

Abstract
Introduction
Skeletal muscle tissue development and regeneration relies on the proliferation, maturation and fusion of muscle progenitor cells (myoblasts), which arise transiently from muscle stem cells (satellite cells). Following muscle damage, myoblasts proliferate and differentiate in response to temporally-varying inflammatory cytokines, growth factors, and extracellular matrix cues, which stimulate a shared network of intracellular signaling pathways. Here we present an integrated data-modeling approach to elucidate synergies and antagonisms among proliferation and differentiation signaling axes in myoblasts stimulated by regeneration-associated ligands.
Methods
We treated mouse primary myoblasts in culture with combinations of eight regeneration-associated growth factors and cytokines in mixtures that induced additive, synergistic, and antagonistic effects on myoblast proliferation and differentiation responses. For these combinatorial stimuli, we measured the activation dynamics of seven signal transduction pathways using multiplexed phosphoprotein assays and scored proliferation and differentiation responses based on expression of myogenic commitment factors to assemble a cue-signaling-response data compendium. We interrogated the relationship between these signals and responses by partial least-squares (PLS) regression modeling.
Results
Partial least-squares data-modeling accurately predicted response outcomes in cross-validation on the training compendium (cumulative R 2 = 0.96). The PLS model highlighted signaling axes that distinctly govern myoblast proliferation (MEK–ERK, Stat3) and differentiation (JNK) in response to these combinatorial cues, and we confirmed these signal-response associations with small molecule perturbations. Unexpectedly, we observed that a negative feedback circuit involving the phosphatase DUSP6/MKP-3 auto-regulates MEK–ERK signaling in myoblasts.
Conclusion
This data-modeling approach identified conflicting signaling axes that underlie muscle progenitor cell proliferation and differentiation.
Electronic supplementary material
The online version of this article (doi:10.1007/s12195-017-0508-5) contains supplementary material, which is available to authorized users.
Keywords: Cue-signal-response modeling, Cytokines, Growth factors, Partial least-squares regression, Skeletal muscle, Systems biology
Introduction
Postnatal skeletal muscle development and regeneration relies upon the coordinated activation, proliferation, and differentiation of muscle stem and progenitor cells.47 In adulthood, muscle stem cells (MuSCs; also known as satellite cells) are retained in a mitotically and metabolically quiescent state and are marked by expression of the transcription factor Pax7.7,28 In response to muscle damage, MuSCs become activated and subsequently undergo multiple rounds of self-renewing divisions that produce both additional stem cells and committed MyoD+ myogenic progenitor cells (myoblasts). Myoblasts rapidly proliferate and then differentiate into fusion-competent, Myogenin+ myocytes. Myocytes fuse into mature Myosin heavy chain (MHC)-expressing myofiber cells, which provide contractile, structural and metabolic function to muscle tissue. Together, MyoD, Myogenin, and MHC serve as a core myogenic cascade, with the transcription factors MyoD and Myogenin expressed successively, followed by MHC, to promote terminal differentiation39 (Fig. S1).
Muscle stem and progenitor cell fates during repair are governed by proliferation- and/or differentiation-inducing ligands secreted by macrophages, myofibers, and myofibroblasts within muscle tissue.47 For example, M1-biased macrophages transiently increase within 2 days post-injury and secrete the cytokines interleukin-1α/β (IL-1α/β), IL-6, and tumor necrosis factor-α (TNF-α) during a myoblast proliferative phase; a second wave of M2-biased macrophages amplify 4–5 days post-injury and secrete IL-4 and insulin-like growth factor (IGF1), which promote myoblast commitment and differentiation.43 Other proliferation and differentiation-inducing cytokines and growth factors are secreted by local myofibroblasts, myofibers, and even myogenic stem/progenitor cells themselves.41,47 Together, these cues present a series of time-varying stimuli with overlapping synergistic and antagonistic effects on myogenic fate regulation, ultimately yielding pools of both committed myogenic progenitors capable of myofiber fusion and quiescent Pax7+ muscle stem cells.
Here, we sought to dissect the cooperating contributions of numerous regeneration-associated growth factors and cytokines in regulating myoblast proliferation and differentiation (Fig. S1). A key group of myoblast survival and mitogenic factors include fibroblast growth factor-2 (FGF2, also known as basic FGF), epidermal growth factor (EGF), and insulin-like growth factor (IGF1).29 FGF family ligands are required for muscle maintenance and regeneration.37 Notably, muscle stem/progenitor cells have reduced responsiveness to FGF stimulation in aged tissues, underscoring their critical role in muscle maintenance.4 FGF ligands bind to FGF receptors-1/4 in muscle cells,37 stimulate the MEK–ERK, p38, PI3K–Akt, and Stat signaling pathways, and promote myoblast proliferation and differentiation.37 ERK1/2 activation, in response to FGF2, is required for myoblast proliferation but is dispensable for FGF-induced expression of myogenic differentiation genes and cell fusion.23 Moreover, FGF2 is secreted by myogenic progenitors and can act as an autocrine factor to self-regulate MEK–ERK signaling in muscle cells.15 Likewise, IGF1, which is produced by resident myofibroblasts, promotes myoblast proliferation and differentiation,43 in part through the activation of PI3K–Akt signaling and MyoD-regulation of myogenic genes,42 and contributes to muscle growth and hypertrophy. Critically, EGF synergizes with insulin ligands to promote myoblast proliferation,33 suggesting that IGF1-induced Akt and EGF-induced ERK signaling cooperatively regulate myoblast proliferation.
A second group of potent myoblast regulatory ligands is the IL-6 family, which includes IL-6, oncostatin M (OSM), and leukemia inhibitor factor (LIF). IL-6 is a pleotropic cytokine that can promote muscle stem cell activation, but also restricts myoblast proliferation while inducing myoblast differentiation.44 These cellular effects underlie the complex tissue-level effects of IL-6 signaling, which has been associated with both muscle wasting and hypertrophy.2,32 IL-6 binds to the IL-6R/gp-130 receptor complex and stimulates the JAK–Stat and p38 pathways, which induce MyoD expression and myogenic target genes.38,44 The dosing and persistence of IL-6–JAK–Stat3 pathway perturbation can yield contradictory results; for example, complete IL-6 ablation impairs muscle stem/progenitor proliferation32 but partial Stat3 inhibition promotes stem cell expansion in vivo.22 Similarly, OSM and LIF, through binding to their cognate receptors OSMR/gp130 and LIFR/gp130, respectively, activate JAK–Stat, PI3K–Akt, and MEK–ERK signaling to varying magnitudes.22 OSM and LIF restrict myoblast differentiation in a MEK-dependent manner and weakly promote proliferation.6 These context-dependent myoblast responses to IL-6 family ligands suggest that differential activation of secondary pathways may explain ligand-specific outcomes.22
TNF-α and IL-1 cytokines promote myoblast differentiation and restrict proliferation. TNF-α binds to TNFR, recruits TRAF6, and stimulates p38, IKK–NFκB, and JNK–cJun pathway activation.14 TNF-α promotes myogenic differentiation through p38, stimulating MyoD activation and epigenetic regulation of myogenic gene programs.35 Inhibition of p38 signaling enhances muscle stem/progenitor cell expansion10 by both antagonizing Myogenin-dependent commitment and promoting the proliferative JNK pathway.14 TNF-α also induces myogenic cell proliferation via JNK-induced expression of the cell cycle promoter Cyclin-D1.14,34 IL-1α/β binds to ILR1 to activate JNK–cJun and IKK–NFκB signaling31 to promote muscle stem/progenitor cell proliferation,16,46 upon co-stimulation with other cytokines and chemokines. Therefore, these regeneration-associated stimuli regulate numerous conflicting pathways governing myoblast proliferation and differentiation responses (Fig. S1).
Interpretation of conflicting signaling pathway regulators of cell fate can be aided by data-driven modeling. A number of approaches have been developed to model the relationships between signal transduction networks and cell-type-specific responses.11,21 When detailed molecular-level understanding of the signaling cross-talk mechanisms is not available, data-driven modeling approaches such as decision trees, fuzzy logic, and partial least-squares (PLS) regression can suggest connections between signaling mediators and cell response outcomes, and even accurately predict cell responses to de novo signaling observations.8,9 Here, we chose to develop a PLS regression-based data-model of myoblast responses to diverse ligand stimuli, given prior successes by PLS models in generating confirmable identification in signaling network-cell fate relationships across diverse cell types.8,9,17,20,21,25,27,30 We calibrated a well-performing PLS data-model from temporally-dense, multi-pathway phosphoprotein signaling data collected under an array of combinatorial growth factor and cytokine conditions. This PLS model generated accurate predictions of myoblast responses in cross-validation-based calibration and also on new test data. Importantly, the PLS model reduced the myoblast signaling-response relationships into a set of four principal component basis axes with time-varying associations to distinct differentiation and proliferation outcomes.
Results
Combinatorial Growth Factor and Cytokine Stimuli Induce Synergistic and Antagonistic Myoblast Response Phenotypes
We established a myoblast differentiation scheme to identify combinatorial ligand stimulations that result in non-additive differentiation outcomes. We seeded mouse primary myoblasts at high-confluency in differentiation medium (DM) with 3% fetal bovine serum (FBS) and stimulated the cells with combinations of three multi-ligand mixtures, which each predominantly reported to activate different signal transduction pathways (Fig. S1). In contrast to most reports using horse serum to induce myoblast differentiation, we chose FBS to allow for co-treatment-induced changes in both differentiation and proliferation. First, we tested Fibroblast Growth Factor-2 (FGF2) + Epidermal Growth Factor (EGF) + Insulin-like Growth Factor (IGF1; together “FEI”), which all activate the PI3K–Akt–mTOR and MAPK (ERK, p38, JNK) signaling pathways.4,15,23,37 Second, we tested Tumor Necrosis Factor-α (TNF-α) + Interleukin-1α (IL-1α; together “TI”), which both activate the IKK–NFκB and JNK pathways.14,16,31,46 Third, we tested Interleukin-6 (IL-6) + Oncostatin-M (OSM) + Leukemia Inhibitor Factor (LIF; together “IOL”), all ligands in IL-6 super-family which predominantly activate the JAK–STAT pathway.2,46
We measured myoblast proliferative index at 72 h post-stimulation and observed that IOL treatment restricted proliferation, but FEI and TI did not have an effect compared to control cultures (Figs. 1a and 1b). We evaluated two-way combinations of these mixtures and compared the observed myoblast proliferation to predictions based on a modified Bliss independence model5 to identify multi-factor treatments that exhibited supra-additive synergy or sub-additive antagonism. We observed FEI/TI to induce supra-additive proliferation, indicating these treatments may lead to a synergistic activation of proliferation-regulating pathways. In contrast, we observed TI/IOL to induce more proliferation than predicted by the additive model, indicating that TI may act to negate the proliferation-restricting effects of the IOL mixture.
Figure 1.
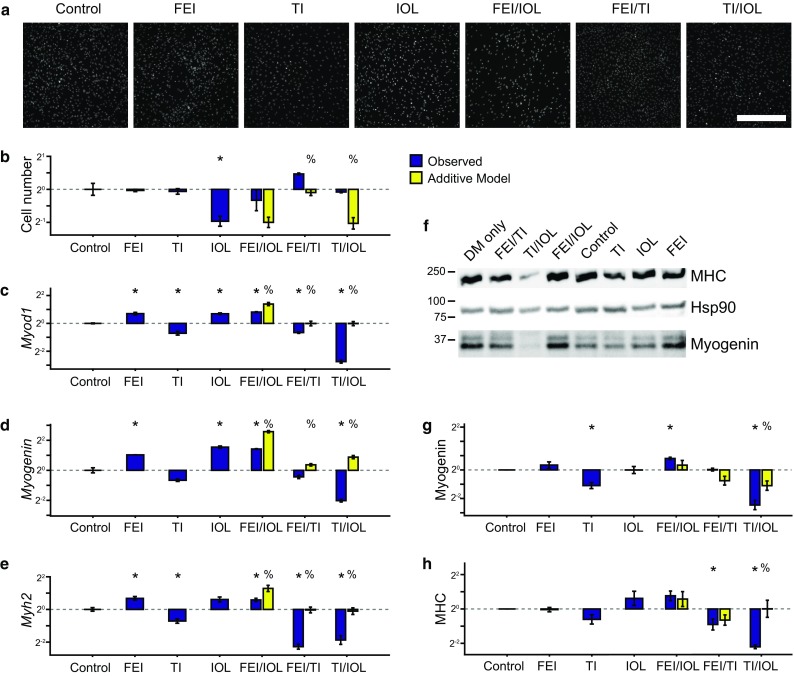
Synergism and antagonism in growth factor and cytokine-stimulated myoblast proliferation and differentiation. Primary mouse myoblasts were cultured in differentiation medium (DM) with 0.1% DMSO (control) and were stimulated with either FGF2 + EGF + IGF1 (FEI), TNF-α + IL-1α (TI), IL-6 + OSM + LIF (IOL), or their two-way combinations for 72 h. (a, b) Proliferative index was determined by nuclei counting from DAPI images. (a) Representative DAPI images. Scale bar, 200 μm. (c–e) RT-qPCR analysis of myogenic differentiation genes Myod1, Myogenin, and Myh2 quantified relative to 36b4 (reference control). (f–h) Immunoblots for MHC, Myogenin, and Hsp90 (as a loading control). Myogenin (g) and MHC (h) densitometry, normalized to Hsp90. In (b–e, g–h), n = 3 replicates are plotted as log2 of mean fold change relative to control ± SEM. For co-treatments, an additive model used to identify synergy or antagonism using a modified Bliss independence model with p < 0.05 by Student’s t test between control and other stimulations (*) or between observed stimulation combinations and the additive model (%).
We then measured myoblast differentiation responses also at 72 h post-stimulation. We assayed expression of three myogenic commitment and differentiation genes, MyoD1 (Myod1), Myogenin (Myog), and Myosin heavy chain (Myh2) using a combination of RT-qPCR and immunoblotting methods (Figs. 1c–1h). Across these assays, we observed that FEI and IOL upregulated and TI downregulated expression of differentiation genes. In the mixture combination conditions, we observed that FEI/IOL synergistically upregulated Myod1 expression. We also found that the FEI/TI and TI/IOL combinations resulted in more downregulation of Myod1, Myogenin, and Myh2 than expected by the additive model, suggesting that TI’s differentiation-restricting effects dominate over IOL and FEI’s differentiation-inducing effects. Collectively, these observations suggested that FEI, TI, and IOL induce varied myoblast proliferation and differentiation responses and their combinations unexpectedly lead to both supra-additive synergies and antagonisms.
Signaling Network Data Compendium Highlights Temporally Patterned Signaling Synergies
To refine how shared and distinct signaling pathways govern the observed myoblast responses, we collected a data-compendium comprised of the activation time-courses of eight phosphoproteins from the pathways predominantly activated (PI3K–Akt–mTOR–p70 S6K, MEK–ERK, p38, IKK–NFκB, JNK–cJun, and JAK–Stat3) by these eight ligands (Figs. S1 and 2a) from mid-passage (p18) primary mouse myoblasts. We used rigorously validated (see Figs. S2 and S3) quantitative Luminex multiplexed assays to measure seven phosphoproteins (Akt pSer473, p70 S6 K pThr389/pThr412, MEK1 pSer222, ERK1/2 pThr185/pTyr187, p38 MAPK pThr180/pTyr182, IκBα pSer32, and cJun pSer73) at six time-points from 0 to 24 h post-stimulation (Figs. 2c and 2d). We complemented these dense time-courses with similarly validated quantitative immunoblots of Akt pSer473 and Stat3 pTyr705 at t = 15 min post-stimulation (Fig. 2b). We collected these data for eight multi-ligand conditions, including all one- and two-way combinations of FEI, TI, and IOL, for which we already collected 72 h myoblast cell response measurements (Fig. 2c). We note that these conditions yielded no significant differences in cellular apoptosis (Fig. S2j) so we generally attribute the observed differences in cell number to proliferative changes.
Figure 2.
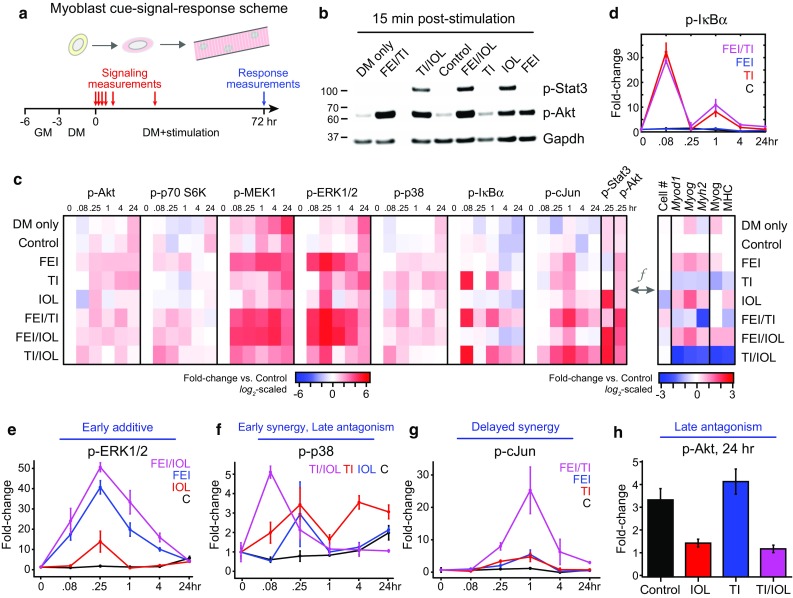
Myoblast cue-signal-response data compendium identifies co-treatment synergies with variable dynamics. (a) A dense time-course of multiple phosphoprotein signals was quantified through a mixture of multiplexed Luminex and immunoblotting assays. Primary mouse myoblasts were cultured in differentiation medium (DM) with 0.1% DMSO (control) and were stimulated with either FGF2 + EGF + IGF1 (FEI), TNF-α + IL-1α (TI), IL-6 + OSM + LIF (IOL), or their two-way combinations for 72 h. Phosphoprotein measurements were quantified at 0, 5 min, 15 min, 1, 4, and 24 h and differentiation responses were quantified at 72 h post-stimulation; (b) Representative immunoblots for p-Stat3, p-Akt, and Gapdh (as a loading control); (c) Heatmap of signaling-response data compendium. Left, Mean values (from n = 3 replicates) of p-Akt, p-p70 S6 kinase, p-MEK1, p-ERK1/2, p-p38 MAP kinase, p-IκBα, and p-cJun Luminex time-courses and phospho-Stat3 and p-Akt immunoblots (15 min time-point only). Signaling data were first normalized to β-tubulin (Luminex) or Gapdh (blots) levels, then were fold-change normalized to the t = 0 Control sample and reported on a log2-scale (see Fig. S2 for full data set). Right, Mean values of proliferation and differentiation (Myod1, Myog, Myh2 by RT-qPCR; Myog, MHC by quantitative immunoblot) responses (from Fig. 1) normalized to the Control sample and reported on a log2-scale; (d) p-IκBα time-course for control, TI, FEI, and FEI + TI conditions demonstrating biphasic activation peaks; (e) Phospho-ERK1/2 time-course for Control, IOL, FEI, and FEI + IOL conditions; (f) Phospho-p38 time-course for Control, TI, IOL, and TI + IOL conditions; (g) p-cJun time-course for Control, TI, FEI, and FEI + TI conditions; (h) p-Akt at 24 h for Control, TI, IOL, and TI + IOL conditions. In (d–h), n = 3 replicates are plotted as mean ± SEM.
For some phosphoproteins, we observed transient activation exclusive to specific single mixtures, such as the induction of p-IκBα by TNF + IL-1α (TI; Fig. 2d). TI-stimulated p-IκBα exhibited both “early” (<1 h) and “late” (~1 h) activation peaks, in agreement with well-observed oscillatory behavior of the IKK–NFκB pathway.24 Furthermore, the p-IκBα oscillations were not perturbed by either FEI or IOL co-treatment (Figs. 2d and S3). Notably, we observed conditions with “early” additive synergy, such as p-ERK1/2 activation by FEI/IOL (Fig. 2e). We observed cases with “early” supra-additive synergy, such as p-p38 activation by TI/IOL (Fig. 2f). We observed cases with more delayed (~1 h) synergy, such as p-cJun by FEI/TI (Fig. 2g). We also observed examples of “late” antagonism at t = 24 h, including p-p38 and p-Akt activation by TI/IOL, in which the repressive effect of the IOL stimulus dominates over the stimulatory effect of TI (Figs. 2f and 2h). These observations suggested that the synergies and antagonisms in myoblast proliferation and differentiation responses might be a function (f) of the underlying combination of signaling combinations.
Calibration of a Partial Least-Squares Data-Model of Myoblast Signaling-Response Compendium
Given the challenge in intuiting the biological connections between signaling (“X”) and response (“Y”) variables in the data-compendium, we reasoned that a data-model might be more successful in identifying these higher-order relationships. Partial least-squares (PLS) regression is a data-modeling approach for relating signaling dynamics to cell response phenotypes made across a set of conditions.8,9,17,20,21,25,27,30 PLS is flexible to data types and scales and, importantly, requires no a priori model of the signaling mechanisms and their net effects on cell phenotypes. Instead, PLS data-modeling provides a reduced dimensionality set of data variable weights and scores in a principal component (PC) space to provide multivariate linear regressions to quantitatively relate the observed signaling dynamics to cell responses. Before training a PLS model, we extracted non-linear signaling dynamics from time-course data in the form of “area-under-the-curve” integrals comprising the early (“E”, 0–1 h) and late (“L”, 1–24 h) peaks and the full (“F”, 0–24 h) time-course (Fig. 3a). Given that we observed both induction and repression of myogenic gene expression (Fig. 2c), we generated two sets of models, using either linearly-scaled signaling and response data or logarithmically-scaled data, which can account for up- and down-regulated variables (Figs. 3c and 3d). After extraction and scaling, all signaling and response variables were unit-variance-transformed into Z-scores to remove bias due to disparate data-range variances.1,17
Figure 3.
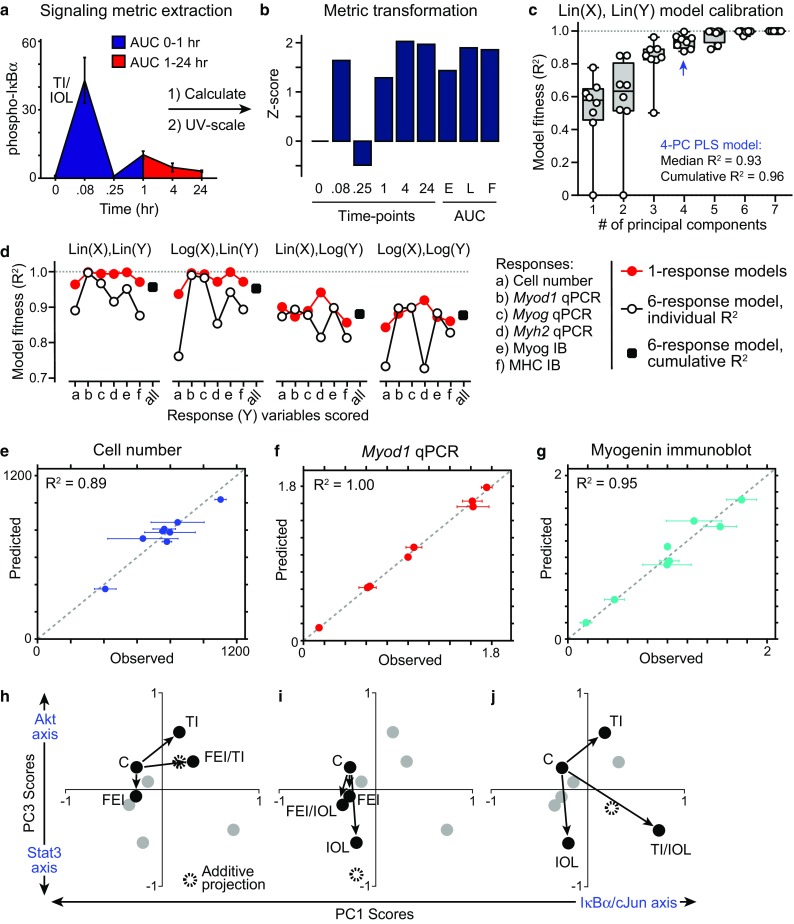
Calibration of a partial least-squares regression model linking signaling dynamics to myoblast responses. (a, b) Signaling metrics were extracted from phosphoprotein activation time-courses, including the mean values at each time-point, the area-under-the-curve (AUC) for the early peak (0–1 h), late peak (1–24 h) and full time-course, and were then unit-variance-transformed to a Z-score prior to data-modeling; (c–f) Partial least-squares (PLS) regression models were calibrated in cross-validation to the signaling-response data set under a number of data-scaling approaches. The fitness of the model to predict response outcomes based on signaling metrics was evaluated for each response; (c) Model fitness for increasing principal components (PC) used in the PLS regression, using unscaled signaling (X) and response (Y) data matrices in a six-response model; (d) Effects of data scaling and number of response variables included on the fitness of 4-PC models (e–g) Observed vs. predicted responses for cell number (e), Myod1 expression by RT-qPCR (f), and Myogenin expression by immunoblot (g; see also Fig. S4a–S4c). Mean ± SEM of n = 3 replicates is reported for observations; (h–j) PLS model “score projections” for co-treatment combinations (black; all other conditions in gray) for PC1 and PC3, relative to the Control (C) condition. Additive projections from two individual treatment conditions are shown.
We trained a set of PLS models on 58 transformed phosphoprotein signaling metrics and six response variables, arrayed over eight conditions into a cue-signal-response (CSR) data compendium, based on the SIMPLS regression algorithm using the PLSREGRESS function in Matlab. We evaluated the quality of each calibrated model using an R 2 fitness metric8,17 calculated individually or cumulatively over all six response variables (Figs. 3c–3g and S4a–S4c). For the non-scaled data model (denoted as “Lin(X),Lin(Y)”), we observed that the cumulative model fitness plateaued at 0.96 by four principal components (Fig. 3c). The additive improvements in model performance from successive PCs up to the fourth PC suggests that four predominant axes of signaling-response covariation20 exist in the myogenic CSR data-compendium. We compared the fitness of PLS models calibrated to unscaled (linear) and logarithmic-scaled data. In one-response and six-response models, we found that the “Lin(X),Lin(Y)” model performed (R 2 = 0.96) better than any log-scaled versions (R 2 = 0.88–0.94; Fig. 3d). Though the one-response models generally had better fitness to the observed response data as expected with fewer regressed variables, the six-response models performed well when the signaling data was not log-scaled. For all further analysis, we focused on the six-response PLS model calibrated to the unscaled signaling-response data given its fitness in calibration.
This unscaled 4-PC calibrated model achieved good cross-validation predictive accuracy for both cell proliferation index (R 2 = 0.89), RT-qPCR gene expression (e.g., Myod1, R 2 = 1.00), and protein immunoblotting measurements (e.g., Myogenin, R 2 = 0.95) of myogenic differentiation, even though the response data were heavily biased towards differentiation measurements (Figs. 3e–3g and S4a and S4b).
PLS Model Loadings and Scores Identify Basis Axes of Cue-Signal-Response Co-variation
Possible connections between stimulation cues, signaling metrics, and cell response outcomes in PLS models can be identified by examining their co-variation in principal component space. We inspected the signaling and response metric PLS model loadings (w*ci) in each PC (Figs. S4d and S4e). We observed that PC1 represented an anti-differentiation axis due to the inclusion of highly-negative PC1 loadings of all five myogenic differentiation measurements (Fig. S4d). We observed that PC2 contains signaling metric co-variation that was not well-correlated with any particular response variable. We observed that PC3 and PC4 are both pro-proliferative axes, and that the proliferation response has negligible loadings in PC1 and PC2. We observed that few signals provided strongly negative PC1 model loadings and, instead, found that strongly-positive, differentiation-antagonizing PC1 loadings, suggesting that specific phosphoprotein signals measured in the CSR compendium do not strongly promote differentiation (Fig. S4d). Instead, we conclude that differentiation is largely a default state of low-serum medium, which agrees with observations that myogenic specification is a result of signaling deprivation.40 In contrast, we identified more signals with either pro- or anti-proliferative correlations due to their strongly-positive or -negative (respectively) loadings in PC3 and PC4 (Fig. S4e).
We examined the model “scores”, which represent how the cues project into the PC axes of the calibrated 4-PC PLS model. Since PC1 represents the strongest differentiation-associated axis and PC3 represents the strongest proliferation-associated axis, we focused model interpretations to these two PCs. We examined how each two-way combination of the stimulation mixtures independently and cooperatively projected on PC1 and PC3 (Figs. 3h–3j). To assess non-additive cooperativity, we generated an additive projection for each two-way condition combination from their PC1/PC3 score vectors. By comparing to these additive projections, we noted that the FEI/TI combination exhibited additive cooperativity, the FEI/IOL combination exhibited antagonism, and the TI/IOL combination exhibited supra-additive cooperativity, within this shared signal-response principal component space.
By examining the cell-response loadings in the PLS model, we identified that PC1 and PC3 provide orthogonal principal axes to delineate signaling mediators that distinguish between myoblast differentiation (PC1) vs. proliferation (PC3) (Fig. 4a). By inspecting all signal metric loadings (Figs. 4a–4d and S4f and S4g), we found that PC1 was dominated by anti-differentiation (positive w*c1) p-IκBα and p-cJun loadings throughout all time-points and pro-differentiation (negative w*c1) p-p38 MAPK loadings from later time-points (Fig. 4b). In contrast, we found that PC3 was dominated by a strongly anti-proliferative (negative w*c3) p-Stat3 15-min loading and pro-proliferative (positive w*c3) late (4–24 h post-stimulation) p-Akt, p-70 S6K, p-MEK1, and p-ERK1/2 loadings (Fig. 4c).
Figure 4.

PLS model reveals distinct signaling axes regulating myoblast proliferation and differentiation. (a) PLS model loadings (w*c) for all signaling metrics (gray) and responses (blue) in PC1 and PC3. All response measurements and the most positive and negative signaling metrics in each PC are noted; (b, c) Heatmaps of signaling loadings in PC1 (b) and PC3 (c); (d) Plots of model loadings in PC1 vs. PC3 as in (a), with each phosphoprotein individually highlighted (black). Early and late AUC metrics excluded due to redundancy. Trajectory of time-points for select phosphoproteins overlaid by red arrow.
Interestingly, we observed that a number of phosphoprotein signaling metrics had loadings that varied in their PC1 and PC3 contributions with respect to their associated post-stimulation time (Fig. 4d, note red arrows represent time axis). Notably, p-MEK1 and p-ERK1/2 metrics had negligible PC1 loadings for all time-points and strongly positive PC3 loadings at late time-points and for the full AUC metric, suggesting MEK–ERK signaling contributing to cell proliferation more so than differentiation. In contrast, p-p38 and p-Akt metrics transition from strongly positive in PC1 at very early time-point (5–15 min) to strongly positive in PC3 at later time-points (4–24 h). These time-varying loadings suggest that even individual signaling pathways could have distinct temporally-encoded contributions to myoblast proliferation and differentiation responses, as has previously been demonstrated for TNF-α-induced colon epithelial cell apoptosis models.20
Confirming Model-Identified Signaling Regulation of Myoblast Fate
We aimed to test the mechanistic roles of these model-identified distinct signal-response associations through a series of small molecule signaling inhibitor studies. First, we examined MEK–ERK signaling, which the model identified as having exclusively a pro-proliferative effect, by treating FEI-stimulated myoblasts with the MEK inhibitor PD0325901. We observed that PD0325901 attenuated FEI-induced p-ERK activation, as expected, and reduced cell numbers below the untreated control, as predicted (Figs. 5a and 5b). Moreover, though FEI weakly induced Myh2 expression, PD0325901 had no effect on myogenic differentiation, confirming the exclusive pro-proliferative role of MEK–ERK signaling (Figs. 5c and 5d). Second, we examined JNK signaling, which the model identified as having exclusively an anti-differentiation role, by treating TI-stimulated myoblasts with the JNK inhibitor SP600125. We observed that SP600125 treatment ameliorated TI-stimulated p-cJun activation and enhanced TI-restricted myoblast differentiation, as evidenced by elevated Myod1 and Myh2 expression, as predicted (Figs. 5e–5g). TI with or without SP600125 co-treatment had no effect on myoblast proliferation, thus confirming the exclusive anti-differentiation role of JNK signaling (Fig. 5h). Third, we examined Stat3 signaling, which notably had a strong anti-proliferative but negligible differentiation association in the model, using the Stat3 inhibitor 5, 15-Diphenylporphyrin (5, 15-DPP). Co-treatment with 5, 15-DPP abrogated the IOL-stimulated proliferative decline, but did not affect differentiation, confirming the exclusively anti-proliferative role of IOL–Stat3 signaling (Figs. 5i–5k). These findings provide evidence that the PLS model gives insight to mechanistic connections between critical signaling mediators and myogenic cell fate regulators, and that orthogonal signaling-fate axes exist within the myoblast signaling network.
Figure 5.
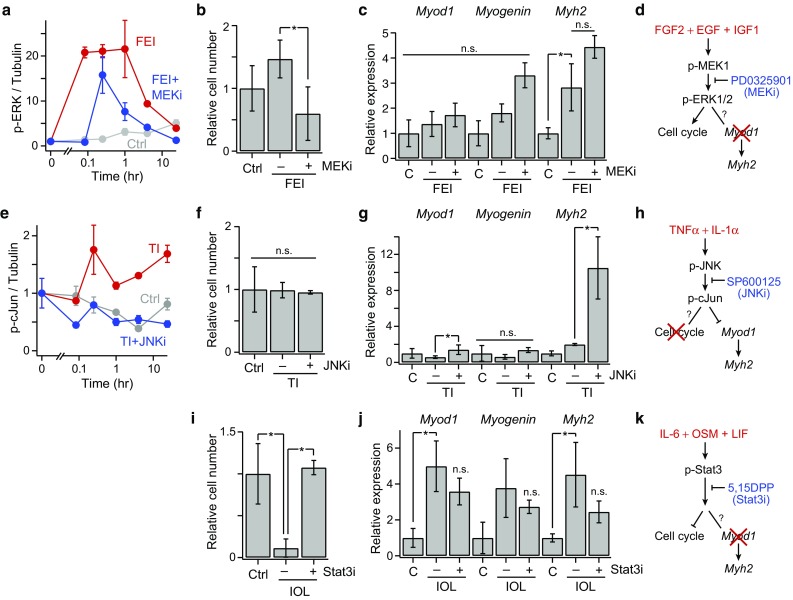
Mechanistic insights into signaling-fate associations. (a) Phospho-ERK1/2 time-courses; (b) relative cell number count; (c) Myod, Myogenin, and Myh2 expression, at 72 h post-stimulation, for control, FEI, and FEI + SB203580 (p38i) conditions; (e) Phospho-cJun time-courses; (f) relative cell number count; (g) Myod, Myogenin, and Myh2 expression for control, TI, and TI + SP600125 (JNKi) conditions; (i) Relative cell number count; (j) Myod, Myogenin, and Myh2 expression control, IOL, and IOL + 5, 15-DPP (Stat3i) conditions. For all plots, n = 3 replicates are plotted as mean ± SEM. In (b, c, f, g, i, j) and (d–f), p < 0.05 (*) by Student’s t tests or not significant (n.s.); (d, h, k) Signaling mechanism conclusions from the data-model and inhibitor studies.
Verifying PLS Model Prediction on Kinase Inhibitor Data Set
We tested the performance of the trained model in predicting the myoblast responses when MEK, p38 and JNK were perturbed by targeted small molecule kinase inhibition. We collected a second “test” CSR data-compendium with eight treatments: control, FEI, FEI with PD0325901 (a MEK inhibitor) or SB203580 pre-treatment (a p38α/β MAPK inhibitor), TI, TI with PD0325901, SB203580 or SP600125 (a JNK inhibitor) in mid-passage (p20) myoblasts (Fig. S5). In this second CSR compendium, we observed that responses to MEK and p38 inhibitor treatments unexpectedly enhanced and/or prolonged the activation of other pathways (such as p-Akt and p-MEK, respectively) that are not biochemically perturbed by these inhibitors. Therefore, we developed a modified version of PLS model trained on the first CSR data set, which did not include any p-MEK signaling metrics due to these confounding effects. We tested the modified model’s ability to predict the proliferation and differentiation responses in the test set using the signaling data alone (Fig. S5). The modified PLS model did not accurately predict myoblast cell fate outcomes across this entire test data set (cumulative R 2 = -0.11) due to a few large prediction inaccuracies associated with inhibitor conditions. These observations suggest that the myoblast signaling network is sufficiently re-wired upon kinase inhibitor treatments to break the linear logic encoded in the PLS model.
Phosphatase-Mediated Pathway Crosstalk
This model fitness result suggested that perhaps these chemical inhibitors exhibited off-target effects on other pathways that were poorly accounted for in the model. For example, SB203580 has been reported to have a ~100-fold-reduced IC50 for JNK, a kinase that phosphorylates cJun, relative to the IC50 for p38α/β.12 We evaluated the signaling activation of multiple pathways in the context of p38 inhibition and observed that a number of untargeted pathways exhibited elevated signaling activation in the presence of the p38 inhibitor SB203580. In particular, p-MEK1 and p-ERK1/2 levels were persistently elevated in the presence of SB203580 (Figs. 6a and 6b). This suggests that p38 inhibition may have unintended consequences through signaling network feedback mechanisms, such as through the downregulation of p38-driven dual-specific phosphatase (DUSP) expression,36 which could act to allow prolonged MEK–ERK pathway signaling. DUSP expression, in particular DUSP1 (MKP-1) and DUSP6 (MKP-3), has been shown to be regulated by ERK and p38 signaling in myoblasts.3,45 We measured Dusp1 and Dusp6 expression in FEI- and TI-treated myoblasts at 3 days post-stimulation (Figs. 6c and 6d). Though Dusp1 expression was not significantly affected, we found that Dusp6 expression was upregulated by TI stimulation and could be abrogated by either MEK or p38 inhibition. Together, these findings suggest an autonomous feedback circuit in which both p38 and ERK signaling is necessary for inducing DUSP6 expression, which may act to dephosphorylate MEK and ERK, thus negatively regulating their activities (Fig. 6e).
Figure 6.
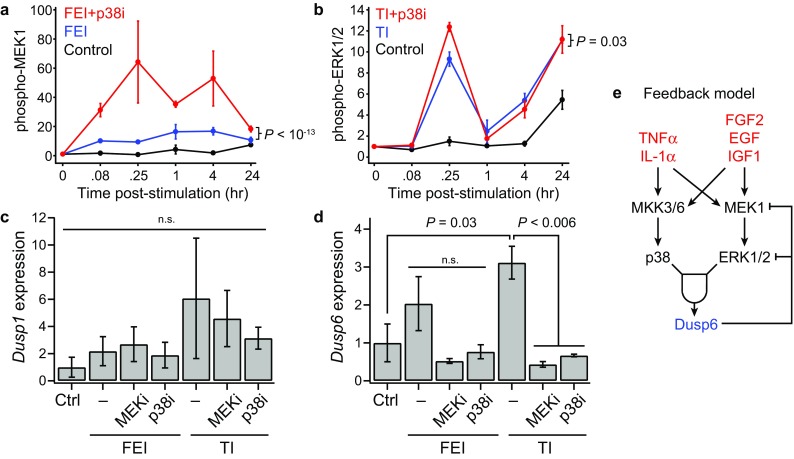
Both MEK and p38 are necessary for stimulation-induced DUSP6 expression in myoblasts. (a, b) Select phosphoprotein time-courses for inhibitor conditions. n = 3 replicates are plotted as mean ± SEM. p values from ANOVA comparisons over the full time-course. (a) Phospho-MEK1 for Control, FEI, and FEI + p38i; (b) Phospho-ERK1/2 for Control, TI, and TI + p38i conditions; (c, d) DUSP1 and DUSP6 expression by RT-qPCR in myoblast following either FEI or TI with either PD0325901 (MEKi), SB203580 (p38i) or 0.1% DMSO-only (–) co-treatment. p values from Student’s t tests or not significant (n.s.); (e) DUSP6-based feedback model.
Discussion and Conclusions
Through a data-modeling approach, we have constructed a predictive cue-signal-response model of myoblast fate decisions pertinent to muscle regeneration within muscle damage-associated cytokine and growth factor microenvironments in vitro. Using a low-serum mouse primary myoblast culture model, in which myoblast proliferation and differentiation are both induced over 3 days, we observed that combinations of the growth factors FGF2, EGF, and IGF1 (FEI) and the cytokines TNF-α and IL-1α (TI) synergistically induce cell proliferation, whereas the IL-6 family ligands IL-6, OSM, and LIF (IOL) together antagonize both basal and FEI/TI-induced proliferation (Figs. 1a and 1b). Furthermore, FEI and IOL induce, and TI restricts, myoblast differentiation as reflected by MyoD1, Myogenin and MHC mRNA and/or protein levels (Figs. 1c–1h). When combined into higher-order mixtures, we observed sub-additive FEI/IOL-induced differentiation and negatively synergistic differentiation effects when either FEI or IOL was combined with TI. Together, these combinatorial effects were not well-predicted by a modified Bliss synergy model suggesting that higher-order pathway synergies and antagonisms govern myoblast responses to these diverse ligand cue mixtures.
To ascertain the important signaling pathway contributors to these conflicting stimuli, we established a cue-signal-response data compendium and used it to calibrate multivariate PLS data-model (Figs. 2 and 3a–3d). Given the balance of up- and down-regulation observed in the CSR training data, we compared log- and linearly-scaled data processing to examine whether log-scale improved model performance. We found that log-scaling the data diminished model fitness (cumulative R 2 = 0.87) relative to the unscaled data (R 2 = 0.96) (Fig. 3d). We note that this modeling approach only utilized the mean values from all signaling and response measurements and did not explicitly take into consideration observed biological variations. Taking into consideration such variation can enable more robust model calibration18 and may improve future modeling implementations.
We found that a single PLS model was capable of accurately relating the myoblast proliferation and differentiation outcomes within four principal components of data-regression loadings. By analyzing PLS loadings associated with the signaling and response variables, we identified a set of principal component axes that distinguish between myoblast differentiation and proliferation outcomes (Figs. 4a–4d and S4d–S4g). PC1 represented an anti-differentiation axis dominated by IκB-α and cJun phosphoprotein signals. PC2 only minimally distinguished between the response variables, and more distinctly separated a subset of signaling metrics (e.g., early and late MEK–ERK variables). Notably, PC2 correctly ordered the myogenic differentiation variables from least committed (MyoD1) to most committed (MHC) in the positive PC2 direction. PC3 and PC4 both represented pro-proliferative axes. In examining the relative contributions of each PC to the overall model fitness (Fig. 3c), we noted that PC1 and PC3 provided the largest incremental improvements in cumulative R 2, suggesting they are the most informative PCs to response variable modeling. Given this and their exclusive associations with the two sets of response variables, we used PC1 and PC3 as differentiation and proliferative axes, respectively, for analyzing model loading variables.
We observed that some phosphoprotein signals (p-Akt, p-p70 S6K, and p-p38) had time-varying contributions to these basis axes of cell-fate regulation (Fig. 4d), which suggests that individual pathways contribute to both myoblast proliferative and differentiation response. In contrast, we focused on signals that predominantly yield model loadings associated with either PC1 (IκB-α and cJun) or PC3 (Stat3, MEK, and ERK), suggesting that they contribute exclusively to one cell-fate outcome.
We confirmed these model-associated signaling-response relationships through small molecule inhibitor studies (Fig. 5) to demonstrate the mechanistic connections suggested by the model. Using the MEK inhibitor PD0325901, we demonstrated that FEI-stimulated MEK–ERK signaling promotes myoblast proliferation but does not affect myogenic differentiation (Figs. 5a–5d). This agrees with prior reports that FGF2, EGF, and IGF1 act as myoblast mitogens,29,37 and notably confirms prior findings that ERK signaling is dispensable for FGF-induced myogenic differentiation23 and suggests that alternative pathways, such as PI3K–Akt–p70, may play a stronger role in FEI-induced myogenic differentiation, as has been reported for IGF1.33,42 Using the JNK inhibitor SP600125, we demonstrated that TI-stimulated JNK–cJun signaling restricts myoblast differentiation but does not perturb proliferation (Figs. 5e–5h). The JNK–cJun pathway has been previously associated with both proliferation-inducing and -restricting effects in horse serum-treated myoblasts.14,31 Our results, using a FBS starvation protocol, further suggest that the contributions of JNK signaling are context-specific in differentiating myoblasts. Further, our findings agree with a recent report34 showing that JNK activation antagonizes myogenic differentiation. Lastly, using the Stat3 inhibitor 5, 15-DPP, we demonstrated that IOL-stimulated Stat3 signaling restricts myoblast proliferation but does not influence the expression of commitment genes (Figs. 5i–5k). These observations agree with reports that Stat3 signaling impairs muscle stem cell proliferation in vivo.22,32 Thus, the ERK, JNK, and Stat3 pathways all regulate distinct myoblast cell fate outcomes with exclusivity.
In testing the PLS model on a second multi-pathway CSR data set, we observed poor model performance due to cross-pathway effects of inhibitor treatments, which we reasoned were due to unexpected changes phosphatase activation. We showed that p38 and MEK–ERK signaling together induce expression the phosphatase DUSP6 (MKP-3), which can dephosphorylate MAP kinases such as JNK, p38, and ERK36 (Fig. 6e). Though DUSP6 has previously been found to be induced in myoblasts,3,45 our findings suggest that DUSP6 induction is dependent on p38 and ERK cooperation and may auto-regulate the activation of these MAP kinases, complicating the effects of targeted inhibitor treatments of cytokine and growth factor stimulation.
Together, our model and experimental observations suggest that, though individual pathways may confer specific cell-fate responses within the context of in vitro myoblast differentiation, the myoblast signaling network is governed by both cooperative, synergistic, and antagonistic pathways that enable cell-fate tuning in response to higher-order cytokine stimulations. Future investigations evaluating time-varying pathway inhibition strategies may prove particularly useful for both more thoroughly validating the PLS model and for optimizing experimental control of myoblast cell-fate outcomes in response to these combinatorial conditions. Moreover, these network-level behaviors argue for the use of an experimental data set that broadly and densely measures key readouts across multiple regulatory pathways for the purpose of model training and testing, as performed here. The cue-signal-response data compendium and PLS data-model presented here provide new insights into the muscle myoblast signaling network-level synergies and antagonisms between a large diversity of muscle regeneration-associated stimuli. In concert with other recent mechanistic models focused on myogenic gene regulatory circuits,13 our findings argue that computational biology models are now poised to contribute to new understanding and hypotheses related to how muscle stem/progenitor cells regulate essential cell-fate decisions at the systems level.
Materials and Methods
Animals Care and Procedures
The Cornell University Institutional Animal Care and Use Committee (IACUC) approved all animal protocols and experiments were performed in compliance with the institutional guidelines of Cornell University. Young adult (4-month-old) C57BL/6 mice were purchased from Jackson Laboratories (#0664, Bar Harbor, ME). Tibialis anterior and gastrocnemius muscles were dissected and subjected to collagenase (0.25%) and dispase (0.04 U mL−1; Roche, Indianapolis, IN) digestion. Non-muscle tissue was removed under a dissection microscope and muscle fibers were dissociated. After 90 min of total digestion, the remaining cell suspension was passed through a nylon 70-µm filter (BD Biosciences, San Jose, CA). Primary myoblasts (PMBs) were isolated as previously described.40
Primary Myoblast Culture
All tissue-culture-treated dishes and plates were coated overnight with type I collagen (Sigma-Aldrich, St. Louis, MO, #C8919, 1:100 dilution in 0.1 M acetic acid) at sufficient volume to cover the entire surface area, then washed once with 1× phosphate buffer saline prior to use. PMBs (up to passage number 20) were expanded in 15-cm collagen-coated dishes at 5000–15,000 cells cm−2 in 20 mL myogenic growth medium (GM). GM is 43% Dulbecco’s Modified Eagle’s Medium (Corning Cellgro, Corning, NY, #10-013), 40% Ham’s F-10 (Corning Cellgro, #10-070-CV), 15% Fetal Bovine Serum (Corning Cellgro, #35-010-CV), 1% Penicillin–Streptomyocin (Corning Cellgro, #30-002-CI), 1% l-glutamine (Corning Cellgro, #25-005), and 2.5 ng mL−1 recombinant mouse bFGF (R&D Systems, Minneapolis, MN, #3139-FB-025). Passage 18 and 20 myoblasts were used, respectively, for the “training” and “test” CSR data compendia.
Primary Myoblast Combinatorial Stimulation
PMBs (passage number 18-20) were seeded at 50,000 cells cm−2 in collagen-coated treated 12-well plates in 1 mL growth media for 2 h, then switched to 1 mL differentiation media (DM: 50% Dulbecco’s Modified Eagle’s Medium, 45% Ham’s F-10, 3% Fetal Bovine Serum, 1% Penicillin–Streptomyocin, 1% l-glutamine) with or without 0.1% DMSO for 3 h. Cells were then stimulated with differentiation media (mock control) or a recombinant mouse protein cocktails consisting of mixtures of the following (final concentrations): 5 ng mL−1 FGF2 (R&D Systems, #3139-FB-025), 10 ng mL−1 EGF (R&D Systems, #2028-EG-200), 10 ng mL−1 IGF1 (R&D Systems, #791-MG-050); 10 ng mL−1 IL6 (R&D Systems, #406-ML-005), 10 ng mL−1 OSM (R&D Systems, #495-MO-025), 10 ng mL−1 LIF (R&D Systems, #8878-LF-025); 10 ng mL−1 TNF-α (R&D Systems, #410-MT-010), 10 ng mL−1 IL-1α (R&D Systems, #400-ML-005). Each of the stimulation conditions (mock, DMSO, FEI, IOL, TI, FEI/IOL, IOL/TI, FEI/TI) had n = 3 biological replicates per assay per time point.
Quantitative Immunoblotting
Lysis. Stimulated cells were lysed for immunoblotting at the following time points post-stimulation: 0, 15 min, 72 h. Cells were washed with cold 1 × PBS and lysed in 50 μL per well of cold NP-40-based lysis buffer (50 mM β-glycerophosphate, 30 mM NaF, 10 mM NaPP, 50 mM Tris–HCl, 0.5% NP-40 substitute, 150 mM NaCl, 1 mM benzamidine, 2 mM EGTA, 400 μM sodium orthovanadate, 200 μM DTT, 2 mM PMSF, 1:200 dilution Phosphatase Inhibitor Cocktail Set III) using minor modifications of previous protocols.17,19 Lysate was scraped, collected, and centrifuged at 4 °C for 10 min at 15,000×g. The lysis supernatant was collected and protein concentration was quantified using a Micro BCA Protein Assay Kit (Thermo Scientific, #23,235) per manufacturer’s protocol. Electrophoresis. Electrophoresis gels (1.5 mm thickness, 10% acryl/bisacrylamide, Tris–HCl, Ammonium Persulfate, TEMED, SDS) were loaded with 25 μg of sample in 25 μL 1× sample buffer (20 mM Tris–HCl, Glycine, 10% SDS, 0.4% β-mercaptoethanol) per lane or 1 μL of strep-tagged unstained protein standards (Bio-Rad, Hercules, CA, #1610363) and run at 100-V for 2 h in Tris–HCl/Glycine/SDS running buffer. Proteins were transferred to a methanol-activated PVDF membrane overnight at 4 °C, 15-V in Tris–HCl/Glycine/Methanol transfer buffer. Immunoblotting. Membranes were blocked in 5% powdered milk in Tris-buffered saline with Tween-20 (TBST) with gentle rocking at room temperature for 1 h. Primary antibodies were diluted in 5% powdered milk in TBST in the following combinations: anti-Myosin heavy chain (223 kDa, MF 20 monoclonal mouse, DSHB, 1:10 dilution of hybridoma supernatant), anti-Myogenin (32/34 kDa, F5D monoclonal mouse, DSHB, Iowa City, IA, 1:2 dilution of hybridoma supernatant), and anti-Hsp90α/β (90 kDa, polyclonal rabbit, Santa Cruz Biotechnology, Dallas, TX, #sc-7947, 1:2500 dilution); p-Stat3 (Tyr705, 79/86 kDa, monoclonal rabbit, Cell Signaling Technology, Danvers, MA, #9131S, 1:1000 dilution), p-Akt (Ser473, 60 kDa, monoclonal rabbit, Cell Signaling Technology, #4060S, 1:1000 dilution), and Gapdh (37 kDa, monoclonal mouse, Thermo Fisher Scientific, #AM4300, 1:2500 dilution). Blots were incubated with primary antibody dilutions with gentle rocking at room temperature for 1 h. Blots were then washed with 1 × TBST three times for 5 min per wash. Secondary antibodies were diluted in 1 × TBST as follows: peroxidase-conjugated goat anti-rabbit (Jackson ImmunoResearch Laboratories, #111-035-144 1:5000 dilution), peroxidase-conjugated goat anti-mouse (Jackson ImmunoResearch Laboratories, West Grove, PA, #115-035-146, 1:5000 dilution), StrepTactin-HRP conjugate (Bio Rad, #1610380, 1:5000 dilution). Blots were incubated with secondary antibody dilutions with gentle rocking at room temperature for 30 min. Blots were then washed with 1 × TBST three times for 5 min per wash. Blots were incubated in ECL substrate (Bio Rad, #1705062, 1:1 mix of luminol/HRP substrate solutions) for 1 min and then imaged for 120 s using the ChemiDoc imaging system (Bio Rad, #17001401). Blots were analyzed using ImageLab software (Bio Rad) for band intensity.
Cell Counting
Stimulated cells were fixed at 72 h post-stimulation. Cells were washed twice with cold 1 × PBS and fixed for 15 min with 4% paraformaldehyde in 1 × PBS. Cells were washed twice with 1 × PBS and then blocked overnight at 4C with 10% donkey serum (Genetex, Irvine, CA, #GTX73245) in 1 × TBST. Cells were then washed three times with 1 × PBS. Cells were incubated with DAPI (Sigma-Aldrich, #32670-25MG-F, 1:2000 dilution) at room temperature in the dark for 15 min. Cells were washed twice with 1 × PBS and kept in 1 × PBS for imaging. Immunofluorescence images were acquired using a Nikon Eclipse Ti-E Microscope (MVI, Avon, MA) with Spectra X light engine (Lumencor, Beaverton, OR) with a DAPI polychroic (Chroma #VCGR-SPX-P01-PC, Bellows Falls, VT), a Nikon NAMC 10× Objective (#MRP60105) and an Andor Zyla 5.5 sCMOS Camera (Belfast, UK). Digital images were captured and nuclear segmentation and automated counting were performed using Nikon Elements software. Images were composed using Illustrator software (Adobe, San Jose, CA).
RT-qPCR
Stimulated cells were lysed with TRK/β-mercaptoethanol lysis buffer (EZNA Total RNA Kit I, Omega Biotek, #R6834-02) 72 h post-stimulation. Lysates were passed through QiaShredder columns (Qiagen, Germantown, MD, #79654) per manufacturer’s protocol, and then RNA was isolated using the EZNA Total RNA Kit I (Omega BioTek, Norcross, GA) per manufacturer’s protocol. cDNA was generated for each sample using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, #4368814) per manufacturer’s protocol. Samples were tested in technical duplicate using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, #4367659) per manufacturer’s protocol. Real-time PCR was performed using a Viia7 Real Time PCR instrument and software (Thermo Fisher). Samples were cycled at 95 °C for 10 min and then 40 cycles of 95 °C for 15 s and 60 °C for 1 min. To quantify relative transcript levels, the method was used with the DM with 0.02% DMSO samples used as reference control. Primer sequences for Myod1, Myog, Myh2, Dusp1, Dusp6, Dusp10, and 36b4 (normalization gene) were obtained from literature or using NIH Primer3. The primer sets for Myod1, Myog, Myh2, and 36b4 are as follows.
| Gene | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Myod | GCCGCCTGAGCAAAGTGAATG | CAGCGGTCCAGGTGCGTAGAAG |
| Myog | TGTTTGTAAAGCTGCCGTCTGA | CCTGCCTGTTCCCGGTATC |
| Myh2 | TTGTGGTGGACCCTAAGGAG | TTCATGGGGAAGACTTGGTC |
| 36b4 | AACGGCAGCATTTATAACCC | CGATCTGCAGACACACACTG |
| Dusp1 | ACCATCTGCCTTGCTTACCTT | AGCACCTGGGACTCAAACTG |
| Dusp6 | TCGGGCTGCTGCTCAAGAAAC | CGGTCAAGGTCAGACTCAATGTCC |
| Dusp10 | ACATCGGCTACGTCATCAAC | CACTGGTGAGCTTCCTCAAT |
Quantitative Multiplex Phosphoprotein Assays by Luminex
Stimulated cells were lysed for Luminex phosphoprotein assays at the following time points post-stimulation: 0, 5, 15, 60 min, 4, 24 h. Cells were washed with cold 1 × PBS and lysed in 50 μL per well of cold Milliplex Lysis Buffer (EMD Millipore, Billerica, MA, #48-602MAG), with 2 mM PMSF and 1:200 dilution Phosphatase Inhibitor Cocktail Set III (EMD Millipore, #524627). Lysate was scraped, collected, and centrifuged at 4 °C for 10 min at 15,000 × g. Supernatants were collected and protein concentrations were quantified using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, #23235) per manufacturer’s protocol. Milliplex MAPmate assays (EMD Millipore) were used to quantify the following phosphoproteins: p-Akt (Ser473, #46-677MAG), p-cJun (Ser73, #46-622MAG), p-IκBα (Ser32, #46-643MAG), p-ERK1/2 (Thr185/Tyr187, #46-602MAG), p-p70 S6 K (Thr389/Thr412, #46-629MAG), p-MEK1 (Ser222, #46-670MAG), and p-p38 (Thr180/Tyr182, #46-610MAG). Assays were performed in multiplex and using the Milliplex MAPmate Cell Signaling Buffer & Detection Kit per the manufacturer’s protocol with 3 μg of sample per well (see Fig. S2 for protein loading optimization and validation), n = 1 technical replicate and n = 3 biological replicates per time point. Background-subtracted fluorescence values for each phosphoprotein were normalized to the background-subtracted fluorescence values for Milliplex MAPmate β-tubulin (#46-713 MAG). Normalized values were averaged across biological replicates, and then fold-change normalized for each phosphoprotein to the 0 min time point.
Cue-Signal-Response Metric Extraction and Data Scaling for Partial Least-Squares Modeling
‘Cue-signal-response’ (CSR) data containing phosphoprotein time-course (Luminex, t = 0–24 h; Immunoblots, t = 15 min), myogenic gene expression (RT-qPCR, t = 72 h), myogenic protein expression (Immunoblot, t = 72 h), and cell proliferative index (Imaging, t = 72 h) data were compiled. Phosphoprotein data were fold-change normalized to untreated samples (t = 0 min) for each phosphoprotein assay. For each phosphoprotein time course, three time-dependent signaling metrics were extracted: (i) integral for 0–24 h post-stimulation (“full AUC”), (ii), integral for 0–1 h post-stimulation (“early AUC”), and (iii) integral for 1–24 h post-stimulation. These were added to the five time points (5, 15, 60 min, 4, and 24 h) to yield eight signaling metrics for each assayed phosphoprotein. Immunoblot phosphoprotein expression data (p-Stat3 and p-Akt) were fold-change normalized to loading control protein Gapdh and then fold-change normalized to the DMSO condition at t = 0. The eight Luminex signaling metrics per phosphoprotein and the two immunoblot phosphoprotein metrics were fused into a signaling network data-matrix (X). Gene expression data (Myod1, Myogenin, and Myh2) were fold-change normalized to loading control gene 36b4 and then fold-change normalized to the DMSO control condition at t = 72 h. Protein expression data (Myogenin and MHC) were fold-change normalized to loading control protein HSP90 and then fold-change normalized to the DMSO control condition at t = 72 h. Nuclei counts from DAPI images were normalized to the DMSO control condition. These six response metrics were then fused into a response data-matrix (Y). To account for both up- and down-regulation of cell signals and responses (see Figs. 1b–1h and 2c), the X and Y data-matrix were tested with and without log2-scaling before model-calibration. Matrices X and Y were arrayed across all treatment conditions for both model-calibration and testing (Fig. 2c).
Partial Least-Squares Modeling of Cue-Signal-Response Data
The relationship between the X signaling data-matrix and the Y response data-matrix was modeled using partial least-squares (PLS) regression data-modeling.8,9,17,20,26 Briefly, we implemented PLS using the SIMPLS algorithm using the PLSREGRESS function in MATLAB (MathWorks, Natick, MA) following standard methods, with some modifications. Signaling and response data were either left unscaled or log2-scaled before transformation (Fig. 3d). Data matrices were then unit-variance-transformed (see Fig. 3b). All models were generated using four principal components under standard optimization criteria (see Fig. 3c). Model calibration was conducted using leave-one-out cross-validation, and model uncertainties were calculated by jack-knifing. Model loadings were calculated using the mean-centered regression coefficients w*ca from the a-th PLSR principal-component. The information content of each signaling metric was assessed by its variable importance of projection (VIP) score, which captures the loss in model fitness upon removal of the individual signaling metric. The accuracy of model predictions was assessed using a model fitness8,17 parameter (R 2), calculated according to the following formula:
where n is the number of observations in the training or test set for the response variable(s) of interest.
Statistical Testing
Cell response measurements were compared to the DMSO control condition (Control) using Student’s t test with α = 0.05. For statistical testing of synergy in combination stimulation conditions (FEI/TI, FEI/IOL, TI/IOL), each combined cell-response measurement (Y) was calculate with a modified Bliss independence5 model:
where Y calc 1,2 is the calculated fold-change value for the combination Y response metric (i.e., FEI + TI cell #), Y 1 and Y 2 are the observed fold-change values for the individual Y response metrics (i.e., FEI cell # and TI cell #), and Y 0 is the observed fold-change value for the Control Y response metric (i.e., Control cell #). Experimental measurement uncertainty was propagated through the calculation. Y response metrics for observed combination stimulation conditions (FEI/TI, FEI/IOL, TI/IOL) were then compared to calculated Y response metrics for combination stimulation conditions (FEI + TI, FEI + IOL, TI + IOL) using Student’s t test with α = 0.05.
Model Test Predictions
Primary myoblasts were seeded for stimulation as described above, with one of the following inhibitors added to the differentiation media prior to stimulation: 10 μM p38 inhibitor (p38i) SB203580 (Cayman Chemical, Ann Arbor, MI, #13067); 50 μM Stat3 inhibitor (Stat3i) 5, 15-diphenylporphyrin (Sigma-Aldrich, #D4071); 50 nM MEK inhibitor (MEKi) PD0325901 (Selleckchem, Houston, TX, #S1036); or 5 μM JNK inhibitor (JNKi) SP600125 (Selleckchem, #S1460). Phosphoprotein assays, RT-qPCR, immunoblotting, immunostaining, and data-normalization were carried out in the same manner as described above for training set conditions. X and Y matrices were assembled as described above. Using a PLS model calibrated to training CSR set (but with all MEK signaling metrics removed, a predicted Y response data-matrix was calculated from the X signaling data-matrix of “test” inhibitor signaling matrix. This predicted Y response data-matrix was compared with the observed Y response data-matrix to generate a model fitness parameter (R 2), calculated as described above.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was financially supported by the National Institute on Aging of the National Institutes of Health under Award R00AG042491 (to B.D.C), a US Department of Education Graduate Assistantship in Areas of National Need under Award P200A150273 (to A.M.L), a Roberta G. and John B. DeVries Graduate Fellowship (to A.M.L.), and Hunter R. Rawlings III Cornell Presidential Research Scholarship (to R.F.K. and J.K.). This work made use of the Nanobiotechnology Center (NBTC) shared research facilities at Cornell University. The authors acknowledge technical assistance from Teresa Porri, Penny Burke, Andrea De Micheli, Hilarie Sit, Muhammad Safwan Jalal, Nancy Mejia, Isabella Mercado, Ryan Ausmus, and Paula Fraczek. The authors thank the anonymous reviewers for their constructive reviews.
Animal Studies
All institutional and national guidelines for the care and use of laboratory animals were followed in a protocol approved by Cornell University’s Institutional Animal Care and Use Committee (IACUC).
Conflicts of interest
A. M. Loiben, S. Soueld-Baumgarten, D. Bhattacharya, R. F. Kopyto, J. C. Kim and B. D. Cosgrove declare that they have no conflicts of interest.
Human Studies
No human studies were carried out by the authors for this article.
Abbreviations
- AUC
Area-under-the-curve
- CSR
Cue-signal-response
- DUSP
Dual specificity phosphatase
- EGF
Epidermal growth factor
- FGF2
Fibroblast growth factor 2
- IGF1
Insulin-like growth factor 1
- IL-1α
Interleukin-1α
- IL-6
Interleukin-6
- LIF
Leukemia inhibitor factor
- MHC
Myosin heavy chain
- OSM
Oncostatin-M
- PC
Principal component
- PLS
Partial-least squares
- TNF-α
Tumor necrosis factor-α
Footnotes
Benjamin D. Cosgrove
is an Assistant Professor in the Meinig School of Biomedical Engineering at Cornell University in Ithaca, NY, where he directs the Laboratory of Regenerative Systems Biology. His research group, which is currently supported by a NIH R00 Pathway-to-Independence Award, develops and implements systems biology and biomaterials engineering approaches to study how cell–cell communication and intracellular signaling networks regulate stem and progenitor cell function in skeletal muscle homeostasis and regeneration, and how these processes become dysfunctional in aging and muscular dystrophies. Dr. Cosgrove earned a Bachelor’s in Biomedical Engineering at the University of Minnesota and a Ph.D. in Bioengineering at the Massachusetts Institute of Technology. His Ph.D. thesis research, under the joint supervision of Dr. Douglas Lauffenburger and Dr. Linda Griffith, which established on experimental and computational systems biology tools to elucidate signaling network mechanisms regulating liver hepatocyte cell-fate decisions, was supported by a Whitaker Foundation Graduate Research Fellowship and a Biomedical Engineering Society Graduate Research Award. His postdoctoral research with Dr. Helen Blau at Stanford University was supported by a Stanford Molecular Imaging Scholars Fellowship and NIH K99 Pathway-to-Independence Award and was recognized by the Cellular and Molecular Bioengineering Special Interest Group of the Biomedical Engineering Society with a Rising Star award in 2015.
This article is part of the 2017 CMBE Young Innovators special issue.
References
- 1.Albeck JG, MacBeath G, White FM, Sorger PK, Lauffenburger DA, Gaudet S. Collecting and organizing systematic sets of protein data. Nat. Rev. Mol. Cell Biol. 2006;7(11):803–812. doi: 10.1038/nrm2042. [DOI] [PubMed] [Google Scholar]
- 2.Belizario JE, Fontes-Oliveira CC, Borges JP, Kashiabara JA, Vannier E. Skeletal muscle wasting and renewal: a pivotal role of myokine il-6. Springerplus. 2016;5:619. doi: 10.1186/s40064-016-2197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997;278(5341):1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 4.Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. P38 mapk signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014;20(3):265–271. doi: 10.1038/nm.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bliss CI. The toxicity of poisins applied jointly. Ann. Appl. Biol. 1939;26(3):585–615. doi: 10.1111/j.1744-7348.1939.tb06990.x. [DOI] [Google Scholar]
- 6.Broholm C, Laye MJ, Brandt C, Vadalasetty R, Pilegaard H, Pedersen BK, Scheele C. LIF is a contraction-induced myokine stimulating human myocyte proliferation. J. Appl. Physiol. (1985) 2011;111(1):251–259. doi: 10.1152/japplphysiol.01399.2010. [DOI] [PubMed] [Google Scholar]
- 7.Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013;14(6):329–340. doi: 10.1038/nrm3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosgrove BD, Alexopoulos LG, Hang TC, Hendriks BS, Sorger PK, Griffith LG, Lauffenburger DA. Cytokine-associated drug toxicity in human hepatocytes is associated with signaling network dysregulation. Mol. BioSyst. 2010;6(7):1195–1206. doi: 10.1039/b926287c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cosgrove, B. D., L. G. Alexopoulos, J. Saez-Rodriguez, L. G. Griffith, and D. A. Lauffenburger. A multipathway phosphoproteomic signaling network model of idiosyncratic drug- and inflammatory cytokine-induced toxicity in human hepatocytes. In: Conf Proc IEEE EMBS, 2009, pp. 5452–5455. [DOI] [PMC free article] [PubMed]
- 10.Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, Blau HM. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat. Med. 2014;20(3):255–264. doi: 10.1038/nm.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosgrove BD, Griffith LG, Lauffenburger DA. Fusing tissue engineering and systems biology toward fulfilling their promise. Cell. Mol. Bioeng. 2008;1(1):33–41. doi: 10.1007/s12195-008-0007-9. [DOI] [Google Scholar]
- 12.Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000;351(Pt 1):95–105. doi: 10.1042/bj3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshpande RS, Spector AA. Modeling stem cell myogenic differentiation. Sci. Rep. 2017;7:40639. doi: 10.1038/srep40639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA. Satellite cells and skeletal muscle regeneration. Compr. Physiol. 2015;5(3):1027–1059. doi: 10.1002/cphy.c140068. [DOI] [PubMed] [Google Scholar]
- 15.Fedorov YV, Rosenthal RS, Olwin BB. Oncogenic ras-induced proliferation requires autocrine fibroblast growth factor 2 signaling in skeletal muscle cells. J. Cell Biol. 2001;152(6):1301–1305. doi: 10.1083/jcb.152.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu X, Xiao J, Wei Y, Li S, Liu Y, Yin J, Sun K, Sun H, Wang H, Zhang Z, Zhang BT, Sheng C, Wang H, Hu P. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 2015;25(9):1082–1083. doi: 10.1038/cr.2015.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaudet S, Janes KA, Albeck JG, Pace EA, Lauffenburger DA, Sorger PK. A compendium of signals and responses triggered by prodeath and prosurvival cytokines. Mol. Cell. Proteom. 2005;4(10):1569–1590. doi: 10.1074/mcp.M500158-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Heinemann T, Raue A. Model calibration and uncertainty analysis in signaling networks. Curr. Opin. Biotechnol. 2016;39:143–149. doi: 10.1016/j.copbio.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Janes KA. An analysis of critical factors for quantitative immunoblotting. Sci. Signal. 2015;8(371):rs2. doi: 10.1126/scisignal.2005966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janes KA, Albeck JG, Gaudet S, Sorger PK, Lauffenburger DA, Yaffe MB. A systems model of signaling identifies a molecular basis set for cytokine-induced apoptosis. Science. 2005;310(5754):1646–1653. doi: 10.1126/science.1116598. [DOI] [PubMed] [Google Scholar]
- 21.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell Biol. 2006;7(11):820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 22.Joanisse S, Parise G. Cytokine mediated control of muscle stem cell function. Adv. Exp. Med. Biol. 2016;900:27–44. doi: 10.1007/978-3-319-27511-6_2. [DOI] [PubMed] [Google Scholar]
- 23.Jones NC, Fedorov YV, Rosenthal RS, Olwin BB. Erk1/2 is required for myoblast proliferation but is dispensable for muscle gene expression and cell fusion. J. Cell. Physiol. 2001;186(1):104–115. doi: 10.1002/1097-4652(200101)186:1<104::AID-JCP1015>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 24.Kellogg RA, Tay S. Noise facilitates transcriptional control under dynamic inputs. Cell. 2015;160(3):381–392. doi: 10.1016/j.cell.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Kemp ML, Wille L, Lewis CL, Nicholson LB, Lauffenburger DA. Quantitative network signal combinations downstream of tcr activation can predict il-2 production response. J. Immunol. 2007;178(8):4984–4992. doi: 10.4049/jimmunol.178.8.4984. [DOI] [PubMed] [Google Scholar]
- 26.Kreeger PK. Using partial least squares regression to analyze cellular response data. Sci. Signal. 2013;6(271):tr7. doi: 10.1126/scisignal.2003849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar N, Wolf-Yadlin A, White FM, Lauffenburger DA. Modeling her2 effects on cell behavior from mass spectrometry phosphotyrosine data. PLoS Comput. Biol. 2007;3(1):e4. doi: 10.1371/journal.pcbi.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lagha M, Sato T, Bajard L, Daubas P, Esner M, Montarras D, Relaix F, Buckingham M. Regulation of skeletal muscle stem cell behavior by pax3 and pax7. Cold Spring Harb. Symp. Quant. Biol. 2008;73:307–315. doi: 10.1101/sqb.2008.73.006. [DOI] [PubMed] [Google Scholar]
- 29.Lawlor MA, Feng X, Everding DR, Sieger K, Stewart CE, Rotwein P. Dual control of muscle cell survival by distinct growth factor-regulated signaling pathways. Mol. Cell. Biol. 2000;20(9):3256–3265. doi: 10.1128/MCB.20.9.3256-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller-Jensen K, Janes KA, Brugge JS, Lauffenburger DA. Common effector processing mediates cell-specific responses to stimuli. Nature. 2007;448(7153):604–608. doi: 10.1038/nature06001. [DOI] [PubMed] [Google Scholar]
- 31.Mueck T, Berger F, Buechsler I, Valchanova RS, Landuzzi L, Lollini PL, Klingel K, Munz B. Traf6 regulates proliferation and differentiation of skeletal myoblasts. Differentiation. 2011;81(2):99–106. doi: 10.1016/j.diff.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Munoz-Canoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 2013;280:4131–4148. doi: 10.1111/febs.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagata Y, Ohashi K, Wada E, Yuasa Y, Shiozuka M, Nonomura Y, Matsuda R. Sphingosine-1-phosphate mediates epidermal growth factor-induced muscle satellite cell activation. Exp. Cell Res. 2014;326(1):112–124. doi: 10.1016/j.yexcr.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 34.Ogura Y, Hindi SM, Sato S, Xiong G, Akira S, Kumar A. TAK1 modulates satellite stem cell homeostasis and skeletal muscle repair. Nat. Commun. 2015;6:10123. doi: 10.1038/ncomms10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, Sartorelli V, Puri PL. TNF/p38alpha/polycomb signaling to pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell. 2010;7(4):455–469. doi: 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem. J. 2009;418(3):475–489. doi: 10.1042/BJ20082234. [DOI] [PubMed] [Google Scholar]
- 37.Pawlikowski B, Orion Vogler T, Gadek K, Olwin B. Regulation of skeletal muscle stem cells by fibroblast growth factors. Dev. Dyn. 2017 doi: 10.1002/dvdy.24495. [DOI] [PubMed] [Google Scholar]
- 38.Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC, Wilson DH, Frenette J, Rudnicki MA. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014;20(10):1174–1181. doi: 10.1038/nm.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins, and post-transcriptional modifications. J. Cell. Physiol. 2000;185(2):155–173. doi: 10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 40.Rando TA, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 1994;125(6):1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudnicki MA, Le Grand F, McKinnell I, Kuang S. The molecular regulation of muscle stem cell function. CSH Symp. Quant. Biol. 2008;73:323–331. doi: 10.1101/sqb.2008.73.064. [DOI] [PubMed] [Google Scholar]
- 42.Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, Jones DR, Du K, Jhala US, Simone C, Puri PL. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol. Cell. 2007;28(2):200–213. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tidball JG. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 2017;17(3):165–178. doi: 10.1038/nri.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, Sacco A. Stat3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014;20(10):1182–1186. doi: 10.1038/nm.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wales S, Hashemi S, Blais A, McDermott JC. Global MEF2 target gene analysis in cardiac and skeletal muscle reveals novel regulation of DUSP6 by p38MAPK-MEF2 signaling. Nucleic Acids Res. 2014;42(18):11349–11362. doi: 10.1093/nar/gku813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao F, Wang H, Fu X, Li Y, Ma K, Sun L, Gao X, Wu Z. Oncostatin m inhibits myoblast differentiation and regulates muscle regeneration. Cell Res. 2011;21(2):350–364. doi: 10.1038/cr.2010.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013;93(1):23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.