

Abstract
Introduction
Kinesin-1 motor is a molecular walking machine constructed with amino acids. The understanding of how those structural elements play their mechanical roles is the key to the understanding of kinesin-1 mechanism.
Methods
Using molecular dynamics simulations, we investigate the role of a helix structure, α4 (also called switch-II helix), of kinesin-1’s motor domain in its processive movement along microtubule.
Results
Through the analysis of the structure and the interactions between α4 and the surrounding residues in different nucleotide-binding states, we find that, mechanically, this helix functions as a shaft for kinesin-1’s motor-domain rotation and, structurally, it is an amphipathic helix ensuring its shaft functioning. The hydrophobic side of α4 consists strictly of hydrophobic residues, making it behave like a lubricated surface in contact with the core β-sheet of kinesin-1’s motor domain. The opposite hydrophilic side of α4 leans firmly against microtubule with charged residues locating at both ends to facilitate its positioning onto the intra-tubulin groove.
Conclusions
The special structural feature of α4 makes for an effective reduction of the conformational work in kinesin-1’s force generation process.
Keywords: Kinesin-1, α4 helix, Tubulin, Molecular dynamics simulation, Hydrophobic contact
Introduction
Kinesin is one kind of motor protein which walks along microtubule and is responsible for a variety of cellular events including but not limited to organelle transportation, mitosis and depolymerizing microtubule.16–19,50 According to the functions and amino acid sequences, kinesin can be divided into 14 subfamilies.28 All the members of kinesin families have a functional domain called “motor domain” or “motor head” which includes kinesin’s nucleotide-binding and microtubule-binding site.8,31 The α4 helix (also called switch-II helix) is the longest helix across the full width of kinesin’s motor domain from nucleotide-binding pocket to neck linker.45 α4 is the structural center of the motor domain with the core β-sheet shaped around it and it also forms one of the key microtubule-binding sites.5,21,26,30,44 In kinesin’s mechanical process, α4 plays multiple roles as a single secondary structure. Here, we investigate how the functions of α4 are achieved through a highly rational structural arrangement.
Kinesin-1 (conventional kinesin) is the most investigated kinesin subfamily and the model protein for the research of N-type kinesin. To efficiently walk along microtubule, two identical kinesin-1 proteins form a dimer and take “hand-over-hand” pattern in their processive movement.1,15,24,50–52 After ~ 30 years intensive investigation, the walking, energy-transformation and inter/intra motor-domain communication mechanism of kinesin-1 is much clearer now.8 It is also realized that the “cytoskeletal track”, microtubule, also plays an important role in the modulation of kinesin-1’s mechanochemical cycle.34,35,41,42 The binding strength of kinesin’s motor domain to microtubule varies with different nucleotide-binding states. For kinesin-1, the motor domain binds tightly to microtubule in nucleotide-free (apo) or ATP-binding state and loosely binds to microtubule in the ADP-binding state.2–4,9 As part of kinesin’s microtubule-binding sites, α4 is considered to play very important role in recognizing and binding microtubule.
The corresponding structure of α4 in myosin is the relay helix.27,49 Similar to myosin’s relay helix, kinesin’s α4 is considered to be responsible for the long-range communication from nucleotide-binding site to motor domain’s C-terminus which triggers the docking movement of neck linker.50 With the progress of cyto-electron microscopy (cryo-EM) and crystallization techniques, high resolution structures of motor domain in different nucleotide-binding states on tubulin are obtained.5,7,14,44 It is realized that, in the conformational change from apo state to ATP-binding state, the α4 helix is stationary on the groove between α- and β-tubulin. In contrast, the motor domain rotates around α4, prepares the environment for ATP hydrolysis,22 detaches the trailing head from microtubule 12 and triggers the neck linker docking.11,13 However, how α4 functions in this process remains unclear.
The experimental structures give us massive knowledge of interactions between α4 and surrounding residues. However, crystal and cryo-EM structures represent the specific conformations of the protein under experimental condition (usually at low temperature). To obtain the stable binding structures of kinesin-1 on tubulin in the environment with thermal fluctuation and statistically investigate the interactions of α4 with microtubule and the inner part of kinesin-1, molecular dynamics (MD) simulations for kinesin-1’s motor domain in different nucleotide-binding states (apo, ATP-binding and ADP-binding state) on tubulin are performed. The analysis of the obtained stable structures shows that α4 is an amphipathic structure to meet the requirements of its mechanical function. In the motor-domain rotation process from the apo state to the ATP-binding state, α4 functions as a lubricated shaft with its hydrophobic side facing the hydrophobic inner surface of the core β-sheet. At the same time, with its hydrophilic side facing the polar surface of microtubule, α4 binds tightly to the intra-tubulin groove through both interactional and geometrical match. This highly rational design of α4 ensures the stability for it as a shaft and a sufficient reduction of the energy consumption in the conformational change. The α4 of the ADP-binding state kinesin-1 has a shorter length and binds weakly to tubulin due to the lost in part of the interactional and geometrical match with tubulin, which is consistent with the experimental fact.
Methods
Model Construction
The kinesin-1-tubulin complex structures (PDB ID: 4HNA 14 and 4LNU 7) and kinesin-1’s motor domain monomer in ADP-binding state (PDB ID: 1BG2 27) are used for modeling. The motor domain of 4HNA is considered in the ATP-binding state and that of 4LNU is in the nucleotide-free state. In the modeling, the ADP-AlF4− of 4HNA is replaced by ATP while keeping the adenosine rings of these two molecules coincided. The designed ankyrin repeat proteins (DARPins) of 4HNA and 4LNU were used to maintain the tubulin structure when crystallization and are deleted in our models. The monomer structure in ADP-binding state, 1BG2, is placed above the tubulin to freely search its binding site on tubulin in the MD simulation and the stable binding structure of kinesin-1 and tubulin in ADP-binding state is obtained. The coordinate water molecules and Mg2+ are maintained. The three kinesin-1-tubulin complexes are buried in a water sphere (the radius is 70 Å). TIP3P 23 is used to model water molecules. Na+ and Cl− ions are added to ensure an ionic concentration of 150 mM and zero net charge. The entire model has ~ 150,000 atoms. During the MD run, the α-carbons of E168, L227, V353 in α-tubulin and T168, L227, T353 in β-tubulin are harmonically constrained to ensure a stable tubulin structure. The software used for modeling and data analysis is VMD (version 1.9.3).20 The MD simulations are performed using NAMD (version 2.12 with CUDA acceleration)39 with force field CHARMM366,32,33,38 at constant temperature of 310 K. The integration time step is 2 fs. The non-bonded coulomb and van der Waals interactions are calculated with a cutoff using a switch function starting at distance of 13 Å and reaching 0 at 15 Å. Before the simulation run, the protein-water systems are minimized for 30,000 steps to remove bad contact. The three systems are all simulated 100 ns and the trajectories are saved every 1 ps. The first 20 ns trajectory is the relaxation process and eliminated in our statistical analysis. Molecular drawing is produced using Discovery studio 3.5 visualizer and Origin 8.5.
Calculation of the Occupancy Rate
The criterion of hydrogen bond is that the maximum distance between acceptor and donor atoms of a hydrogen bond is 3 Å.37 For the hydrogen bond associated with charged group, the criterion is 4.5 Å because the hydrogen-bonding interaction of charged group with other residues is stronger than normal hydrogen bond. The criterion for salt bridge is that the maximum distance of the central atoms (Cζ of arginine, Nζ of lysine, Cγ of aspartic acid and Cδ of glutamic acid) of two charged groups is 8 Å. For the hydrophobic interaction, two hydrophobic residues with distance between central atoms of hydrophobic groups smaller than 8 Å is considered having hydrophobic interaction.29 Using these criteria, the total interactions between α4 and the surrounding residues are statistically analyzed. The effective strength of each interaction is quantitatively indicated by the occupancy rate which is the percentage of the binding time vs. the total production run (80 ns) and the average distance between interacting atoms.
Results and Discussion
Kinesin’s α4 is an Amphipathic Helix
Kinesin’s motor domain has ~ 340 amino acids. The α4 helix locates at kinesin’s interface with microtubule. Fig. 1 shows the conformations of α4 in kinesin-1’s different nucleotide states. The α4 of kinesin-1 consists of 25 residues (from A246 to E270 in human kinesin; herein, we use the residue number of human kinesin with PDB ID: 4HNA 14). The geometrical locations of the 25 residues in α4 are shown in Fig. 2a. In these 25 residues, there are 5 charged residues and 13 hydrophobic residues. The arrangement of these residues reveals a marked amphipathic feature of α4: the upper half, which points to the inner part of the motor domain, mainly consists of hydrophobic residues and the lower half, which points to the microtubule surface, consists mainly of charged and polar residues. The residue sequences of kinesin’s different subfamilies are listed in Fig. 2b. From the comparison of sequences, it is seen that some residues are highly conservative (E250, I254, N255, S257, L258, L261, I265 and L268). Most of the hydrophobic residues locate toward the core β-sheet of kinesin and form hydrophobic contact with residues in kinesin’s inner part. These residues play very important role in kinesin’s functional movement along microtubule as shown in the following sections.
Figure 1.

Stable complex structures of kinesin’s motor domain (blue) with tubulin in different nucleotide-binding states obtained from molecular dynamics simulations. (a) ATP-binding state. (b) apo state. (c) ADP-binding state. Motor domain is shown in blue, α-tubulin in light brown and β-tubulin in dark brown. The binding nucleotides are explicitly shown. α4s of these structures are shown in red.
Figure 2.

Residues of kinesin’s α4 helix. (a) Locations of α4’s residues in the helix structure. (b) Sequence alignments of α4s of different kinesin subfamilies (2KIN43 (from Rattus), 4HNA14 (from human) and 1GOJ47 (from N. crassa) belong to kinesin-1 subfamily; 1I5S25 belongs to kinesin-3 subfamily; 1II648 belongs to kinesin-5 subfamily; 1T5C10 belongs to kinesin-7 subfamily; 2NR8 belongs to kinesin-9 subfamily and 1V8J36 belongs to kinesin-13 subfamily). The residues toward motor domain’s core β-sheet (the upper half) and toward tubulin (the lower half) are colored in green and red respectively at the top of the alignments.
α4 has different conformations in different nucleotide-binding states (apo, ATP-binding and ADP-binding state) and microtubule-binding states (with or without microtubule). As seen in Figs. 1a and 1b, the MD simulation results show that α4 has similar conformations (7-turn helix) in kinesin-1’s ATP-binding and apo state on microtubule. While in the ADP-binding state, α4 is a 4-turn helix as shown in Fig. 1c. The cryo-EM study of kinesin-1 in ADP-binding state on microtubule showed a consistent structure.46 From the ADP-binding state to apo state, α4 will change from a 4-turn helix to a 7-turn helix. When and how this conformational change of α4 take place are still unclear. It is rational to speculate that, when binding to microtubule, the N-terminal part of α4 (from A246 to K256) will turn from a loop structure to three extra helix structure (transform from a 4-turn helix to a 7-turn helix). This conformational change might be related with the catalytic role of microtubule in kinesin-1’s ATPase cycle.
To statistically analyze the interactions between α4 and the surrounding residues, three MD simulations are performed based on the crystal structures of kinesin-1 in different nucleotide-binding states (see “Methods”). All the interactions between residues of α4 and surrounding residues are listed in Tables 1 and 2. The occupancy rate of each interaction in the MD simulation is calculated to quantitatively describe the effective strength of the interaction. The analysis of following sections is based on these statistical calculations and the stable structures obtained from MD simulations.
Table 1.
Occupancy rates of interactions between α4 and tubulin, the average distance between interacting atoms and the corresponding distance in the crystal structures.
| Interactions between α4 and tubulin: occupancy rate (average distance) (distance in crystal structures) | |||
|---|---|---|---|
| Interacting residues | Apo state | ATP-binding state | ADP-binding state |
| Leu248–Thr109 (α-tubulin) | 100% (5.21 Å) (5.09 Å) | 96.94% (6.53 Å) (7.48 Å) | 0% (14.59 Å) (null) |
| Leu248–Tyr108 (α-tubulin) | 100% (6.76 Å) (5.65 Å) | 100% (5.12 Å) (6.55 Å) | 6.37% (11.25Å) (null) |
| Leu248–Lys112 (α-tubulin) | 99.75% (6.78 Å) (6.21 Å) | 95.15% (7.17 Å) (4.88 Å) | 32.32% (10.02 Å) (null) |
| Lys252–Glu411 (α-tubulin) | 92.95% (6.04 Å) (4.99 Å) | 99.98% (5.12 Å) (4.65 Å) | 0% (16.15 Å) (null) |
| Asn255–Gly412 (α-tubulin) | 98.94% (3.10 Å) (3.63 Å) | 63.44% (3.85Å) (3.52 Å) | 0% (12.39 Å) (null) |
| Ser259–Glu415 (α-tubulin) | 26.40% (6.43 Å) (4.05 Å) | 84.96% (3.93 Å) (5.29 Å) | 12.27% (7.40 Å) (null) |
| Glu270–Lys401 (α-tubulin) | 84.35% (6.72 Å) (8.20 Å) | 95.53% (5.65 Å) (8.01 Å) | 91.44% (5.62 Å) (null) |
| Asn263–Arg402 (α-tubulin) | 63.73% (6.42 Å) (6.23 Å) | 98.47% (4.92 Å) (6.13 Å) | 26.57% (7.40 Å) (null) |
Table 2.
Occupancy rates of interactions between α4 and core β-sheet, the average distance between interacting atoms and the corresponding distance in the crystal structures.
| Interactions between α4 and core β-sheet: occupancy rate (average distance) (distance in crystal structures) | |||
|---|---|---|---|
| Interacting residues | Apo state | ATP-binding state | ADP-binding state |
| Val247–Ala243 | 90.97% (7.45 Å) (4.23 Å) | 100% (4.69 Å) (4.42 Å) | 21.04% (11.78 Å) (13.26 Å) |
| Leu248–Val238 | 99.67% (6.56 Å) (5.82 Å) | 100% (5.34 Å) (8.71 Å) | 76.92% (8.17 Å) (13.79 Å) |
| Leu248–Lys252 | 100% (5.05 Å) (4.59 Å) | 100% (5.26 Å) (10.44 Å) | 31.50% (10.64 Å) (7.20 Å) |
| Asp249–Lys252 | 90.37% (6.29 Å) (6.25 Å) | 100% (4.47 Å) (6.38 Å) | 4.70% (12.46 Å) (8.44 Å) |
| Asp249–Asn253 | 78.09% (3.40 Å) (5.49 Å) | 31.93% (4.60 Å) (4.52 Å) | 0% (13.04 Å) (12.11 Å) |
| Glu250–Tyr138 | 99.81% (2.71 Å) (4.49 Å) | 99.53% (2.76 Å) (4.08 Å) | 0% (16.48 Å) (20.64 Å) |
| Glu250–Arg203 | 100% (3.79 Å) (4.71 Å) | 99.55% (6.74 Å) (5.20 Å) | 0.29% (14.17 Å) (15.35 Å) |
| Ala251–Val238 | 100% (4.95 Å) (4.67 Å) | 100% (5.26 Å) (6.85 Å) | 63.17% (7.77 Å) (10.78 Å) |
| Ala251–Ala243 | 7.61% (10.01Å) (4.66 Å) | 100% (5.19 Å) (8.56 Å) | 0.32% (18.40 Å) (12.00 Å) |
| Ala251–Glu236 | 100% (4.10 Å) (4.55 Å) | 100% (4.88 Å) (4.04 Å) | 0% (10.39 Å) (15.34 Å) |
| Ile254–Leu139 | 99.23% (7.51 Å) (8.97 Å) | 97.35% (7.30 Å (8.56 Å) | 0% (14.15 Å) (16.43 Å) |
| Ile254–Ala233 | 100% (4.57 Å) (3.95 Å) | 100% (4.23 Å) (3.97 Å) | 26.27% (8.54 Å) (11.93 Å) |
| Ile254–Ala233 | 100% (4.22 Å) (4.13 Å) | 100% (4.00 Å) (3.66 Å) | 3.09% (10.18 Å) (9.19 Å) |
| Asn255–Glu236 | 99.55% (2.93 Å) (2.74 Å) | 81.70% (3.58 Å) (3.26 Å) | 0% (8.07 Å) (9.33 Å) |
| Asn255–Glu236 | 71.85% (2.71 Å) (2.03 Å) | 76.59% (2.76 Å) (2.09 Å) | 0% (8.51 Å) (9.40 Å) |
| Lys256–Asp140 | 63.31% (7.58 Å) (6.85 Å) | 97.81% (5.16 Å) (8.74 Å) | 62.28% (7.51 Å) (3.94 Å) |
| Ser257–His205 | 96.62% (2.08 Å) (3.10 Å) | 66.55% (2.73 Å) (2.80 Å) | 14.78% (4.00 Å) (4.45 Å) |
| Lys256–Asp279 | 74.13% (6.99 Å) (10.01 Å) | 7.77% (9.96 Å) (9.69 Å) | 77.31% (5.86 Å) (8.80 Å) |
| Leu258–Leu232 | 100% (6.13 Å) (5.94 Å) | 100% (5.45 Å) (6.00 Å) | 100% (5.89 Å) (5.68 Å) |
| Leu258–Ala233 | 100% (6.01 Å) (6.73 Å) | 100% (6.65 Å) (6.28 Å) | 100% (5.89 Å) (5.22 Å) |
| Leu258–Met282 | 99.58% (6.28 Å) (6.24 Å) | 85.48% (7.47 Å) (9.56 Å) | 99.24% (6.86 Å) (6.65 Å) |
| Ala260–Thr283 | 100% (5.05 Å) (4.54 Å) | 99.99% (5.72 Å) (4.05 Å) | 100% (4.15 Å) (4.28 Å) |
| Ala260–Ser280 | 100% (3.88 Å) (3.44 Å) | 100% (4.17 Å) (3.73 Å) | 100% (3.83 Å) (3.40 Å) |
| Leu261–Met282 | 100% (4.87 Å) (4.20 Å) | 99.98% (5.88 Å) (7.95 Å) | 100% (5.02 Å) (6.84 Å) |
| Leu261–Leu286 | 100% (7.90 Å) (7.70 Å) | 100% (7.23 Å) (6.65 Å) | 100% (6.86 Å) (8.03 Å) |
| Leu261–Phe82 | 100% (4.74 Å) (4.58 Å) | 100% (5.10 Å) (4.88 Å) | 100% (4.74 Å) (4.80 Å) |
| Leu261–Ile298 | 99.94% (6.47 Å) (6.20 Å) | 95.14% (8.05 Å) (7.24 Å) | 97.38% (7.80 Å) (5.78 Å) |
| Leu261–Ala322 | 91.16% (7.93 Å) (7.54 Å) | 65.62% (8.87Å) (8.22 Å) | 22.08% (9.50 Å) (9.96 Å) |
| Leu261–Leu232 | 100% (6.12 Å) (6.09 Å) | 100% (6.99 Å) (6.34 Å) | 100% (6.03 Å) (6.24 Å) |
| Leu261–Leu290 | 100% (7.34 Å) (7.14 Å) | 100% (6.69 Å) (6.63 Å) | 100% (7.36 Å) (6.65 Å) |
| Leu261–Phe318 | 100% (5.07 Å) (4.82 Å) | 99.84% (4.80 Å) (4.09 Å) | 91.69% (6.98 Å) (4.12 Å) |
| Leu261–Val230 | 100% (8.15 Å) (7.84 Å) | 80.87% (9.64 Å) (9.57 Å) | 100% (8.01 Å) (8.24 Å) |
| Asn263–Arg321 | 99.35% (4.18 Å) (3.93 Å) | 0% (10.02 Å) (7.65 Å) | 78.13% (5.43 Å) (5.93 Å) |
| Asn263–Asp279 | 79.78% (6.04 Å) (4.03 Å) | 99.95% (3.69 Å) (5.29 Å) | 62.39% (6.63 Å) (5.11 Å) |
| Val264–Ile9 | 90.46% (8.36 Å) (8.93 Å) | 0.26% (10.31 Å) (9.38 Å) | 27.53% (9.35 Å) (7.49 Å) |
| Val264–Val275 | 100% (5.81 Å) (5.88 Å) | 100% (6.68 Å) (5.69 Å) | 100% (5.78 Å) (5.61 Å) |
| Val264–Leu290 | 100% (6.19 Å) (6.08 Å) | 100% (6.47 Å) (5.90 Å) | 100% (6.37 Å) (5.42 Å) |
| Val264–Leu286 | 98.32% (9.25 Å) (9.02 Å) | 88.08% (9.13 Å) (8.38 Å) | 99.86% (8.59 Å) (9.19 Å) |
| Val264:CB-Pro276 | 100% (5.25 Å) (5.09 Å) | 96.77% (7.92 Å) (5.38 Å) | 100% (5.29 Å) (4.83 Å) |
| Ile265–Ile9 | 99.86% (4.82 Å) (7.29 Å) | 96.03% (6.53 Å) (4.95 Å) | 77.77% (6.47 Å) (5.40 Å) |
| Ile265–Phe318 | 54.19% (7.64 Å) (5.34 Å) | 99.91% (4.24 Å) (4.57 Å) | 80.88% (6.45 Å) (3.54 Å) |
| Ile265–Val11 | 62.44% (8.83 Å) (8.35 Å) | 2.77% (9.88 Å) (8.90 Å) | 48.86% (8.95 Å) (7.09 Å) |
| Ile265–Ala322 | 99.93% (6.16 Å) (4.42 Å) | 100% (4.92 Å) (4.30 Å) | 99.91% (5.99 Å) (6.28 Å) |
| Ile265–Ile298 | 100% (4.27 Å) (3.70 Å) | 100% (5.48 Å) (4.64 Å) | 100% (4.27 Å) (3.49 Å) |
| Ile265–Leu290 | 99.79% (5.46 Å) (6.79 Å) | 100% (4.81 Å) (4.50 Å) | 99.97% (6.38 Å) (6.76 Å) |
| Ile265–Phe82 | 100% (5.07 Å) (5.62 Å) | 97.65% (6.92 Å) (5.24 Å) | 99.73% (5.46 Å) (6.43 Å) |
| Ile265–Leu232 | 21.11% (9.41 Å) (8.27 Å) | 3.82% (10.44 Å) (10.50 Å) | 57.98% (8.85 Å) (8.77 Å) |
| Ile265–Leu286 | 59.63% (8.94 Å) (9.57 Å) | 68.90% (8.75 Å) (8.47 Å) | 42.25% (9.21 Å) (10.52 Å) |
| Ser266–Arg321 | 99.91% (3.85 Å) (3.85 Å) | 84.82% (5.30 Å) (3.52 Å) | 99.04% (4.01 Å) (4.03 Å) |
| Ala267–Val275 | 100% (4.40 Å) (4.24 Å) | 100% (4.70 Å) (4.12 Å) | 100% (4.41 Å) (4.57 Å) |
| Ala267–Pro276 | 100% (4.33 Å) (3.68 Å) | 99.83% (6.16 Å) (3.84 Å) | 100% (4.21 Å) (3.59 Å) |
| Leu268–Val275 | 100% (5.21 Å) (4.80 Å) | 100% (5.03 Å) (4.48 Å) | 100% (5.07 Å) (4.74 Å) |
| Leu268–Leu290 | 100% (6.24 Å) (5.84 Å) | 100% (6.66 Å) (6.44 Å) | 100% (6.08 Å) (5.96 Å) |
| Leu268–Ile9 | 99.98% (4.83 Å) (5.06 Å) | 99.55% (7.65 Å) (7.02 Å) | 100% (5.13 Å) (4.89 Å) |
| Leu268–Pro276 | 66.18% (8.96 Å) (7.21 Å) | 1.90% (9.72 Å) (7.07 Å) | 76.95% (8.77 Å) (6.96 Å) |
| Leu268–Ala294 | 19.79% (10.30 Å) (9.27 Å) | 0% (12.33 Å) (12.28 Å) | 67.70% (8.89 Å) (9.32 Å) |
| Ala269–Ile9 | 99.01% (5.42 Å) (4.14 Å) | 0.05% (9.44 Å) (8.81 Å) | 100% (4.79 Å) (6.18 Å) |
| Ala269–Ala322 | 78.96% (7.15 Å) (6.72 Å) | 95.61% (7.38 Å) (6.93 Å) | 100% (5.47 Å) (5.02 Å) |
α4 Binds Tightly to MT Through Both Geometric and Interactional Match in Apo and ATP States
It is known that kinesin binds to microtubule strongly in the ATP-binding and apo state and weakly in the ADP-binding state.2–4 α4 is one of the major microtubule binding sites on kinesin. The way of α4 binding to microtubule surface plays an important role in stabilizing kinesin’s motor domain on microtubule.
Binding of one molecule to another is mainly determined by two factors, i.e., geometric match and interactional match. In the ATP-binding and apo state, kinesin’s α4 is a long 7-turn helix that, together with loop L12, fits perfectly into the groove between α- and β-tubulin (Fig. 3). As the longest α helix in kinesin, α4 strides across the tubulin protein. The C-terminal part of α4 locates between the C-terminal part of H12 helix of β-tubulin and the C-terminal part of H11 helix of α-tubulin. The N-terminal part of α4 (mainly the second and third turns) makes a tight contact with H11’ of α-tubulin. In Fig. 3, we depict kinesin-1’s α4 and loop L12 (red) and the contacting part on tubulin (yellow). The matched contact of the two partners ensures a geometric stability for the binding of α4 to microtubule, i.e., the geometric constraint could effectively resist perturbations on α4.
Figure 3.

Binding position of α4 on tubulin. (a) α4 (red helix) locates in the groove between α-tubulin (light brown) and β-tubulin (dark brown) when motor domain (omitted here) binding stably to tubulin. (b) Front view, (c) side view and (d) top view of the geometrical match between α4 and tubulin. The contact atoms (α4 in red and tubulin in yellow) are explicitly shown. The structure used here is the kinesin-1-tubulin complex structure in kinesin-1’s apo state obtained from molecular dynamics simulation.
The binding of α4 to microtubule is also enhanced and stabilized by the matched interactions between them. The interactions between α4 and tubulin are shown in Table 1 and Fig. 4. Comparison of averaged distance of interacting atoms from MD simulations and the corresponding distance in the crystal structures shows that the interaction strength between α4 and tubulin is overestimated from the crystal structure in kinesin-1’s apo state. Seven of the nine interaction distances show larger value in the MD simulation than that in the crystal structure. On the contrary, six interaction distances in the MD simulation are smaller than that in the crystal structure. Thus, the α4-tubulin interaction strength is underestimated from the crystal structure in kinesin-1’s ATP-binding state. As shown in Fig. 4, α4’s lower part, which faces to microtubule, consists mostly of charged and polar residues. These residues have specific interactions with the residues on tubulin, including two salt bridges (formed between K252 of α4 and E411 of α-tubulin, E270 of α4 and K401 of α-tubulin) and three hydrogen bonds (formed between N255 of α4 and G412 of α-tubulin, S259 of α4 and E415 of α-tubulin, N263 of α4 and R402 of α-tubulin) (Fig. 4). The two salt bridges locate at the two ends of α4, which could play an important role in positioning α4 to the right binding site. The three hydrogen bonds locate at the middle part of α4, which effectively enhances the tight binding of α4 to tubulin. The interactions between α4 and tubulin are mostly identical in the apo and ATP-binding state. This is consistent with the experimental results that both apo and ATP-binding states are strong microtubule-binding states.
Figure 4.
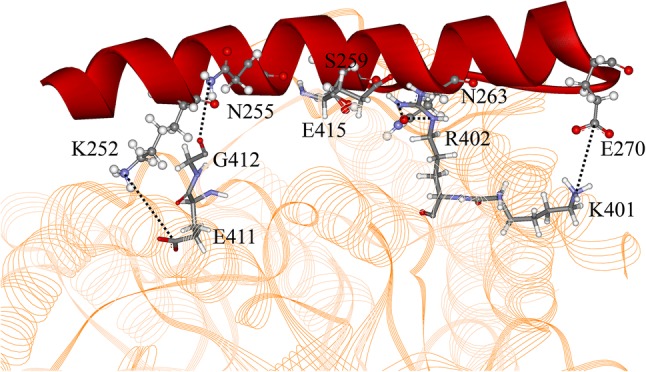
Interactions between α4 (red) and α-tubulin (yellow). Residues in α4 are shown in “Ball and Stick” mode and that in α-tubulin are shown as “Stick” mode. Hydrogen bonds and salt bridges are shown as dotted lines.
In the ADP-binding state, kinesin-1’s α4 has only four turns that largely weakens its geometrical match and interactions with microtubule. As shown above, the long α4s in the apo and ATP-binding states have a tight contact with H11’ of α-tubulin by its second and third turns, which forms a stable geometrical constraint for α4. In the ADP-binding state, this geometrical constraint is lost since the first three turns of α4 resolves into a loop structure. At the same time, the salt bridge and the hydrogen bond formed between the N-terminal part of α4 and tubulin are also broken. The weakened binding of α4 should be the main reason of the weakened binding of the ADP-binding kinesin-1 to microtubule, since the other microtubule binding sites on kinesin-1 (including β5, L12 and α6) remain unchanged.
The stability difference of structures in different nucleotide-binding states can be clearly described with the root mean square deviations (RMSDs) of protein backbone in MD simulations. Figure 5a shows the RMSDs of α4 in different states. It is clearly seen that the RMSD of α4 in ADP state is much larger than that in the other two states, indicating that the stability of α4 in ADP state is much weaker than that in the other two states. The difference of the RMSDs of kinesin-1 heads in different states (Fig. 5b) is highly consistent with that of the α4s. The ATP-binding state motor domain is the most stable one, the stability of the apo state motor domain is a little weaker and the stability of the ADP-binding motor domain is the weakest, that is consistent with the experimental results.2,3
Figure 5.

Root mean square deviations (RMSDs) of (a) α4 helix and (b) kinesin’s motor domain in the molecular dynamics simulations.
α4 Functions Like a Lubricated Shaft in Kinesin-1’s Force-Generation Process
Kinesin is a molecular machine that generates the driving force for cargo movement along microtubule.50 Kinesin’s force-generation process is initiated by ATP binding to its nucleotide-binding site on motor domain.4,40 The key mechanical process from ATP binding to force generation is the rotation of the motor domain.45 In the rotation process, the core β-sheet of the motor domain functions as a mechanical element that amplifies the effect of ATP binding to the forward displacement of the β-domain against the backward resistance from the cargo.8,11,12,26 The rotation process is the main conformational change of kinesin-1’s motor domain from the apo state to the ATP-binding state. In Fig. 6, the structures of the two states (ATP state in yellow and apo state in green) are superimposed. It is seen that the core β-sheet together with α6 of the ATP-binding state structure has an apparent rotation relative to the apo state structure, while the α4 helix and loop L12 remain unchanged. The core β-sheet has a geometrically matched shape around α4 and α4 functions as a fixed shaft for the rotation.
Figure 6.
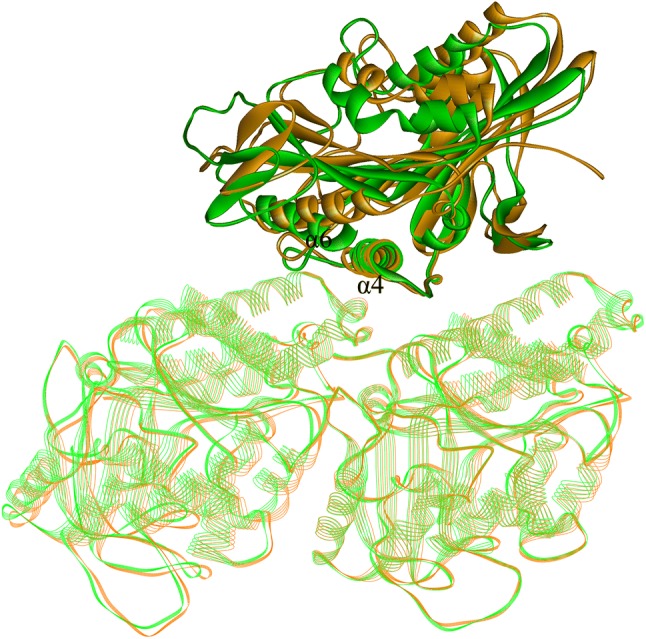
Superposition of crystal structures 4LNU7 (green, nucleotide-free state) and 4HNA7 (brown, ATP-binding state). The tubulins and α4s of two structures are perfectly coincided but the other parts of the two motor domains have an apparent relative rotation. Readers can also refer to Fig. 5 of Ref. 22 which shows that the rotation angle of core β-sheet rotation around α4 is ~ 17.8°.
The relative displacement of different secondary structures in the rotational movement of core β-sheet around α4 can be partially reflected through the differences of occupancy rate and average distance of the interactions between them. From Table 2, interactions between α4 and I9 (β1), R203 (β6), P276 (L12), A294, I298 (β8) and R321 (α6) are weakened in the rotation, and that between α4 and D140 (β4), A243 (L11), F318 and A322 (α6) are enhanced in the rotation of core β-sheet around α4. The largest displacement appears in the C-terminal part of α6 helix. The occupancy rate of the hydrogen bond between N263 and R321 (α6) drops from 99.35% (apo state) to 0% (ATP-binding state). From our previous work, this conformational change of α6 is not directly induced by motor domain rotation. The rotation is transmitted from β1 to β0, and to kinesin-1’s neck linker through its interactions with β0, and finally transmitted to the C terminus of α6. Consistent with this pathway, the occupancy rate of interaction between α4 and I9 which belongs to β1 drops from 99.01% in apo state to 0.05% in ATP-binding state.
From the above section, it is known that α4 binds tightly to microtubule in the apo and ATP-binding states so that it provides a stable support and constraint to ensure that the motor domain rotates to the right position. However, it is known from engineering that a shaft must be well lubricated in order to reduce the energy consumption due to the interfacial friction. Kinesin-1 meets this engineering requirement with a highly rational sequence arrangement. α4 has an amphipathic structure with its lower hydrophilic side facing and binding to the microtubule surface and its upper hydrophobic side facing the inner part of the core β-sheet (Fig. 2). All the residues of the upper side have hydrophobic sidechains (V247, A251, I254, L258, L261, I265 and A269) and most of them are conservative residues in kinesin family (I254, L258, L261 and I265) (Fig. 2). Correspondingly, most of the residues of kinesin’s core β-sheet on the interface with α4 are hydrophobic residues. These residues are full of methyl and methylene groups (the main content of oil molecules) that make the two contact regions behave effectively like structural composite materials with well lubricated surface layers and firm inner frames (Fig. 7). In addition, careful inspection of the simulation structures shows that there is no hydrogen bond formed between the two surfaces and no water molecules could get into the hydrophobic contact region. All these make kinesin-1 minimize the energy consumption for conformational change during the rotation process and export a large enough useful work. Therefore, rather a relay helix, α4 is more like a stationary shaft. One side of α4 “stands” on the groove between α- and β-tubulin through geometric fit and polar interactions. Another side of α4 is hydrophobic and contact with kinesin-1’s core β-sheet through hydrophobic interaction. These hydrophobic interactions ensure the compact structure of kinesin-1’s motor domain and the reduction of energy consumption required for kinesin-1’s efficient walking process.
Figure 7.
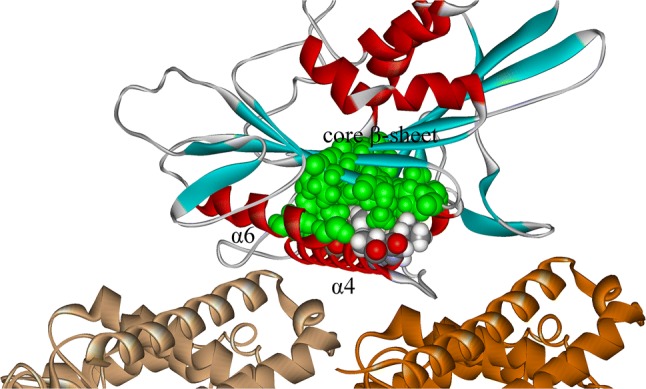
Hydrophobic contact of α4 and core β-sheet. α-tubulin is shown in light brown and β-tubulin is shown in dark brown. The α-helices in kinesin-1 are shown in red and β-sheets are shown in blue. The residues which form hydrophobic contacts between α4 and core β-sheet are explicitly shown in “CPK” mode. The residues from core β-sheet are shown in green color and those belong to α4 are shown in multiple colors (oxygen in red, carbon in grey, hydrogen in white and nitrogen in blue). The structure is obtained from molecular dynamics simulation in kinesin-1’s apo state.
Conclusion
Kinesin is an exquisite protein machine. The α4 helix is an amphipathic structure to play multiple roles in kinesin-1’s mechanical process. First, α4 is a major microtubule binding site. In the apo and ATP-binding states, it is a long 7-turn helix with its hydrophilic side facing the microtubule and binds tightly to the intra-tubulin groove through both geometrical and interactional match. In the ADP-binding state, it binds weakly to microtubule due to partial loss of interactions caused by the shortening of its helix length (4-turn). Statistical analysis based on MD simulation shows that the binding stability of α4 in kinesin-1’s ADP-binding state is much weaker than that in the apo and ATP-binding states, which should be the main reason for the weak microtubule binding of kinesin-1 in the ADP-binding state, as revealed by experiments. Second, α4 functions like a lubricated shaft for the motor domain rotation. The arrangement of the hydrophobic residues on the interface between α4 and the core β-sheet make kinesin-1 effectively reduce the conformational work in the force-generation process.
Relative rotation of protein subdomains is one of the major forms of conformational change in protein functioning. The design mechanism of kinesin-1’s α4 as revealed here should be of heuristic significance for the understanding of protein mechanics.
Acknowledgments
This work was supported by the National Natural Science Foundation of China under Grant Nos. 11605038, 11545014, and 11847120.
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical Approval
No human studies were carried out by the authors for this article; no animal studies were carried out by the authors for this article.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yi-Long Ma and Tie Li have contributed equally to this work.
Contributor Information
Yi-Zhao Geng, Phone: +86-22-60435643, Email: gengyz@hebut.edu.cn.
Qing Ji, Phone: +86-22-60435643, Email: jiqingch@hebut.edu.cn.
References
- 1.Asbury CL, Fehr AN, Block SM. Kinesin moves by an asymmetric hand-over-hand mechanism. Science. 2003;302:2130–2134. doi: 10.1126/science.1092985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asenjo AB, Krohn N, Sosa H. Configuration of the two kinesin motor domains during ATP hydrolysis. Nat. Struct. Mol. Biol. 2003;10:836–842. doi: 10.1038/nsb984. [DOI] [PubMed] [Google Scholar]
- 3.Asenjo AB, Sosa H. A mobile kinesin-head intermediate during the ATP-waiting state. Proc. Natl. Acad. Sci. USA. 2009;106:5657–5662. doi: 10.1073/pnas.0808355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asenjo AB, Weinberg Y, Sosa H. Nucleotide binding and hydrolysis induces a disorder-order transition in the kinesin neck-linker region. Nat. Struct. Mol. Biol. 2006;13:648–654. doi: 10.1038/nsmb1109. [DOI] [PubMed] [Google Scholar]
- 5.Atherton J, Farabella I, Yu I-M, Rosenfeld SS, Houdusse A, et al. Conserved mechanisms of microtubule-stimulated ADP release, ATP binding, and force generation in transport kinesins. eLife. 2014;3:e03680. doi: 10.7554/eLife.03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Best RB, Zhu X, Shim J, Lopes P, Mittal J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and sidechain chi1 and chi2 dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao L, Wang W, Jiang Q, Wang C, Knossow M, et al. The structure of apo-kinesin bound to tubulin links the nucleotide cycle to movement. Nat. Commun. 2014;5:5364. doi: 10.1038/ncomms6364. [DOI] [PubMed] [Google Scholar]
- 8.Cochran JC. Kinesin motor enzymology: chemistry, structure, and physics of nanoscale molecular machines. Biophys. Rev. 2015;7:269–299. doi: 10.1007/s12551-014-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crevel I, Lockhart A, Cross RA. Weak and strong states of kinesin and ncd. J. Mol. Biol. 1996;257:66–76. doi: 10.1006/jmbi.1996.0147. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Saez I, Yen T, Wade RH, Kozielski F. Crystal structure of the motor domain of the human kinetochore protein CENP-E. J. Mol. Biol. 2004;340:1107–1116. doi: 10.1016/j.jmb.2004.05.053. [DOI] [PubMed] [Google Scholar]
- 11.Geng Y, Ji Q, Liu S, Yan S. Initial conformation of kinesin’s neck linker. Chin. Phys. B. 2014;23:108701. doi: 10.1088/1674-1056/23/10/108701. [DOI] [Google Scholar]
- 12.Geng Y, Liu S, Ji Q, Yan S. Mechanical amplification mechanism of kinesin’s β-domain. Arch. Biochem. Biophys. 2014;543:10–14. doi: 10.1016/j.abb.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 13.Geng Y, Zhang H, Lyu G, Ji Q. Initiation mechanism of kinesin’s neck linker docking process. Chin. Phys. Lett. 2017;34:028701. doi: 10.1088/0256-307X/34/2/028701. [DOI] [Google Scholar]
- 14.Gigant B, Wang W, Dreier B, Jiang Q, Pecqueur L, et al. Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat. Struct. Mol. Biol. 2013;20:1001–1007. doi: 10.1038/nsmb.2624. [DOI] [PubMed] [Google Scholar]
- 15.Guo S-K, Wang P-Y, Xie P. A model of processive movement of dimeric kinesin. J. Theor. Biol. 2017;414:62–75. doi: 10.1016/j.jtbi.2016.11.023. [DOI] [PubMed] [Google Scholar]
- 16.Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 17.Hirokawa N, Noda Y. Intracellular transport and kinesin superfamily proteins, kifs: structure, functions, and dynamics. Physiol. Rev. 2008;88:1089–1118. doi: 10.1152/physrev.00023.2007. [DOI] [PubMed] [Google Scholar]
- 18.Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell. Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- 19.Hirokawa N, Tanaka Y. Kinesin superfamily proteins (KIFs): various functions and their relevance for important phenomena in life and diseases. Exp. Cell. Res. 2015;334:16–25. doi: 10.1016/j.yexcr.2015.02.016. [DOI] [PubMed] [Google Scholar]
- 20.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 21.Hwang W, Lang MJ, Karplus M. Kinesin motility is driven by subdomain dynamics. eLife. 2017;6:e28948. doi: 10.7554/eLife.28948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin Y, Geng Y, Lyu L, Ma Y, Lyu G, et al. Anchor effect of interactions between kinesin’s nucleotide-binding pocket and microtubule. Cell Mol. Bioeng. 2017;10:162–173. doi: 10.1007/s12195-017-0477-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. doi: 10.1063/1.445869. [DOI] [Google Scholar]
- 24.Kaseda K, Higuchi H, Hirose K. Alternate fast and slow stepping of a heterodimeric kinesin molecule. Nat. Cell Biol. 2003;5:1079–1082. doi: 10.1038/ncb1067. [DOI] [PubMed] [Google Scholar]
- 25.Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, et al. Switch-based mechanism of kinesin motors. Nature. 2001;411:439–445. doi: 10.1038/35078000. [DOI] [PubMed] [Google Scholar]
- 26.Krukau A, Knecht V, Lipowsky R. Allosteric control of kinesin’s motor domain by tubulin: a molecular dynamics study. Phys. Chem. Chem. Phys. 2014;16:6189–6198. doi: 10.1039/c3cp53367k. [DOI] [PubMed] [Google Scholar]
- 27.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrence CJ, Dawe RK, Christie KR, Cleveland DW, Dawson SC, et al. A standardized kinesin nomenclature. J. Cell. Biol. 2004;167:19–22. doi: 10.1083/jcb.200408113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J-L, Car R, Tang C, Wingreen NS. Hydrophobic interaction and hydrogen-bond network for a methane pair in liquid water. Proc. Natl. Acad. Sci. USA. 2007;104:2626–2630. doi: 10.1073/pnas.0610945104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M, Zheng W. All-atom structural investigation of kinesin-microtubule complex constrained by high-quality cryo-electron-microscopy maps. Biochemistry. 2012;51:5022–5032. doi: 10.1021/bi300362a. [DOI] [PubMed] [Google Scholar]
- 31.Liu D, Liu X, Shang Z, Sindelar CV. Structural basis of cooperativity in kinesin revealed by 3D reconstruction of a two-head-bound state on microtubules. eLife. 2017;6:e24490. doi: 10.7554/eLife.24490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.MacKerell AD, Jr, Feig M, Brooks CL. Imporved treatment of the protein backbone in empirical force field. J. Am. Chem. Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 34.Naber N, Minehardt TJ, Rice S, Chen X, Grammer J, et al. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 2003;300:798–801. doi: 10.1126/science.1082374. [DOI] [PubMed] [Google Scholar]
- 35.Naber N, Rice S, Matuska M, Vale RD, Cooke R, et al. EPR spectroscopy shows a microtubule-dependent conformational change in the kinesin switch 1 domain. Biophys. J. 2003;84:3190–3196. doi: 10.1016/S0006-3495(03)70043-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogawa T, Nitta R, Okada Y, Hirokawa N. A common mechanism for microtubule destabilizers: M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell. 2004;116:591–602. doi: 10.1016/S0092-8674(04)00129-1. [DOI] [PubMed] [Google Scholar]
- 37.Pan Z, Chen J, Lyu G, Geng Y, Zhang H, et al. An ab initio molecular dynamics study on hydrogen bonds between water molecules. J. Chem. Phys. 2012;136:164313. doi: 10.1063/1.4705371. [DOI] [PubMed] [Google Scholar]
- 38.Pavelites JJ, Gao J, Bash PA, MacKerell AD., Jr A molecular mechanics force field for NAD+ NADH, and the pyrophosphate groups of nucleotides. J. Comput. Chem. 1997;18:221–239. doi: 10.1002/(SICI)1096-987X(19970130)18:2<221::AID-JCC7>3.0.CO;2-X. [DOI] [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, et al. Scalable molecular dynamics with namd. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rice S, Lin AW, Safer D, Hart CL, Naber N, et al. A structural change in the kinesin motor protein that drives motility. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 41.Rosenfeld SS, Fordyce PM, Jefferson GM, King PH, Block SM. Stepping and stretching: how kinesin uses internal strain to walk processively. J. Biol. Chem. 2003;278:18550–18556. doi: 10.1074/jbc.M300849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenfeld SS, Xing J, Jefferson GM, Cheung HC, King PH. Measuring kinesin’s first step. J. Biol. Chem. 2002;277:36731–36739. doi: 10.1074/jbc.M205261200. [DOI] [PubMed] [Google Scholar]
- 43.Sack S, Müller J, Marx A, Thormählen M, Mandelkow E-M, et al. X-ray structure of motor and neck domains from rat brain kinesin. Biochemistry. 1997;36:16155–16165. doi: 10.1021/bi9722498. [DOI] [PubMed] [Google Scholar]
- 44.Shang Z, Zhou K, Xu C, Csencsits R, Cochran JC, et al. High-resolution structures of kinesin on microtubules provide a basis for nucleotide-gated force-generation. eLife. 2014;3:e04686. doi: 10.7554/eLife.04686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sindelar C. A seesaw model for intermolecular gating in the kinesin motor protein. Biophys. Rev. 2011;3:85–100. doi: 10.1007/s12551-011-0049-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sindelar CV, Downing KH. An atomic-level mechanism for activation of the kinesin molecular motors. Proc. Natl. Acad. Sci. USA. 2010;107:4111–4116. doi: 10.1073/pnas.0911208107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song Y-H, Marx A, Müller J, Woehlke G, Schliwa M, et al. Structure of a fast kinesin: implications for ATPase mechanism and interactions with microtubules. EMBO J. 2001;20:6213–6225. doi: 10.1093/emboj/20.22.6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tuner J, Anderson R, Guo J, Beraud C, Fletterick R, et al. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J. Biol. Chem. 2001;276:25496–25502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 49.Vale RD. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J. Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/S0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- 51.Xie P. Stepping behavior of two-headed kinesin motors. Biochim. Biophys. Acta. 2008;1777:1195–1202. doi: 10.1016/j.bbabio.2008.04.040. [DOI] [PubMed] [Google Scholar]
- 52.Yildiz A, Tomishige M, Vale RD, Selvin PR. Kinesin walks hand-over-hand. Science. 2004;303:676–678. doi: 10.1126/science.1093753. [DOI] [PubMed] [Google Scholar]