

ABSTRACT
Interleukin (IL)-26, known as a Th17 cytokine, acts on various cell types and has multiple biological functions. Although its precise role still remains to be elucidated, IL-26 is suggested to be associated with the pathology of diverse chronic inflammatory diseases such as psoriasis, inflammatory bowel diseases and rheumatoid arthritis. To develop novel neutralizing anti-human IL-26 monoclonal antibodies (mAbs) for therapeutic use in the clinical setting, we immunized mice with human IL-26 protein. Hybridomas producing anti-IL-26 mAbs were screened for various in vitro functional assays, STAT3 phosphorylation and antibiotic assays. Although the IL-20RA/IL-10RB heterodimer is generally believed to be the IL-26 receptor, our data strongly suggest that both IL-20RA-dependent and -independent pathways are involved in IL-26-mediated stimulation. We also investigated the potential therapeutic effect of anti-IL-26 mAbs in the imiquimod-induced psoriasis-like murine model using human IL-26 transgenic mice. These screening methods enabled us to develop novel neutralizing anti-human IL-26 mAbs. Importantly, administration of IL-26-neutralizing mAb did not have an effect on the antimicrobial activity of IL-26. Taken together, our data strongly suggest that our newly developed anti-human IL-26 mAb is a potential therapeutic agent for the treatment of diverse chronic inflammatory diseases including psoriasis.
KEYWORDS: Interleukin-26, monoclonal antibody, neutralization, chronic inflammatory diseases, psoriasis
Introduction
Originally discovered in herpesvirus saimiri-transformed T cells,1 human interleukin (IL)-26 is a 171-amino acid protein that belongs to the IL-10 family of cytokines, a family that includes IL-10, IL-19, IL-20, IL-22 and IL-24.2,3 Human IL-26 protein is encoded by the IL26 gene located on chromosome 12q15 between the genes for interferon (IFN)-γ and IL-22, and is conserved in several vertebrate species but not found in mice and rats.4 Production of IL-26 was first reported from memory CD4+ T cells and natural killer (NK) cells,5 and IL-26 is now known as a Th17 cytokine.6 Recently, production of IL-26 by various cell types, such as synoviocytes from rheumatoid arthritis patients, alveolar macrophages and bronchial epithelial cells, has been reported.7–9 It is generally believed that the IL-20RA/IL-10RB heterodimer is the IL-26 receptor. Binding of IL-26 to IL-20RA/IL-10RB results in functional activation via STAT3 phosphorylation.10 While IL-10RB is ubiquitously expressed in various cells, expression of IL-20RA, the key IL-26 receptor subunit that mediates IL-26 signaling, is observed in epithelial cell types such as keratinocytes, intestinal and lung epithelial cells, strongly suggesting that IL-26 likely plays important roles in cutaneous and mucosal immunity.
Studies on the effect of IL-26 on IL-20RA-expressing cells have shown that IL-26 regulates production of IL-8 from keratinocytes, and enhances the production of IL-10, IL-8, and tumor necrosis factor (TNF) and the surface expression of CD54 (ICAM-1) on intestinal epithelial cells.10,11 Although the expression of IL-20RA is not observed in human peripheral blood T cells, B cells, NK cells and monocytes,5 IL-26 effects on human monocytes and NK cells have also been reported recently. IL-26 acts on human monocytes and NK cells to induce the production of inflammatory cytokines and to enhance cell surface TRAIL expression.7,12 Moreover, we have recently shown that IL-26 directly acts on vascular endothelial cells to promote proliferation and tube formation at a similar level as vascular endothelial growth factor (VEGF) regardless of the deficiency of IL-20RA expression in vascular endothelial cells.13 These findings strongly implicate the existence of a distinct IL-26 receptor other than the IL-20RA/IL-10RB heterodimer. In addition to its immunological effects, IL-26 exerts antimicrobial activity and contributes to host defense against both extracellular and intracellular bacteria.14,15 However, the antimicrobial activity of IL-26 seems to be inefficient in hidradenitis suppurativa patients, a chronic inflammatory skin disorder accompanied by severe and recurrent skin infections, suggesting that cutaneous antimicrobial incompetence in hidradenitis suppurativa may be related to IL-26.16
Due to the deficiency of the gene encoding IL-26 in mice, the precise functions and identification of target cells of IL-26 in inflammatory disorders remain to be elucidated. Meanwhile, recent studies have demonstrated that IL-26 is locally expressed at inflammatory sites, and its expression level is increased in serum, sputum, synovial fluid, bronchoalveolar lavage fluid and cerebrospinal fluid of patients with diverse chronic inflammatory diseases such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis, spondyloarthritis, multiple sclerosis, pediatric asthma, Behcet’s disease, allergic contact dermatitis and chronic obstructive pulmonary disease.6,7,11,17–23 Our group has used human IL-26 bacterial artificial chromosome (BAC) transgenic (hIL-26Tg) mice24,25 or human T cell-transplanted immuno-deficient mice to elucidate the role of IL-26 in inflammatory disorders. We recently showed that IL-26 activates both human and murine fibroblasts, leading to increased collagen production, and that human IL-26-producing CD4 T cells are deeply involved in the pathophysiology of pulmonary chronic graft-versus-host disease (GVHD).26,27 More recently, we found that vascularization and immune cell infiltration were dramatically enhanced in hIL-26Tg mice in the imiquimod (IMQ), a potent agonist of Toll-like receptor (TLR) 7 and TLR8, -induced psoriasis-like murine model.13 These findings strongly suggest that IL-26 may represent a novel promising therapeutic target for refractory chronic inflammatory diseases, for which currently available drugs cannot yet achieve the desired therapeutic outcome.
Although several anti-human IL-26 monoclonal antibodies (mAbs) are commercially available, these mAbs are designated as research reagents for Western blotting, flow cytometry or enzyme-linked immunosorbent assay (ELISA), not for neutralization. We and other groups showed that polyclonal antibodies (pAb) purchased from R&D Systems completely blocked IL-26 stimulation in vitro while also having in vivo functions.10,12,26 However, pAbs are not the ideal reagents for therapeutic use in the clinical setting. Selection of the appropriate screening methods is crucially important for the development of novel mAbs designed to serve specific purposes. In contrast with the mAbs that are only suitable for flow cytometry or ELISA, anti-human IL-26 mAbs useful for neutralization can recognize specific regions of IL-26 that are essential for binding with its receptor. Although the number of uncharacterized receptors involved in IL-26-mediated activation is not yet known, the binding regions of IL-26 to these receptors would be different from the IL-26 binding domain for IL-20RA/IL-10RB, and the inhibitory effect of anti-IL-26 neutralizing mAbs on the binding of IL-26 to its receptors would be different, depending on the kind of receptor.
For these reasons, to accurately evaluate the neutralizing capacity of mAbs in the this study, our chosen screening methods included both in vitro functional assays utilizing various cell types and in vivo evaluation of the potential therapeutic effect, resulting in the successful development of novel neutralizing anti-human IL-26 mAbs. Our data strongly suggest that the newly developed anti-human IL-26 mAbs may represent a novel promising therapeutic strategy for the treatment of diverse chronic inflammatory diseases including psoriasis and chronic GVHD.
Results
Development of novel neutralizing anti-human IL-26 mAbs
To develop novel anti-human IL-26 neutralizing mAbs for therapeutic use, we immunized mice with recombinant human IL-26 protein. After the fusion of splenocytes of immunized mice and P3U1 myeloma cells, the culture supernatant was first screened by ELISA for selective reactivity with human IL-26. In the first screening, 40 ng of recombinant human IL-26 was coated per well. To exclude the possibility of non-specific binding to the blocking proteins, we prepared the wells coated with carbonate bicarbonate buffer alone (without recombinant human IL-26), subsequently blocked with blocking proteins and incubated with hybridoma supernatants. From our examination of various blocking proteins such as bovine serum albumin, fetal bovine serum (FBS) and milk proteins, BlockAce containing milk proteins was the best blocking buffer to reduce the background absorbance. If the absorbance to human IL-26 was higher than 0.5, the clone was judged to be sufficiently reactive to human IL-26. As a result of the first screening, more than 80 clones were selected for the next screening.
Binding with the IL-20RA/IL-10RB heterodimer resulting in functional activation via STAT3 phosphorylation is a well-known characteristic of IL-26. Therefore, to screen for anti-human IL-26 mAbs suitable for neutralization of IL-26, in vitro functional assays utilizing IL-20RA-expressing cells were conducted for the next round of screening. We examined mRNA and cell surface protein expression of IL-20RA and IL-10RB in several cell lines by real-time RT-PCR and flow cytometry, respectively. As reported previously,5,10,11,28 mRNA expression of IL-20RA was detected in the human keratinocyte cell line HaCaT, human colon cancer cell lines COLO205, LoVo, HT-29 and human gastric cancer cell line MKN45 (Figure S1(a)). Among them, the expression of IL-20RA in COLO205 was the most prominent at both mRNA and cell surface protein levels. In contrast, neither mRNA nor cell surface protein expression was detected in human umbilical vein endothelial cells (HUVEC) (Figure S1). The expression of IL-10RB was detected at both mRNA and cell surface protein levels in all the cells. Although multiple biological effects of IL-26 on various cell types have been reported, we focused on ICAM-1 expression on COLO205 following IL-26 stimulation. COLO205 was then stimulated with IL-26 in the presence of the culture supernatant from more than 80 clones, and cell surface ICAM-1 expression on COLO205 was analyzed by flow cytometry. The clones that had the potential to inhibit ICAM-1 expression on IL-26-stimulated COLO205 were screened, and the neutralizing capacity of each clone was reexamined following purification of mAbs from culture supernatants. As a result of the second screening utilizing COLO205, we obtained 4 clones with the potential to neutralize human IL-26.
The reactivity of the representative 4 clones to human IL-26 was confirmed by ELISA. As shown in Figure 1(a), all the novel anti-IL-26 mAbs reacted well to the immobilized IL-26, and the absolute value of absorbance increased in an antigen dose-dependent manner. Among them, the absolute value of absorbance to IL-26 incubated with 20–3 mAb or 69–10 mAb was particularly higher than the absorbance to IL-26 incubated with the commercially available anti-IL-26 mAb (Figure 1(a)). Since human IL-26 has been categorized into the IL-10 family, with approximately 25% homology and 47% similarity to human IL-10,29 the reactivity of the novel anti-IL-26 mAbs to immobilized human IL-10 was analyzed. No non-specific binding to human IL-10 was observed in the 4 mAbs and commercially available anti-IL-26 mAb (Figure 1(b)).
Figure 1.
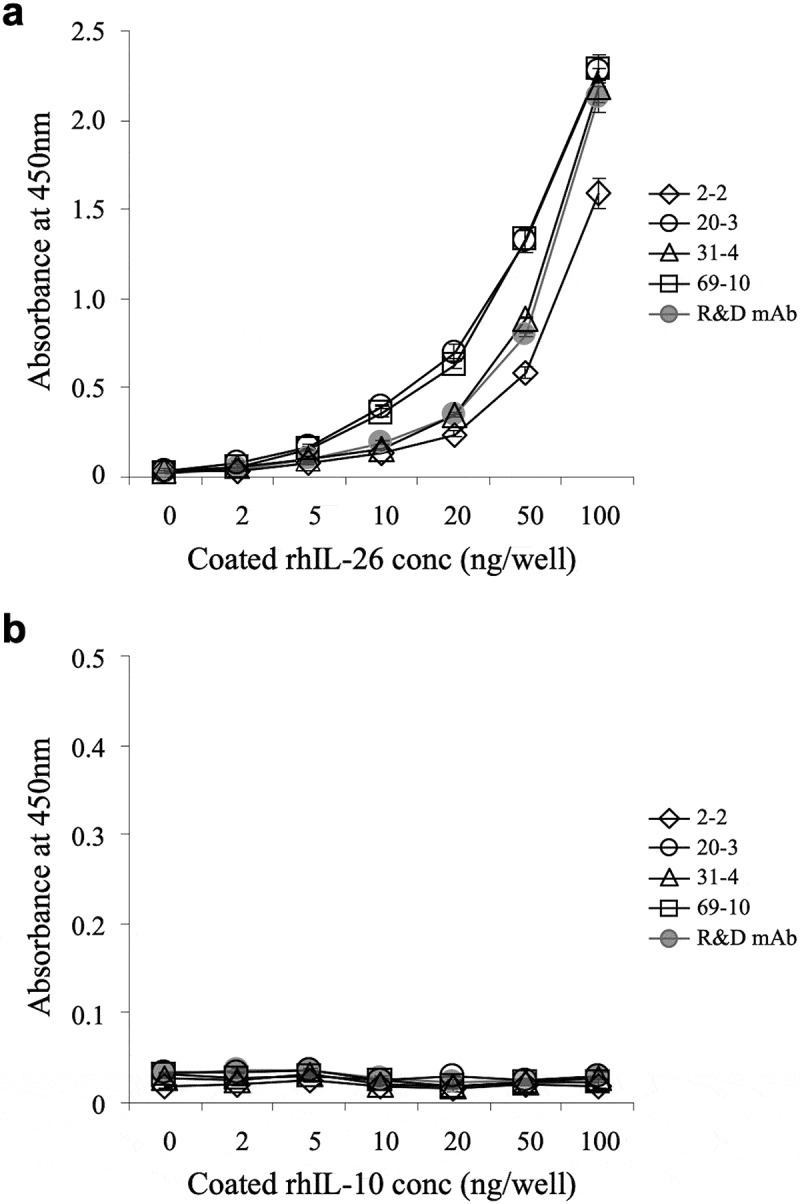
Representative results of ELISA analysis with novel anti-IL-26 mAbs.
Immobilized recombinant human IL-26 (rhIL-26) (a) or IL-10 (rhIL-10) (b) was incubated with purified novel mouse anti-human IL-26 mAb (2–2, 20–3, 31–4 or 69–10) or commercial mouse anti-human IL-26 mAb (R&D Systems). The absorbance at 450 nm/570 nm was measured. Representative data of three independent experiments are shown as mean ± S.D. of triplicate samples, and similar results were obtained in each experiment.
The inhibitory effect of the novel anti-IL-26 mAbs on cell surface ICAM-1 expression on IL-26-stimulated COLO205 cells
Although we could not obtain mAb that completely suppressed ICAM-1 expression on IL-26-stimulated COLO205, the addition of 2–2 mAb, 20–3 mAb, 31–4 mAb or 69–10 mAb partially inhibited ICAM-1 expression on IL-26-stimulated COLO205 (Figure 2(a)). Of note, while addition of the commercially available anti-IL-26 pAb completely inhibited ICAM-1 expression on IL-26-stimulated COLO205 to a level similar to unstimulated COLO205, addition of the commercially available anti-IL-20RA pAb only partially inhibited ICAM-1 expression on IL-26-stimulated COLO205 to a level similar to 69–10 mAb (Figure 2(a)). Addition of the commercially available anti-IL-10RB pAb hardly affected ICAM-1 expression on IL-26-stimulated COLO205 (Figure 2(a)).
Figure 2.
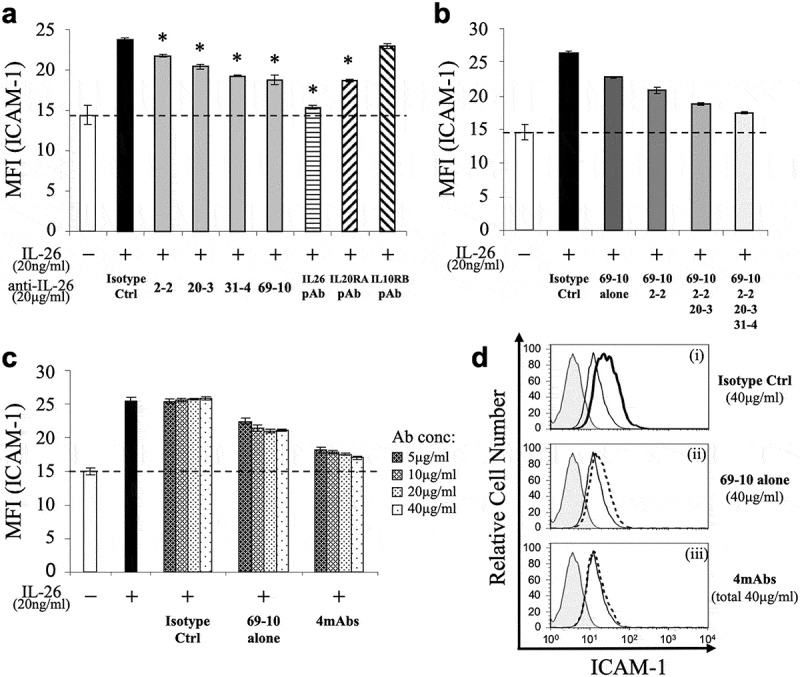
Addition of novel anti-IL-26 mAbs inhibits cell surface ICAM-1 expression on IL-26-stimulated COLO205 cells.
COLO205 cells were stimulated with recombinant human IL-26 (20 ng/ml) for 24 hr. Cell surface ICAM-1 gated for viable cells was detected by flow cytometry. (a) Prior to the onset of stimulation, the indicated Ab or isotype control Ab (isotype ctrl) was added to the culture wells to give a final concentration of 20 μg/ml each. (b) Prior to the onset of stimulation, isotype control mAb (5 μg/ml), 69–10 mAb alone (5 μg/ml) or the combination of 69–10 mAb and 2–2 mAb, 20–3 mAb, 31–4 mAb (5 μg/ml, respectively) was added to the culture wells. (c) Prior to the onset of stimulation, the indicated concentrations of isotype control mAb, 69–10 mAb alone or the combination of 4 mAbs were added to the culture wells. (a-c) The dashed line is the standard value of unstimulated cells (vehicle). Representative data of five independent experiments are shown as mean ± S.D. of mean fluorescence intensity (MFI) from triplicate samples, comparing values in each Ab to those in isotype control (* p < 0.01), and similar results were obtained in each experiment. (d) Data are shown as histogram of ICAM-1, and are representative of five independent experiments. The thin black lines in each histogram show the data of ICAM-1 expression on unstimulated cells. The bold black line (i) and the dotted lines (ii, iii) in each histogram show the data of ICAM-1 expression on IL-26-stimulated cells in the presence of the indicated Abs (40 μg/ml each). The gray areas in each histogram show the data of the PE-labeled isotype control.
We also examined the inhibitory effect of a combination of novel anti-IL-26 mAbs on ICAM-1 expression. As shown in Figure 2(b), a combination of 69–10 mAb and 2–2 mAb additively inhibited ICAM-1 expression on IL-26-stimulated COLO205 compared with 69–10 mAb alone. Moreover, addition of 20–3 mAb and 31–4 mAb further suppressed ICAM-1 expression on IL-26-stimulated COLO205. (Figure 2(b)). As shown in Figure 2(c) (representative histograms are shown in Figure 2(d)), ICAM-1 expression on IL-26-stimulated COLO205 was inhibited in an antibody dose-dependent manner, and the combination of 4 mAbs almost completely inhibited ICAM-1 expression on IL-26-stimulated COLO205 to a level nearly identical to the commercially available anti-IL-26 pAb.
The inhibitory effect of the novel anti-IL-26 mAbs on STAT3 phosphorylation in IL-26-stimulated COLO205 cells
We next analyzed the inhibitory effect of the novel anti-IL-26 mAbs on STAT3 phosphorylation in IL-26-stimulated COLO205. We first examined the phosphorylation level of STAT3 in COLO205 following IL-26 stimulation by Western blotting, with recombinant human IL-22 and IL-6 used as positive controls. As shown in Figure 3(a), stimulation with IL-26 resulted in the prominent phosphorylation of STAT3 in COLO205 with a peak around 10 min. The peak intensity of STAT3 phosphorylation in COLO205 following IL-26 stimulation was stronger than IL-6 stimulation, a well-known inducer of STAT3 phosphorylation, although the intensity was further amplified following IL-22 stimulation for 10 min (Figure 3(a)). We then examined the effect of anti-IL-26 mAbs on STAT3 phosphorylation in COLO205 following IL-26 stimulation for 10 min. Similar to the results obtained with the ICAM-1 expression assays, addition of 2–2 mAb, 20–3 mAb, 31–4 mAb or 69–10 mAb partially inhibited STAT3 phosphorylation in IL-26-stimulated COLO205, while the commercially available anti-IL-26 pAb completely blocked STAT3 phosphorylation to a level seen in unstimulated COLO205 (Figure 3(b)). The combination of 4 mAbs resulted in a deeper level of inhibition compared with anti-IL-26 mAb alone, one which was similar to that observed with the commercially available anti-IL-20RA pAb (Figure 3(b)). Considering that the combination of 4 mAbs markedly suppressed ICAM-1 expression on IL-26-stimulated COLO205 as compared with anti-IL-20RA pAb (as shown in Figure 2(a,c)), our data strongly suggest that IL-20RA and STAT3-independent pathways are involved in ICAM-1 expression on COLO205 following IL-26 stimulation.
Figure 3.
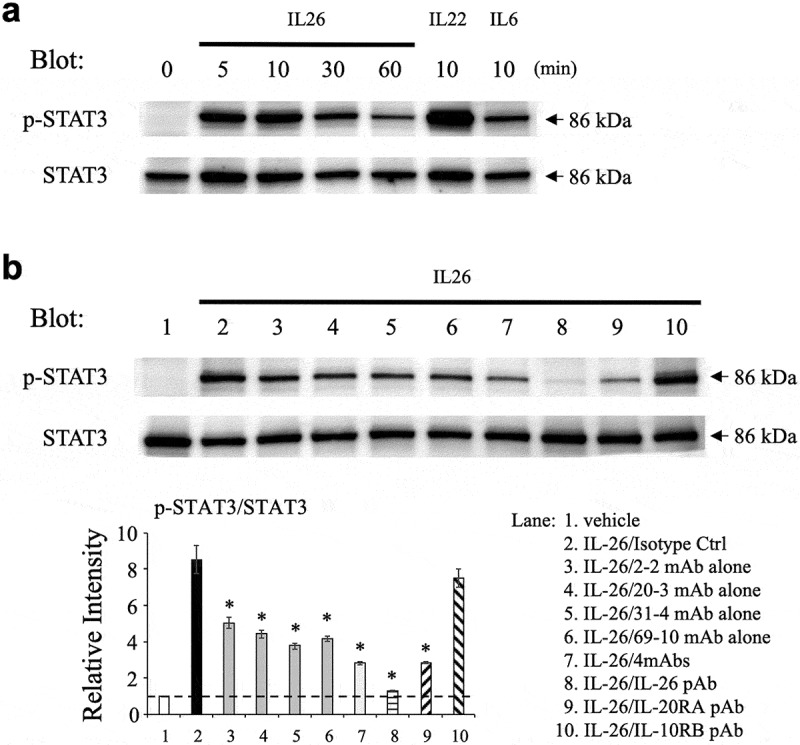
Addition of novel anti-IL-26 mAbs inhibits phosphorylation of STAT3 in IL-26-stimulated COLO205 cells.
(a) COLO205 cells were stimulated with recombinant human IL-26 (20 ng/ml) for the indicated times, or stimulated with recombinant human IL-6 (20 ng/ml) or IL-22 (20 ng/ml) for 10 min. (b) COLO205 cells were stimulated with recombinant human IL-26 (20 ng/ml) for 10 min in the presence of the indicated Ab or isotype control Ab (isotype ctrl) (20 μg/ml, respectively). (a, b) Whole cell lysates were separated by SDS-PAGE (each, 25 μg), and phospho(p)-STAT3 was detected by immunoblotting. The same blots were stripped and reprobed with antibodies specific for pan STAT3. Data shown are representative of three independent experiments with similar results. Band intensity of p-STAT3 was normalized to pan STAT3, and relative intensity compared with unstimulated cells was indicated in the bottom graph (b). The dashed line is the standard value of unstimulated cells (vehicle). Data are shown as mean ± S.E. of relative intensity from three independent experiments, comparing values in each Ab to those in isotype control (* p < 0.01).
The inhibitory effect of the novel anti-IL-26 mAbs on FGF7 and VEGF expression in IL-26-stimulated HaCaT cells
Since keratinocytes expressing IL-20RA are thought to be one of the primary targets of IL-26, we examined the inhibitory effect of the novel anti-IL-26 mAbs on keratinocytes activation following IL-26 stimulation. We have recently shown that IL-26 upregulates the expression of fibroblast growth factor (FGF)-1 and FGF2 in primary human keratinocytes.13 Considering the uniformity of the assay, the human keratinocyte cell line HaCaT was used in this study. Although we examined mRNA expression levels of FGF1 and FGF2 in HaCaT following IL-26 stimulation, significant increase was not observed as compared with unstimulated HaCaT (data not shown). On the other hand, IL-26 enhanced mRNA expression levels of FGF7 and VEGF in HaCaT (Figure 4(a,b)). We then examined the effect of anti-IL-26 mAbs on FGF7 and VEGF expression in HaCaT following IL-26 stimulation. As shown in Figure 4(a), addition of 2–2 mAb, 20–3 mAb, 31–4 mAb or 69–10 mAb inhibited FGF7 expression in IL-26-stimulated HaCaT. Of note, the inhibitory effect of each mAb, particularly 31–4 mAb and 69–10 mAb, on FGF7 expression in HaCaT was much more prominent compared with ICAM-1 expression or STAT3 phosphorylation in IL-26-stimulated COLO205 (as shown in Figures 2 and 3). Addition of 31–4 mAb or 69–10 mAb almost completely inhibited FGF7 expression in IL-26-stimulated HaCaT to a level seen in unstimulated HaCaT, whereas the commercially available anti-IL-20RA pAb only slightly inhibited FGF7 expression in IL-26-stimulated HaCaT (Figure 4(a)). Similar results were also obtained regarding VEGF expression in IL-26-stimulated HaCaT (Figure 4(b)).
Figure 4.
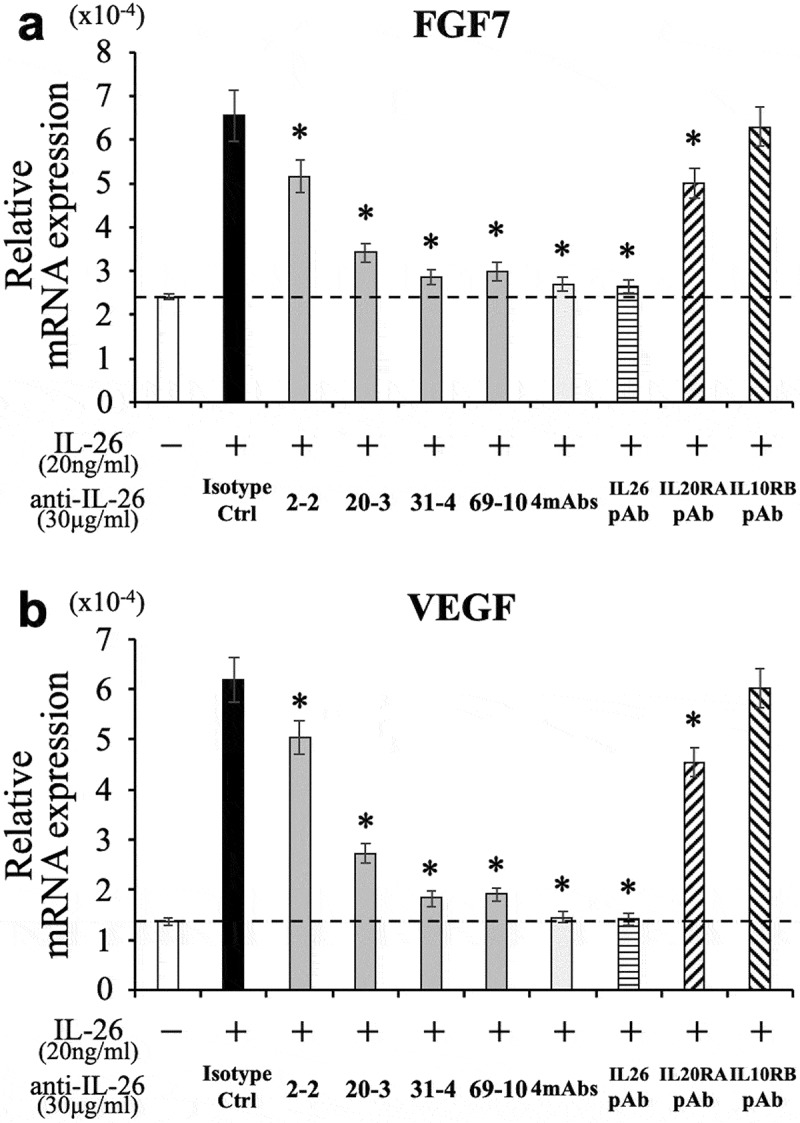
Addition of novel anti-IL-26 mAbs inhibits the expression of FGF7 and VEGF in IL-26-stimulated HaCaT cells.
HaCaT cells were stimulated with recombinant human IL-26 (20 ng/ml) for 6 hr. Prior to the onset of stimulation, the indicated Ab or isotype control Ab (isotype ctrl) was added to the culture wells to give a final concentration of 30 μg/ml each. mRNA expression of FGF7 (a) and VEGF (b) was quantified by real-time RT-PCR. Each expression was normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT1). The dashed line is the standard value of unstimulated cells (vehicle). Representative data of three independent experiments are shown as mean ± S.D. of triplicate samples, comparing values in each Ab to those in isotype control (* p < 0.01), and similar results were obtained in each experiment.
The inhibitory effect of the novel anti-IL-26 mAbs on proliferation and tube formation of IL-26-stimulated HUVEC
Given the fact that IL-20RA may be differentially involved in IL-26-mediated stimulation depending on cell types, we next examined the inhibitory effect of the novel anti-IL-26 mAbs on vascular endothelial cells activation following IL-26 stimulation. We recently found that IL-26 directly acts on HUVEC to promote proliferation and tube formation at a level similar to VEGF regardless of the lack of IL-20RA expression.13 We therefore examined the effect of the anti-IL-26 mAbs on proliferation and tube formation of HUVEC following IL-26 stimulation. As shown in Figure 5(a,b), addition of 2–2 mAb, 20–3 mAb, 31–4 mAb or 69–10 mAb inhibited both proliferation and tube formation of IL-26-stimulated HUVEC. Similar to the results obtained with IL-26-stimulated HaCaT shown in Figure 4, addition of the anti-IL-26 mAb, particularly 31–4 mAb and 69–10 mAb, strongly inhibited proliferation and tube formation of IL-26-stimulated HUVEC to a level nearly identical to that seen in unstimulated HUVEC. In contrast, the commercially available anti-IL-20RA pAb and anti-IL-10RB pAb hardly affected proliferation and tube formation of IL-26-stimulated HUVEC (Figure 5(a,b)). Taken together, our data indicate that both IL-20RA-dependent and -independent pathways are involved in IL-26-mediated stimulation, and strongly suggest the existence of distinct receptor(s) other than the IL-20RA/IL-10RB heterodimer.
Figure 5.
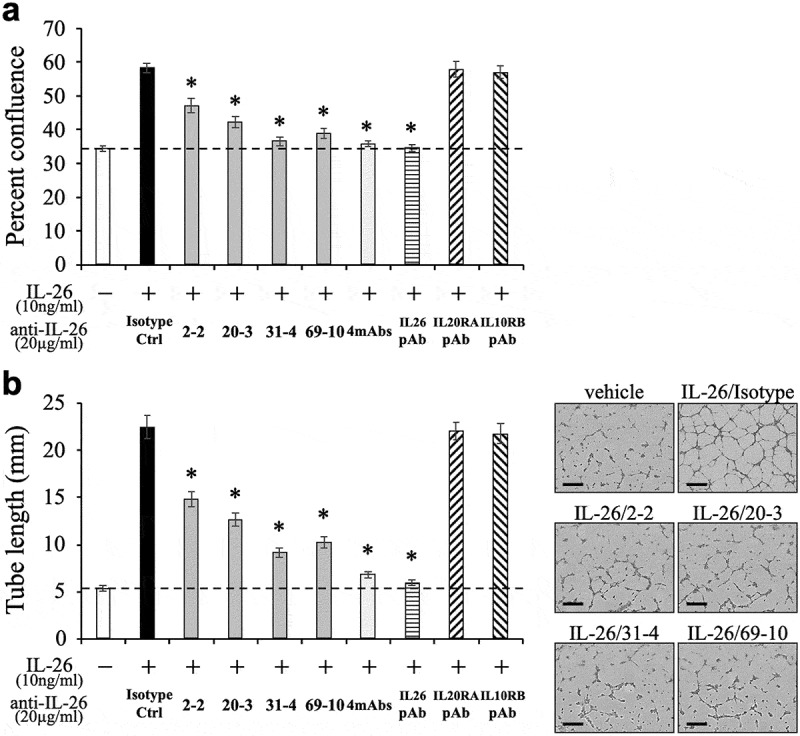
Addition of novel anti-IL-26 mAbs inhibits proliferation and tube formation of IL-26-stimulated HUVEC.
HUVEC were stimulated with recombinant human IL-26 (10 ng/ml) for 48 hr (a) or 9 hr (b). Prior to the onset of stimulation, the indicated Ab or isotype control Ab (isotype ctrl) was added to the culture wells to give a final concentration of 20 μg/ml each. (a) Proliferation was assessed by cell confluence. (b) Tube formation was assessed by cell sprouts formation. Representative images are shown in the right panels. Scale bar, 300 μm. Quantification of tube length is demonstrated in the graph in the left panel. (a, b) The dashed line is the standard value of unstimulated cells (vehicle). Representative data of three independent experiments are shown as mean ± S.D. of triplicate samples, comparing values in each Ab to those in isotype control (* p < 0.01), and similar results were obtained in each experiment.
Lack of effect on the antimicrobial activity of IL-26 by the novel anti-IL-26 mAbs
IL-26 has been reported to have a multitude of biological functions. Due to its cationic amphipathic feature, similar to that seen with antimicrobial peptides, IL-26 exerts its killing effect on extracellular bacteria via membrane-pore formation and appears to have an important role in host defense.14 Therefore, the development of novel anti-IL-26 mAb-targeted therapy for chronic inflammatory diseases should also include investigation into its effect on IL-26-mediated antimicrobial activity. Binding assays revealed that both lipopolysaccharide (LPS) from gram-negative bacteria (Escherichia coli O111:B4) and lipoteichoic acid (LTA) from gram-positive bacteria (Staphylococcus aureus) strongly bound to recombinant human IL-26 in a dose-dependent manner as compared with recombinant human IL-10 and IL-22 (other IL-10 family cytokines) (Figure 6(a)).
Figure 6.
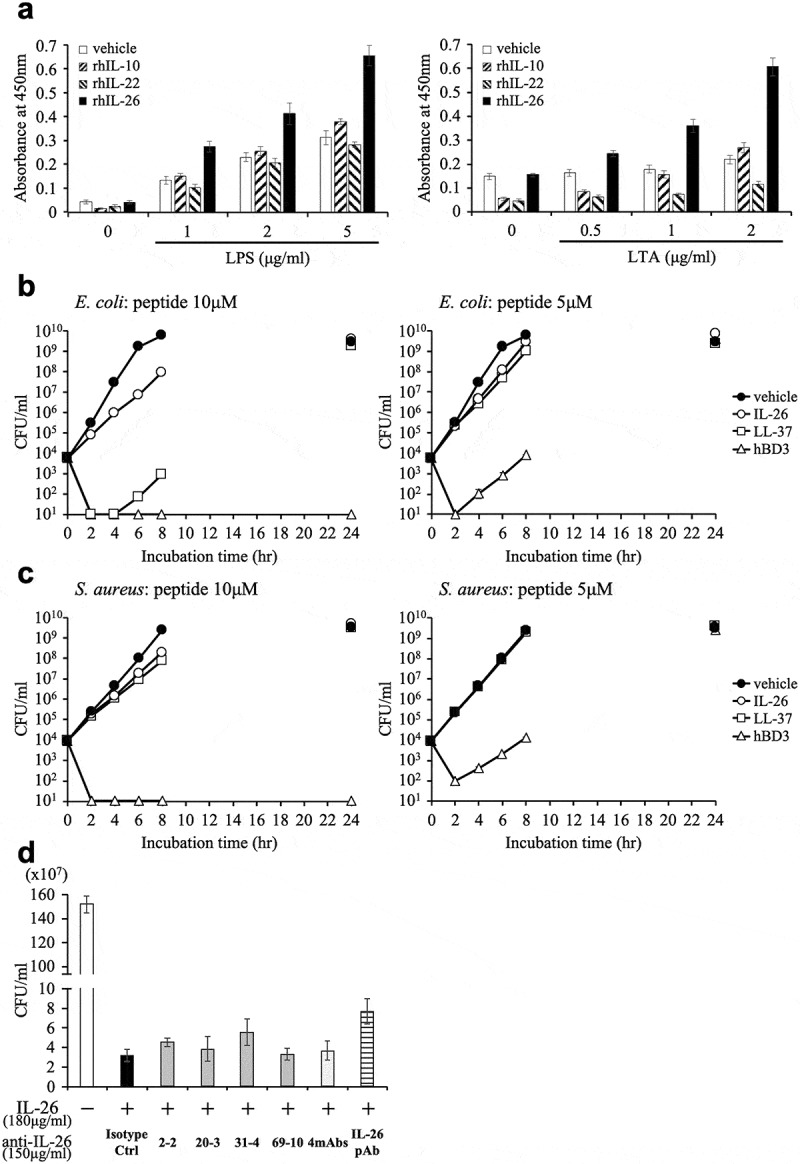
Addition of novel anti-IL-26 mAbs hardly affects the antimicrobial activity of IL-26.
(a) Immobilized recombinant human IL-10 (rhIL-10), IL-22 (rhIL-22), IL-26 (rhIL-26) or vehicle (PBS) alone was incubated with the indicated concentrations of purified LPS (left panel) or LTA (right panel). The absorbance at 450 nm/570 nm was measured. Representative data of three independent experiments are shown as mean ± S.D. of triplicate samples, and similar results were obtained in each experiment. (b, c) Growth kinetics of Escherichia coli (b) and Staphylococcus aureus (c) cultured with 10 μM (left panels) or 5 μM (right panels) rhIL-26, LL-37 or human β-defensin 3 (hBD3). (d) Escherichia coli was cultured with 10 μM rhIL-26 for 6 hr. Prior to the onset of culture, the indicated Ab or isotype control Ab (isotype ctrl) was added to the culture wells to give a final concentration of 150 μg/ml each. (b-d) Serial dilutions of bacterial cultures were plated onto agar plates, and the number of colonies was counted. Representative data of three independent experiments are shown as mean ± S.D. of triplicate samples, and similar results were obtained in each experiment.
We next examined the antimicrobial capacity of IL-26 through the use of Escherichia coli (ATCC 8739), and Staphylococcus aureus (ATCC 29213) as representative gram-negative and gram-positive bacteria, respectively. Representative antimicrobial peptides LL-37 and human β-defensin 3 (hBD3) were used for comparison. Growth kinetics analysis showed that 10 μM human IL-26 substantially inhibited and delayed the growth of Escherichia coli, although this antimicrobial activity was not as prominent as that seen with hBD3 and LL-37 in our assay system (Figure 6(b)). hBD3 exhibited the strongest antimicrobial capacity, with 10 μM hBD3 being able to completely eradicate both Escherichia coli and Staphylococcus aureus, while 10 μM human IL-26 or LL-37 only slightly delayed the growth of Staphylococcus aureus, and no inhibition was observed with 5 μM human IL-26 or LL-37 (Figure 6(c)).
Since the growth of Escherichia coli incubated with vehicle reached saturation level from 6 to 8 hrs of incubation, the difference in the number of bacteria incubated with vehicle and 10 μM human IL-26 was most prominently observed after 6 hrs of culture. We thus investigated the effect of anti-IL-26 mAbs on antimicrobial activity of IL-26 under the experimental conditions described in the Materials and methods section. Considering the clinically effective dose range of therapeutic antibody in the blood, we examined the effect of anti-IL-26 Ab up to 150 μg/ml, equivalent to 1 μM. As shown in Figure 6(d), addition of 2–2 mAb, 20–3 mAb, 31–4 mAb or 69–10 mAb hardly affected the antimicrobial activity of IL-26. Even the commercially available anti-IL-26 pAb showed little effect on the antimicrobial activity of IL-26, strongly suggesting that 1 μM of antibody is insufficient to neutralize 10 μM human IL-26. Taken together, these results strongly suggest that at least at physiological doses, the novel anti-human IL-26 mAb does not inhibit the antimicrobial activity of IL-26.
Anti-IL-26 mAb administration suppresses skin inflammation by inhibiting angiogenesis and infiltration of inflammatory cells in psoriatic lesions of IMQ-treated hIL-26Tg mice
Since the IL-26 gene is absent in rodents,4 we recently examined the role of IL-26 in angiogenesis and skin inflammation utilizing hIL-26Tg mice.13 In this study, we conducted in vivo experiments to determine the potential therapeutic effect of the newly developed anti-IL-26 mAbs. Importantly, although the IL-26 gene is absent in mice, the IL-20RA and IL-10RB subunits that are part of the receptor complex of the other IL-10 cytokine family members are also expressed in mice.10 In addition, our recent report showed that human IL-26 functions in both human and murine fibroblasts and vascular endothelial cells.13,26 For the in vivo studies, Δconserved noncoding sequence (CNS)-77 Tg mice (control BAC Tg mice with deleting human IL-26 transcription) were used as controls. The therapeutic effect of anti-IL-26 mAbs was assessed in the low dose (20 mg) IMQ-induced psoriasis model, since scaling and thickness of the back skin were prominent even in the human IL-26 transcription-lacking ΔCNS-77 Tg mice treated with high dose (more than 40 mg) IMQ.13 We previously confirmed that the expression of human IL-26 was increased at both mRNA and protein levels in the skin lesions of hIL-26Tg mice following daily application of IMQ cream, while no expression of human IL-26 was detected in the skin of ΔCNS-77 Tg mice.13
As shown in Figure 7(a,b), the back skin of hIL-26Tg mice injected with isotype control mAb appeared markedly affected as compared with ΔCNS-77 Tg mice, and the scores of erythema, scaling and thickness in isotype control-injected hIL-26Tg mice were all higher than those of ΔCNS-77 Tg mice. In contrast, the clinical symptoms of erythema, scaling and thickening of the skin lesion were all remarkably suppressed in the hIL-26Tg mice treated with 69–10 mAb or the combination of 4 mAbs, as compared with the hIL-26Tg mice injected with isotype control mAb, with Psoriasis Area and Severity Index (PASI) score of the hIL-26Tg mice treated with anti-IL-26 mAb being at almost the same level as the ΔCNS-77 Tg mice (Figure 7(a,b)). We also examined the potential therapeutic effect of 31–4 mAb, and similar results with those obtained with 69–10 mAb were observed (data not shown).
Figure 7.
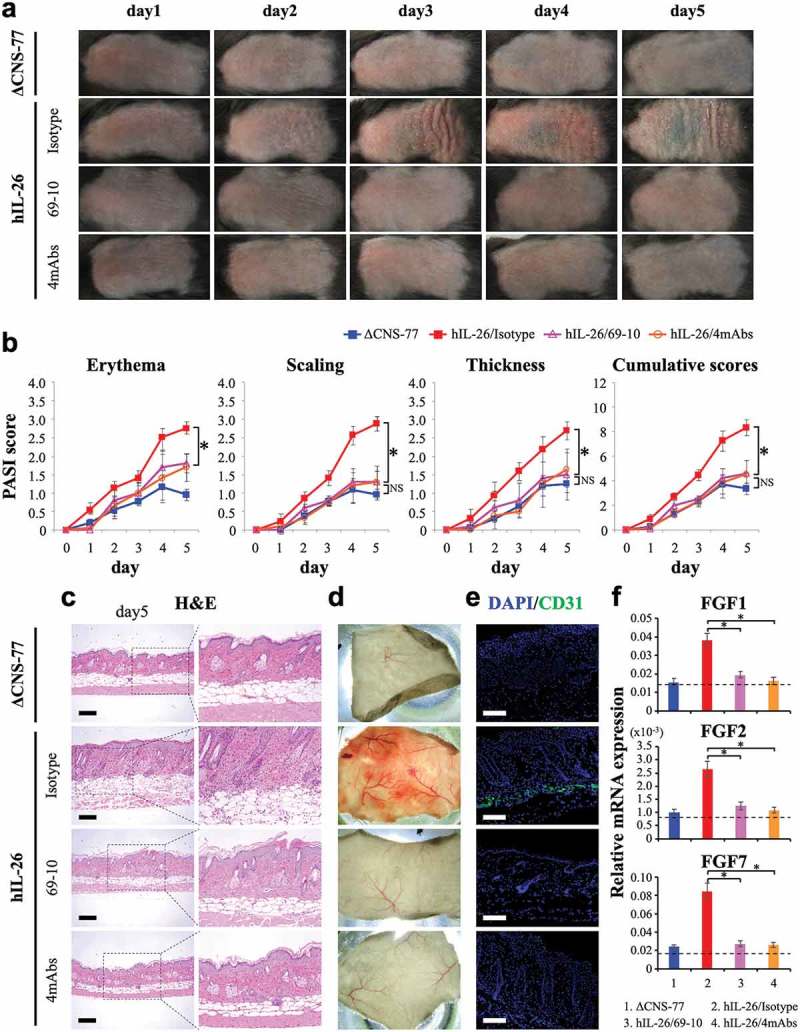
Anti-IL-26 mAb treatment suppresses IMQ-induced skin inflammation via inhibition of angiogenesis and infiltration of inflammatory cells.
Data are shown from IMQ (20 mg)-treated back skin in ΔCNS-77 Tg mice, hIL-26Tg mice treated with anti-IL-26 mAb (69–10 mAb alone or the combination of 4 mAbs) or isotype control mAb. (a) Phenotypical representation of each group. Representative images are shown (n = 6 mice for each group at each time point). (b) Time course of PASI scores (erythema, scaling and thickness were scored daily on a scale from 0 to 4, respectively) in each group (n = 6 mice for each group at each time point). Data are shown as mean ± S.D. of each group, comparing values in hIL-26Tg mice treated with anti-IL-26 mAb to those in hIL-26Tg mice injected with isotype control mAb or those in ΔCNS-77 Tg mice (* p < 0.01). NS denotes ‘not significant’. Data represent the combined results of two independent experiments. (c) H&E staining of skin lesions from each group on day-5. Higher magnification images show inflammatory cell infiltration. Original magnification x100. Scale bar, 200 μm. (d) Subcutaneous vascular formation of each group on day-5. (e) Immunofluorescence staining of skin lesions from each group on day-5 using anti-CD31 pAb. Positive cells were shown in green. All sections were stained with DAPI to mark the nuclei (blue). Original magnification x100. Scale bar, 200 μm. (c-e) Representative images are shown with similar results (for each, n = 6 mice). (f) mRNA expression levels of FGF1, FGF2 and FGF7 in skin lesions from each group on day-3 (n = 6 mice for each group). Each expression was normalized to hypoxanthine phosphoribosyltransferase (HPRT). The dashed line is the mean value of non-treated mice. Data are shown as mean ± S.D. of each group, comparing values in hIL-26Tg mice treated with anti-IL-26 mAb to those in hIL-26Tg mice injected with isotype control mAb (* p < 0.01).
Histologic studies of the skin showed the development of psoriasis-like skin inflammation characterized by acanthosis, parakeratosis, papillomatosis, infiltration of inflammatory cells and altered dermal vascularity,30 in the hIL-26Tg mice injected with isotype control mAb, while anti-IL-26 mAb-treated mice had similar appearance as ΔCNS-77 Tg mice (Figure 7(c)). Excessive blood vessel formation and vascular invasion were observed in subcutaneous tissues of isotype control-injected hIL-26Tg mice compared to ΔCNS-77 Tg mice, whereas blood vessel formation was significantly suppressed in the subcutaneous tissue of anti-IL-26 mAb-treated hIL-26Tg mice (Figure 7(d)). To further examine the vascular invasion, we conducted immunofluorescence staining for CD31 in the back skin sections of each mouse. As shown in Figure 7(e), levels of CD31-positive cells in the lower dermis were clearly decreased in the hIL-26Tg mice treated with anti-IL-26 mAb as compared with isotype control-injected mice. Moreover, expression levels of FGF1, FGF2 and FGF7, key angiogenic factors in this model,13 were all increased in the skin of isotype control-injected hIL-26Tg mice, and they were remarkably suppressed in the subcutaneous tissue of anti-IL-26 mAb-treated hIL-26Tg mice, resulting in expression levels similar to those seen in ΔCNS-77 Tg mice (Figure 7(f)).
We also examined the kinetics of mRNA expression of the major effector cytokines involved in psoriasis pathogenesis. The mRNA expression levels of IL-1β and IL-6 were significantly increased in the skin lesions from hIL-26Tg mice as compared with ΔCNS-77 Tg mice, while there was no marked difference in the expression levels of TNF, IL-17A, IL-23 p19 and IL-12 p40 between hIL-26Tg mice and control mice (Figure S2(a)). Similar with the results of FGF1, FGF2 and FGF7, expression levels of IL-1β and IL-6 were prominently suppressed in the subcutaneous tissue of anti-IL-26 mAb-treated hIL-26Tg mice to levels nearly identical to those seen in ΔCNS-77 Tg mice (Figure S2(b)). Taken together, our results strongly suggest that administration of novel neutralizing anti-IL-26 mAb (31–4 mAb and 69–10 mAb) provides a novel therapeutic approach for psoriasis by regulating angiogenesis and skin inflammation.
Discussion
Although its precise function in inflammatory disorders is not fully understood, IL-26 is thought to play a role in the pathology of diverse chronic inflammatory diseases such as psoriasis, inflammatory bowel diseases, rheumatoid arthritis and chronic GVHD. In this study, we used precise screening methods that incorporated both IL-20RA-expressing and -deficient cells, as well as assays examining potential in vivo therapeutic effects, to develop novel neutralizing anti-human IL-26 mAbs that may be of future use as therapeutic agents for the treatment of diverse chronic inflammatory diseases.
Selection of the appropriate screening methods is crucially important for the development of novel mAb designed to serve specific purposes. Although multiple biological effects of IL-26 on various cell types have been reported, we focused on ICAM-1 expression on COLO205 following IL-26 stimulation. The key points of this assay were its reproducibility and simplicity. Cell surface expression of ICAM-1 on COLO205 was enhanced following IL-26 stimulation at similar levels in all of the independent experiments. Moreover, when COLO205 was stimulated with IL-26 in triplicates or more, the standard deviation of the mean ICAM-1 expression in each group was extremely small, which is critical for the evaluation of the neutralizing capacity of each mAb. Furthermore, COLO205 can be collected without trypsin treatment and the expression level of ICAM-1 can be easily and promptly analyzed shortly after acquisition of data, which is crucially important for evaluating the neutralizing capacity of a multitude of candidate clones. From these reasons, we chose this screening method as the first in vitro functional assay.
IL-20RA/IL-10RB heterodimer is generally believed to be the IL-26 receptor. Previous work has demonstrated that IL-26 binding to IL-20RA/IL-10RB results in functional activation via STAT3 phosphorylation.10 However, although commercially available anti-IL-20RA pAb and the combination of 4 novel anti-IL-26 mAbs strongly suppressed STAT3 phosphorylation in IL-26-stimulated COLO205 at similar levels (Figure 3(b)), anti-IL-20RA pAb only partially inhibited ICAM-1 expression on IL-26-stimulated COLO205, a much less inhibitory effect than that seen with the combination of 4 mAbs (Figure 2(a,c)). In addition, anti-IL-20RA pAb exhibited only a slight or no inhibitory effect on IL-26-stimulated HaCaT or HUVEC, respectively (Figures 4 and 5). These results strongly suggest that both IL-20RA-mediated signals and the hitherto uncharacterized receptor(s)-mediated signals are involved in IL-26-mediated stimulation, and that the relative involvement of each receptor in IL-26-mediated stimulation would depend on cell types due to differences in expression levels and binding affinities of those receptors. Our data indicate that blockade of IL-20RA with anti-IL-20RA antibody is insufficient to inhibit IL-26-mediated stimulation. Moreover, IL-20RA not only forms IL-20RA/IL-10RB heterodimer, but also is able to form IL-20RA/IL-20RB heterodimer, a receptor for IL-19, IL-20 and IL-24,27 suggesting that blockade of IL-20RA may interfere with these signaling pathways. These findings indicate that IL-20RA may not be an appropriate target for novel IL-26-directed therapeutic approaches, and neutralization of IL-26 itself may be more effective than blockade of IL-20RA. To better understand the precise biological functions of IL-26 and the exact mechanisms of action of anti-IL-26 mAb, it is essential to identify the uncharacterized second receptor of human IL-26, and we are currently examining this important topic.
IL-26 is an unusual cationic and amphipathic cytokine, with the predominance of cationic residues on one side and hydrophobic amino acids on the opposite side.14 These characteristics closely resemble the structure of antimicrobial peptides. In fact, Meller et al.14 first reported that IL-26 disrupted bacterial membranes via pore forming and exhibited direct bacterial killing effects, similar to other antimicrobial peptides such as LL-37 and human β-defensin. Their group showed that recombinant human IL-26 markedly inhibited the growth of several gram-negative bacterial strains, including Pseudomonas aeruginosa (ATCC 27853, PA14), Escherichia coli (O1:K1:H7, O18:K1:H7, O111:B4, O111:K58:H2) and Klebsiella pneumoniae (O1:K2), as well as gram-positive Staphylococcus aureus (ATCC 6538), while no inhibition was observed with Enterococcus faecalis or Candida albicans.14 Our data showed that although 10 μM human IL-26 certainly inhibited and delayed the growth of Escherichia coli (ATCC 8739), the number of bacteria gradually increased and finally reached the same saturation level as seen with bacteria cultured with vehicle only (Figure 6(b)). The difference in the antimicrobial activity of IL-26 between the previous report14 and this study may be due to the difference in the bacterial strains. In addition, there is also a critical difference in the bacterial culture conditions. Meller et al.14 examined the antimicrobial activity of IL-26 in relatively unconventional culture conditions. Bacteria were incubated for 24 hr at 37°C under low-ionic-strength conditions (water only containing 10 mM NaCl without any nutrients), and then IL-26 or other antimicrobial peptides were added to these cultures. Under that culture condition, growth of bacteria other than Pseudomonas aeruginosa even cultured with vehicle only was slightly decreased during the culture period.14 In this study, bacteria was cultured in Mueller-Hinton broth, the nutrient-rich international standard culture medium to evaluate antimicrobial activity of agents, and rapid bacterial growth was observed during the period of assay (Figure 6(b,c)). An important point is that 5–10 μM (considering that the molecular weight of human IL-26 is 19.8 kDa, 5–10 μM is therefore equivalent to 100–200 μg/ml), which is an extremely high concentration, of IL-26 is needed to exert antimicrobial activity. For in vitro neutralization assay, to sufficiently block 1–50 ng/ml of cytokines, much higher levels (5–50 μg/ml) of antibodies are usually used. While the relative local concentrations of human IL-26 in such tissues as skin, intestine or lung are unknown, considering the serum dose range of therapeutic antibody in the clinical setting, it is reasonable to project from our findings that administered anti-human IL-26 mAb may hardly affect the antimicrobial activity of IL-26, as shown in Figure 6(d).
IL-17 is a proinflammatory cytokine that plays a key role in the pathology of various autoimmune diseases including psoriasis,30–32 inducing hyperproliferation of keratinocytes and stimulating production of psoriasis-associated molecules such as CCL20, CXCL8, matrix metalloproteinase 3, and angiogenic factors such as VEGF, which lead to the chronic skin inflammation of psoriasis.33,34 MAbs directly targeting IL-17, such as secukinumab (Cosentyx®), have shown very promising results in the treatment for psoriasis.35,36 However, IL-17 plays a pivotal role in host defense by enhancing the production of antimicrobial peptides from keratinocytes and mucosal epithelial cells, and recruiting neutrophils to inflammation/infection sites.37 As expected, cases of adverse effects of IL-17-targeting therapy for psoriasis involving neutropenia and fungal infection, especially Candida infection, have been reported.38,39 Our study showed that anti-IL-26 mAb markedly suppressed the pathognomonic symptoms of angiogenesis and infiltration of inflammatory cells in IMQ-induced psoriatic skin of hIL-26Tg mice (Figure 7). Invasion of blood vessels in the dermis, which is one of the histological hallmarks of psoriatic skin lesions, induces the deterioration of psoriatic skin lesions.40 In addition, mRNA expression levels of IL-1β and IL-6, key cytokines for inducing and enhancing Th17 response, were significantly increased in the skin lesions from hIL-26Tg mice as compared with ΔCNS-77 Tg mice (Figure S2(a)). Scala et al.16 have recently shown similar results through different experimental approaches. We analyzed the expression levels of cytokines in the skin lesions of hIL-26 BAC Tg mice, whereas Scala et al. used skin lesions obtained from hidradenitis suppurativa patients. Addition of commercial anti-human IL-26 pAb to an ex vivo culture of human skin samples significantly decreased the expression levels of IL-1β and IL-6 in the skin, while IL-23 and IL-17A were not affected. Although we have not identified the main source of IL-1β and IL-6 in the skin lesion of IMQ-treated hIL-26Tg mice, monocytes and fibroblasts may be possible candidates from recent findings.7,17,41
In contrast to IL-1β and IL-6, there was no significant difference in the expression levels of IL-17A and TNF between hIL-26Tg mice and control mice, at least in the total skin samples (Figure S2(a)). To analyze the precise effect of IL-26 on the T cell phenotype in inflammatory diseases, detailed analyses of T cell subsets at the inflammatory sites and gene expression profiles following purification of T cells would be essential for future studies. Moreover, multiple functions of human IL-26 have been reported. Due to clustered cationic charges, IL-26 can bind extracellular DNA/RNA released from bacteria or dying cells to form complexes that are highly protected from degradation by extracellular DNase/RNase and promote DNA/RNA immunogenicity. These IL-26-DNA complexes trigger IFN-α production from plasmacytoid dendritic cells via activation of TLR9, and enhance the expression of IL-1β, IL-6 and IFN-β in monocytes and CXCL8 in neutrophils in a STING- and inflammasome-dependent manner.14,41 In addition, IL-26 enhances the chemotactic response of neutrophils toward bacterial stimuli and IL-8.8 Moreover, IL-26 is associated with monocyte and NK cell activation.7,12 Since IL-26 is broadly associated with innate immune activation, the effects of neutralizing anti-IL-26 mAb on these functions of IL-26 need to be investigated in future studies.
In conclusion, our current work indicates that IL-20RA is differentially involved in IL-26-mediated stimulation depending on cell types, and anti-IL-20RA blocking antibody is insufficient to inhibit IL-26-mediated stimulation. Our screening methods enabled us to develop 4 novel neutralizing anti-human IL-26 mAbs. Among them, both 31–4 mAb and 69–10 mAb clearly suppressed the excessive angiogenesis and inflammation in the skin lesions in a IMQ-induced psoriasis-like murine model. Our data strongly suggest that IL-26-targeted therapy may be an effective novel therapeutic approach for diverse chronic inflammatory diseases, including psoriasis and chronic GVHD.
Materials and methods
Cell culture
The human colon cancer cell line COLO205 was purchased from RIKEN Bioresource Center (Ibaraki, Japan) and grown in RPMI 1640 medium supplemented with 10% FBS. The human keratinocyte cell line HaCaT was a kind gift from Dr. Nobuhiro Nakano (Juntendo University, Tokyo, Japan) and grown in DMEM medium supplemented with 10% FBS. HUVEC were purchased from LONZA (Walkersville, MD) and grown in EGM-2 medium (LONZA). Cells were cultured at 37°C in a humidified 5% CO2 incubator.
Antibodies and reagents
To characterize the novel mouse anti-human IL-26 mAbs, purified mouse anti-human IL-26 mAb (clone 510414), goat anti-human IL-26 pAb (AF1375), goat anti-human IL-20RA pAb (AF1176), or goat anti-human IL-10RB pAb (AF874) purchased from R&D Systems (Minneapolis, MN) were used for comparison. Mouse IgG1,κ isotype control mAb (clone MG1-45) purchased from BioLegend (San Diego, CA) or goat IgG isotype control pAb (AB-108-C) purchased from R&D Systems was used as a negative control. For Western blot analysis, rabbit anti-human phospho-STAT3 (Tyr705) pAb (AF4607) was purchased from R&D Systems. Rabbit anti-human/mouse/rat/monkey STAT3 mAb (clone 79D7) was purchased from Cell Signaling Technology (Danvers, MA). Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG was purchased from GE Healthcare (Buckinghamshire, UK). For immunofluorescence staining of mouse skin sections, rabbit anti-mouse CD31 pAb (ab28364) was purchased from Abcam (Cambridge, U.K.). Alexa Fluor 488-conjugated donkey anti-rabbit IgG pAb (A21206) was purchased from Thermo Fisher Scientific (Waltham, MA). Recombinant human IL-26 monomer and dimer were purchased from R&D Systems. Recombinant human IL-6, IL-10 and IL-22 were purchased from BioLegend. For binding assay, ultra-pure LPS from Escherichia coli O111:B4 and purified LTA from Staphylococcus aureus were purchased from InvivoGen (San Diego, CA). Mouse anti-E. coli and LPS mAb (clone 1C6) purchased from Chondrex, Inc. (Redmond, WA) and mouse anti-native LTA mAb (clone 55) purchased from Novus Biologicals (Centennial, CO) were biotinylated utilizing Biotin Labeling Kit-NH2 (Dojindo Laboratories, Kumamoto, Japan). For analyzing antimicrobial activity of IL-26, LL-37 fragment (18–37 amino acids) purchased from KareBay Biochem, Inc. (Monmouth Junction, NJ) and hBD3 purchased from the Peptide Institute, Inc. (Osaka, Japan) were used for comparison.
Mice
Female BALB/c mice were purchased from CLEA Japan (Tokyo, Japan). C57BL/6 mice carrying a 190-kb BAC transgene with human IFNG and IL26 gene (hIL-26Tg) and a BAC Tg-deleting CNS positioned at 77 kb upstream of the IFNG transcription start site, an enhancer element required for IL-26 mRNA expression (ΔCNS-77 Tg) were developed in Thomas Aune’s laboratory.24,25 The hIL-26Tg mice exhibited production of human IL-26 by CD4 T cells under Th1- or Th17-polarizing conditions, whereas expression of human IL-26 was completely abrogated in ΔCNS-77 Tg mice carrying human IFNG transgene with deleting IL26 transcription.25 All mice used in this study were housed in a specific pathogen-free facility in micro-isolator cages, and used at 8–12 weeks of age.
Development of hybridomas and monoclonal anti-human IL-26 antibodies
Forty μg of recombinant human IL-26 monomer per 50 μl of phosphate-buffered saline (PBS) was emulsified with 50 μl of adjuvant, TiterMax Gold (TiterMax USA, Norcross, GA). A 6-wk-old female BALB/c mouse was immunized via subcutaneous administration with 100 μl of the emulsion five times every two weeks and finally intravenously injected with half volume of the emulsion. Three days after the final immunization, the spleen was removed and 1 × 108 spleen cells were fused with 1 × 108 P3U1 myeloma cells by using polyethylene glycol 4000 (Merck, Darmstadt, Germany) and were cultured in serum-free GIT medium (Wako Pure Chemicals, Osaka, Japan), 5% BriClone (NICB, Dublin, Ireland) and HAT (Invitrogen, Carlsbad, CA) in 96-well flat-bottom plates (Costar, Corning Incorporated, Corning, NY). Hybridoma supernatants were first screened for selective reactivity with human IL-26 by ELISA. The supernatants containing mAbs that were reactive to human IL-26 were next screened for neutralization assay. The hybridomas were cloned by limiting dilution. MAbs were purified from the supernatants using Protein A IgG Purification Kit (Pierce, Rockford, IL).
ELISA
The 96-well immunoplates (NUNC, Roskilde, Denmark) were coated with recombinant human IL-26 or IL-10 in carbonate bicarbonate buffer (2, 5, 10, 20, 50, 100 ng/well) or buffer alone as a negative control at 4°C overnight. Each well of the plate was blocked with 2% BlockAce (DS Pharma Biomedical, Osaka, Japan) in deionized distilled water for 1 hr at room temperature (RT), and then incubated with 10 μg/ml of purified novel mouse anti-human IL-26 mAb (2–2, 20–3, 31–4 or 69–10) or commercial mouse anti-human IL-26 mAb in RPMI 1640 medium for 1 hr at RT, and subsequently incubated with HRP-conjugated goat anti-mouse Ig pAb (BD Biosciences) in 1% BlockAce solution for 1 hr at RT. Tetramethylbenzidine (TMB) Peroxidase Substrate (KPL, Gaithersburg, MD) was finally added to each well and the reaction was stopped by 2 N H2SO4. The absorbance at 450 nm/570 nm was measured in a Microplate Reader (Bio-Rad, Hercules, CA) and data were analyzed with Microplate Manager 6 software (Bio-Rad).
Flow cytometry
COLO205 cells (5 x 104) were stimulated with recombinant human IL-26 (20 ng/ml) in the presence or absence of neutralizing or blocking antibody in 120 μl of RPMI 1640 medium in 96-well flat-bottom plates for 24 hr at 37°C. After stimulation, cells were collected and washed in PBS containing 1% FBS and 0.1% sodium azide (FCM buffer), and stained with phycoerythrin (PE)-labeled mouse anti-human ICAM-1 mAb (clone 15.2, Bay Bioscience, Kobe, Japan) or PE-labeled mouse IgG1,κ isotype control (clone MOPC-21, BioLegend) for 25 min at 4°C. Acquisition was performed using FACSCalibur (BD Biosciences) and data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Western blotting
To analyze phosphorylation of STAT3, COLO205 cells (1 x 106) were stimulated with recombinant human IL-26 (20 ng/ml), IL-6 (20 ng/ml) or IL-22 (20 ng/ml) in 1.5 ml of RPMI 1640 medium in 12-well plates (Corning) for 5, 10, 30 or 60 min at 37°C. After stimulation, cells were collected and lysed in RIPA buffer supplemented with 2% protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and 1x PhosSTOP (Roche Diagnostics, Tokyo, Japan), being resolved by SDS-PAGE in reducing condition (25 μg/Lane) and immunoblotted using anti-phosphorylated STAT3 antibody. The methods for Western blotting were detailed previously.42 For reprobing, the membranes were submerged in a stripping buffer. After a stripping procedure, the membranes were reprobed with anti-pan STAT3 antibody. The images were taken using luminescent image analyzer LAS 4000 (GE Healthcare, Pittsburgh, PA), and data were analyzed with image reader LAS 4000 and Multi Gauge software (GE Healthcare).
Quantitative real-time RT-PCR
HaCaT cells (3 x 105) were incubated overnight at 37°C in DMEM medium in 24-well plates (Corning). The next day, cells were stimulated with recombinant human IL-26 (20 ng/ml) in the presence or absence of neutralizing antibody in 750 μl of DMEM medium for 6 hr at 37°C. After stimulation, cells were lysed and total RNA was extracted by the use of RNeasy Mini kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). The methods for cDNA synthesis and quantitative real-time RT-PCR were detailed previously.43 Sequences of primers used in quantitative real-time RT-PCR analysis are shown in Table S1.
Proliferation assay for HUVEC
HUVEC (5 x 103) were incubated overnight at 37°C in EGM-2 medium containing 2% FBS and half volume of adjunctive growth factors in 96-well flat-bottom plates. The next day, cells were stimulated with recombinant human IL-26 (10 ng/ml) in the presence or absence of neutralizing antibody in EGM-2 medium containing 2% FBS and no growth factors for 48 hr at 37°C. Cell growth was measured as cell confluence using IncuCyte ZOOM (Essen Biosciense, Ann Arbor, MI).
Tube formation assay
HUVEC (1 x 106) were incubated overnight at 37°C in EGM-2 medium containing 2% FBS and no growth factors in 100-mm dish. The next day, cells (1.5 x 104) were seeded on 50 μl of Cultex Basement Membrane Extract (R&D Systems) in 96-well flat-bottom plates. Seeded cells were stimulated with recombinant human IL-26 (10 ng/ml) in the presence or absence of neutralizing antibody for 9 hr at 37°C. Cell growth was observed as cell sprouts formation using IncuCyte ZOOM. Tube form length was measured using the MetaMorph image analysis system (Molecular Device, Sunnyvale, CA).
Binding assay
The 96-well immunoplates were coated with 2 μg/ml of recombinant human IL-10, IL-22 or IL-26 in PBS or PBS alone as a negative control at 4°C overnight. Each well of the plate was blocked with 2% BlockAce in deionized distilled water for 1 hr at RT, and then incubated with ultra-pure LPS (1, 2, 5 μg/ml) or purified LTA (0.5, 1, 2 μg/ml) in PBS for 2 hr at RT, and next incubated with 1 μg/ml of biotinylated anti-LPS mAb or 0.2 μg/ml of biotinylated anti-LTA mAb in PBS for 1 hr at RT, and subsequently incubated with Streptavidin-HRP (BD Biosciences) in PBS for 1 hr at RT. Colorimetric methods and data analysis were described in the ELISA section.
CFU assay
Escherichia coli (ATCC 8739), and Staphylococcus aureus (ATCC 29213) were cultured overnight at 37°C in trypticase soy broth (BD Biosciences) in 14-ml polystyrene round-bottom tube (Corning) to achieve mid-logarithmic phase growth. Bacterial concentrations were measured by spectrophotometry at 600 nm and diluted in Mueller-Hinton broth (BD Biosciences) to a final concentration of 1–2 × 104 CFU/ml. Fifty μl of the diluted bacterial suspension was cultured in 96-well round-bottom plates (Corning), and then 50 μl of recombinant human IL-26, LL-37 or hBD3 diluted in Mueller-Hinton broth was added to these cultures. After 2, 4, 6, 8 or 24 hrs of incubation at 37°C, serial dilutions of bacterial cultures were plated onto lysogeny broth (LB) agar plates. The number of colonies formed after overnight incubation was counted by two independent investigators.
IMQ-induced psoriasis model
Mice received a daily topical dose of 20 mg 5% IMQ cream (Beselna Cream; Mochida Pharmaceutical, Tokyo, Japan) on their shaved back for 5 consecutive days. For mAb treatment, IL-26 mAb (31–4 mAb alone, 69–10 mAb alone or combination of 4 mAbs) or mouse IgG1 isotype control was diluted in sterile PBS at 1 mg/ml and 200 μl (200 μg) was injected intraperitoneally on day 0 and day 3. The severity of inflammation of the back skin was measured by an objective scoring system based on the clinical PASI. Erythema, scaling, and thickness were scored independently on a score from 0 to 4: 0, none; 1, slight; 2, moderate; 3, marked; 4, very marked. The cumulative score (erythema plus scaling plus thickness) served as a measure of the severity of inflammation (score 0–12).44 H&E staining and immunofluorescence staining of skin lesions, and RNA isolation from skin lesions were conducted as described previously.13
Statistics
Data were analyzed by two-tailed Student t test for two-group comparison or by one-way ANOVA test with Tukey’s for multiple comparison testing. The assay was performed in triplicates, and data are presented as mean ± S.D. of triplicate samples of the representative experiment, or mean ± S.E. of triplicate samples of independent experiments. Significance was analyzed using GraphPad Prism 6 (GraphPad Software, San Diego, CA) and values of p < 0.01 were considered significant and are indicated in the corresponding figures and figure legends.
Study approval
Animal experiments were conducted following protocols approved by the Animal Care and Use Committees at Juntendo University (Tokyo, Japan).
Funding Statement
This study was supported in part by a grant of the Ministry of Health, Labor, and Welfare, Japan (Grant Number 180101-01 (C.M.)), JSPS KAKENHI Grant Numbers JP16H05345 (C.M.), JP18H02782 (K.O.), JP17K10008 (R.H.), JP18H06169 (T.I.). This work was also supported by a research grant from the Japanese Society of Hematology (R.H.), and a research grant from the Japan Research Institute of Industrial Science (K.O.).
Abbreviations
- BAC
bacterial artificial chromosome
- CNS
conserved noncoding sequence
- ELISA
enzyme-linked immunosorbent assay
- FBS
fetal bovine serum
- FGF
fibroblast growth factor
- GVHD
graft-versus-host disease
- hIL-26Tg
human IL-26 transgenic
- HRP
horseradish peroxidase
- IFN
interferon
- IL
interleukin
- IMQ
imiquimod
- hBD3
human β-defensin 3
- HUVEC
human umbilical vein endothelial cells
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- mAb
monoclonal antibody
- NK
natural killer
- pAb
polyclonal antibodies
- PBS
phosphate-buffered saline
- RT
room temperature
- TLR
Toll-like receptor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Acknowledgments
The authors thank members of Atopy (Allergy) Research Center (Juntendo University Graduate School of Medicine, Japan), members of the Laboratory of Morphology and Image Analysis, Research Support Center (Juntendo University Graduate School of Medicine, Japan), and members of the Laboratory of Cell Biology, Research Support Center (Juntendo University Graduate School of Medicine, Japan) for technical assistance and for the use of the experimental apparatus.
Disclosure statement
Ryo Hatano, Takumi Itoh, Kei Ohnuma and Chikao Morimoto are inventors of the novel anti-human IL-26 mAbs 2-2, 20-3, 31-4 and 69-10, and are now applying for a patent regarding their invention (Anti-human IL-26 antibody. Application 2017-12-01 JP2017231439). Other authors declare no competing financial interests associated with this manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
References
- 1.Knappe A, Hor S, Wittmann S, Fickenscher H.. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J Virol. 2000;74:3881–87. doi: 10.1128/jvi.74.8.3881-3887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotenko SV. The family of IL-10-related cytokines and their receptors: related, but to what extent? Cytokine Growth Factor Rev. 2002;13:223–40. [DOI] [PubMed] [Google Scholar]
- 3.Donnelly RP, Sheikh F, Dickensheets H, Savan R, Young HA, Walter MR. Interleukin-26: an IL-10-related cytokine produced by Th17 cells. Cytokine Growth Factor Rev. 2010;21:393–401. doi: 10.1016/j.cytogfr.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–42. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol. 2002;168:5397–402. doi: 10.4049/jimmunol.168.11.5397. [DOI] [PubMed] [Google Scholar]
- 6.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–57. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 7.Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, Hoppe E, Barre B, Audran M, Bouvard B, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012;10:e1001395. doi: 10.1371/journal.pbio.1001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Che KF, Tengvall S, Levanen B, Silverpil E, Smith ME, Awad M, Vikstrom M, Palmberg L, Qvarfordt I, Skold M, et al. Interleukin-26 in antibacterial host defense of human lungs. Effects on neutrophil mobilization. Am J Respir Crit Care Med. 2014;190:1022–31. doi: 10.1164/rccm.201404-0689OC. [DOI] [PubMed] [Google Scholar]
- 9.Che KF, Kaarteenaho R, Lappi-Blanco E, Levanen B, Sun J, Wheelock A, Palmberg L, Skold CM, Linden A. Interleukin-26 production in human primary bronchial epithelial cells in response to viral stimulation: modulation by Th17 cytokines. Mol Med. 2017:23. doi: 10.2119/molmed.2016.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hor S, Pirzer H, Dumoutier L, Bauer F, Wittmann S, Sticht H, Renauld JC, de Waal Malefyt R, Fickenscher H. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. 2004;279:33343–51. doi: 10.1074/jbc.M405000200. [DOI] [PubMed] [Google Scholar]
- 11.Dambacher J, Beigel F, Zitzmann K, De Toni EN, Goke B, Diepolder HM, Auernhammer CJ, Brand S. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58:1207–17. doi: 10.1136/gut.2007.130112. [DOI] [PubMed] [Google Scholar]
- 12.Miot C, Beaumont E, Duluc D, Le Guillou-Guillemette H, Preisser L, Garo E, Blanchard S, Hubert Fouchard I, Creminon C, Lamourette P, et al. IL-26 is overexpressed in chronically HCV-infected patients and enhances TRAIL-mediated cytotoxicity and interferon production by human NK cells. Gut. 2015;64:1466–75. doi: 10.1136/gutjnl-2013-306604. [DOI] [PubMed] [Google Scholar]
- 13.Itoh T, Hatano R, Komiya E, Otsuka H, Narita Y, Aune TM, Dang NH, Matsuoka S, Naito H, Tominaga M, et al. Biological effects of IL-26 on T cell-mediated skin inflammation, including psoriasis. J Invest Dermatol. 2019;139:878–89. doi: 10.1016/j.jid.2018.09.037. [DOI] [PubMed] [Google Scholar]
- 14.Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, Conrad C, Gregorio J, Le Roy D, Roger T, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. 2015;16:970–79. doi: 10.1038/ni.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dang AT, Teles RM, Weiss DI, Parvatiyar K, Sarno EN, Ochoa MT, Cheng G, Gilliet M, Bloom BR, Modlin RL. IL-26 contributes to host defense against intracellular bacteria. J Clin Invest. 2019;130:1926–39. doi: 10.1172/JCI99550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scala E, Di Caprio R, Cacciapuoti S, Caiazzo G, Fusco A, Tortorella E, Fabbrocini G, Balato A. A new T helper 17 cytokine in hidradenitis suppurativa: antimicrobial and proinflammatory role of interleukin-26. Br J Dermatol. 2019. doi: 10.1111/bjd.17854. [DOI] [PubMed] [Google Scholar]
- 17.Fujii M, Nishida A, Imaeda H, Ohno M, Nishino K, Sakai S, Inatomi O, Bamba S, Kawahara M, Shimizu T, et al. Expression of Interleukin-26 is upregulated in inflammatory bowel disease. World J Gastroenterol. 2017;23:5519–29. doi: 10.3748/wjg.v23.i30.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heftdal LD, Andersen T, Jaehger D, Woetmann A, Ostgard R, Kenngott EE, Syrbe U, Sieper J, Hvid M, Deleuran B, et al. Synovial cell production of IL-26 induces bone mineralization in spondyloarthritis. J Mol Med (Berl). 2017;95:779–87. doi: 10.1007/s00109-017-1528-2. [DOI] [PubMed] [Google Scholar]
- 19.Esendagli G, Kurne AT, Sayat G, Kilic AK, Guc D, Karabudak R. Evaluation of Th17-related cytokines and receptors in multiple sclerosis patients under interferon beta-1 therapy. J Neuroimmunol. 2013;255:81–84. doi: 10.1016/j.jneuroim.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Konradsen JR, Nordlund B, Levanen B, Hedlin G, Linden A. The cytokine interleukin-26 as a biomarker in pediatric asthma. Respir Res. 2016;17:32. doi: 10.1186/s12931-016-0351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaabachi W, Bouali E, Berraies A, Dhifallh IB, Hamdi B, Hamzaoui K, Hamzaoui A. Interleukin-26 is overexpressed in Behcet’s disease and enhances Th17 related -cytokines. Immunol Lett. 2017;190:177–84. doi: 10.1016/j.imlet.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 22.Caiazzo G, Di Caprio R, Lembo S, Raimondo A, Scala E, Patruno C, Balato A. IL-26 in allergic contact dermatitis: resource in a state of readiness. Exp Dermatol. 2018;27:681–84. doi: 10.1111/exd.13521. [DOI] [PubMed] [Google Scholar]
- 23.Savchenko L, Mykytiuk M, Cinato M, Tronchere H, Kunduzova O, Kaidashev I. IL-26 in the induced sputum is associated with the level of systemic inflammation, lung functions and body weight in COPD patients. Int J Chron Obstruct Pulmon Dis. 2018;13:2569–75. doi: 10.2147/COPD.S164833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins PL, Chang S, Henderson M, Soutto M, Davis GM, McLoed AG, Townsend MJ, Glimcher LH, Mortlock DP, Aune TM. Distal regions of the human IFNG locus direct cell type-specific expression. J Immunol. 2010;185:1492–501. doi: 10.4049/jimmunol.1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins PL, Henderson MA, Aune TM. Lineage-specific adjacent IFNG and IL26 genes share a common distal enhancer element. Genes Immun. 2012;13:481–88. doi: 10.1038/gene.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnuma K, Hatano R, Aune TM, Otsuka H, Iwata S, Dang NH, Yamada T, Morimoto C. Regulation of pulmonary graft-versus-host disease by IL-26+CD26+CD4 T lymphocytes. J Immunol. 2015;194:3697–712. doi: 10.4049/jimmunol.1402785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohnuma K, Hatano R, Itoh T, Iwao N, Dang NH, Morimoto C. Role of IL-26+CD26+CD4 T cells in pulmonary chronic graft-versus-host disease and treatment with caveolin-1-Ig Fc conjugate. Crit Rev Immunol. 2016;36:239–67. doi: 10.1615/CritRevImmunol.2016018772. [DOI] [PubMed] [Google Scholar]
- 28.You W, Tang Q, Zhang C, Wu J, Gu C, Wu Z, Li X. IL-26 promotes the proliferation and survival of human gastric cancer cells by regulating the balance of STAT1 and STAT3 activation. PloS One. 2013;8:e63588. doi: 10.1371/journal.pone.0063588. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Stephen-Victor E, Fickenscher H, Bayry J. IL-26: an emerging proinflammatory member of the IL-10 cytokine family with multifaceted actions in antiviral, antimicrobial, and autoimmune responses. PLoS Pathog. 2016;12:e1005624. doi: 10.1371/journal.ppat.1005624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boehncke WH, Schon MP. Psoriasis. Lancet. 2015;386:983–94. doi: 10.1016/S0140-6736(14)61909-7. [DOI] [PubMed] [Google Scholar]
- 31.Lowes MA, Bowcock AM, Krueger JG. Pathogenesis and therapy of psoriasis. Nature. 2007;445:866–73. doi: 10.1038/nature05663. [DOI] [PubMed] [Google Scholar]
- 32.Golden JB, McCormick TS, Ward NL. IL-17 in psoriasis: implications for therapy and cardiovascular co-morbidities. Cytokine. 2013;62:195–201. doi: 10.1016/j.cyto.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki T, Hirakawa S, Shimauchi T, Ito T, Sakabe J, Detmar M, Tokura Y. VEGF-A promotes IL-17A-producing gammadelta T cell accumulation in mouse skin and serves as a chemotactic factor for plasmacytoid dendritic cells. J Dermatol Sci. 2014;74:116–24. doi: 10.1016/j.jdermsci.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 34.Honorati MC, Neri S, Cattini L, Facchini A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2006;14:345–52. doi: 10.1016/j.joca.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Reszke R, Szepietowski JC. Secukinumab in the treatment of psoriasis: an update. Immunotherapy. 2017;9:229–38. doi: 10.2217/imt-2016-0128. [DOI] [PubMed] [Google Scholar]
- 36.Ariza ME, Williams MV, Wong HK. Targeting IL-17 in psoriasis: from cutaneous immunobiology to clinical application. Clin Immunol. 2013;146:131–39. doi: 10.1016/j.clim.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Interleukin VM. 17 is a chief orchestrator of immunity. Nat Immunol. 2017;18:612–21. doi: 10.1038/ni.3742. [DOI] [PubMed] [Google Scholar]
- 38.Gordon KB, Blauvelt A, Papp KA, Langley RG, Luger T, Ohtsuki M, Reich K, Amato D, Ball SG, Braun DK, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345–56. doi: 10.1056/NEJMoa1512711. [DOI] [PubMed] [Google Scholar]
- 39.Esfahani K, Miller WH Jr.. Reversal of autoimmune toxicity and loss of tumor response by interleukin-17 blockade. N Engl J Med. 2017;376:1989–91. doi: 10.1056/NEJMc1703047. [DOI] [PubMed] [Google Scholar]
- 40.Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol. 2009;90:232–48. doi: 10.1111/j.1365-2613.2009.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poli C, Augusto JF, Dauve J, Adam C, Preisser L, Larochette V, Pignon P, Savina A, Blanchard S, Subra JF, et al. IL-26 confers proinflammatory properties to extracellular DNA. J Immunol. 2017;198:3650–61. doi: 10.4049/jimmunol.1600594. [DOI] [PubMed] [Google Scholar]
- 42.Hatano R, Yamada T, Madokoro H, Otsuka H, Komiya E, Itoh T, Narita Y, Iwata S, Yamazaki H, Matsuoka S, et al. Development of novel monoclonal antibodies with specific binding affinity for denatured human CD26 in formalin-fixed paraffin-embedded and decalcified specimens. PloS One. 2019;14:e0218330. doi: 10.1371/journal.pone.0218330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatano R, Ohnuma K, Otsuka H, Komiya E, Taki I, Iwata S, Dang NH, Okumura K, Morimoto C. CD26-mediated induction of EGR2 and IL-10 as potential regulatory mechanism for CD26 costimulatory pathway. J Immunol. 2015;194:960–72. doi: 10.4049/jimmunol.1402143. [DOI] [PubMed] [Google Scholar]
- 44.Alrefai H, Muhammad K, Rudolf R, Pham DA, Klein-Hessling S, Patra AK, Avots A, Bukur V, Sahin U, Tenzer S, et al. NFATc1 supports imiquimod-induced skin inflammation by suppressing IL-10 synthesis in B cells. Nat Commun. 2016;7:11724. doi: 10.1038/ncomms11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.