

Abstract
Here, we report the neuroimaging findings and neurological changes in 17 unpublished patients with Macrocephaly–Capillary Malformation (M–CM). This syndrome has been traditionally known as Macrocephaly–Cutis Marmorata Telangiectatica Congenita (M–CMTC), but we explain why M–CM is a more accurate term for this overgrowth syndrome. We analyzed the 17 patients with available brain MRI or CT scans and compared their findings with features identified by a comprehensive review of published cases. White matter irregularities with increased signal on T2-weighted images were commonly observed findings. A distinctive feature in more than half the patients was cerebellar tonsillar herniation associated with rapid brain growth and progressive crowding of the posterior fossa during infancy. In four such cases, we confirmed that the tonsillar herniation was an acquired event. Concurrently, with the development of these findings, ventriculomegaly (frequently obstructive) and dilated dural venous sinuses were observed in conjunction with prominent Virchow–Robin spaces in many of those in whom cerebellar tonsil herniation had developed. We postulate that this constellation of unusual features suggests a dynamic process of mechanical compromise in the posterior fossa, perhaps initiated by a rapidly growing cerebellum, which leads to congestion of the venous drainage with subsequently compromised cerebrospinal fluid reabsorption, all of which increases the posterior fossa pressure and leads to acquired tonsillar herniation. We make a distinction between congenital Chiari I malformation and acquired cerebellar tonsil herniation in this syndrome. We also observed numerous examples of abnormal cortical morphogenesis, including focal cortical dysplasia, polymicrogyria which primarily involved the perisylvian and insular regions, and cerebral and/or cerebellar asymmetric overgrowth. Other findings included a high frequency of cavum septum pellucidum or vergae, thickened corpus callosum, prominent optic nerve sheaths and a single case of venous sinus thrombosis. One patient was found to have a frontal perifalcine mass resembling a meningioma at age 5 years. This is the second apparent occurrence of this specific tumor in M–CM.
Keywords: Macrocephaly–Cutis Marmorata Telangiectatica Congenita (M–CMTC), white matter irregularities with increased signal on T2-weighted images, acquired cerebellar tonsil herniation, congenital Chiari I malformation, rapidly growing cerebellum, progressive posterior fossa crowding, ventriculomegaly, obstructive ventriculomegaly, dural venous sinuses, prominent Virchow–Robin spaces, venous drainage congestion, compromised cerebrospinal fluid reabsorption, abnormal cortical morphogenesis, focal cortical dysplasia, polymicrogyria, cerebral and/or cerebellar asymmetric overgrowth, cavum septum pellucidum or vergae, thickened corpus callosum, prominent optic nerve sheaths, venous sinus thrombosis, meningioma
INTRODUCTION
Macrocephaly–Capillary Malformation (M–CM) is a well-described syndrome characterized by prenatal overgrowth, variable somatic and cerebral asymmetry, primary megalencephaly, characteristic facial features, an abnormal neurocognitive profile, lax joints, thickened subcutaneous tissue which often has a dough-like feel, cutaneous vascular malformations and/or cutis marmorata, and digital anomalies including syndactyly or polydactyly (Fig. 1A,B).
Fig. 1.

Patient 5 (left) and his unaffected twin sister at ages 12 months (A) and 40 months (B). The boy has classic features of M–CM including left sided hemihyperplasia, typical facial features, toe syndactyly, and connective tissue involvement with loose, redundant skin. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Problems With Nomenclature
Traditionally, this syndrome has been called Macrocephaly–Cutis Marmorata Telangiectatica Congenita (M–CMTC) [Moore et al., 1997; Lapunzina et al., 2004]. However, Toriello and Mulliken [2007] renamed the disorder as Macrocephaly–Capillary Malformation (M–CM) because cutis marmorata telangiectatica congenita is not a feature of this disorder. The term cutis marmorata is the common cutaneous marbling found in normal white infants. The term cutis marmorata telangiectatica congenita (CMTC) is a distinct vascular lesion of the limbs and trunk. It has a serpiginous appearance, is reticulated, and commonly ulcerates The lesion improves during infancy, but atrophy and discoloration are characteristic. Macrocephaly–Capillary Malformation (M–CM) is a more accurate name for this disorder because the lesions are patchy, reticular, and represent a type of capillary malformation [Toriello and Mulliken, 2007]. We have adopted this term.
Brain Abnormalities and Neuroimaging
Ever since the delineation of this disorder in 1997 [Clayton-Smith et al., 1997; Moore et al., 1997] there has been an increasing appreciation of distinctive structural and functional neurologic anomalies in this condition. Indeed, analysis of approximately 70 published cases [see Lapunzina et al., 2004 for review] demonstrates that neonatal hypotonia or developmental delays in the mild-to-severe range are present in most patients with a firm diagnosis of M–CM. Sufficient delineation has occurred to allow a classic case to be readily recognizable on a clinical basis. However, this syndrome appears rare enough that many aspects of the natural history and issues of medical management remain poorly understood. Similarly, despite numerous clinical reports describing a variety of structural brain abnormalities, a thorough understanding of what truly defines the central nervous system phenotype and its alterations over time is lacking.
Several challenges have made characterization of a CNS phenotype in this syndrome difficult. Specific neurologic abnormalities are not usually the focus of most reports. Moreover, some case descriptions fail to mention any neuroimaging studies. In those instances where neuroimaging data are available, some patients received only a single head imaging study, so data about how the brain may change over time in individuals with M–CM are lacking. In most instances, literature cases received neuroimaging at a relatively young age, so less is known about the brains of adolescents and adults with this syndrome compared to infants and young children. For some case reports, the exact age at which the study was performed is not stated, and this is a critical omission because some observations, such as irregularities in myelination, are age-dependent. Among the earlier literature reports, patients often received head CTs rather than MRI scans, so details about cortical abnormalities or descriptions of the posterior fossa contents are generally lacking in those particular descriptions. While most reports provide pertinent positive findings, pertinent negatives are often not clarified. Lastly, it is not always clear if the same neuroradiologist is interpreting the findings on multiple patients in any given article, or if the literature simply incorporates neuroimaging data by report into each clinical description. This raises the possibility of variability in interpretation.
To further clarify the neurologic structural and functional abnormalities of this syndrome, we undertook a systematic longitudinal analysis of neuroimaging studies performed on 17 unpublished patients. Analysis was provided by a single experienced neuroradiologist (BP), an expert on genetic cortical abnormalities (BD), and a pediatric neurosurgeon (MD). To our knowledge, this cohort represents the largest group of patients with M–CM studied in detail by such a team of experts, and this is the first report specifically looking at how brain abnormalities may evolve with patient age in this syndrome.
Volumetric analysis of quantitative changes in the posterior fossa over time was performed when digital serial imaging studies were available.
NEUROIMAGING FINDINGS IN THE LITERATURE
Literature Review
Though there have been isolated reports of patients with probable M–CM that presented as unknown disorders or unusual examples of rare known disorders prior to formal delineation of this syndrome, the clinical phenotype was first described through the report of 22 patients in two different papers published in 1997 [Clayton-Smith et al., 1997; Moore et al., 1997]. To date, these initial patient reports represent nearly one third of all reported patients with M–CM. However, even from these seminal studies it was clear that a distinctive and recurrent brain phenotype was present. Eight patients from the Clayton-Smith study had reported neuroimaging; although the Moore study did not explicitly state that all patients received neuroimaging, it did identify all patients as having the presence or absence of hydrocephalus, and additional neuroimaging findings were reported in particular cases. All patients in the Clayton-Smith report who underwent neuroimaging had enlarged ventricles (variably described as ventriculomegaly, ventricular dilatation/enlargement, or hydrocephalus); in the Moore report, eight patients had similar findings. Furthermore, two cases in the Clayton-Smith study had asymmetric ventricular dilatation. Other findings among these 22 patients included hemimegalencephaly in three cases, cavum septum pellucidum or vergae in three cases, white matter or myelination defects in two cases, and singular cases of Chiari I malformation, diffuse frontal lobe atrophy, sagittal sinus thrombosis, localized arteriovenous malformation, “cortical dysplasia” in the area of perisylvian fissure, and a case of a parietal defect resembling distal cerebral infarction.
Carcao et al. [1998] reported a male infant with a detailed description of MRI findings at about 1 year of age. Neuroimaging disclosed multiple abnormalities, many of which had been observed in the Moore and Clayton-Smith reports including left hemimegalencephaly, asymmetrically enlarged ventricles, bilateral cortical abnormalities consisting of dysgenetic, thickened frontal cortex, and “bulky” white matter with increased T2 signal. This child had left-sided hemihyperplasia involving the body and face, and the left cerebral cortex demonstrated a greater degree of cortical irregularity. In addition, the MRI showed many other distinctive findings, including a thick-appearing corpus callosum, “hydropic” optic nerves with thin optic chiasma, and a cerebellum which appeared “disproportionately large” for the size of the posterior fossa, along with inferior displacement of the cerebellar tonsils (Chiari I). That same year, Vogels et al. [1998] reported one patient with M–CM who also had Chiari I malformation and another who had asymmetric ventriculomegaly.
Adding to the interesting findings of Carcao et al. [1998], Thong et al. [1999] provided an extensive description of a patient with macrocephaly, right hemihyperplasia of the body and face, cutis marmorata and numerous cutaneous capillary malformations, hemimegalencephaly, dysmorphic facial features, bilateral 2–3 toe syndactyly with a wide gap between the first and second toes, hypoplastic toenails, hypotonia, global developmental delay, thickened subcutaneous tissue, streaky pigmentation along Blaschko’s lines, hypermobile knee joints, and intestinal lymphangiectasia. While M–CM was considered, the authors thought their patient represented a “diagnostic dilemma” similar to a patient reported by Reardon et al. [1996]. However, by considering the features in Thong’s patient, and studying the clinical photographs provided, M–CM appears to be the most appropriate diagnosis. This patient had multiple brain MRI scans. The first was performed early in life and showed right ventriculomegaly, right cortical dysplasia with abnormal sulcation suggestive of polymicrogyria, and enlargement of the white matter of the right cerebral hemisphere. A repeat head MRI showed superior sagittal and transverse sinus thrombosis for which no cause was identified. A third MRI at 6 months of age showed a new and unexpected finding of Chiari I to the level of the C2/C3 intervertebral disc. An MRI of the brain at 21 months disclosed minimal progression of the cerebellar tonsillar herniation from the 6- month scan, though there was now clearly abnormal periventricular and deep white matter T2 hyperintense signal alteration suggestive of a dysmyelinating process.
Three patients with M–CM were reported by Yano and Watanabe [2001] who had severe clinical courses. Patient 1 had a brain MRI at 11 months which showed “delayed” myelination and a disproportionately large-appearing cerebellum with Chiari I. She died suddenly at 33 months of age during sleep without clear explanation. Patient 2 had atrial flutter in the newborn period that was incompletely responsive to medication. Interestingly, this patient had congenital macrosomia and macrocephaly, though subsequently had failed to thrive (weight and height below 5th centile) with the head circumference at the 50th centile. This patient developed cardiomegaly, cardiomyopathy and heart block, and later died at 19 months. No head imaging was reported for patient 2. Patient 3, a female, also developed atrial flutter suddenly after a respiratory illness at 13 months, and this was eventually responsive to procainamide. Brain MRI showed ventriculomegaly, increased T2 white matter signal, and protrusion of the cerebellar tonsils. This patient also died, though after the report was published (Yano, personal communication, 2007).
More recent reports have identified M–CM patients with ectopic cerebellar tonsils, most commonly described as Chiari I malformation by the various authors. Giuliano et al. [2004] reported seven patients, two of whom had a Chiari I (patients 2 and 5). Ventricular asymmetry was seen in two (patients 2 and 4) and symmetric “hydrocephalus” was noted in a third (case 6). One patient (case 3) suffered an early demise in conjunction with complex congenital heart disease; this patient had polymicrogyria, cavum septum pellucidum, and gray matter alterations. Three other patients had hemimegalencephaly (cases 1, 4, and 7), one of whom also had cortical dysplasia (case 7). Garavelli et al. [2005] reported 10 patients with head MRI scans. Chiari I was seen in 7 patients (with cerebellar crowding of the posterior fossa in 1), increased white matter signal in 5, dilated ventricles or hydrocephalus in 5 (4 had received ventricular shunting), cerebral asymmetry or hemimegalencephaly in 6, and cavum septum pellucidum, polymicrogyria, and thick optic nerves noted in 1 patient each. While some of these patients may have received more than one brain MRI, it was not explicitly stated and so it is not clear if any of these findings changed over time. Nevertheless, the authors clearly demonstrated that Chiari I appeared to be much more common in M–CM than noted in earlier reports.
The report of Robertson et al. [2000] described five patients with neuroimaging studies, four of whom had multiple studies. Four of five had hydrocephalus or dilated ventricles, and in two of these patients (cases 2 and 4) there was confirmed absence of ventriculomegaly in the newborn period with this finding developing later in life. A third patient (case 3) did not have ventriculomegaly, but also had a normal CT scan in the newborn period; follow up study at 4 years of age demonstrated new observations: “small posterior fossa causing distortion of the brainstem, cerebellum, and pituitary stalk,” along with dilated perivascular spaces in the periventricular white matter. Patients reported by Brnicoat et al. [1996] and Stoll [2003] demonstrated progressive ventriculomegaly during infancy compared to studies performed in the newborn period. As with the above case described by Thong et al. [1999], these reports demonstrated that there appear to be certain structural findings on head imaging in M–CM that are age-dependent, and that these changes usually occur during infancy and early childhood, the time of maximal postnatal brain growth.
To determine the scope of described neuroimaging abnormalities in M–CM, we reviewed in the English language all published patients who had received neuroimaging. Table I shows the type of imaging study together with the sex of each reported patient. The patients of Reardon et al. [1996], Barnicoat et al. [1996], and patient 1 from Cristaldi et al. [1995] were included as examples of highly probable cases of M–CM. Two relatively recent cases of “atypical” M–CM [Schwartz et al., 2002; Megarbane et al., 2003] were not included because of different phenotypes and alternative diagnoses. Also excluded was the patient of Meyer [1979]; M–CM is possible, but clinical description does not provide sufficient detail to confirm the diagnosis.
TABLE I.
Review of Literature Cases
| Cristaldi | Barnicoat | Reardon | Moore | Clayton-Smith | Vogels | Carcao | Thong | Franceschini | Baralle and Firth, 2000 | Robertson | Yano | Stoll | Lapunzina | Giuliano | Ackar | Nyberg et al., 2005 | Garavelli | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Date | 1995 | 1996 | 1996 | 1997 | 1997 | 1998 | 1998 | 1999 | 2000 | 2000 | 2000 | 2001 | 2003 | 2004 | 2004 | 2004 | 2005 | 2005 | |
| Total number | 1 | 1 | 1 | 13 | 8 | 3 | 1 | 1 | 2 | 1 | 5 | 2 | 1 | 6 | 7 | 1 | 1 | 10 | 65 |
| Patient sex | F | F | M | 8M/5F | 6M/2F | 1M/2F | M | M | 1M/1F | F | 2M/3F | 2F | M | 4M/2F | 2M/5F | M | F | 3M/7F | 32M/33F |
| Type of study | C, M | C, M | C | NS | C (4–6;8–9), M (3;5), U (7) NS (1) | C (2); M (1;4) | M | M | C(1); M(1;2) | U | C (1–5); M (4;5) | M | C | M (1); NS (2–6) | M (1–3;5) NS (4;6;7) | M | NS | M |
=CT scan, head.
=MRI, head.
=ultrasound, head.
Numbers in parantheses=Case number in report.
The compiled neuroimaging features from our literature review are shown in Table II. While many CNS findings are found in the literature, only a few abnormalities were reported with high frequency. The most common abnormality observed in over 50% of cases was ventriculomegaly. Other commonly reported features included cerebral asymmetry, increased white matter signal, and cerebellar tonsillar herniation or Chiari I. Less frequent anomalies include asymmetric ventricles, cavum septum pellucidum, cortical dysgenesis/dysplasia, and polymicrogyria. Numerous other features, including “hydropic” appearing optic nerve sheaths, venous sinus thrombosis, thickened corpus callosum, and dilated perivascular spaces of the cortical veins, were noted rarely or as singular findings.
TABLE II.
Neuroimaging Findings in the Literature
| Totals | |
|---|---|
| Total number of literature cases | 65 |
| Hydrocephalus/ventriculomegaly | 37/65 |
| Assymetric lateral ventricle size | 12/65 |
| Hemimegalencephaly/assymetric brain | 21/65 |
| White matter defects (increased signal, myelination defects, etc.) | 17/65 |
| “Bulky” or “Thick” appearing white matter | 2/65 |
| Chiari I/CTH | 15/65 |
| Large cerebellum | 3/65 |
| Crowded posterior fossa | 3/65 |
| Hypoplastic cerebellum/cerebellar vermis | 2/65 |
| Polymicrogyria | 5/65 |
| Cortical dysgenesis/dysplasia | 5/65 |
| Pachygyria | 1/65 |
| Cavum septum pellucidum | 7/65 |
| Cavum vergae | 2/65 |
| ”Bifid” Septum pellucidum | 1/65 |
| Thick corpus callosum | 2/65 |
| Thin/hypoplastic corpus callosum | 1/65 |
| Ischemic injury/stoke | 2/65 |
| Venous sinus thrombosis | 2/65 |
| Hemorrhagic lesions | 2/65 |
| Structural Vascular anomaly | 2/65 |
| Hydropic/globular optic nerves | 2/65 |
| Dilated Perivascular spaces of white matter | 1/65 |
| Heterotopic gray matter | 2/65 |
| Calvarial hemangioma | 1/65 |
| Nodular White matter lesion | 1/65 |
| Cystic white matter lesion | 1/65 |
| Porencephalic lesion | 1/65 |
| Choroid plexus cysts | 1/65 |
| Atrophy | 1/65 |
| Nodular heterotopia | 1/65 |
| Schizencephaly | 1/65 |
| Arachnoid cyst | 1/65 |
Comments on CNS Terminology
Not unexpectedly, we found that different reports utilized various terms to describe apparently similar findings. We attempted to address discrepancies of terminology in this review, and we made an effort to avoid implicating the etiopathogenesis in the descriptive terms that we used for our cohort. Rather than hydrocephalus, which may imply a pathologic and obstructive process, we opted for the term “ventriculomegaly” and clarified whether the radiographic picture was obstructive or non-obstructive. We also noted whether asymmetric ventricular dilatation was observed in a given case. If a patient had undergone ventricular shunting, then we assumed obstructive ventriculomeglay (either known or presumed by the treating physicians). While Chiari I malformation was associated with M–CM with some frequency in the literature, we preferred the term “cerebellar tonsil herniation” (CTH), as Chiari I often implies ectopic cerebellar tonsils secondary to a congenitally small posterior fossa. We used the term “cerebral (or cerebellar) asymmetry” to describe brain hemispheres that were discrepant in size rather than hemimegalencephaly; this latter term, which is often used to describe a primary brain malformation with a unilaterally enlarged, dysplastic cerebral cortex, is often associated with a severe seizure disorder and a poor neurologic prognosis [Flores-Sarnat, 2002; Tinkle et al., 2005; Di Rocco et al., 2006]. Likewise, we consolidated white matter disease, leukoencephalopathy, myelination defects, and similar irregularities of white matter as white matter signal abnormalities. We considered polymicrogyria as multiple small gyrations along the margins of the brain, identified on at least two contiguous imaging cuts, which may or may not have disclosed cortical thickening, depending on the density of gyral approximation. We judged cortical dysplasia to be abnormal cortical invagination into the white matter or cortical enlargement and thickening approximating the white matter.
COHORT ANALYSIS
Cohort Identiftcation, Diagnosis of M–CM, and Clinical Features
We undertook a systematic longitudinal review of the neuroimaging findings in 17 patients with M–CM. Patients were identified and collected through collaborative efforts and discussions between the contributing authors. All patients had received a diagnosis of M–CM/M–CMTC by a clinical geneticist. Since the underlying genetic cause of this syndrome remains unknown, the diagnosis is made by recognizing a distinctive pattern of features. Two articles have proposed clinical diagnostic criteria for M–CM/M–CMTC. Robertson et al. [2000] suggested that the diagnosis can be made with confidence in a patient who has cutis marmorata telangiectatica congenita and congenital macrocephaly as major features, together with at least four of the following supportive features: neonatal hypotonia, developmental delay, connective tissue defect of the skin or joints, frontal bossing, midline facial capillary malformation, cutaneous toe syndactyly, partial asymmetric overgrowth, and hydrocephalus. Franceschini et al. [2000], who reported two patients, one of whom appeared to have M–CM/M–CMTC without cutis marmorata, offered a less restrictive set of criteria—macrocephaly being a required major feature along with at least two of the following to make the diagnosis: overgrowth, asymmetry, cutis marmorata, cutaneous “angiomas,” and polydactyly/syndactyly. Hydrocephalus was mentioned by this author as a helpful feature in cases which are otherwise unclear.
While neither set of proposed clinical criteria have yet to be accepted as diagnostically definitive, and noting that the criteria of Robertson et al. [2000] are more specific, both lists incorporate principal clinical features found in most affected subjects and provide an objective way to assess a suspected case. To corroborate the expert clinical diagnostic opinion in each patient, we applied both diagnostic checklists to our cohort, the results of which are shown in Tables IIIa and IIIb. All patients satisfied the diagnostic criteria of Franceschini et al. [2000] and all but two (patients 15 and 16) satisfied the criteria of Robertson et al. [2000]. These two did not have a history of cutis marmorata or CMTC, a mandatory feature of Robertson et al. [2000], but did have facial capillary malformations. Moreover, all the minor criteria of Robertson et al. [2000] were present in both “atypical” patients. They may represent uncommon examples of M–CM/M–CMTC without cutis marmorata, similar to the patient reported by Franceschini et al. [2000] or perhaps cutis marmorata was present at one time and not appreciated clinically, but had faded by the time of a later assessment. Fading with time is consistent with the natural history of this vascular phenomenon.
TABLE IIIa.
Robertson Diagnostic Criteria
| Patient | Macrocephaly (mandatory) | CMTC (mandatory) | Hypotonia | Developmental delay | Connective tissue involvement | Frontal bossing | Facial capillary malformation | Toe syndactyly | Asymmetry | Hydrocephalus | Meets Robertson Criteria |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | + | + | + | + | + | + | Yes |
| 2 | + | + | + | + | + | + | + | − | + | − | Yes |
| 3 | + | + | + | + | ND | + | + | + | + | − | Yes |
| 4 | + | + | + | + | + | + | − | − | + | + | Yes |
| 5 | + | + | + | + | + | + | + | + | + | + | Yes |
| 6 | + | + | ND | + | + | + | + | − | + | − | Yes |
| 7 | + | + | + | + | + | + | − | − | + | + | Yes |
| 8 | + | + | − | + | − | − | + | + | + | − | Yes |
| 9 | + | + | + | + | ND | + | − | + | + | + | Yes |
| 10 | + | + | + | + | + | + | + | − | + | + | Yes |
| 11 | + | + | + | + | + | + | + | + | + | + | Yes |
| 12 | + | + | + | + | ND | + | ND | − | + | + | Yes |
| 13 | + | + | ND | + | ND | + | + | + | + | + | Yes |
| 14 | + | + | + | + | + | + | − | + | − | + | Yes |
| 15 | + | − | + | + | + | + | + | + | + | + | No |
| 16 | + | − | + | + | + | + | + | + | + | + | No |
| 17 | + | + | + | + | + | + | + | − | + | + | Yes |
, feature present
, feature absent
, no data/unknown.
TABLE IIIb.
Franceschini Diagnostic Criteria
| Patient | Macrocephaly (mandatory) | CMTC | Cutaneous angiomata | Syndactyly/ polydactyly | Overgrowth | Asymmetry | Hydrocephalus* | Meets Franceschini criteria |
|---|---|---|---|---|---|---|---|---|
| 1 | + | + | + | + | + | + | + | Yes |
| 2 | + | + | + | − | − | + | − | Yes |
| 3 | + | + | + | + | ND | + | − | Yes |
| 4 | + | + | − | − | − | + | + | Yes |
| 5 | + | + | + | + | + | + | + | Yes |
| 6 | + | + | + | − | + | + | − | Yes |
| 7 | + | + | − | + | + | + | + | Yes |
| 8 | + | + | + | + | − | + | − | Yes |
| 9 | + | + | + | + | ND | + | + | Yes |
| 10 | + | + | + | − | − | + | + | Yes |
| 11 | + | + | + | + | + | − | + | Yes |
| 12 | + | + | + | − | − | + | + | Yes |
| 13 | + | + | + | + | + | + | + | Yes |
| 14 | + | + | − | + | + | − | + | Yes |
| 15 | + | − | + | + | + | + | + | Yes |
| 16 | + | − | + | − | + | + | + | Yes |
| 17 | + | + | + | − | + | + | + | Yes |
, feature present
, feature absent
, no data/unknown.
Supportive feature.
The clinical diagnostic features in our cohort are summarized in Table IVa. Laboratory and genetic studies, including chromosome analysis, are summarized in Table IVb. Cytogenetic and molecular testing, when performed, was normal in all patients investigated. All cases were sporadic and no consanguinty was found. Occasionally reported were prenatal abnormalities such as large or asymmetric ventricles (patients 1, 4, 7, and 17), mild polyhydramnios (patient 17), and third trimester gestational diabetes (patient 11) although prenatal records and histories were not always sufficient to determinecomplications of pregnancy accurately. Several patients had low frequency findings not found in Table IVa. Two patients had relative hemiatrophy of the right arm (patients 7 and 17, more significant in the former), which is an occasional finding in nonsyndromic cutis marmorata telangiectatica congenita, but is rarely mentioned in M–CM. Other unusual findings included: right-sided aortic arch with anomalous right and left subclavian arteries and a mesenteric nodule filled with blood vessels, identified as a mesenteric hemangioma (patient 11, autopsy); double superior vena cava (patient 17); Meckel diverticulum (patient 15); moderate-sized pericardial cyst (patient 14, found incidentally on echocardiogram at 5 years); history of small subcutaneous masses, which, when examined by ultrasonography, appeared to be cystic in nature and self resolving over time (patient 5); splenic cyst examined by abdominal ultrasonography (same patient 5); right pararenal vascular mass stable over several months (patient 10); and left paraspinous mass, stable during interval assessments, with a mixed solid and cystic appearance thought to be of neurogenic origin (patient 16).
TABLE IVa.
Cohort Clinical Features
| Patient | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 |
|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Male | Male | Female | Male | Female | Male |
| Age at last assessment | 3 y 7 m | 3 y 1 m | 10 m | 3 y1 m | 2 y | 14 y | 29 m | 14 y 6 m | 5 y |
| Gestational age (weeks) | 40 | 36 | FT | 39 | 35 | 40 | 37 | 39 | ND |
| Birth Wt (g) | 4,848 | 3,033 | 3,685 | 3,600 | 3,827 | 3,898 | 3,742 | 3,620 | ND |
| SD from the mean* | +3 SD | +1 to +2 SD | 0 to +1 SD | 0 to +1 SD | +5 to +6 SD | +1 SD | +2 SD | ~+1 SD | ND |
| Birth Length (cm) | 54 | 48.3 | ND | ND | 50.3 | 50 | ND | 53 | ND |
| SD from the mean* | +1 to +2 SD | 0 to +1 SD | ND | ND | +1 to +2 SD | 0 to 1 SD | ND | ~+1 SD | ND |
| Birth OFC (cm) | 43.2 | 32 | ND | ND | 35.2 | 36 | ND | 39 | ND |
| SD from the mean* | +6 to +7 SD | 0 to 1 SD | ND | ND | ~+2 SD | +1 SD | ND | +4 SD | ND |
| Hypotonia | + | + | + | + | + | ND | + | − | ND |
| Dev delay/mental retardation | + | + | + | + | + | + | + | + | + |
| Neonatal hypoglycemia | − | − | ND | − | + | − | − | − | ND |
| Hemihyperplasia | + (R; leg, body, face) | + (L; leg) | − | + (L; face, leg) | + | + | + | + (L; face, arm, leg) | + |
| Toe syndact | + (B, 2–3) | + | + (B, 2-3-4) | − | + (L, 2-3-4; R, 2–3) | − | − | + (B, 2–3) | + (B, 2–3) |
| Finger syndact | − | − | − | − | + (L, 2-3-4) | − | − | − | − |
| Toe polydact | − | − | − | − | − | − | + (B) | − | − |
| Finger polydact | − | − | − | − | + (L) | − | − | − | − |
| Toe Sandle Gap | − | − | − | − | + | ND | + | − | − |
| Cutis Marmorata or CMTC | + | + | + | + | + | + | + | + | + |
| Philtrum/Lip cap malformation | + | + | + | − | + | ND | − | + | − |
| Other cutaneous vascular anomalies | +1 | +1,2 | ND | +3,4 | +4 | +1 | − | +1,5 | +1 |
| Other Derm findings | − | ND | ND | ND | − | ND | − | + multiple dark nevi | − |
| Soft/loose/thick skin | + | + | ND | ND | + | − | + | + | ND |
| Lax joints | + | − | ND | + | + | + | ND | − | ND |
| Face/head | Broad forehead | Broad forehead | Broad forehead | Broad forehead | Broad forehead | Midfacial hypoplasia | Broad forehead | Broad forehead | Broad forehead |
| Nose | Flattened nasal bridge | Flattened bridge, broad nasal tip | ND | Flattened nasal bridge and tip | Flat bridge and broad tip | Flat nasal bridge | Flat nasal bridge | Broad tip | Flat nasal bridge |
| Eyes | Hypertelorism | Downslanting palpebral fissues; mild hypertelorism | ND | Epicanthal folds, telecanthus | Hypertelorism and downslanted palpebral fissures | Epicanthal folds, downslanted palpebral fissures | Hypertelorism, upslanted palpebral fissures | Esotropia | ND |
| Mouth | Smooth philtrum | Long, smooth philtrum | ND | Mild micrognathia, smooth philtrum | Narrow, arched palate | Narrow, high palate | Arched palate | Left tongue and gum hemihyperplasia | ND |
| Ears | Fleshy lobes | ND | ND | Normal | Fleshy pinnae, full lobes | ND | Left larger | Normal | ND |
| Internal vascular malformations | − | − | ND | − | − | − | − | − | − |
| Other | Constipation | Feeding problems as newborn | ADHD | Seizures; larygomalacia; feeding problems | Laryngotracheomalacia | ||||
| Maternal age at birth | 37 | 26 | 23 | 35 | 26 | 33 y | 19 y | 26 | 38 |
| Paternal age at birth | 38 | 28 | ND | 37 | ND | ND | 19 y | ND | 36 |
| Sex | Female | Female | Female | Male | Male | Male | Male | Female | 10M/7F |
| Age at last assessment | 9 m | 2 y 6 m | 11 m | 33 m | 5 y 6 m | 3 y 6 m | 6 y 4 m | 6 m | |
| Gestational age (weeks) | 39 | 35 | 37 | 35 | 37 | 37 | FT | 36 | |
| Birth Wt (g) | 3,685 | 3,560 | 3,355 | 3,289 | 5,268 | 3,569 | 5,216 | 4,210 | |
| SD from the mean* | +1 SD | +3–4 SD | ~+1 SD | +2 to +3 SD | +7 to +8 SD | +1 to 2 SD | +7 to +8 | +4 to +5 SD | |
| Birth Length (cm) | 50.8 | 51 | 46.8 | 53.3 | 58 cm | ND | 53 | 52.5 | |
| SD from the mean* | +0 SD | +2 to +3 SD+L42 | rv-1 SD | +4 SD | +5 SD | ND | +4 SD | +3 SD | |
| Birth OFC (cm) | 37 | 38.5 | 39 | ND | 40.5 cm | 40 cm | 41 | 38.5 | |
| SD from the mean* | +2 SD | ND | +4 to +5 SD | ND | ~+6 SD | +5 to +6 SD | ~+6 SD | +4 to +5 SD | |
| Hypotonia | + | + | + | ND | + | ND | + | + | 12/13 |
| Dev delay/mental retardation | + | + | + | + | + | + | + | + | 17/17 |
| Neonatal hypoglycemia | ND | − | ND | − | + | + | − | − | 3/13 |
| Hemihyperplasia | + | + (R; arm, leg) | + | + | − | + | + (R; body, face) | + (R; leg, face, body) | 15/17 |
| Toe syndact | − | + (B, 2–3) | − | + (B, 2–3) | + (B, 2–3) | + (B, 2–3) | + (R, 2–3) | − | 11/17 |
| Finger syndact | − | + (L, 3–4) | − | − | − | − | − | − | 2/17 |
| Toe polydact | − | − | − | − | − | − | − | − | 1/17 |
| Finger polydact | − | − | − | − | − | − | − | − | 1/17 |
| Toe Sandle Gap | − | ND | + | − | − | + | − | + | 5/15 |
| Cutis Marmorata or CMTC | + | + | + | + | + | − | − | + | 15/17 |
| Philtrum/Lip cap malformation | − | + | − | + | − | + | +6 | +6 | 10/16 |
| Other cutaneous vascular anomalies | +1,3 | − | +1 | ND | − | +1,3 | − | +1 | 11/15 |
| Other Derm findings | − | ND | − | ND | − | − | + dry texture | − | |
| Soft/loose/thick skin | + | + | + | ND | + | ND | + thick | + thick, doughy | 10/12 |
| Lax joints | ND | ND | + | ND | + | + | ND | + | 8/9 |
| Face/head | Broad forehead | Broad forehead | Broad forehead | Broad forehead | Broad forehead | Frontal bossing | Round face, broad forehead | Broad forehead, full cheeks | |
| Nose | Flat bridge, broad tip | ND | Broad nasal tip | flattened bridge | Flat bridge with flat tip | ND | Flat bridge | Flat bridge | |
| Eyes | Upslanted palpebral fissures | Tortuous retinals vessels | ND | Mild hypertelorism | Hypertelorism | Downslanted palpebral fissures | Large cornea, long lashes | Upslanted palpebral fissures | |
| Mouth | Arched palate | Delayed dental development | ND | Flat philtrum | Long philtrum | High palate | Normal | High palate | |
| Ears | Fleshy, creased lobules | Lowset, absent superior crus | ND | Fleshy lobes | Fleshy lobes | Lowset | Fleshy, dimpled lobes | Low-set, rotated | |
| Internal vascular malformations | + pararenal vascular mass | + mesenteric hemangioma | − | − | − | − | − | − | 2/16 |
| Other | Atrial flutter in newborn period; Died at 2 y 6 m of seizure | Seizures | Seizure, laryngomalacia, pericardial cyst | Epistaxis | Seizures, umbilical hernia | ||||
| Maternal age at birth | 29 | 27 | 28 | 28 | 34 | 23 | 26 | 34 | |
| Paternal age at birth | 31 | ND | 29 | 28 | 38 | ND | 28 | 34 |
(1) Capillary malformation, (2) Streaky hyperpigmentation, (3) Hemangioma, (4) Prominent veins, (5) Lower extremity varicose veins, and (6) Capillary malformation on lower lip.
: Right.
: Left.
: Bilateral.
: No data/not described.
: Standard deviations.
Per reference curves of Saul et al., 1998.
TABLE IVb.
Cytogenetic and Molecular Tests Performed on Cohort
| Patient # | Karyotype | Subtelomere probes | CGH microarray | PTEN | LIT 1 methylation | NSD1 |
|---|---|---|---|---|---|---|
| 1 | X | X | X | |||
| 2 | X | X | ||||
| 3 | X | |||||
| 4 | X | X | ||||
| 5 | X | X | ||||
| 6 | ||||||
| 7 | X | |||||
| 8 | ||||||
| 9 | X | |||||
| 10 | X | X | X | X | ||
| 11 | X | |||||
| 12 | X | X | ||||
| 13 | X | X | ||||
| 14 | X | X | ||||
| 15 | X | |||||
| 16 | X | X | X | |||
| 17 | X |
COHORT NEUROIMAGING ANALYSIS
Cohort Characteristics, Methods, and Summary of Neuroimaging Studies
The cohort is comprised of 10 males and 7 females. Each patient received between 1 and 7 separate neuroimaging studies with a total of 47 separate studies reviewed (Table V). All neuroimaging studies were head MRIs with the exception of patient 8, for whom we only had a single head CT. Findings from literature review (Table II) were used as a checklist in evaluating the presence or absence of specific features reported in M–CM in addition to assessment for novel features.
TABLE V.
Neuroimaging Performed on Cohort
| Patient | Number of imaging studies | Age at first study | Age at last study |
|---|---|---|---|
| 1 | 7 | 2 days | 43 months |
| 2 | 1 | 39 months | — |
| 3 | 2 | 2 months | 10 months |
| 4 | 1 | 13 months | — |
| 5 | 5 | 11 months | 38 months |
| 6 | 1 | 14 years | — |
| 7 | 2 | 6 months | 35 months |
| 8 | 1* | 3 days | — |
| 9 | 1 | 11 months | — |
| 10 | 2 | 7 months | 18 months |
| 11 | 3 | 23 days | 5 months |
| 12 | 2 | 2 months | 11 months |
| 13 | 4 | 7 months | 33 months |
| 14 | 7 | 8 days | 6 years |
| 15 | 4 | 5 months | 3 years |
| 16 | 1 | 28 months | — |
| 17 | 3 | 3 days | 5 months |
| Totals | 47 |
Study only included CT scan.
No patient in our cohort had normal neuroimaging. We found a high frequency of brain asymmetry (Fig. 2), polymicrogyria and cortical dysgenesis (Fig. 3), ventriculomegaly, white matter abnormalities with increased T2 signal (Fig. 4), and CTH (Figs. 5 and 6). Twelve patients had cavum septum pellucidum or cavum vergae. In addition, several patients were identified with enlarged or dilated venous sinuses (Fig. 7) together with dilated perivascular spaces (Fig. 8) in many (the latter was previously a unique finding), most of whom had concurrent CTH. The corpus callosum was often unusually thick and thickened optic nerve sheaths were seen in multiple patients (Fig. 9). Also found were two cases of unexpected vascular events (Fig. 10), one possible case of heterotopia (Fig. 11), and a single case of a probable meningioma (Fig. 12). Neuroimaging findings in the cohort are summarized in Table VI.
Fig. 2.
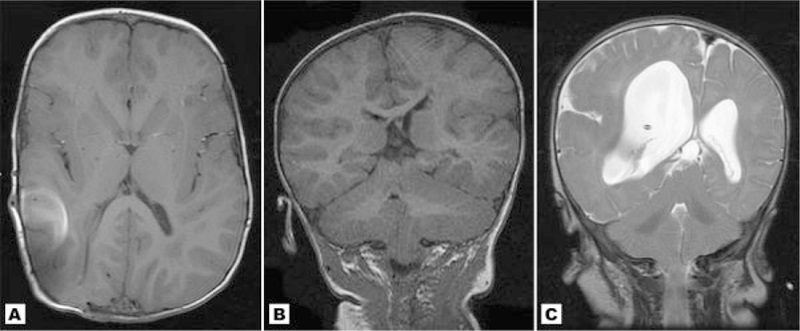
A: Study of patient 1 at 27 months showing mild cerebral asymmetry with focal enlargement of the occipital-temporal region, which was less noticeable when younger. The bright spot on the right hemisphere represents artifact from a shunt. B: Coronal MRI of Patient 5 showing moderate cerebral and cerebellar asymmetry. This patient also has total left-sided hyperplasia from the midline. C: T2 weighted coronal image of patient 17 showing marked right sided brain asymmetry similar to true hemimegalencephaly. The enlarged hemisphere shows an abnormal gyral pattern with a deep sylvian fissure, perisylvian polymicrogyria and focal cortical dysplasia resembling pachygyria.
Fig. 3.
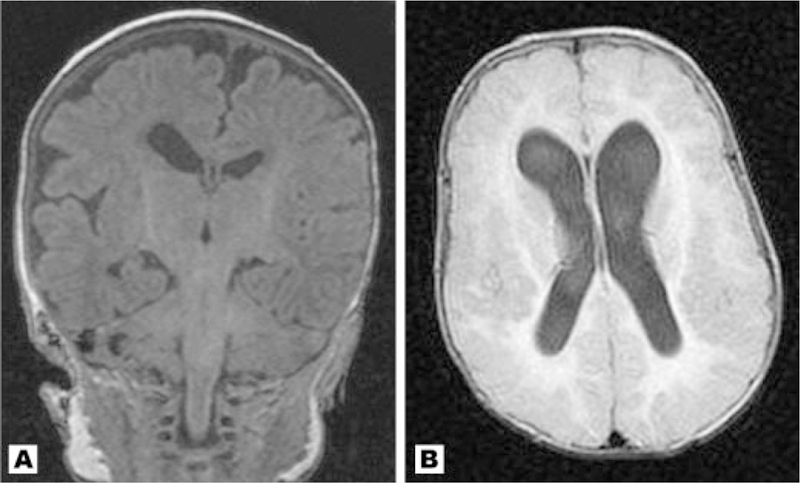
A: Coronal T1 MRI of Patient 12 at 2 months with right sided incomplete opercularization and perisylvian polymicrogyria with cortical irregularity. In addition, there is ventricular asymmetry. B: Axial image of Patient 14 demonstrating extensive cortical dysplasia with thick and irregular cortex in the bilateral frontal and parietal regions. This patient suffers from severe mental retardation and a partially refractive seizure disorder. Cavum septum pellucidum with mild cavum vergae are also seen.
Fig. 4.

A,B: Patient 1 at different ages. A: 9 months with demonstration of increased T2 weighted signal in deep and periventricular white matter; (B) 22 months with improved but persistent white matter abnormalities in same distribution; (C) Patient 5. The asymmetrically enlarged left hemisphere demonstrates more prominent white matter signal changes than the right; (D) Patient 3 with dramatic bilateral white matter signal abnormalities. A diagnosis of leukodystrophy was initially considered, but the patient was neurologically stable and remained mildly and statically delayed.
Fig. 5.

A–D: Sagittal images of patient 14 at different ages: (A) At 8 days old there is a normal cerebellum, a noncrowded posterior fossa, and no CTH; (B) At approximately 1 month there is now early growth of the cerebellum. The brainstem and midbrain appears to be mildly compressed anteriorly; (C) The cerebellum appears bulkier with crowded posterior fossa and further anterior compression of the brainstem. There is now mild to moderate ventriculomegaly and mild CTH; (D) At age 15 months, after posterior fossa decompression, the cerebellum has continued to grow and there is further crowding of the posterior fossa and compression of the brainstem, which now appears kinked at the foramen magnum. There is clear worsening of the CTH.
Fig. 6.

A–C: Sagittal images of Patient 11 at different ages showing acquired cerebellar tonsil herniation; (A) at 4 weeks. The cerebellum appears normal without posterior fossa crowding. A sinus thrombosis is present; (B) at 3 months, there appears to be interval enlargement of the cerebellum with a new finding of ventriculomegaly. The brainstem appears anteriorly deviated but motion artifact limits the study; (C) at 5.5 months, the cerebellum appears further enlarged. There is now inferior displacement of the cerebellar tonsils and herniation through the foramen magnum. The cerebellum is pushing anteriorly on the brainstem and midbrain, and the cerebellum appears to ramp up toward the tentorium and now rises as high as the superior margin of the midbrain. D–F: Sagittal scans of Patient 1 taken at different ages: (D) at 2 days there is a normal-sized cerebellum and no CTH. The brainstem is not compressed anteriorly; (E) by 6 months there is interval rapid growth of the cerebellum with new ventriculomegaly and early CTH. The straight sinus is enlarged; (F) after ventricular shunting the ventriculomegaly has resolved but the posterior fossa remains crowded by a large cerebellum. The CTH is still present.
Fig. 7.
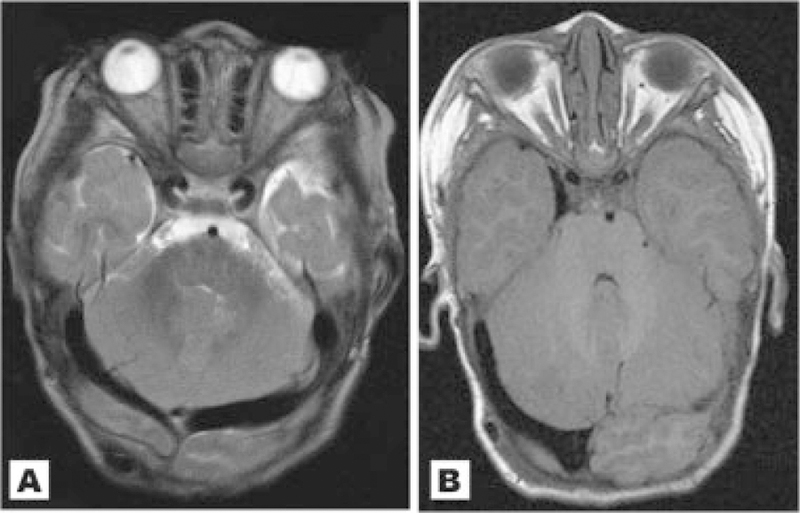
A: Patient 14 at 22 months. Both transverse sinuses are dilated. B: Axial image through the posterior fossa of Patient 1 at 22 months showing a unilaterally enlarged right transverse venous sinus.
Fig. 8.
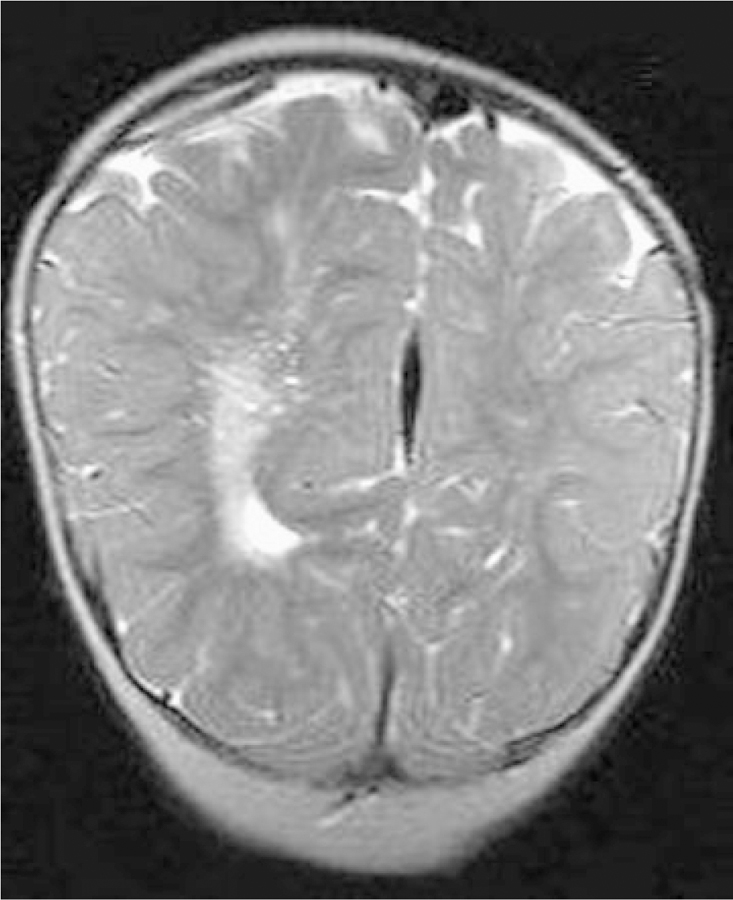
T2 weighted coronal image through the occipital region of Patient 12. Prominent Virchow–Robin spaces are more apparent in the ipsilateral enlarged cerebral hemisphere.
Fig. 9.
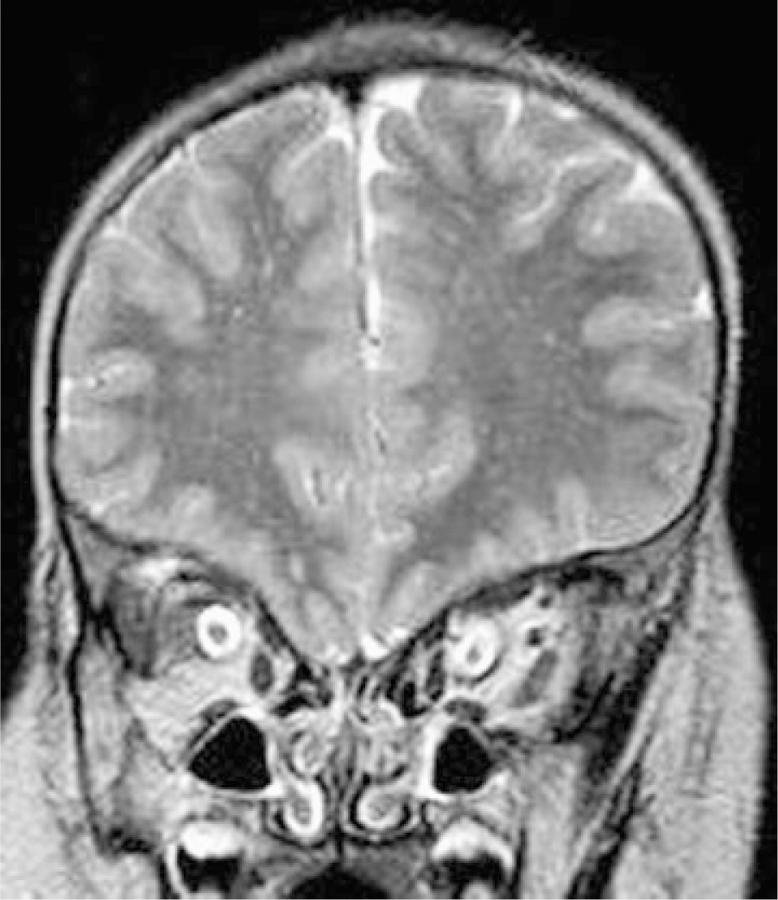
Coronal image of Patient 7 at approximately 3 years. Bilateral optic nerve sheaths are abnormally thick.
Fig. 10.
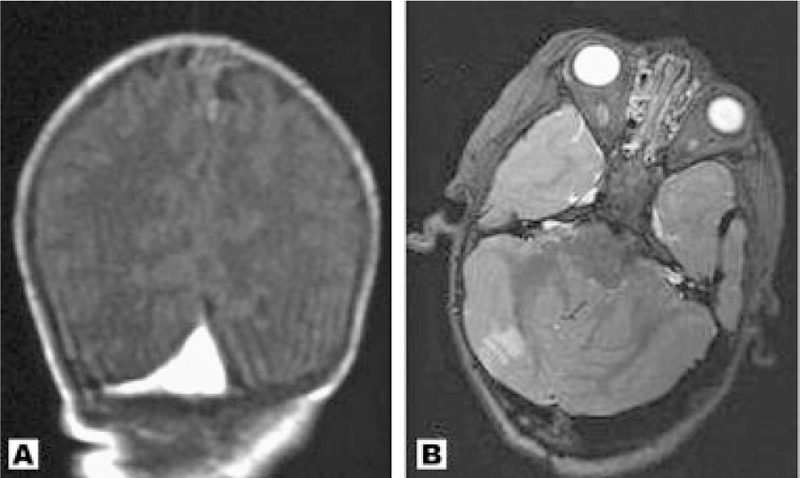
(A) Coronal image of Patient 11 taken at 4 weeks. There is a venous thrombosis at the junction of the transverse and sagittal sinuses; (B) Axial image of Patient 13 at 33 months showing an unexpected and asymptomatic right posterior inferior cerebellar artery infarction. There is also dilation of the transverse sinus.
Fig. 11.
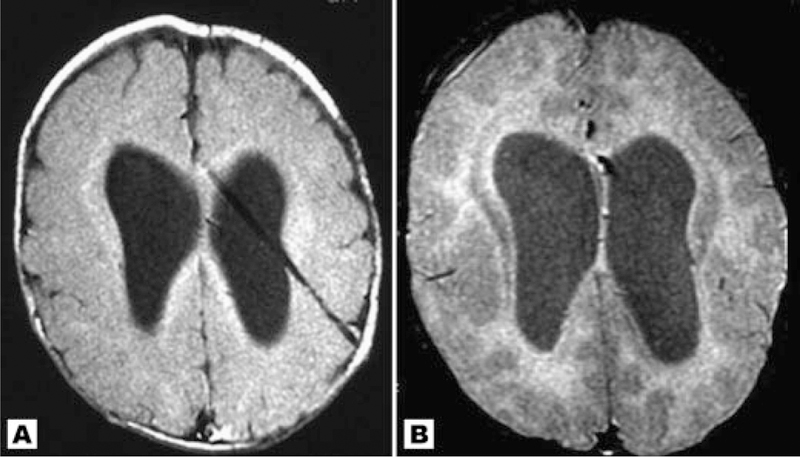
Axial images of patient 11 at different ages. A: Age 1 month with normal gray-white matter differentiation for age; (B) age 5.5 months with appearance of possible band heterotopia.
Fig. 12.
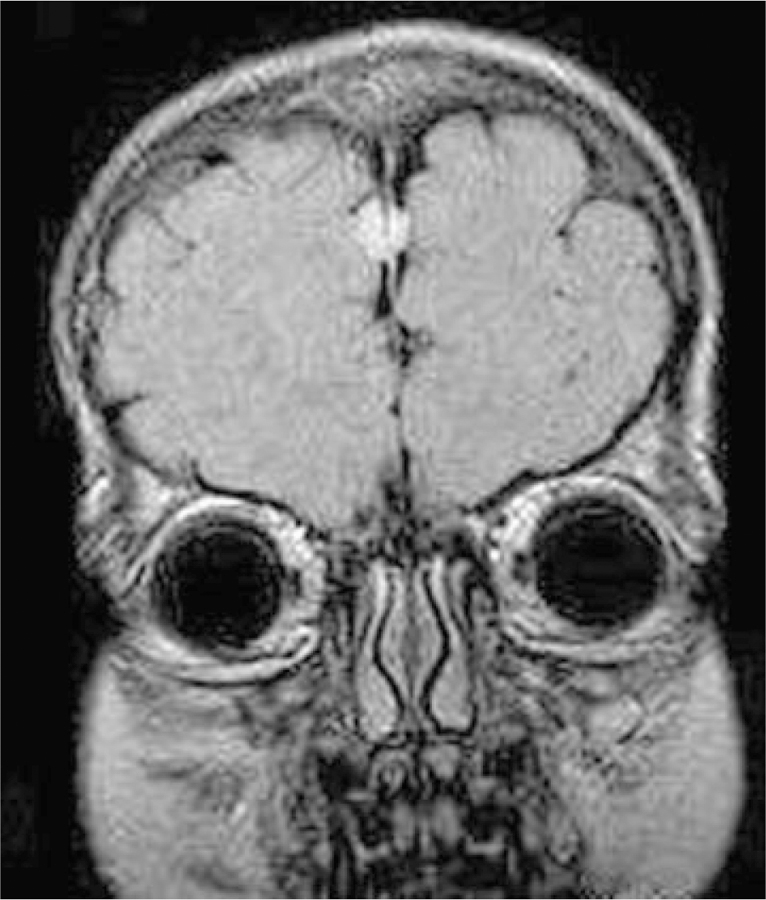
Coronal scan of Patient 14 at age 5 years demonstrating a frontal perifalcine mass consistent with meningioma.
TABLE VI.
Neuroimaging Findings in Cohort
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | Totals |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ventriculomegaly (obstructive) | + | − | − | − | − | − | + | − | − | + | + | − | + | + | + | − | + | 8/17 |
| Ventriculomegaly (non-obstructive) | − | − | − | + | − | + | − | − | + | − | − | + | − | − | − | + | − | 5/17 |
| Asymmetric ventricles (L/R) | +L | − | − | +L | − | − | −L (4) | +L | +R | − | − | +R | − | − | − | +L | +R | 7/17 |
| Increased T2 white matter signal | + | + | + | + | +L | − | + | 0 | +R | + | + | +R | + | + | + | +L | − | 14/16 |
| Brain asymmetry (L/R) | +R(1) | − | − | − | +L | − | +L | +L | +R | − | − | +R | +L | − | − | +L | +R | 9/17 |
| Cerebellar tonsil herniation | + | + | − | + | + | + | − | 0 | − | + | + | − | + | + | + | + | − | 11/16 |
| Large cerebellum | + | − | − | + | + | − | + | 0 | + | + | + | +R | + | + | + | + | − | 12/16 |
| Crowded/compressed posterior fossa | + | − | − | + | + | − | +(5) | 0 | + | + | + | + | + | + | + | + | + | 13/16 |
| Dilated/large venous sinuses | + | − | − | − | + | + | +(6) | − | +(8) | + | − | − | + | + | − | + | − | 9/17 |
| Dilated Virchow–Robin spaces | + | − | − | + | + | − | − | 0 | − | + | − | +R | + | + | + | + | − | 9/16 |
| Thickened optic nerve sheath | − | 0 | − | − | +L | − | + | 0 | − | − | − | +R | − | + | + | − | − | 5/15 |
| Polymicrogyria | − | − | − | − | − | − | + | +(7) | − | − | + | +(10) | − | + | − | − | + | 6/17 |
| Cortical dysplasia | − | + | − | − | − | − | − | − | − | − | − | + | − | + | − | − | +(12) | 4/17 |
| Wide sylvian fissure (L/R/B) | − | − | +B | − | − | − | +L | +L | − | − | − | − | − | +B | − | − | +R | 5/17 |
| Cavum septum pellucidum | + | − | − | − | − | + | + | + | − | − | − | − | + | − | − | + | − | 6/17 |
| Cavum Vergae | − | + | − | − | − | − | − | − | − | − | + | + | − | + | + | − | + | 6/17 |
| Thick corpus callosum | − | + | + | − | − | − | + | 0 | − | − | − | − | + | + | + | + | − | 7/16 |
| Sinus thrombosis | − | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − | 1/17 |
| Ischemic event | − | − | − | − | − | − | − | − | − | − | − | − | + | − | − | − | − | 1/17 |
| Structural vascular anomaly | − | − | − | − | +(2) | − | − | 0 | +(9) | − | − | − | − | − | − | − | − | 2/16 |
| Other | +(3) | +(11) | +(13,14) | |||||||||||||||
| Surgical Interventions | VS/PFD | VS/PFD | VS | VS | VS | PFD | VS/PFD | V | VS |
, feature present
, feature absent
, feature could not be assessed
, ventricular shunt
, ventriculostomy
, posterior fossa decompression
, feature more prominent on right
, feature more prominent on left
, feature present bilaterally.
, Involves right occipital region only
, Absent left transverse sinus; left carotid artery thicker than right
, Prominent pineal gland
, Ventricular asymmetry appreciable due to shunting
, Posterior fossa less crowded on 35-month scan compared to 6-month scan
, Right transverse sinus
, Probable PMG, but not definitive
, Enlarged left transverse sinus
, Absent right internal jugular
, Probable PMG, but not definitive
, Perifalcine frontal mass, suspicious of meningioma
, Frontoparietal pachygyria
, Bilateral periventricular calcifications
, Hypoplastic corpus callosum.
Ventriculomegaly
Ventriculomegaly was present in 13/17 cases. It was regarded as obstructive if the ventricular dilation appeared to be under pressure at any point on longitudinal assessment, or if the patient had received ventricular shunt or ventriculostomy. Eight patients had obstructive ventriculomegaly, and five had a nonobstructive pattern. Seven patients had received ventricular shunting (cases 1, 5, 7, 10, 11, 14, and 17) while one patient was treated with ventriculostomy (case 16). Of the eight patients with obstructive ventriculomegaly, six also had CTH and a large-appearing cerebellum with posterior fossa crowding (cases 1, 10, 11, 13, 14, and 15; Figs. 5 and 6); patients 7 and 17 were the exceptions. Two patients had CTH with nonobstructive ventriculomegaly (cases 6 and 16) and another two (cases 2 and 5) had CTH without any ventricular dilation, suggesting that obstructive hydrocephalus is not necessarily the primary cause of CTH in this syndrome. Three patients had normal or minimally dilated ventricles on scans in the newborn period, and subsequently developed obstructive ventriculomegaly (cases 1, 15, and 16). All these patients also developed CTH about the same time as obstructive ventriculomegaly occurred.
Asymmetrically sized ventricles (not due to shunting) wereseen in seven patients. In five of these (cases 8, 9, 12, 16, and 17), there was concurrent cerebral asymmetry with enlargement on the ipsilateral side. Of note, in patient 8, although the ventricles were asymmetric in size, there was no appreciable ventriculomegaly.
Defects of Cortical Morphogenesis
The literature clearly indicates that some patients with M–CM have cortical dysgenesis, with polymicrogyria and cortical dysplasia being the most commonly observed features (⁓5–10% of reported cases). In addition, the literature identifies one case of pachygyria, one case of schizencephaly, two cases of gray matter heterotopy and one case of nodular heterotopia. [Cristaldi et al., 1995; Clayton-Smith, 1997; Carcao et al., 1998; Thong et al., 1999; Franceschini et al., 2000; Robertson et al., 2000; Ackar et al., 2004; Giuliano et al., 2004; Garavelli et al., 2005].
Polymicrogyria and focal cortical dysplasia were the most common cortical defects we observed (Fig. 3). We identified four patients with definite polymicrogyria (cases 7, 11, 14, and 17) and two with probable or suggestive polymicrogyria (8 and 12). In the majority, the polymicrogyria typically affected the perisylvian and/or insular regions more than other areas. It was generally asymmetric in its degree of severity, with the left side being more prominent in patients 7 and 8, the right side being more affected in patients 12 and 17, and both sides affected in patients 11 and 14. In patients 7, 8, 14, and 17 we saw wide sylvian fissures associated with the regions of polymicrogyria, suggesting a possible perisylvian syndrome in these cases. Patient 3 had incomplete opercularization of the sylvian fissures but no associated polymicrogyria. Of note, four patients who had cerebral asymmetry (patients 7, 8, 12, and 17) had more significant perisylvian polymicrogyria and cortical irregularities on the ipsilaterally enlarged side, suggesting that the mechanisms for brain overgrowth and cortical malformation may be related in this syndrome.
Focal cortical dysplasia was found in patients 2, 12, 14, and 17. In Patient 17, the changes were mostly in the frontoparietal region and resembled focal pachygyria. In patient 2 there was mild bilateral cortical thickening in the frontal lobes. Patient 12 had focal areas of poor gyral formation and reduced sulcation bilaterally in the inferior frontal regions, more noticeable on the larger right hemisphere. There was also apparent cortical thickening in the posterior-occipital regions and mild dysplasia of the vertical striatum in the posterior perisylvian region. Of interest, both these patients demonstrated comparatively mild developmental delays. For instance, patient 2 sat at 7 months, rolled at 8 months, crawled at 12 months, and walked well at 22 months, though he was not yet demonstrating meaningful language at 3 years. Patient 12 (Fig. 3A) was younger, and by clinical assessment at 11 months exhibited only mild gross motor delays. This is in contrast to patient 14 (Fig. 3B), who had more extensive cortical dysplasia of the frontal and parietal regions with an irregular, nodular appearing cortical ribbon and subtle frontal polymicrogyria-like changes in addition to the perisylvian region but without dramatically thickened cortex. There was particular sparing of the posterior parietal, temporal, and occipital regions. Patient 14 also had a severe neurocognitive phenotype. Although a playful and social child, he was severely retarded and at 6 years of age he required an adaptive walker to ambulate and bear weight. His language development was markedly impacted because he required a tracheostomy, but he demonstrated limited evidence of emerging vocabulary and used only a few signs. This patient also had a refractory seizure disorder which was difficult to control with medication. Seizures also occurred in patients 7, 11 (which was a contributing causeof deathatage 2 years), 12 (onsetat 18 months), and 17 (onset at 4 months). All of these patients had co-existing cortical malformations, and no patient without cortical defects suffered from seizures. Based on the experience in our cohort, it appears that cortical defects represent a significant risk factor for seizures in this syndrome.
Although the literature mentions a few patients with M–CM who have neuronal heterotopia, we did not see radiographic evidence of heterotopia as a common feature in our cohort. However, patient 11 did have what appeared to be abnormal gray matter in a periventricular distribution, reminiscent of band heterotopia (Fig. 11A,B) as seen on MRI T1 and T2 weighted scans obtained at 5.5 months. This finding was not evident on this patient’s MRI scans obtained at 1 and 3 months, though it could be that myelination was not adequate at these ages to permit adequate gray-white matter differentiation. This patient died at 2 years of age without subsequent neuroimaging. The postmortem neuropathologic analysis of this patient’s brain was limited, as only certain blocks of brain tissue were retained, but in one parietal section, a narrow subcortical band of gray matter was discernible (see Neuropathology Section below).
With the exception of asymmetric brain growth, other reported brain malformations such as lissencephaly and schizencephaly were not observed.
Brain Asymmetry
We found brain asymmetry in 9/17 patients; all had co-existing ipsilateral facial hemihyperplasia with the exception of patient 7, who had no apparent facial asymmetry (although this patient had relatively mild cerebral asymmetry). Of note, patient 6 had hemihyperplasia of the lower right face, but had no brain asymmetry. Generally, the degree of brain asymmetry was not severe, and in patient 1, the asymmetry was focal with principal involvement of the right posterior cerebral hemisphere (Fig. 2A). Patient 17 was the most dramatic example of cerebral asymmetry (Fig. 2C). Patient 5 demonstrated ipsilateral cerebral and cerebellar asymmetry (Fig. 2B). In four patients with an asymmetrically enlarged brain, we also observed primary cortical developmental abnormalities, such as focal polymicrogyria (cases 7, 8, 12, and 17) or cortical dysplasia (cases 12 and 17). Of interest was the fact that in two cases (patients 1 and 13) the asymmetry was not evident on initial scans but became evident at 9 and 33 months, respectively. The mechanism for the acquired asymmetry is not clear, but it suggests the possibility of disproportionate growth.
In contrast to many patients with true hemimegalencephaly, the enlarged brain hemisphere in these patients was not universally associated with a dysplastic cortex with the exception of the two cases mentioned above. Concurrent seizures occurred in three with brain asymmetry (cases 8, 12, and 17). Only one patient (case 17) had a clinically apparent contralateral hemiplegia to the enlarged cerebral hemisphere. While some patients with M–CM may indeed have true hemimegalencephaly (patient 17 is the most suggestive case in our series for this particular anomaly), the brain asymmetry in this syndrome may more typically reflect a process of generalized overgrowth that is characteristic of the syndrome, and hence an example of brain hemihyperplasia rather than “true hemimegalencephaly” in such instances.
White Matter Abnormalities
One finding we observed at a much higher frequency than what is apparent from the literature review is white matter abnormality with increased T2 signal intensity, seen in 14/16 patients. Analysis for patient 8 was limited during our survey because our only images were from a neonatal CT scan; however, medical records on this patient indicated that she had a follow-up MRI at age 5 years, which showed bilateral white matter signal defects with more significant involvement on the ipsilaterally enlarged left side (15/17 cases had white matter signal irregularities if these data are considered). We found white matter signal abnormalities in M–CM to be age-dependent and related to the progression of myelination such that MRI examinations prior to 6 months of age generally did not display irregular white matter for age, although these defects became more apparent during the later half of infancy. As we did not have any MRI scans after 5 months of age for patient 17, this may explain the lack of apparent white matter involvement in this particular case.
The white matter signal abnormalities tended to involve the deep white matter and/or the periventricular regions. In situations of co-existent obstructive ventriculomegaly requiring shunting, it was not clear if periventricular white matter signal alterations represented pressure-related gliotic changes, as such periventricular involvement was seen in addition to the deep white matter changes in patients 1, 10, and 13, all of whom had received decompression through ventricular shunting or posterior fossa decompression. The evolution of the white matter changes was evident in those patients whom we were able to study with sufficient longitudinal imaging and tended to follow a predicable pattern. In general, white matter abnormalities seemed to improve but persist through early childhood (Fig. 4A,B). This seems to suggest a process of dysmyelination in addition to possible delayed myelination as an explanation for these findings. Cases with cerebral asymmetry had abnormal white matter disease bilaterally, but it tended to be more marked on the larger side (Fig. 2C), suggesting that the processes leading to brain overgrowth and white matter signal hyperintensities may be related. Case 3 developed rather striking bilateral white matter signal enhancement in the interval between scans at 2 and 10 months. The appearance was so dramatic that initially a metabolic leukodystrophy was considered as a possible secondary diagnosis (Fig. 2D). However, this patient had no evidence of neurodegeneration, had comparatively mild developmental delays, and had no cortical anomalies besides primary megalencephaly. Of importance, no cases in this series demonstrated a relentless and progressive worsening of white matter signal defects through later childhood, and presence of the white matter defects was not associated with development of new neurologic symptoms or neurocognitive decline in any patient.
There is a possibility that with enough time, these white matter irregularities may largely resolve, as in patient 6, a 14-year-old female with no significant white matter lesions. However, as we have no early neuroimaging on this patient, it could be that by this time myelination had essentially normalized.
Cerebellar Tonsil Herniation and Posterior Fossa Findings
We were able to confirm that CTH frequently occurs in M–CM, being present in 11/16 of our cases. However, of great significance was the observation that CTH was not present in several patients in the newborn period but developed during infancy. In patients 1, 11, and 14 we confirmed a normal-sized cerebellum with an unremarkable posterior fossa before 1 month of age (Figs. 5 and 6). In all these individuals, there was noticeable overgrowth of the cerebellum during the first few months of life with progressive crowding of the posterior fossa and eventual descent of the cerebellar tonsils. Consequently, patient 1 demonstrated CTH by age 6 months, patient 11 was affected at age 5 months, and patient 14 by age 2 months. Patients 1 and 14 both developed neurologic signs and symptoms that were later relieved by posterior fossa decompression with tonsillectomy and dural patch graft. A third patient from the cohort (case 5) also developed neurologic symptoms of lower extremity weakness at around 1 year of age in association with severe CTH that was also successfully treated with posterior fossa decompression and he subsequently experienced improvement in these symptoms. The clinical course and surgical management of these patients 1, 5, and 14 are described more fully in a separate report (Conway et al., 2007). A fourth patient (case 15) had no CTH by MRI scans at 5, 12, and 24 months, but at age 3 years, MRI showed CTH in association with a progressively crowded posterior fossa and secondary anterior compression of the brainstem. This child was treated with ventriculostomy with good outcome. Lastly, patient 13 also had posterior fossa decompression for CTH, but did not undergo fulguration of the cerebellar tonsils. All patients who have received posterior fossa decompression are currently alive though with developmental delays consistent with the underlying diagnosis.
Few of these patients have had spinal cord MRIs to look for syringomyelia, which has been previously been associated with Chiari Malformation in other patients. In those who have undergone this study (cases 5, 10, 14, and 16), no cord tethering has been found, although case 14 did have a slight cystic appearance in the lower thoracic cord that was felt to represent either a mild syrinx or a normal variant.
The occurrence of either a large-appearing cerebellum or crowded posterior fossa in the majority of the cohort was a notable finding. Posterior fossa crowding was assessed radiographically by looking for either apparent interval changes in the size and shape of the cerebellar hemispheres relating to surrounding structures in the posterior fossa over time, and by the presence of mechanical impingement on structures around the cerebellum, such as compression or pinching of the brainstem, upward bowing of the tentorium, or abnormal angulation of the adjacent dural sinuses. Patient 14, who had marked CTH and anterior compression of the brainstem from the large cerebellum at 15 months, was found to have gliosis at the pons on follow-up scan at 26 months. Patient 15 showed progressive but mild brainstem compression resulting from posterior compression from the cerebellum. Fourteen of 16 patients had a large appearing cerebellum or crowded posterior fossa at some point in time. In only one patient (case 7) was the posterior fossa crowding more significant at a younger age (6 months vs. 35 months).
Not every case in the cohort with CTH demonstrated progressive descent of the tonsils or secondary symptoms. Patient 6 had a mild degree of CTH at adolescence (about 4 mm below the foramen magnum), a point when brain growth ought to be nearly complete. Patient 2 had mild CTH at 3 years of age and showed no significant progression of the lesion on follow-up assessment 1 year later. Patient 16 also had stable CTH over several years and had no neurologic sequelae to suggest brain stem impingement at age 6 years. None of these patients have required any type of neurosurgical intervention.
Patients 10, 13, and 15 all had stable CTH after ventricular shunting, and this may have prevented progression in these specific instances.
Longitudinal Measure of Head Circumference and Posterior Fossa Volume
Many patients with this syndrome seem to experience a markedly increased occipitofrontal circumference growth velocity which is most significant in infancy. We present the standardized OFC growth curves based on CDC data from 0 to 36 months for all patients in whom we had data during this age range. These curves show a general tendency to cross centiles during the first 12–24 months of life, after which the patients generally adopt a growth curve parallel to the standard curves (Fig. 13A,B). To investigate whether a congenitally small posterior fossa might be the explanation for the development of CTH in this syndrome, we performed volumetric analysis on available digital images using the Analyze, versions 6.0 and 7.0, volume rendering programs (Table VII, Fig. 14A,B). Studied posterior fossa volumes were traced using sequential MRI images of the region of interest in the sagittal plane and calculated volumes from planar cuts were summed by the program to generate the full volume of the posterior fossa. We were able to perform such measurements on Patients 1, 3, 7, 10, 12, 13, and 14. We did not perform volumetric analysis on any patients after they received posterior fossa decompression, as the posterior fossa volume would then reflect postsurgical changes.
Fig. 13.

Head circumference curves for male (A) and female (B) cohort patients from age 0 to 36 months plotted against the normal standards. The graphs show the typical tendency for head circumference to cross centiles during infancy and early toddler years in this syndrome.
TABLE VII.
Measured Posterior Fossa Volumes
| Patient | Age at study | Volume (cc) |
|---|---|---|
| 1 | 2 d | 57.5 |
| 6 m | 159.5 | |
| 9 m | 186.6 | |
| 15 m | 212.6 | |
| 3 | 2 m | 80 |
| 10 m | 140.8 | |
| 7 | 5 m | 115.3 |
| 35 m | 191.8 | |
| 10 | 7 m | 144 |
| 18 m | 154.1 | |
| 12 | 2 m | 77.7 |
| 11 m | 168.1 | |
| 13 | 7 m | 140.7 |
| 12 m | 170.1 | |
| 14 | 8 d | 53.9 |
| 1 m | 71.8 | |
| 2 m | 113.7 |
Fig. 14.

Graphic plots of digitally rendered posterior fossa volumes in 4 male (A) and three female (B) patients with M–CMTC compared to published normative data. The posterior fossa volumes tend to cross centiles over time in most patients.
Meaningful volumetric analysis of the posterior fossa is somewhat difficult due to relatively limited data on intracranial compartment volumes in children, including the posterior fossa. In addition, because the posterior fossa is a complex three dimensional space, variations of measurement methodologies from different studies can lead to variance in normative data. In an attempt to correlate our findings to available normal posterior fossa volume curves, we plotted our patients’ calculated posterior fossa measures on an extrapolated curve of posterior fossa volumes as measured from late fetal life to childhood that was obtained from data provided by the studies of Chen et al. [2006] and Prassopoulos et al. [1996]. A curve was generated using the Stata statistical analysis computer program [StataCorp, 2005] to connect the fetal growth curve of Chen et al. [2006], which provided data up to approximately 35 weeks gestation, and the childhood growth curve of Prassopoulos et al. [1996], which had no data on children less than 3 months of age. Chen et al. [2006] applied summed volumes of traced planar images on fetal MRI, similar to the methods employed in this study, whereas Prassopoulos et al. [1996] generated their data from a calculated algorithm incorporating linear measures from landmarks of the posterior fossa on head CT scans. Moreover, while the curve provided by Chen et al. [2006] incorporates data from both sexes, Prassopoulos et al. [1996] generated sex-specific posterior fossa volume curves. We therefore differentiated the posterior fossa volume measures between our male and female patients on separate graphs. Although the two studies utilized different methods for posterior volume calculation, once the data points were entered into the Stata program, the prenatal and postnatal curves overlapped nicely and the Stata program was able to generate a postulated curve between 35 weeks gestation and 3 months that was in accordance with the data from both articles [Prassopoulos et al., 1996; Chen et al., 2006].
Applying our combined extrapolated curves, none of the patients had a small posterior fossa volume, and in fact all patients in whom we had data in early infancy or the newborn period plotted in the normal range (though this may imply that the posterior fossa is relatively small at birth compared to overall head size). All patients demonstrated an increased or stable posterior fossa volume size with age, with most crossing centiles. The exception was patient 10, who dropped from about 2 standard deviations at 7 months to 1 standard deviation at 18 months. Interestingly, this leveling off of growth correlated to the growth velocity of this child’s head circumference, which also saw a leveling of growth in the same time period (Figs. 13B and 14B).
Of those with measured posterior fossa volumes, patients 1, 7, 10, 13, and 14 had CTH. As previously mentioned, patients 1 and 14 had no CTH in the newborn period, and acquired it later in life. While we were able to show that none of the children in the study had a truly small posterior fossa, the question of whether or not the posterior fossa may be relatively small has yet to be definitively determined. It is clear that CTH develops at a time when head size and posterior fossa volumes tend to cross centiles, and that the growth pattern may be a contributing factor to this particular phenomenon.
Large Venous Sinuses and Prominent Perivascular Spaces
An intriguing discovery, previously not well-appreciated, was the occurrence of large or dilated venous sinuses (Fig. 7). These were seen in 9/17 patients, and preferentially involved the sinuses adjacent to the posterior fossa (transverse, posterior sagittal, and straight sinuses). Similar to other findings such as the ventriculomegaly and white matter defects, these were never present at birth but demonstrated progression throughout early life in association with brain growth. In some cases, the venous engorgement was markedly asymmetric; Patient 1 had a dramatically engorged right transverse sinus but a normal sized left transverse sinus. In Patient 13, we could observe an obvious decrease in the size of the venous sinuses after posterior fossa decompression between 12- and 33-month scans, indicating that the enlarged venous sinuses may be due to a compromise of cerebral venous return and/or venous hypertension that is partially relieved through surgical enlargement of the posterior fossa. This reduction in venous sinus size was less evident in Patients 1, 5, and 14 who also underwent posterior fossa decompression.
In addition, we noted prominent perivascular spaces of the penetrating vessels of the white matter (Virchow–Robin spaces or VRS) in 9/16 patients (Fig. 8). This was previously reported as a singular finding by Robertson et al. [2000]. As with other observations in this study, there was a positive correlation between patient age and the appearance of the dilated VRS, as they were never observed in the young infant. The earliest we observed them was at age 11 months (case 12). Two patients had asymmetric distribution of prominent VRS with ipsilateral cerebral enlargement (patients 5 and 12).
The arterioles and veins passing through the white matter are lined with extensions from the pia mater, with the potential space between this layer and the vessel comprising the Virchow–Robin space. These spaces are thus continuous with the pial compartment but not the subarachnoid space. They appear to play a primary role in lymphatic flow or drainage paths of interstitial fluids from the brain and eventually drain to the nasal lymphatics through the cribriform plate. [Groeschel et al., 2006]. Given that the perivascular spaces also seem to play a role in intracerebral fluid homeostasis and transport, it is of interest to know whether occurrence of VRS, dilated venous sinuses, and even ventriculomegaly might be causally related in this syndrome.
Six of the patients who had prominent VRS also had large-appearing venous sinuses (patients 1, 5, 10, 13, 14, and 16), though in all cases in whom we had serial scans which demonstrated both features, the venous enlargement preceded the development of dilated perivascular spaces. In the case of patient 1, there was appreciable enlargement and stabilization of the venous sinuses between ages 6 and 22 months and the dilated VRS didnot appear until 27 months. Patient 5 showed large venous sinuses at age 11 months, but first developed prominent VRS at age 18 months. Patient 10 had large venous sinuses at age 7 months andevident VRS at age 18 months. Patient 13 hadlarge sinuses at age 7 months and apparent VRS at age 12 months. Patient 14 had signs of early sinus enlargement at age 1–2 months, which later became marked on MRI scan at age 15 months along with prominent VRS at that time. Patient 16 had only a single study, so we could not come to a conclusion about the temporal relationship between these findings. Three patients who had prominent VRS did not have any appreciable enlargement of the venous sinuses (Patients 4, 12, and 15). In three cases (Patients 6, 7, and 9), there was enlargement of the venous sinuses without prominent VRS, though in these particular patients the degree of venous sinus dilation was comparatively milder than in the other patients.
Looking at the relation between venous sinus enlargement and changes of the posterior fossa contents, seven patients who had dilated venous sinuses also had CTH. One patient had a large left transverse sinus without CTH (patient 9), whereas five patients had CTH without dilation of venous sinuses (patients 2, 4, 6, 11, and 15). All patients who had large venous sinuses except one (patient 6) also had a large appearing cerebellum and crowded posterior fossa. All patients who had prominent VRS were found to have a large-appearing cerebellum or crowded posterior fossa. Patients 10 and 17 had a large appearing cerebellum or a compressed posterior fossa but did not have any prominent VRS, although Patient 17 was significantly younger than when VRS typically developed in the cohort. The association of these features suggests that posterior fossa crowding, CTH, and the presence of large venous sinuses may indeed be mechanistically related. The fact that several patients had CTH without large venous sinuses suggests that the former may be more likely the primary event rather than the latter, but the fact that so many patients in the cohort demonstrated most of these features makes drawing firm conclusions difficult at this time, as their concurrence might be explained by distinct but related mechanisms in the same patients. Prominent VRS may be more evident in this circumstance simply because more fluid has shunted its way into the interstitial space due to the postulated venous congestion and altered CSF reabsorption patterns.
Thick Corpus Callosum
Seven of 16 patients had a thick corpus callosum, all confirmed by measurement. As with the white matter hyperintensities, this was never a prominent feature in the newborn period or early infancy but became apparent in certain patients during the toddler period (between 1 and 3 years of age). Measurements of the corpus callosum at the genu, mid-body, and splenium taken at mid-sagittal cuts from the most recent study of each patient are provided in Table VIII together with published reference range data. As with our analysis of the posterior fossa volumes, a sparseness of normative data for corpus callosum measurement in a large sampling of pediatric patients limits definitive analysis. The data from the study of Barkovich and Kjos [1988], documenting corpus callosum growth during the infant period (0–12 months), and that of Ng et al. [2005], which ascertained 100 Chinese children of the school age population (ages 6.5–10 years) were used as the reference ranges. While measurements are presented for patient 6, they were not included in the analysis as we did not have normative values of corpus callosum size in pediatric patients over 10 years of age. Moreover, measurements were not taken in patients 9 or 11 because we did not have access to digital imaging in these patients, and accurate measurements could not be confidently obtained, although by rough inspection neither of them had a large corpus callosum. We could not assess the corpus callosum in patient 8 since we only had CT scans for this case. The corpus callosum was considered thick if it measured large in all parameters, though interestingly, all but two patients had at least one measured region of the corpus callosum above the normal range.
TABLE VIII.
Corpus Callosum Measurements of Cohort (in mm)
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 10 | 12 | 13 | 14 | 15 | 16 | 17 | Totals |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at study | 43 months | 39 months | 10 months | 13 months | 38 months | 14 years | 35 months | 18 months | 11 months | 33 months | 6 years | 3years | 28 months | 5 months | |
| Genu/Thick? | 14/Y | 16/Y | 9/Y | 8/N | 10/N | 14 | 17/Y | 9/N | 10/Y | 15/Y | 15/Y | 11/Y | 15/Y | 6/N | 9/13 |
| Body/Thick? | 5/N | 9/Y | 6/Y | 5/N | 6/N | 7 | 10/Y | 7/Y | 7/Y | 8/Y | 9/Y | 8/Y | 9/Y | 3/N | 9/13 |
| Splenium/Thick? | 9/N | 14/Y | 12/Y | 9/N | 11/Y | 11 | 13/Y | 12/Y | 8/N | 14/Y | 11/Y | 11/Y | 16/Y | 3/N | 9/13 |
| Thick at all locations? | N | Y | Y | N | N | Y | N | N | Y | Y | Y | Y | N | 7/13 | |
| Age (months) | Genu | Midbody | Splenium | ||||||||||||
| Normal values | (mm) | (mm) | (mm) | ||||||||||||
| 0–2 | 5.1 ± 1.0 | 2.3 ± 0.5 | 3.7 ± 0.6 | ||||||||||||
| 2–4 | 5.0 ± 1.3 | 2.5 ± 0.5 | 4.5 ± 0.9 | ||||||||||||
| 4–6 | 7.0 ± 1.3 | 3.0 ± 0.8 | 5.8 ± 1.3 | ||||||||||||
| 6–8 | 6.3 ± 1.0 | 2.8 ± 0.4 | 6.6 ± 0.6 | ||||||||||||
| 8–10 | 7.7 ± 1.3 | 4.2 ± 1.0 | 7.6 ± 1.6 | ||||||||||||
| 10–12 | 7.8 ± 1.1 | 4.2 ± 0.8 | 8.3 ± 1.2 | ||||||||||||
| Childhood (<10 years) | 9.35 ± 1.47 | 5.49 ± 0.92 | 8.88 ± 1.47 |
Structural Vascular Anomalies and Thrombovascular Events
Two patients had structural vascular anomalies: absence of the right internal jugular in patient 9, and absent left transverse sinus together with an enlarged left carotid artery (consistent with left-sided hemihyperplasia) in patient 5. Similar vascular defects have been reported sporadically [Clayton-Smith et al., 1997; Franceschini et al., 2000] with one patient having an absent branch of the right carotid artery and another having an aberrant left superior vena cava with absent innominate vein.
Two patients in our cohort had unexpected thrombovascular or ischemic events. Patient 11 was found to have a sagittal and transverse venous sinus thrombosis on initial MRI in the newborn period and patient 13 had a nonacute posterior inferior cerebellar artery stroke between 12 and 33 months (Fig. 10A,B). There was no definitive cause for these events in either patient. Both venous thrombosis and cerebral arterial vascular accidents at young age have been documented as infrequent events by several authors [Clayton-Smith et al., 1997; Moore et al., 1997; Moffitt et al., 1999; Thong et al., 1999; Giuliano et al., 2004].
In Patient 1, a small singular white matter lesion was seen in the right centrum semiovale at 2 days of age. However, this lesion had resolved by the 6-month scan and may have been a small hemorrhage related to perinatal events.
Other Findings
Cavum septum pellucidum or cavum vergae (dilation of the potential spaces between the frontal horns and lateral ventricles, respectively) were each present in 6/17 patients. While isolated cavum septum pellucidum is often regarded as a normal variant in children less than 12 months of age, all patients with this finding had persistence of the lesion past this age except for patient 8. While cavum septum pellucidum and cavum vergae usually have little direct clinical consequence for patients, their presence may indicate underlying aberrant brain morphogenesis [Piatt, 2004].
Prior to this report, two cases were known to have abnormal-appearing optic nerves, described as “hydropic” in one report and “globular” in another [Carcao et al., 1998; Garavelli et al., 2005]. In our cohort, we were able to assess the appearance of the optic nerves in 15 patients. Five of these patients were noted to have wide or thick-appearing optic nerves (Fig. 9). Similar to the above-mentioned thick-appearing corpus callosum, this was never evident in the newborn period or early infancy. The youngest patients we found with optic nerve thickening were both 11 months old (cases 5 and 12). In one case, widening of the nerve sheaths was relatively mild (case 12) and in another, it was clearly asymmetric, corresponding to this patient’s ipsilateral hemihyperplasia of the brain and head (case 5).
Patient 17 had multiple bilateral cerebral calcifications in a mostly periventricular distribution. Of note, they were more prominent in the enlarged, dysplastic right cortical hemisphere. This patient’s mother had a history of cytomegalovirus conversion with a prior pregnancy and recalled a history of labyrinthitis with this pregnancy at 4 months gestation. A workup for congenital infection was not performed on this patient in the newborn period. It is not clear if these lesions are secondary to an environmental event, such as intrauterine infection, or represent a feature of a primary cortical dysgenesis.
Patient 6 had a prominent-appearing pineal gland. As this was the oldest patient in the cohort with neuroimaging, it is uncertain if this finding has any age-related significance in M–CM or if it is an incidental finding.
Lastly, patient 14 was found to have an incidental 1 cm frontal perifalcine mass on routine neuroimaging follow up. The pathology of this lesion has not yet been definitively confirmed by biopsy but its appearance and size has remained stable for over 1 year since initially noted at 5 years of age, suggesting the nature and appearance of meningioma. One patient in the series of Moore et al. [1997] also had a report of a meningioma. This patient represents the first time a specific tumor type has possibly occurred in more than one patient with M–CM (Fig. 12).
NEUROPATHOLOGIC FINDINGS
Case 11 died of circumstances related to an acute seizure and cardiovascular decompensation. Autopsy was performed on this patient. The fixed brain of this 2-year-old child weighed 1,450 g (normal 1,100 g). Initial macroscopic examination suggested only slight asymmetry with a short left frontal lobe and right occipital lobe, while at the base the cranial nerves, the cerebellum and brainstem appeared normal with no obvious macroscopic evidence of tonsillar herniation. On coronal sectioning, many parts of the cortex had broad convolutions with a striated appearance suggesting polymicrogyria, while the right occipital cortex appeared pale and its underlying white matter was yellowish and firm. A ventriculoatrial shunt was present in the right lateral ventricle, which was mildly decreased in size, while the contralateral ventricle appeared slightly dilated. Central gray and white matter was unremarkable. The hindbrain was also unremarkable.
Limited microscopic examination on some blocks of brain tissue by one of our co-authors (BH) showed extensive migration defects. In both hemispheres, the cortex showed coarse macrogyric convolutions comprising unlayered polymicrogyria, including branching fingers of an aberrantly directed molecular layer simulating fused sulci. The cortex from the frontal, parietal insular, and temporal lobes was affected, and the overlying leptomeninges contained heterotopic glioneuronal elements. In places, over-migrated neurons straddled the molecular layer and continued into the leptomeningeal heterotopia. Heterotopic gray matter was also present in the subjacent white matter, notably in the inferior parietal area as a thin crescentic band of heterotopic gray matter. The right occipital cortex was entirely replaced by gliotic tissue, which was the region adjacent to the venous sinus thrombosis that occurred in early infancy. In the hippocampus, the granule cells were depleted and their ribbon in the dentate fascia was splayed. In the medulla, the arcuate nuclei appeared unusually large, and the inferior olives were slightly simplified, while in the cerebellum, the dentate ribbon was dysplastic and fragmented into islands of gray matter (Fig. 15A,B).
Fig. 15.
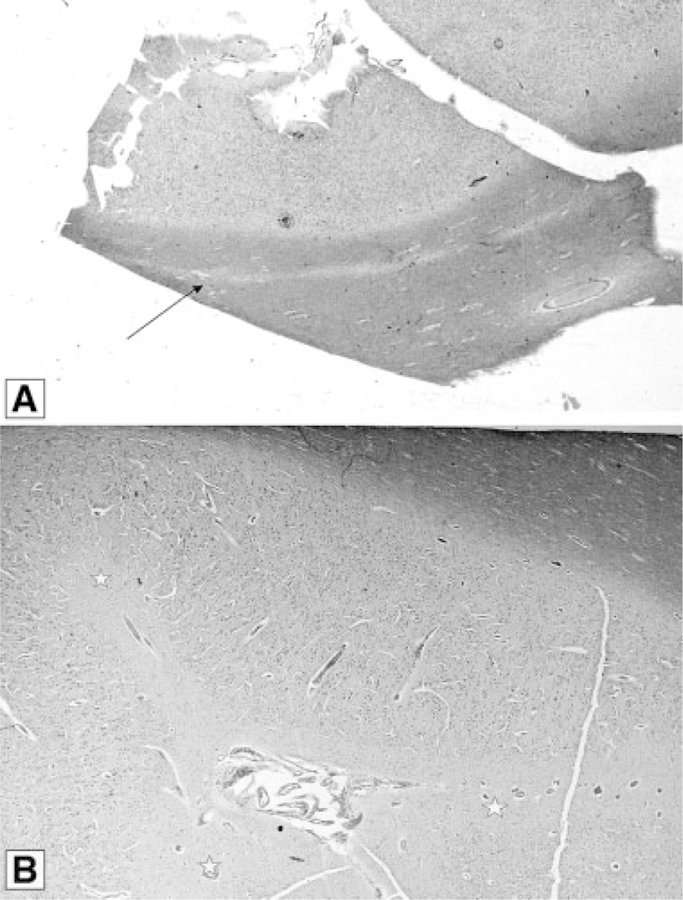
Patient 11 autopsy findings. A: The arrow indicates a thin lamina of heterotopic grey matter within the white matter and well separated from overlying cortex (luxol fast bluecresyl violet stain). B: Abortive sulci (stars) with median vessels suggesting gyral fusion in polymicrogyric cerebral cortex. These fingers of molecular layer point perpendicularly away from the surface.
In comparing the findings of case 11 with the case of Reardon et al. [1996] (also examined by BH) some similarities of neuromigration defects are evident. That patient died at 10 months. The autopsy demonstrated a large brain of 1,488 g (normal for age 1,080 g), right hemimegalencephaly, including the cerebellum and brainstem, a normal left hemisphere, and right-sided broad gyral convolutions with a cobblestone appearance over the frontal and temporal lobes and around the sylvian fissure. The right optic nerve was twice the size of the left, and the enlarged right cerebellar tonsil had a bifid appearance that was herniated to the C3 level. On re-review of this patient’s neuropathology taking into account the findings of case 11, there was extensive unlayered polymicrogyria in the right hemisphere, leptomeningeal heterotopia and band heterotopia anteriorly. The only abnormality in the brainstem was an enlarged right corticospinal tract.
DISCUSSION
Just as the clinical phenotype for M–CM is readily recognizable, there is often a recurrent and recognizable CNS phenotype ascertainable by MRI brain findings that can support the clinical diagnosis of the syndrome. This cohort was notable for the fact that all patients had some type of abnormality on head imaging and many of the abnormalities occurred at high frequency (>50% of cases). This pattern of neuroimaging anomalies appears to have enough consistency that a completely normal neuroimaging study in a patient with suspected M–CM should prompt the clinician to re-assess the diagnosis. In addition, this report supports the clinical observations of Robertson et al. [2000] and Thong et al. [1999] that neuroradiographic findings such as development of ventriculomegaly and CTH are dynamic events and are age-dependent.
In our opinion, the most striking observation was the frequency of CTH and the fact that in several of these patients this was an acquired rather than a congenital event. CTH appears to be a very common abnormality in M–CM, occurring in approximately 70% of our cohort. Noting that Garavelli et al. [2005] also identified CTH in 7/10 patients in their study, it may be that M–CM has one of the highest associations of CTH of any specific syndrome. In our four patients who exhibited acquired CTH, we were struck by the fact that this developed over a time span ranging from the first few months to 3 years of age, together with a marked increase in the occipitofrontal circumference and crossing of head growth centiles. This leads us to suspect that excessive brain growth is a contributing factor to CTH in this syndrome and indicates that patients with M–CM should be monitored with appropriate imaging studies during infancy and childhood.
Yano and Watanabe [2001] suggested that cardiac arrhythmia can occur in M–CM and may be life threatening, although this appears to be a relatively infrequent complication. We note that one of our patients suffered from atrial flutter in the newborn period (case 11), but after appropriate cardioversion, the problem did not recur and was not a factor in this patient’s death at 2 years of age. These authors suggested that arrhythmia is a potential cause of an early demise in this condition. While this may be a valid conclusion, we note that patient 1 in their report died unexpectedly during sleep at 33 months after being previously well, without any documented arrhythmia but with known CTH. Occasional patients with this disorder have also died unexpectedly during childhood [Stoll, 2003]. A dramatic example was the case of Reardon et al. [1996] that probably had M–CM, and who died suddenly at 10 months of age after an acute seizure. Autopsy showed right-sided brain overgrowth, polymicrogyria, and severe CTH to the C3 level. Since CTH in M–CM may be acquired and progressive, it represents a significant potential risk for sudden death due to brainstem compression. In cases with severe CTH, neurosurgical management could be a life-saving intervention. In our cohort, seven patients underwent ventricular shunting, one underwent ventriculostomy, and four received posterior fossa decompression for CTH (all of whom were also shunted). As has been noted by previous authors, since this is a disorder of primary megalencephaly, shunting does not improve the macrocephaly of this disorder. Nevertheless, indications for intervention become apparent when there is obvious obstructive ventriculomegaly. Whether or not adequate and early posterior fossa decompression decreases the risk of developing obstructive ventriculomegaly and the need for ventricular shunting by enlarging the infratentorial space and making room for the growing hindbrain remains unclear at this time.
We emphasize that acquired CTH can be a surprisingly rapid process in M–CM. In patient 11, CTH developed between the 3- and 5-month scans; in patient 14 the MRI at 8 days of age was normal, although by 2 months of age, there was clear crowding of the posterior fossa with compression on the brainstem and early CTH and with further progression over the next several months. This indicates that in the event of new neurologic findings in this syndrome, a high level of surveillance must be maintained. However, even if cerebellar overgrowth is the initial event leading to the dynamic changes of the posterior fossa, it is still unclear if there are secondary events contributing to the development of CTH or other observed abnormalities, such as venous dilatation or ventriculomegaly.
Since there is no report of basilar skull malformation or craniosynostosis in this syndrome, one might expect that the calvaria would remodel itself to accommodate brain growth. However, in the cases where cerebellar crowding of the posterior fossa is observed concurrently with CTH, this clearly does not occur. There may be additional causes for increased intracranial pressure in addition to increase of brain mass. We suspect that as the cerebellum grows, there might be secondary impingement of venous sinus drainage, and this may explain some of the neuroimaging findings in our study.
There is precedent for this hypothesis. Dilation of the CSF space can be impacted by obstructed venous drainage through basilar foramina in achondroplasia [Steinbok et al., 1989; Lundar et al., 1990; Rollins et al., 2000a,b; Moritani et al., 2006] Similar concerns may exist in syndromes of craniosynostosis [Martinez-Perez et al., 1996; Rollins et al., 2000a,b; Rich et al., 2003]. Thus, the dural sinus engorgement and perhaps even the ventriculomegaly of this syndrome might be due to impaired intracranial venous drainage. In achondroplasia and craniosynostotic disorders, venous congestion is due to external compression by abnormally small cranial foramina and a narrowed foramen magnum. However, in M–CM, we theorize that internal compression of the draining sinuses from the contents of the posterior fossa is the primary event that culminates in cerebral venous hypertension. If static pressure in the large draining sinuses builds, reabsorption of cerebrospinal fluid into the circulation through the arachnoid villi might be affected due to decreased CSF-venous pressure gradients. Reduced CSF absorption could thereby cause progressive ventriculomegaly and would explain why these findings are often seen concurrently in M–CM. If there is any impingement of CSF drainage, either via compression of the aqueduct due to a rapidly growing cerebellum, or congestion of CSF flow at the level of the foramen magnum from ectopic cerebellar tonsils, then this might lead to obstructive ventriculomegaly in some cases. Future investigations, perhaps utilizing magnetic resonance venography and cerebrospinal fluid flow studies, would be of value in investigating these issues.
Some individual findings in our cohort may be nonspecific, such as ventriculomegaly (which is reported in over 450 syndromes in Pictures of Standard Syndromes & Undiagnosed Malformations (POSSUM); http://www.possum.net.au/pub/manual.doc) and prominent Virchow–Robin spaces, which can be found in numerous disorders and syndromes, including autism, benign macrocephaly, lysosomal storage diseases, adrenoleukodystrophy, myotonic dystrophy, CADASIL, multiple sclerosis, Parkinson disease, and others. [Artigas et al., 1999; Laitinen et al., 2000; Di Costanzo et al., 2001; Patlas et al., 2001; Achiron and Faibel, 2002; Taber et al., 2004; Patankar et al., 2005; Cumurciuc et al., 2006; Groeschel et al., 2007]. Recent studies have demonstrated that some normal people may have dilated perivascular spaces, which raises questions concerning the pathologic significance of these anomalies in isolation [Groeschel et al., 2006]. However, since Virchow–Robin spaces occur in such a large percentage of our cohort and are often found together with CTH, ventriculomegaly, and large venous sinuses, this suggests that they are not coincidental occurrences.
The aberrant brain development in M–CM may explain much of the hypotonia, developmental delay, mental retardation, and seizure risk observed in this syndrome. However, comprehensive neuropathology of this syndrome has yet to be reported. The literature suggests that hemimegalencephaly is associated with M–CM, though the cerebral asymmetry in this syndrome probably represents brain hyperplasia caused by the underlying process of abnormal brain morphogenesis and overgrowth. It may differ from true hemimegalencephaly, as most patients in our cohort who have brain asymmetry do not have a severe seizure disorder, hemiplegia, or severe ipsilateral cortical dysplasia. In fact, in our cohort the presence of seizures seemed to correlate more definitively with cortical dysgenesis than with brain asymmetry. Whether the underlying defect of overgrowth is due to excessive growth factors, dysregulated cell cycle arrest, defective apoptosis, or another mechanism remains to be clarified. Clinicians caring for children with this syndrome should anticipate the development of white matter signal changes that resemble a leukoencephalopathy. Oneof the patients in our study was observed to have abnormal white matter in late infancy, which caused the treating physicians to initially consider a possible metabolic disorder despite the fact that there were no new neurologic symptoms or signs. We feel that these white matter changes are a common central nervous system marker of M–CM. It is still uncertain if they have pathologic significance outside of representing an abnormal myelination pattern. Importantly, they do not seem to herald or predict a poor prognosis and may improve during childhood. Concern for a neurodegenerative process affecting the white matter or metabolic syndrome causing leukodystrophy should be tempered if these changes are observed in a child with a secure diagnosis of M–CM.
While a significant percentage of patients in our cohort had cortical irregularities, they were not present in the majority. In the cases of polymicrogyria, a perisylvian distribution predominated. A similar pattern of perisylvian polymicrogyria has been documented in other genetic conditions [Barkovich et al., 1999; Guerreiro et al., 2000; Villard et al., 2002]. It has also been seen in the rare overgrowth disorder, Megalencephaly-Perisylvian polymicrogyria-Postaxial polydactyly-Hydrocephalus (MPPH) syndrome, a condition which shares some clinical overlap with, but is clearly distinct from, M–CM [Mirzaa et al., 2004; Colombani et al., 2006]. Patient 14, who had the most severe degree of cortical dysgenesis in the cohort, also had one of the most severe neurologic phenotypes with marked developmental delay and recurrent seizures that were partially responsive to medications. There is clearly more work that needs to be done on the molecular mechanisms that lead to the primary megalencephaly and cortical dysplasia in this disorder. While neuropathologic findings obtained at autopsy are extremely limited to date, available evidence suggests that this syndrome may be characterized by aberrant neuromigration during fetal development, though more investigations are required to determine the extent of possible cortical malformations on a cellular and molecular level.
The observations of a possible meningioma in one patient (case 14), and another with a paraspinal mass of possible neurogenic origin (case 16), are intriguing, given that meningioma has been seen before in M–CM. Meningiomas are extremely rare in the pediatric population, with incidence at 0–19 years of age of approximately 1:1,000,000 [Abeloff et al., 2004]. If the diagnosis of meningioma can be confirmed in Patient 14, it could be an important clue into possible causative pathways or responsible genes.
It is not at all clear why a significant minority of children with M–CM develop strokes or thrombotic events. Given that aberrant vascular morphogenesis seems nearly universal in the disorder, such events could relate to extreme examples of vascular compromise such as focal vascular dysplasia or malformations. Limited thrombophilia workups in affected patients do not appear to explain these problems [Moffitt et al., 1999].
MANAGEMENT RECOMMENDATIONS AND CONCLUSIONS
This study clearly demonstrates that neuroimaging with MRI should be part of the evaluation of a patient with M–CM. Principal neuroimaging abnormalities include white matter abnormalities, ventriculomegaly (obstructive or nonobstructive), cerebral asymmetry, cortical dysplasia and polymicrogyria-like changes, and CTH. We have demonstrated that CTH can be an acquired phenomenon in this disorder. In addition, there may be a mildly increased risk of meningioma or other benign neurogenic tumors. Because many of these findings seem to develop at a young age in affected patients and significant neurologic sequelae can result from compromise at the medullocervical junction, we strongly advocate serial brain imaging with MRI in patients with this syndrome, paying particular attention to assessing the posterior fossa.
In an otherwise stable patient, based on the age at which symptoms occurred in affected patients in our cohort, we suggest a baseline brain MRI at the time of diagnosis, with follow up studies every 6 months until age 2 years, and then interval assessment at 3 years. By this time most brain growth should be complete and we suspect that the clinical picture should become stable. Additional studies specifically designed to assess cerebrospinal fluid or venous flow dynamics should be considered on an individual basis and may help clarify contributing factors for some of the neuroimaging abnormalities in M–CM.
An alternative regimen to the one proposed may be indicated, depending on the specific neuroradiographic findings and the clinical situation of the individual patient. Physicians should maintain a very low threshold to order additional or interval neuroimaging studies based on neurologic signs and symptoms. Imaging assessments or referral to neurosurgery should be considered if there is a rapidly enlarging head that is crossing growth centiles; acute or progressive paresis; focal neurologic signs; seizures; apnea, swallowing problems, oculomotor difficulties, and other brain stem signs; or lethargy, irritability, headache, or other symptoms of increased intracranial pressure. Prompt neurosurgical evaluation should be requested when there is progressive ventriculomegaly or CTH in conjunction with symptoms of concern. Cautious surveillance for neurologic symptoms might help prevent significant mortality or co-morbidity, improve long-term outcomes for affected patients, and decrease the risk of early or sudden death in M–CM.
ACKNOWLEDGMENTS
We appreciate support from SHARE’s Childhood Disability Center, the Steven Spielberg Pediatric Research Center, the Cedars-Sinai Burns and Allen ResearchInstitute,theSkeletalDysplasiasNIH/NICHD Program Project Grant (HD22657), and the Medical Genetics NIH/NIGMS Training Program Grant (GM08243). In addition, we extend our acknowledgment and thanks to Dr. Charis Eng of the Genomic Medicine Institute at the Cleveland Clinic, who performed PTEN analysis on many of our patients.
Grant sponsor: NIH/NICHD Program Project; Grant number: HD22657; Grant sponsor: Medical Genetics NIH/NIGMS Training Program; Grant number: GM08243.
REFERENCES
- Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, Gillies McKenna W. 2004. Clinical oncology. 3rd edition. New York: Churchill Livingstone. [Google Scholar]
- Achiron A, Faibel M. 2002. Sandlike appearance of Virchow-Robin spaces in early multiple sclerosis: A novel neuroradiologic marker. Am J Neuroradiol 23:376–380. [PMC free article] [PubMed] [Google Scholar]
- Ackar N, Adapinar B, Dinleyici C, Durak B, Ozkan IR. 2004. A case of macrocephaly-cutis marmorata telangiectatica congenita and review of neuroradiologic features. Ann Genet 47:261–265. [DOI] [PubMed] [Google Scholar]
- AnalyzeDirect. Analyze Versions 6.0 and 7.0.
- Artigas J, Poo P, Rovira A, Cardo E. 1999. Macrocephaly and dilated Virchow-Robin spaces in childhood. Pediatr Radiol 29:188–190. [DOI] [PubMed] [Google Scholar]
- Baralle D, Firth H. 2000. A case of the new overgrowth syndrome—Macrocephaly with cutis marmorata, haemangioma and syndactyly. Clin Dysmorphol 9:209–211. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Kjos BO. 1988. Normal postnatal development of the corpus callosum as demonstrated by MR imaging. Am J Neuroradiol 9:487–491. [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Hevner R, Guerrini R. 1999. Syndromes of bilateral symmetrical polymicrogyria. Am J Neuroradiol 20:1814–1821. [PMC free article] [PubMed] [Google Scholar]
- Barnicoat A, Salman M, Chitty L, Baraitser M. 1996. A distinctive overgrowth syndrome with polysyndactyly. Clin Dysmorphol 5:339–346. [PubMed] [Google Scholar]
- Carcao M, Blaser SI, Grant RM, Weksberg R, Siegel-Bartelt J. 1998. MRI findings in macrocephaly-cutis marmorata telangiectatica congenita. Am J Med Genet 76:165–167. [PubMed] [Google Scholar]
- Chen SC, Simon EM, Haselgrove JC, Bilaniuk LT, Sutton LN, Johnson MP, Shera DM, Zimmerman RA. 2006. Fetal posterior fossa volume: Assessment with MR imaging. Radiology 238: 997–1003. [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J, Kerr B, Brunner H, Tranebjaerg L, Magee A, Hennekam RCM, Mueller RF, Brueton L, Super M, Steen-Johnsen J, Donnai D. 1997. Macrocephaly with cutis marmorata, haemangioma and syndactyly—A distinctive overgrowth syndrome. Clin Dysmorphol 6:291–302. [DOI] [PubMed] [Google Scholar]
- Colombani M, Chouchane M, Pitelet G, Morales L, Callier P, Pinard JP, Lion-Francois L, Thauvin-Robinet C, Mugneret F, Huet F, Guibaud L, Faivre L. 2006. A new case of megalencephaly and perisylvian polymicrogyria with post-axial polydactyly and hydrocephalus: MPPH syndrome. Eur J Med Genet 49:466–471. [DOI] [PubMed] [Google Scholar]
- Conway RL, Danielpour M, Graham JM Jr. 2007. Surgical management of cerebellar tonsillar herniation in three patients with macrocephaly-cutis marmorata telangiectatica congenita. Report of three cases. J Neurosurg 106:296–301. [DOI] [PubMed] [Google Scholar]
- Cristaldi A, Vigevano F, Antoniazzi G, di Capua M, Andreuzzi A, Morselli G, Iorio F, Fariello G, Trasimeni G, Gualdi GF. 1995. Hemimegalencephaly, hemihypertrophy and vascular lesions. Eur J Pediatr 154:134–137. [DOI] [PubMed] [Google Scholar]
- Cumurciuc R, Guichard JP, Reizine D, Gray F, Bousser MG, Chabriat H. 2006. Dilation of Virchow-Robin spaces in CADASIL. Eur J Neurol 13:187–190. [DOI] [PubMed] [Google Scholar]
- Di Costanzo A, Di Salle F, Santoro L, Bonavita V, Tedeschi G. 2001. Dilated Virchow-Robin spaces in myotonic dystrophy: Frequency, extent and significance. Eur Neurol 46:131–139. [DOI] [PubMed] [Google Scholar]
- Di Rocco C, Battaglia D, Pietrini D, Piastra M, Massimi L. 2006. Hemimegalencephaly: Clinical implications and surgical treatment. Childs Nerv Syst 22:852–866. [DOI] [PubMed] [Google Scholar]
- Flores-Sarnat L. 2002. Hemimegalencephaly: Part 1. Genetic, clinical, and imaging aspects. J Child Neurol 17:373–384. [DOI] [PubMed] [Google Scholar]
- Franceschini P, Licata D, Di Cara G, Guala A, Franceschini D, Genitori L. 2000. Macrocephaly-Cutis marmorata telangiectatica congenita without cutis marmorata? Am J Med Genet 90:265–269. [PubMed] [Google Scholar]
- Garavelli L, Leask K, Zanacca C, Pedori S, Albertini G, Della Giustina E, Croci GF, Magnani C, Banchini G, Clayton-Smith J, Bocian M, Firth H, Gold JA, Hurst J. 2005. MRI and neurological findings in macrocephaly-cutis marmorata telangiectatica congenita syndrome: Report of ten cases and review of the literature. Genet Counsel 16:117–128. [PubMed] [Google Scholar]
- Giuliano F, David A, Edery P, Sigaudy S, Bonneau D, Cormier-Daire V. 2004. Macrocephaly-cutis marmorata telangiectatica congenita: Seven cases including two with unusual cerebral manifestations. Am J Med Genet Part A 126A:99–103. [DOI] [PubMed] [Google Scholar]
- Groeschel S, Chong WK, Surtees R, Hanefeld F. 2006. Virchow-Robin spaces on magnetic resonance images: Normative data, their dilatation, and a review of the literature. Neuroradiology 48:745–754. [DOI] [PubMed] [Google Scholar]
- Groeschel S, Brockmann K, Hanefeld F. 2007. Virchow-Robin spaces on magnetic resonance images of children with adrenoleukodystrophy. Eur J Paediatr Neurol 11:142–145. [DOI] [PubMed] [Google Scholar]
- Guerreiro MM, Andermann E, Guerrini R, Dobyns WB, Kuzniecky R, Silver K, Van Bogaert P, Gillain C, David P, Ambrosetto G, Rosati A, Bartolomei F, Parmeggiani A, Paetau R, Salonen O, Ignatius J, Borgatti R, Zucca C, Bastos AC, Palmini A, Fernandes W, Montenegro MA, Cendes F, Andermann F. 2000. Familial perisylvian polymicrogyria: A new familial syndrome of cortical mal development. Ann Neurol 48:39–48. [PubMed] [Google Scholar]
- Laitinen LV, Chudy D, Tengvar M, Hariz MI, Bergenheim AT. 2000. Dilated perivascular spaces in the putamen and pallidum in patients with Parkinson’s disease scheduled for pallidotomy: A comparison between MRI findings and clinical symptoms and signs. Mov Disord 15:1139–1144. [DOI] [PubMed] [Google Scholar]
- Lapunzina P, Gairi A, Delicado A, Mori MA, Torres ML, Goma A, Navia M, Pajares IL. 2004. Macrocephaly-cutis marmorata telangiectatica congenita: Report of six new patients and a review. Am J Med Genet Part A 130A:45–51. [DOI] [PubMed] [Google Scholar]
- Lundar T, Bakke SJ, Nornes H. 1990. Hydrocephalus in an achondroplastic child treated by venous decompression at the jugular foramen. Case report. J Neurosurg 73:138–140. [DOI] [PubMed] [Google Scholar]
- Martinez-Perez D, Vander Woude DL, Barnes PD, Scott RM, Mulliken JB. 1996. Jugular foraminal stenosis in Crouzon syndrome. Pediatr Neurosurg 25:252–255. [DOI] [PubMed] [Google Scholar]
- Megarbane A, Haddad J, Lyonnet S, Clayton-Smith J. 2003. Child with overgrowth, pigmentary streaks, polydactyly, and intestinal lymphangiectasia: Macrocephaly-cutis marmorata telangiectatica congenita syndrome or new disorder? Am J Med Genet Part A 116A:184–187. [DOI] [PubMed] [Google Scholar]
- Meyer E. 1979. Neurocutaneous syndrome with excessive macrohydrocephalus. (Sturge-Weber/Klippel-Trenaunay syndrome). Neuropädiatrie 10:67–75. [DOI] [PubMed] [Google Scholar]
- Mirzaa G, Dodge NN, Glass I, Day C, Gripp K, Nicholson L, Straub V, Voit T, Dobyns WB. 2004. Megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus: A rare brain malformation syndrome associated with mental retardation and seizures. Neuropediatrics 35:353–359. [DOI] [PubMed] [Google Scholar]
- Moffitt DL, Kennedy CT, Newbury-Ecob R. 1999. What syndrome is this? Macrocephaly with cutis marmorata, hemangioma, and syndactyly syndrome. Pediatr Dermatol 16:235–237. [DOI] [PubMed] [Google Scholar]
- Moore CA, Toriello HV, Abuelo DN, Bull MJ, Curry CJ, Hall BD, Higgins JV, Stevens CA, Twersky S, Weksberg R, Dobyns WB. 1997. Macrocephaly-cutis marmorata telangiectatica congenita: A distinct disorder with developmental delay and connective tissue abnormalities. Am J Med Genet 70:67–73. [PubMed] [Google Scholar]
- Moritani T, Aihara T, Oguma E, Makiyama Y, Nishimoto H, Smoker WR, Sato Y. 2006. Magnetic resonance venography of achondroplasia: Correlation of venous narrowing at the jugular foramen with hydrocephalus. Clin Imaging 30:195–200. [DOI] [PubMed] [Google Scholar]
- Ng WH, Chan YL, Au KS, Yeung KW, Kwan TF, To CY. 2005. Morphometry of the corpus callosum in Chinese children: Relationship with gender and academic performance. Pediatr Radiol 35:565–571. [DOI] [PubMed] [Google Scholar]
- Nyberg RH, Uotila J, Kirkinen P, Rosendahl H. 2005. Macrocephaly-cutis marmorata telangiectatica congenita syndrome—Prenatal signs in ultrasonography. Prenat Diagn 25:129–132. [DOI] [PubMed] [Google Scholar]
- Patankar TF, Mitra D, Varma A, Snowden J, Neary D, Jackson A. 2005. Dilatation of the Virchow-Robin space is a sensitive indicator of cerebral microvascular disease: Study in elderly patients with dementia. Am J Neuroradiol 26:1512–1520. [PMC free article] [PubMed] [Google Scholar]
- Patlas M, Shapira MY, Nagler A, Sheffer R, Gomori JM. 2001. MRI of mannosidosis. Neuroradiology 43:941–943. [DOI] [PubMed] [Google Scholar]
- Piatt JH Jr. 2004. Unexpected findings on brain and spine imaging in children. Pediatr Clin North Am 51:507–527. [DOI] [PubMed] [Google Scholar]
- Prassopoulos P, Cavouras D, Golfinopoulos S. 1996. Developmental changes in the posterior cranial fossa of children studied by CT. Neuroradiology 38:80–83. [DOI] [PubMed] [Google Scholar]
- Reardon W, Harding B, Winter R, Baraitser M. 1996. Hemihypertrophy, hemimegalencephaly, and polydactyly. Am J Med Genet 66:144–149. [DOI] [PubMed] [Google Scholar]
- Rich PM, Cox TC, Hayward RD. 2003. The jugular foramen in complex and syndromic craniosynostosis and its relationship to raised intracranial pressure. Am J Neuroradiol 24:45–51. [PMC free article] [PubMed] [Google Scholar]
- Robertson SP, Gattas M, Rogers M, Ades LC. 2000. Macrocephalycutis marmorata telangiectatica congenita: Report of five patients and a review of the literature. Clin Dysmorphol 9:1–9. [PubMed] [Google Scholar]
- Rollins N, Booth T, Shapiro K. 2000a. MR venography in children with complex craniosynostosis. Pediatr Neurosurg 32:308–315. [DOI] [PubMed] [Google Scholar]
- Rollins N, Booth T, Shapiro K. 2000b. The use of gated cine phase contrast and MR venography in achondroplasia. Childs Nerv Syst 16:569–575. [DOI] [PubMed] [Google Scholar]
- Saul RA, Seaver LH, Sweet KM, Geer JS, Phelan MC, Mills CM. 1998. Growth References: Third Trimester to Adulthood. Greenwood Genetic Center.
- Schwartz IV, Felix TM, Riegel M, Schuler-Faccini L. 2002. Atypical macrocephaly-cutis marmorata telangiectatica congenita with retinoblastoma. Clin Dysmorphol 11:199–202. [DOI] [PubMed] [Google Scholar]
- StataCorp. 2005. Stata Statistical Software: Release 9. College Station, TX: StataCorp LP. [Google Scholar]
- Steinbok P, Hall J, Flodmark O. 1989. Hydrocephalus in achondroplasia: The possible role of intracranial venous hypertension. J Neurosurg 71:42–48. [DOI] [PubMed] [Google Scholar]
- Stoll C. 2003. Macrocephaly-cutis marmorata telangiectatica congenita: Report of a patient with a translocation. Genet Counsel 14:173–179. [PubMed] [Google Scholar]
- Taber KH, Shaw JB, Loveland KA, Pearson DA, Lane DM, Hayman LA. 2004. Accentuated Virchow-Robin spaces in the centrum semiovale in children with autistic disorder. J Comput Assist Tomogr 28:263–268. [DOI] [PubMed] [Google Scholar]
- Thong MK, Thompson E, Keenan R, Simmer K, Harbord M, Davidson G, Haan E. 1999. A child with hemimegalencephaly, hemihypertrophy, macrocephaly, cutaneous vascular malformation, psychomotor retardation and intestinal lymphangiectasia—A diagnostic dilemma. Clin Dysmorphol 8:283–286. [PubMed] [Google Scholar]
- Tinkle BT, Schorry EK, Franz DN, Crone KR, Saal HM. 2005. Epidemiology of hemimegalencephaly: A case series and review. Am J Med Genet Part A 139A:204–211. [DOI] [PubMed] [Google Scholar]
- Toriello HV, Mulliken JB. 2007. Accurately renaming macrocephaly-cutis marmorata telangiectatica congenita (M-CMTC) as macrocephaly-capillary malformation (M-CM). Am J Med Genet Part A (in press). [DOI] [PubMed]
- Villard L, Nguyen K, Cardoso C, Martin CL, Weiss AM, Sifry-Platt M, Grix AW, Graham JM Jr, Winter RM, Leventer RJ, Dobyns WB. 2002. A locus for bilateral perisylvian polymicrogyria maps to Xq28. Am J Hum Genet 70:1003–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogels A, Devriendt K, Legius E, Decock P, Marien J, Hendrickx G, Fryns JP. 1998. The macrocephaly-cutis marmorata telangiectatica congenita syndrome; Long-term follow-up data in 4 children and adolescents. Genet Counsel 9:245–253. [PubMed] [Google Scholar]
- Yano S, Watanabe Y. 2001. Association of arrhythmia and sudden death in macrocephaly-cutis marmorata telangiectatica congenita syndrome. Am J Med Genet 102:149–152. [DOI] [PubMed] [Google Scholar]