid chromatography performed using reverse phase (RP),17 size exclusion,20,21 or ion exchange22,23 columns. Each LC-MS/MS run takes several minutes, only one fragmentation method is used per run generally, and multiple injections are needed to maximize sequence coverage.14 Furthermore, liquid chromatography instruments add expense, with elevated operational costs depending on the columns and extent of method development required. Front-end separation based on liquid chromatography also raises issues of sample carryover, contamination, and potential sample losses via irreversible adsorption.
In sharp contrast to liquid-phase separation, a new high-field asymmetric waveform ion mobility spectrometry (FAIMS) device with cylindrical electrodes and improved transmission that allows rapid and effective gas-phase separation of molecules after they are ionized and prior to the mass spectrometer entrance has recently been described.24,25 FAIMS devices operate at atmospheric pressure, conducting ions among an inner and an outer electrode under a high or low electric field.26 The electric fields are generated from an asymmetric waveform, and the ion separation is based on ion differential mobility. Ions with moderate to no difference in mobility between the high and low fields are conducted to the MS, while ions with a large mobility difference are deflected to the electrodes and are lost. Adding a direct current (DC) voltage, termed the compensation voltage (CV), to the system alters ion trajectories, which provides a compensation for the drift of specific ions and permits those ions to pass through the electrodes and be analyzed.27,28 Ions above ~30 kDa show a strong increase of mobility at high fields, which agrees with expected ion dipole alignment and expands the useful FAIMS separation power.29-31 Changes in CV will thus favor different groups of ions and function as a filter, as observed for peptides32 and proteins.29 The use of gas-phase fractionation can exclude the liquid separation step for middle-down mAb analysis, making it fast, less expensive, and more robust.
Herein, we present a novel method for fast middle-down analysis of reduced Lc and Hc chains of mAbs without liquid-phase pre-fractionation, using only FAIMS Pro™ coupled to an Orbitrap Eclipse™ Tribrid™ mass spectrometer (FAIMS-MS/MS) capable of performing multiple ion fragmentation techniques.
NIST Monoclonal Antibody Reference Material 8671 (National Institute of Standards and Technology), 300 µg, was denatured in 6 M guanidium chloride and reduced using 30 mM tris (2-carboxyethyl) phosphine hydrochloride for 90 min at 37°C under agitation. The sample was desalted using 3 kDa molecular weight cutoff Amicon Ultra-0.5 (Millipore Sigma), and the solution was buffer exchanged to LC/MS grade water (Fisher Scientific) for over 10 cycles in a refrigerated centrifuge at 4°C applying 8,000 × g. Reduced polypeptide chains were resuspended in 50% acetonitrile containing 0.4% of formic acid for an ~2 µM final concentration.
The polypeptide mixture was sprayed using a Nanospray Flex™ static source (Thermo Fisher Scientific) and medium-length borosilicate-coated emitters (Thermo Fisher Scientific) on an Orbitrap Eclipse™ Tribrid™ (Thermo Fisher Scientific) equipped with FAIMS Pro™ (Thermo Fisher Scientific). The spray voltage was set between 1.5 and 2.5 kV, and, for MS experiments, the acquisition range was set between m/z 800–2,000 using a resolving power of 7,500 (at m/z 200), 2 microscans/spectrum, an average of 20 spectra, 100 ms of maximum injection time, automatic gain control (AGC) target of 5 × 105 charges, and source collision-induced dissociation (CID) of 10 V; the instrument was operated in “intact protein” mode (pressure of 2 mTorr). FAIMS Pro™ was run at an N2 carrier gas flow of 0 L/min, an inner electrode temperature of 100°C, an outer electrode temperature of 100°C, a dispersion voltage (DV) of −5,000 V for the asymmetric waveform, an entrance plate voltage of 250 V, and the CV ranged from −30 to +40 V in 10 V steps.
MS/MS experiments for Lc were carried out at FAIMS CV −20 V using the following parameters: 2 microscans/spectrum, resolving power 120,000 (at m/z 200), source CID of 10 V, isolation window of 20 Th centered at m/z 1,102 (charge state +21), maximum injection time of 100 ms, AGC target of 5 × 106 charges, acquisition range set between m/z 500–2,000, average of 20 spectra, and ion transfer tube temperature set at 300°C. For higher-energy collisional dissociation (HCD) normalized collision energy (NCE) was set at 10% for charge state 1, CID NCE at 25% for charge state 1, ultraviolet photodissociation (UVPD) was performed using a 213 nm laser and irradiation time of 70 ms, electron transfer dissociation (ETD) AGC target value for fluoranthene radical anions was set to 7–8 × 105 charges, default charge state of 3, and ETD reaction times of 5 and 7 ms.
MS/MS experiments for Hc were carried out at FAIMS CV +40 V using the following parameters: 1 microscan/spectrum, resolving power 60,000 of (at m/z 200), source CID of 20 V, isolation window of 100 Th centered at m/z 1,000 or 1,200 (charge states +49-53 or +41-44, respectively), maximum injection time of 100 ms, AGC target of 5 × 106 charges, acquisition range set between m/z 500–2,000, average of 20 spectra, ion transfer tube temperature set at 300°C; for HCD, NCE was set at 15% for charge state 1; CID was performed using 10% of NCE for charge state 1; ETD AGC target value for fluoranthene radical anions was set to 7–8 × 105 charges, default charge state of 3, using ETD reaction times of 2, 5, and 10 ms; electron-transfer/higher-energy collision dissociation (EThcD) was performed using the same ETD conditions with 2 ms reaction time and 15% of NCE for HCD at charge state 1.
The data were analyzed using Thermo XCalibur Qual Browser v4.0.27.10 (Thermo Fisher Scientific) to average spectra and manipulate raw files. Mass deconvolution of low-resolution data was performed on UniDec GUI v3.0.0.33 Fragmentation peak fitting and annotation were performed with TDValidator v1.014 (Proteinaceous) using the following parameters: signal-to-noise (S/N) cutoff of 20 for Lc and 2 for Hc data, max ppm tolerance 20 ppm, sub ppm tolerance 15 ppm, cluster tolerance 0.35, minimum score of 0.7, charge range 1–15, and distribution generator Mercury7. S/N was calculated according to the expression: S/N = (S – B)/(N – B) where S is the signal intensity, B is the spectrum baseline intensity, and N is the spectrum noise intensity.
A mixture of reduced Lc and Hc, from NIST mAb reference material, was directly sprayed into an Orbitrap Eclipse™ Tribrid™ mass spectrometer; the obtained MS spectrum was dominated by the charge state envelope of the Lc with the Hc charge state distribution below 20% of relative intensity (Figure 1a). The constitutional ratio between Lc and Hc for NIST mAb is 1:1. However, S/N for Lc and Hc were 119 (m/z 1,052, charge state +22) and 14.4 (m/z 1,043, charge state +49), respectively, and signal intensities were not equivalent. The lower signal and S/N observed for Hc is due to the signal splitting into more charge states than Lc, the presence of more proteoforms (glycosylation), and differences in ionization efficiency. The deconvoluted spectrum (Figure 1b) confirms the abundance discrepancy, with Lc representing ~90% of peak intensities and Hc only ~10%. Looking at Hc proteoforms, G1F was the most intense glycoform observed, G0F represented one-third of its intensity, and no other Hc glycoform masses were detected.
Figure 1.
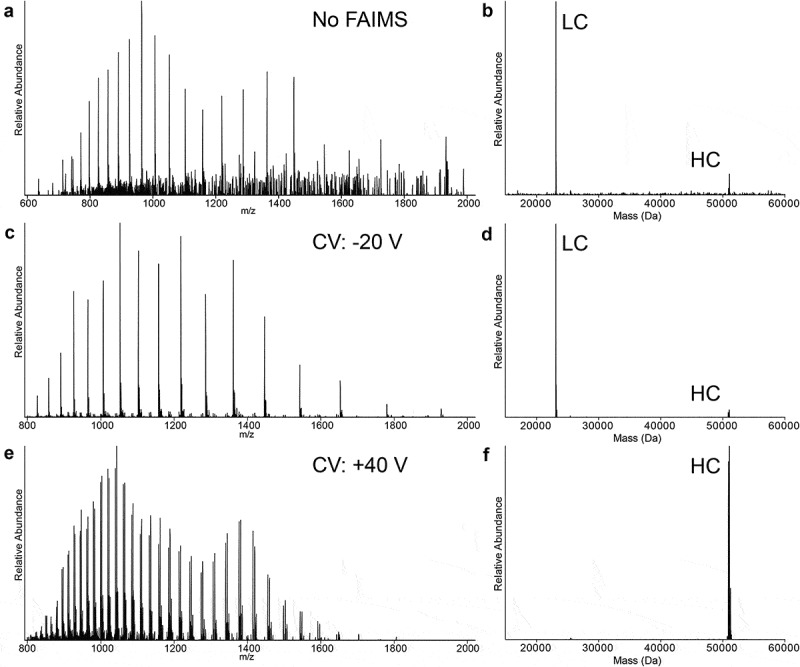
Mass spectra of reduced mAb (obtained from NIST) for two different settings of the FAIMS compensation voltage (CV) compared to no FAIMS. Spectra from direct injection (no FAIMS) of the mixture containing reduced light (Lc) and heavy (Hc) chains with the resultant spectra displayed in the m/z domain (a) and the deconvoluted spectra in the mass domain (b). Spectra obtained using FAIMS and applying −20 V of CV and displayed in the m/z domain (c) or deconvoluted into the mass domain (d) spectra. Spectra obtained using FAIMS and applying +40 V of CV and displayed in the m/z domain (e), or deconvoluted into the mass domain (f).
Spraying the same mixture into the instrument equipped with FAIMS Pro™ and stepping the compensation voltage (CV) by 10 V from −30 V to +40 V allowed the gas-phase separation of Lc and Hc based on their ion mobilities across the generated high-field asymmetric waveforms (Supplementary Figures S1 and S2). Acquisition was performed for 3 min in each CV 10 V steps, and the −20 V step presented the clearest spectrum for Lc (Figure 1c). The most abundant charge state showed an S/N of 2,390 for m/z 1,052 (charge state +22), which is equal to an increase of 20-fold compared to the no FAIMS spectrum. Moreover, the Hc charge distribution observed was below 3% of total ion relative intensity. The deconvoluted spectra are composed of 95.2% of Lc and 4.8% of Hc based on peak intensities (Figure 1d), and the Lc observed average mass of 23,127.31 Da is −11.2 ppm off the theoretical mass 23,127.57 Da. Adducts of sodium (22 Da), guanidine (60 Da), their combination, and the double guanidine species were also observed, as well as a 162 Da mass shift that corresponds to the addition of one hexose (Hex) to Lc (Figure 2a). The non-enzymatic, but covalent, adduction of a Hex sugar molecule on a lysine or on a protein N-terminus is called “glycation”, and 22 distinct glycated peptides for the Lc of NIST mAb have been reported.34 Lc+Hex are reported as trace level post-translational modifications (PTMs).35 Glycated Lc proteoforms corresponded to ~4% of the total ion intensity of the non-modified Lc, indicating that FAIMS-MS is suitable to detect low stoichiometry mAbs PTMs.
Figure 2.
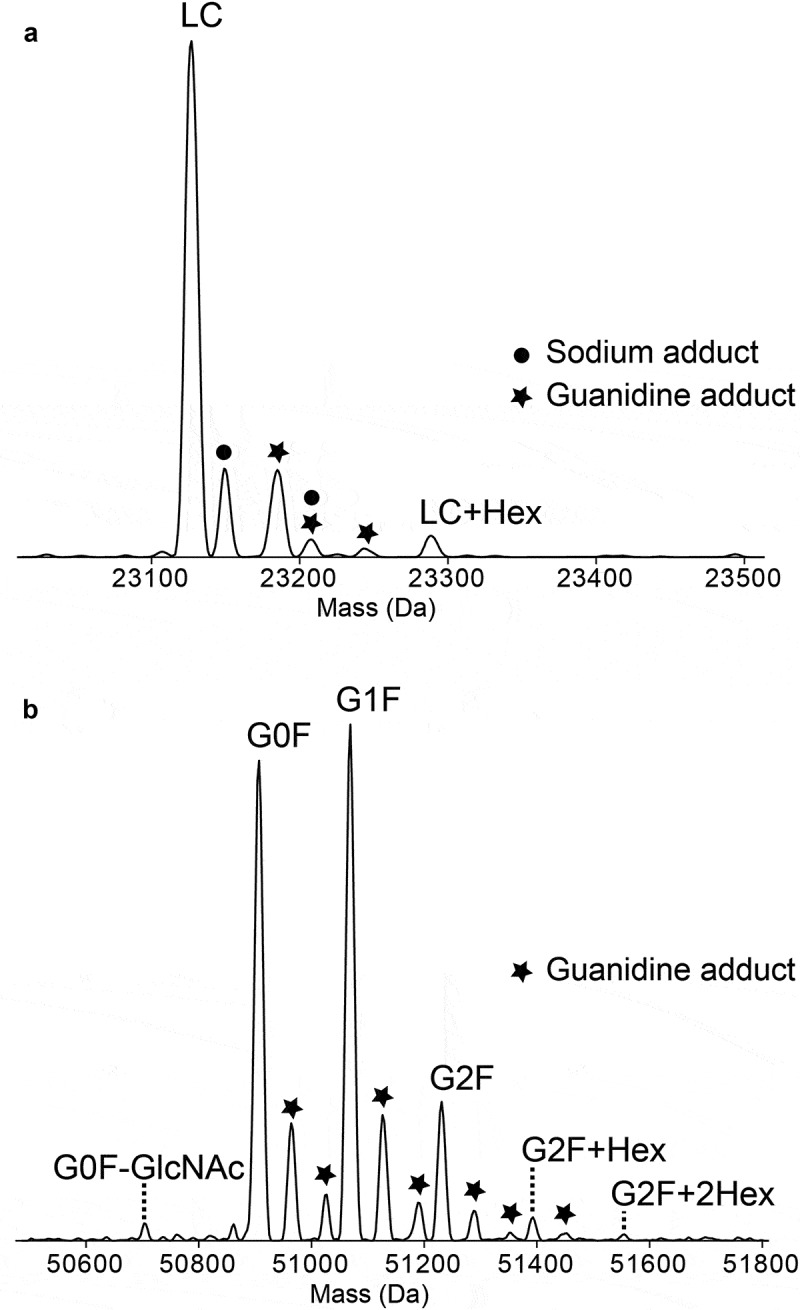
Expanded section of deconvoluted spectrum of light (Lc) and heavy (Hc) chains. Spectra obtained applying −20 V and +40 V of CV were deconvoluted and zoomed-in to show Lc (a) and Hc (b) proteoforms. Sodium adducts (+22 Da) are represented by a circle (●) and guanidine adducts (+60 Da) by a star (★). Addition of hexose (+162 Da) is characterized by +Hex and the loss of N-acetylglucosamine (−203 Da) as –GlcNAc.
The CV of +40 V generated the cleanest spectrum for Hc (Figure 1e) with no detection of Lc and an S/N of 477 for m/z 1,043 (charge state +49), which corresponds to a 34-fold increase compared to the spectrum recorded without FAIMS. The observed deconvoluted average mass for Hc G1F was 51,068.59 Da, −10.4 ppm off from the theoretical mass 51,069.12 Da. In the deconvoluted spectrum only Hc was detected (Figure 1f), and it presented 6 glycoforms (Figure 2b): G0F-GlcNAc, G0F, G1F, G2F, G2F+Hex, and G2F+2Hex. Their relative abundances based on ion intensity were 1.5%, 40.5%, 43.6%, 11.8%, 2.1%, and 0.5%, respectively. Single- and double-guanidine (60 Da) adducts were also observed. The observed ratios of the glycoforms are in accordance with the literature findings.36,37 The low-abundant proteoforms G0F-GIcNAc, G2F+Hex, and G2F+2Hex were only observed using FAIMS, and their detection and accurate relative quantitation are a good indicator of the heightened sensitivity afforded by FAIMS-MS.
Preventing the overlap of Lc and Hc charge state envelopes in the m/z space permits the isolation of a single charge state of each polypeptide chain or the isolation of multiple charge states of the same polypeptide chain without co-isolation with the other chain. Avoiding co-isolation is important for successful fragmentation of a single species, generating non-chimeric spectra, which are subsequently easier to correctly interpret and match against the polypeptide primary chain sequence. A single charge state of the Lc chain was quadrupole-isolated using a 20 Th window and fragmented by HCD, CID, ETD, and UVPD. MS/MS data were acquired for only 3 min in each dissociation method, and Lc graphical fragmentation maps were generated using TDValidator (Supplementary Figure S3). The combination of all the quickly acquired dissociation maps (resulting from 12 min total instrument time) yielded 65% sequence coverage for Lc (Figure 3a). For Hc, multiple charge states were isolated in a 100 Th isolation window, and ions were fragmented using HCD, CID, ETD, and EThcD (Supplementary Figure S4). Acquisition time was comparable to the one needed for the Lc, and the combination of fragmentation maps resulted in 34.4% of sequence coverage for the most abundant proteoform Hc G1F (Figure 3b). No changes in the fragmentation maps were observed considering G0F or G2F glycans.
Figure 3.

Graphical fragmentation maps obtained from middle-down tandem MS of light (Lc) and heavy (Hc) chains. Cumulative fragmentation maps obtained from HCD, CID, ETD, and UVPD dissociation methods applied on Lc (a) and from HCD, CID, ETD, and EThcD applied to Hc (b). The red brackets represent c- and z- ions, blue brackets b- and y- ions, and green brackets a- and x- ions. The gray rectangle denotes a pyroglutamic acid post-translational modification and the orange rectangle represent the addition of the N-linked glycan G1F mass (the most abundant proteoform observed for the standard mAb obtained from NIST).
FAIMS-MS/MS was capable of separation and analysis of a mixture of reduced Lc and Hc from mAb in an unprecedented, easy, and fast fashion without the use of liquid chromatography or any other liquid fractionation technique. All fragmentation data obtained corresponded to a minimum of four regular LC-MS/MS runs, which would take ~10–15 min each, totaling 40–60 min of analysis not considering loading times, blanks, washes, and standards. The FAIMS-MS/MS method presented here required only 24 min to acquire all fragmentation data on Lc and Hc using 4 different dissociation methods, which is 40-60% faster than regular LC-MS/MS analysis.
In comparing liquid chromatography with FAIMS-based separations of biomolecules for targeted MS analysis, both resolution and analysis time must be considered. The fundamental resolution (Rs) equation for liquid chromatography is:
where n is the number of theoretical plates, α the selectivity factor, and k the retention factor.38 For optimal characterization of mAb chains targeted LC-MS/MS, full separation (high selectivity) between species is desirable, but this is typically associated with narrow elution peaks (high efficiency), limiting the number of MS/MS scans that can be acquired. Under ideal conditions for ultra-high-pressure liquid chromatography, Lc and Hc are separated in about 6–7 min,39 although generally 10–15 min gradients are necessary for higher quality MS/MS datasets to be acquired during peak elution off the column. Further, multiple LC runs are needed to obtain good fragmentation data using different ion-activation techniques.
Conversely, in FAIMS the resolving power is calculated as the ratio of the CV that corresponds to maximal signal for an analyte relative to its full width at half maximum (FWHM) across the CV range:
The maximum resolving power for FAIMS is limited to about 50040 and typically ranges between 30 and 10041,42 depending on the analytes. We were able to completely separate Lc and Hc, as their maxima differ in CV by 60 V. The total time for stepping through the CVs and acquiring intact Lc and Hc mass spectra was only ~20 s, with a total of 3 min of instrument time required for collection of high-quality fragmentation spectra. Considering that 10–15 min are required for regular liquid-chromatography separations, the acquisition of Lc and Hc intact mass spectra using FAIMS offers, at a minimum, ~30-45 times faster run times. Moreover, MS/MS analysis can be performed as long as needed for applications where the species of interest can be resolved via ion mobility.
The FAIMS device used in this study has high transmission efficiency comparable to standard LC-MS/MS analysis.24 The same equipment was successfully used for quantitative LC-FAIMS-MS/MS bottom-up proteomics experiments with similar high-quality quantitative results.32,43 In addition, the use of FAIMS ProTM was shown to lower limits of quantitation by ~3-10 fold for peptide peaks limited by chemical noise.24 Based on previous studies, the FAIMS-based approach presented here has potential to be used in semi- and quantitative experiments with reduced mAbs accessing low abundant mAbs proteoforms and determining their ratios in the sample.
The method presented here should accelerate the assessment of new products (in the context of both research and formulation) and has longer-term prospects for use in MAM-type QC monitoring for mature biopharmaceutical products in the industry. Further improvements in the acquisition routine can make the method even faster for the characterization of mAb and mAb conjugates. In this work, samples were manually sprayed, and data were acquired directly from Tune (instrument controller software). However, it is possible to use automated nanospray or microspray to run FAIMS-MS/MS in a high throughput manner that is far simpler than current LC-MS/MS analyses. The analysis of mAbs is a growing field that attracts the attention of many researchers and biopharmaceutical companies, and the new FAIMS-MS/MS method presented can improve speed, limit artifacts, and reduce costs of middle-down mAb analysis.
Funding Statement
This work was supported by the National Resource for Translational and Developmental Proteomics [Grant P41 GM108569]; University of Oklahoma (US); Sherman Fairchild Foundation.
Abbreviations
| mAb | Monoclonal antibody |
| IgGs | Immunoglobulins G |
| Lc | Light chain |
| Hc | Heavy chain |
| MS | Mass spectrometry |
| LC–MS/MS | Liquid chromatography tandem mass spectrometry |
| S/N | Signal to noise |
| MS/MS | Tandem mass spectrometry |
| RP | Reverse phase |
| FAIMS | High-field asymmetric waveform ion mobility spectrometry |
| DC | Direct current |
| CV | Compensation voltage |
| AGC | Automatic gain control |
| CID | Collision induced dissociation |
| DV | Dispersion Voltage |
| HCD | Higher-energy collisional dissociation |
| NCE | Normalized collision energy |
| UVPD | Ultraviolet photodissociation |
| ETD | Electron transfer dissociation |
| EThcD | Electron-transfer/higher-energy collision dissociation |
| Hex | Hexose |
| PTMs | Post-translational modifications |
| FWHM | Full width at half maximum |
Acknowledgments
This research was carried out in collaboration with the National Resource for Translational and Developmental Proteomics under Grant P41 GM108569 from the National Institute of General Medical Sciences (NLK) and supported by the Sherman Fairchild Foundation. LF would like to thank the University of Oklahoma, Department of Biology, for start-up funds.
Disclosure of potential conflicts of interest
KS and RH are Thermo Fisher Scientific employees, and NLK declares a COI with Proteinaceous.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S.. Characterization of therapeutic antibodies and related products. Anal Chem. 2013;85(2):715–36. doi: 10.1021/ac3032355. [DOI] [PubMed] [Google Scholar]
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11(2):219–38. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol. 2010;10(5):345–52. doi: 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- 4.Singh S, Kumar NK, Dwiwedi P, Charan J, Kaur R, Sidhu P, Chugh VK. Monoclonal antibodies: a review. Curr Clin Pharmacol. 2018;13(2):85–99. doi: 10.2174/1574884712666170809124728. [DOI] [PubMed] [Google Scholar]
- 5.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. Middle-down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal Chem. 2014;86(6):3005–12. doi: 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee R, Adhikary L, Khedkar A, Iyer H. Probing deamidation in therapeutic immunoglobulin gamma (igg1) by ‘bottom-up’ mass spectrometry with electron transfer dissociation. Rapid Commun Mass Sp. 2010;24(7):879–84. doi: 10.1002/rcm.4464. [DOI] [PubMed] [Google Scholar]
- 7.Tsybin YO, Fornelli L, Stoermer C, Luebeck M, Parra J, Nallet S, Wurm FM, Hartmer R. Structural analysis of intact monoclonal antibodies by electron transfer dissociation mass spectrometry. Anal Chem. 2011;83(23):8919–27. doi: 10.1021/ac201293m. [DOI] [PubMed] [Google Scholar]
- 8.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of intact monoclonal antibody igg1 by electron transfer dissociation orbitrap ftms. Mol Cell Proteomics. 2012;11(12):1758–67. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mao Y, Valeja SG, Rouse JC, Hendrickson CL, Marshall AG. Top-down structural analysis of an intact monoclonal antibody by electron capture dissociation-fourier transform ion cyclotron resonance-mass spectrometry. Anal Chem. 2013;85(9):4239–46. doi: 10.1021/ac303525n. [DOI] [PubMed] [Google Scholar]
- 10.Fornelli L, Ayoub D, Aizikov K, Liu XW, Damoc E, Pevzner PA, Makarov A, Beck A, Tsybin YO. Top-down analysis of immunoglobulin g isotypes 1 and 2 with electron transfer dissociation on a high-field orbitrap mass spectrometer. J Proteomics. 2017;159:67–76. doi: 10.1016/j.jprot.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He L, Anderson LC, Barnidge DR, Murray DL, Hendrickson CL, Marshall AG. Analysis of monoclonal antibodies in human serum as a model for clinical monoclonal gammopathy by use of 21 tesla ft-icr top-down and middle-down ms/ms. J Am Soc Mass Spectrom. 2017;28(5):827–38. doi: 10.1007/s13361-017-1602-6. [DOI] [PubMed] [Google Scholar]
- 12.Srzentic K, Fornelli L, Laskay UA, Monod M, Beck A, Ayoub D, Tsybin YO. Advantages of extended bottom-up proteomics using sap9 for analysis of monoclonal antibodies. Anal Chem. 2014;86(19):9945–53. doi: 10.1021/ac502766n. [DOI] [PubMed] [Google Scholar]
- 13.Cotham VC, Brodbelt JS. Characterization of therapeutic monoclonal antibodies at the subunit-level using middle-down 193 nm ultraviolet photodissociation. Anal Chem. 2016;88(7):4004–13. doi: 10.1021/acs.analchem.6b00302. [DOI] [PubMed] [Google Scholar]
- 14.Fornelli L, Srzentic K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, Kelleher NL. Accurate sequence analysis of a monoclonal antibody by top-down and middle-down orbitrap mass spectrometry applying multiple ion activation techniques. Anal Chem. 2018;90(14):8421–29. doi: 10.1021/acs.analchem.8b00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srzentic K, Nagornov KO, Fornelli L, Lobas AA, Ayoub D, Kozhinov AN, Gasilova N, Menin L, Beck A, Gorshkov MV, et al. Multiplexed middle-down mass spectrometry as a method for revealing light and heavy chain connectivity in a monoclonal antibody. Anal Chem. 2018;90(21):12527–35. doi: 10.1021/acs.analchem.8b02398. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Lin Z, Xu Q, Fu C, Zhang Z, Zhang Q, Pritts WA, Ge Y. Comprehensive characterization of monoclonal antibody by fourier transform ion cyclotron resonance mass spectrometry. MAbs. 2019;11(1):106–15. doi: 10.1080/19420862.2018.1525253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rehder DS, Dillon TM, Pipes GD, Bondarenko PV. Reversed-phase liquid chromatography/mass spectrometry analysis of reduced monoclonal antibodies in pharmaceutics. J Chromatogr A. 2006;1102(1–2):164–75. doi: 10.1016/j.chroma.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 18.von Pawel-Rammingen U, Johansson BP, Bjorck L. Ides, a novel streptococcal cysteine proteinase with unique specificity for immunoglobulin g. Embo J. 2002;21(7):1607–15. doi: 10.1093/emboj/21.7.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sjogren J, Olsson F, Beck A. Rapid and improved characterization of therapeutic antibodies and antibody related products using ides digestion and subunit analysis. Analyst. 2016;141(11):3114–25. doi: 10.1039/c6an00071a. [DOI] [PubMed] [Google Scholar]
- 20.Liu H, Gaza-Bulseco G, Chumsae C. Analysis of reduced monoclonal antibodies using size exclusion chromatography coupled with mass spectrometry. J Am Soc Mass Spectrom. 2009;20(12):2258–64. doi: 10.1016/j.jasms.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Hong P, Koza S, Bouvier ES. Size-exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J Liq Chromatogr Relat Technol. 2012;35(20):2923–50. doi: 10.1080/10826076.2012.743724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fekete S, Beck A, Fekete J, Guillarme D. Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, part i: salt gradient approach. J Pharm Biomed Anal. 2015;102:33–44. doi: 10.1016/j.jpba.2014.08.035. [DOI] [PubMed] [Google Scholar]
- 23.Fekete S, Beck A, Veuthey JL, Guillarme D. Ion-exchange chromatography for the characterization of biopharmaceuticals. J Pharm Biomed Anal. 2015;113:43–55. doi: 10.1016/j.jpba.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 24.Prasad S, Belford MW, Dunyach JJ, Purves RW. On an aerodynamic mechanism to enhance ion transmission and sensitivity of faims for nano-electrospray ionization-mass spectrometry. J Am Soc Mass Spectrom. 2014;25(12):2143–53. doi: 10.1007/s13361-014-0995-8. [DOI] [PubMed] [Google Scholar]
- 25.Purves RW, Prasad S, Belford M, Vandenberg A, Dunyach JJ. Optimization of a new aerodynamic cylindrical faims device for small molecule analysis. J Am Soc Mass Spectrom. 2017;28(3):525–38. doi: 10.1007/s13361-016-1587-6. [DOI] [PubMed] [Google Scholar]
- 26.Buryakov IA, Krylov EV, Nazarov EG, Rasulev UK. A new method of separation of multi-atomic ions by mobility at atmospheric-pressure using a high-frequency amplitude-asymmetric strong electric-field. Int J Mass Spectrom. 1993;128(3):143–48. doi: 10.1016/0168-1176(93)87062-W. [DOI] [Google Scholar]
- 27.Guevremont R. High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J Chromatogr A. 2004;1058:3–19. [PubMed] [Google Scholar]
- 28.Cooper HJ. To what extent is faims beneficial in the analysis of proteins? J Am Soc Mass Spectrom. 2016;27(4):566–77. doi: 10.1007/s13361-015-1326-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shvartsburg AA, Bryskiewicz T, Purves RW, Tang K, Guevremont R, Smith RD. Field asymmetric waveform ion mobility spectrometry studies of proteins: dipole alignment in ion mobility spectrometry? J Phys Chem B. 2006;110(43):21966–80. doi: 10.1021/jp062573p. [DOI] [PubMed] [Google Scholar]
- 30.Shvartsburg AA. Ultrahigh-resolution differential ion mobility separations of conformers for proteins above 10 kda: onset of dipole alignment? Anal Chem. 2014;86(21):10608–15. doi: 10.1021/ac502389a. [DOI] [PubMed] [Google Scholar]
- 31.Shvartsburg AA, Andrzejewski R, Entwistle A, Giles R. Ion mobility spectrometry of macromolecules with dipole alignment switchable by varying the gas pressure. Anal Chem. 2019;91:8176–83. doi: 10.1021/acs.analchem.9b00525. [DOI] [PubMed] [Google Scholar]
- 32.Hebert AS, Prasad S, Belford MW, Bailey DJ, McAlister GC, Abbatiello SE, Huguet R, Wouters ER, Dunyach JJ, Brademan DR, et al. Comprehensive single-shot proteomics with faims on a hybrid orbitrap mass spectrometer. Anal Chem. 2018;90(15):9529–37. doi: 10.1021/acs.analchem.8b02233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, Robinson CV. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem. 2015;87(8):4370–76. doi: 10.1021/acs.analchem.5b00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong Q, Liang Y, Yan X, Markey SP, Mirokhin YA, Tchekhovskoi DV, Bukhari TH, Stein SE. The nistmab tryptic peptide spectral library for monoclonal antibody characterization. MAbs. 2018;10(3):354–69. doi: 10.1080/19420862.2018.1436921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Formolo T, Ly M, Levy M, Kilpatrick L, Lute S, Phinney K, Marzilli L, Brorson K, Boyne M, Davis D, et al. Determination of the nistmab primary structure In: Schiel JE, Davis DL, Borisov OV, editors. State-of-the-art and emerging technologies for therapeutic monoclonal antibody characterization volume 2 biopharmaceutical characterization: the nistmab case study. Washington (DC): American Chemical Society; 2015. p. 1–62. [Google Scholar]
- 36.Prien JM, Stockmann H, Albrecht S, Martin SM, Varatta M, Furtado M, Hosselet S, Wang MY, Formolo T, Rudd PM, et al. Orthogonal technologies for nistmab n-glycan structure elucidation and quantitation. Acs Sym Ser. 2015;1201:185–235. [Google Scholar]
- 37.Chen CH, Feng HT, Guo R, Li PJ, Laserna AKC, Ji Y, Ng BH, Li SFY, Khan SH, Paulus A, et al. Intact nist monoclonal antibody characterization-proteoforms, glycoforms-using ce-ms and ce-lif. Cogent Chem. 2018;4(1). doi: 10.1080/23312009.2018.1480455. [DOI] [Google Scholar]
- 38.Dong MW. Modern hplc for practicing scientists. Hoboken (N.J.): Wiley-Interscience; 2006. [Google Scholar]
- 39.Fekete S, Rudaz S, Fekete J, Guillarme D. Analysis of recombinant monoclonal antibodies by rplc: toward a generic method development approach. J Pharm Biomed Anal. 2012;70:158–68. doi: 10.1016/j.jpba.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 40.Shvartsburg AA, Seim TA, Danielson WF, Norheim R, Moore RJ, Anderson GA, Smith RD. High-definition differential ion mobility spectrometry with resolving power up to 500. J Am Soc Mass Spectrom. 2013;24(1):109–14. doi: 10.1007/s13361-012-0517-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shvartsburg AA, Tang K, Smith RD. Modeling the resolution and sensitivity of faims analyses. J Am Soc Mass Spectrom. 2004;15(10):1487–98. doi: 10.1016/j.jasms.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 42.Shvartsburg AA, Smith RD. Scaling of the resolving power and sensitivity for planar faims and mobility-based discrimination in flow- and field-driven analyzers. J Am Soc Mass Spectrom. 2007;18(9):1672–81. doi: 10.1016/j.jasms.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 43.Pfammatter S, Bonneil E, McManus FP, Prasad S, Bailey DJ, Belford M, Dunyach JJ, Thibault P. A novel differential ion mobility device expands the depth of proteome coverage and the sensitivity of multiplex proteomic measurements. Mol Cell Proteomics. 2018;17(10):2051–67. doi: 10.1074/mcp.TIR118.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.