

Abstract
Introduction
The goal of this study was to investigate how concurrent shear stress and tensile strain affect endothelial cell biomechanical responses.
Methods
Human coronary artery endothelial cells were exposed to concurrent pulsatile shear stress and cyclic tensile strain in a programmable shearing and stretching device. Three shear stress–tensile strain conditions were used: (1) pulsatile shear stress at 1 Pa and cyclic tensile strain at 7%, simulating normal stress/strain conditions in a healthy coronary artery; (2) shear stress at 3.7 Pa and tensile strain at 3%, simulating pathological stress/strain conditions near a stenosis; (3) shear stress at 0.7 Pa and tensile strain at 5%, simulating pathological stress/strain conditions in a recirculation zone. Cell morphology was quantified using immunofluorescence microscopy. Cell surface PECAM-1 phosphorylation, ICAM-1 expression, ERK1/2 and NF-κB activation were measured using ELISA or Western blot.
Results
Simultaneous stimulation from pulsatile shear stress and cyclic tensile strain induced a significant increase in cell area, compared to that induced by shear stress or tensile strain alone. The combined stimulation caused significant increases in PECAM-1 phosphorylation. The combined stimulation also significantly enhanced EC surface ICAM-1 expression (compared to that under shear stress alone) and transcriptional factor NF-κB activation (compared to that under control conditions).
Conclusion
Pulsatile shear stress and cyclic tensile strain could induce increased but not synergistic effect on endothelial cell morphology or activation. The combined mechanical stimulation can be relayed from cell membrane to nucleus. Therefore, to better understand how mechanical conditions affect endothelial cell mechanotransduction and cardiovascular disease development, both shear stress and tensile strain need to be considered.
Electronic supplementary material
The online version of this article (10.1007/s12195-019-00585-0) contains supplementary material, which is available to authorized users.
Keywords: Shear stress, Tensile strain, Endothelial cells, Mechanotransduction, Shearing–stretching device
Introduction
Every year about 720,000 people suffer from heart attacks, and nearly 380,000 people die from coronary artery disease.27 Atherosclerosis, a major cause for coronary artery disease, is a process that gradually hardens and narrows the arteries. The presence of the atherosclerotic plaque can change the mechanical environment, especially local shear stress and tensile strain, within the coronary arteries greatly.
It is well established that blood flow-induced shear stress plays important roles in hemostasis. In most blood vessels, blood flow is laminar, which helps to maintain the anti-inflammatory and anti-thrombotic properties of the endothelium.86 Altered shear stress, caused by changes in blood vessel geometry (such as bifurcation), large blood vessel motion (such as the bending motion of the coronary arteries), or disease conditions (such as atherosclerosis), can affect the normal functions of vascular wall endothelial cells (EC) greatly. Physiological flow and shear stress make EC align their cytoskeleton proteins with the blood flow direction to reduce the effective resistance of the flow.15 Pathological flow and shear stress can cause EC activation.31 Activated EC can change their phenotype from anti-thrombotic to pro-thrombotic, promoting platelet adhesion. Many have reported that low pathological shear stress can cause EC inflammation, followed by reduced nitric oxide production, increased LDL oxidation and apoptosis.8,20,33
Blood pressure exerted on the blood vessel wall can cause blood vessels to stretch. Physiological stretch has been reported to be athero-protective,58 while excessive stretch can increase EC permeability5 and superoxide production,83 causing inflammation.
EC are sensitive to their mechanical environment, and multiple mechano-sensors are involved in shear stress– and/or tensile stretch-induced EC biomechanical responses, such as integrins, PECAM-1, VE-cadherin, and VEGF receptor 2.75 Even though there are common mechano-sensors that can be activated by both shear stress and tensile stretch, it is generally accepted that these two forms of mechanical stimulations cause EC biomechanical responses through different pathways. Among many mechano-transduction pathways, shear stress is known to act through PECAM-1 phosphorylation, while tensile strain is thought to induce direct changes in actin polymerization in cytoskeleton and extracellular matrix.49 The activation of EC junctional mechano-transducers (such as PECAM-1, VE-cadherin, VEGF receptors) is known to activate Src, Rho, and PI3K, which can mediate cytoskeletal remodeling and basal integrin activation, and eventually result in endothelial alignment change (to flow direction) and nuclear adaptation.17,50 Changes in transcriptional factors (such as NF-κB, which plays important roles in inflammatory regulation) affect EC phenotype (pro-atherogenic or anti-atherogenic). Pedrigi et al. reported that normal-level (8%) multidirectional tensile stretch can significantly reduce NF-κB activation in EC, compared to pathological low stretch (2%) or no stretch.58 It has been shown that low stretch (1%) can lead to increased oxygen species production in porcine carotid artery,73 while large strain (> 9%) can enhance ICAM-1 (intercellular adhesion molecule-1) expression,10 causing apoptosis41,61 and many inflammatory responses.21 On the other hand, many studies have shown that fluid shear stress can transiently activate NF-κB26; and pathological low shear stress can cause significant increase in EC NF-κB activation for a longer duration.54
It’s evident that both fluid shear stress and tensile strain can affect vascular EC mechano-transduction, leading to various pathophysiological responses. Moore et al. used calf pulmonary artery endothelial cells to study how shear stress and tensile hoop strain affected cell morphology (aspect ratio and alignment). They found that shear stress at 8 ± 4 dyne/cm2 significantly increased endothelial cell aspect ratio and alignment (to the flow/shear stress direction), which were further increased when a 10% tensile hoop strain was applied at the same time.55 Breen et al. reported that shear stress at 25 dyne/cm2 could cause ICAM-1 upregulation on human umbilical vein endothelial cells (HUVEC), which was downregulated by the addition of cyclic tensile hoop strain between 4 and 12%.6 Tarbell’s group reported that stenosis in a blood vessel can alter the stress phase angle (i.e., temporal phase angle) between vascular wall fluid shear stress and circumferential strain. They observed that a large negative stress phase angle (such as that in the coronary artery, which is about -180°) could induce proatherogenic gene expression and attenuate NO production, compared to synchronous shear stress and tensile strain.1,2,14 Zhao et al. developed an apparatus to apply concurrent shear stress and hoop stretch to bovine aortic endothelial cells, and reported that shear stress (2–6 dynes/cm2) and hoop stretch (2–5%) could synergistically induce morphological changes (cytoskeleton) in endothelial cells, and endothelial cell sensitivity to shear stress was modulated by circumferential strain.87
Results obtained from the above-mentioned shear stress–tensile strain studies were not consistent. Overall, how concurrent shear stress and tensile strain affect EC mechanotransduction was under investigated. We recently developed a programmable shearing–stretching device that can apply pulsatile shear stress and cyclic tensile strain concurrently to cultured EC monolayer.51 We hypothesized that pulsatile shear stress and cyclic tensile strain had a synergistic effect on endothelial cell activation, and the combined mechanical signal could be relayed from cell membrane to nucleus, and cause transcriptional changes. Using the programmable shearing–stretching device, how shear stress and tensile strain collectively affect EC morphology, ICAM-1 expression (EC activation), PECAM-1 phosphorylation (mechano-transduction), ERK1/2 (MAPK pathway activation) and NF-κB activation (transcriptional changes), was investigated, to test our hypothesis.
Materials and Methods
Endothelial Cell Culture
Human coronary artery endothelial cells (HCAEC) were purchased from ScienCell Research Laboratory (Carlsbad, CA). EC were maintained in endothelial cell culture medium supplemented with 5% fetal bovine serum, endothelial cell growth factors, and penicillin/streptomycin.53 All cell culture media and reagents were purchased from ScienCell Research Laboratory and used as recommended by the vendor. Cells were grown to confluence in 6-well Flexcell plates pre-coated with fibronectin (2 µg/mL, Sigma Aldrich, St. Louis, MO). The Flexcell plates were purchased from Flexcell International Corporation (Burlington, NC) and the well bottom material was silicone elastomer. EC were used between passages 2 and 7. EC were seeded at the same seeding density of 1000 cells/mm2 (about 600,000 cells per well) and used immediately when the cells reached confluence in a monolayer (The average density of untreated confluent EC in Flexcell wells was 2685 ± 180 cells/mm2, mean ± standard deviation, n = 17).
The Shearing–Stretching Device, Pulsatile Shear Stress, and Cyclic Tensile Strain
A custom-designed shearing–stretching device was used to apply concurrent fluid shear stress and tensile strain to EC monolayer (Fig. 1). This device was developed based on a cone-and-plate shearing device and a vacuum-driven membrane stretching system. The design and operation of the device was reported previously.51 It can generate dynamic fluid shear stress and tensile strain simultaneously in a 6-well Flexcell plate. As illustrated in Fig. 1, circular-shaped supporting plates were placed underneath the flexible membrane. The area of the membrane that was in contact with the supporting plate is referred to as the “in-plane area.” Fibronectin was coated in the “in-plane area” of the membrane for endothelial cells to grow. When vacuum pressure was applied, the membrane was pulled and stretched, causing tensile strain in the endothelial cells grown on the in-plane area of the membrane. It was demonstrated in our previous report that the device can produce uniform biaxial strain.51 Shear flow/stress was generated by a rotating cone sitting above the cell monolayer in the well. Both the cone speed and vacuum pressure (magnitude, frequency, duration, etc.) were controlled by a computer. The fluid shear stress and tensile strain waveforms used in the present study were obtained from a fluid structure interaction model (FSI) of a human left coronary artery.52 Incorporating patient specific geometry, transient blood flow, cyclic bending motion of the artery, and myocardial contraction, this numerical model effectively estimated dynamic fluid shear stress and tensile strain on the vascular wall of a left coronary artery during one cardiac cycle, under physiological and pathological conditions. Tensile strains calculated using this numerical model included the axial tensile strain (induced by the bending motion of left coronary artery as the heart contracts and relaxes) and the circumferential tensile strain (due to changes in blood pressure). During diastole, the magnitudes of axial strain and circumferential strain were similar; while during systole, the magnitudes of axial strain and circumferential strain could be quite different (the difference could be as large as 4–5 folds). Since our shearing–stretching device can only generate equal-axial tensile strain, it could not be used to replicate the real strain conditions (axial strain and circumferential strain) within the left coronary artery. Instead, in the present study, only coronary artery circumferential tensile strain was applied to the cultured endothelial cells. As depicted in Fig. 2, three shear stress–tensile strain (circumferential strain) waveforms were generated using the shearing–stretching device to simulate different physiological and pathological shear stress/tensile strain conditions: (1) pulsatile shear stress varying between 0 and 1 Pa16,53,66,69 and cyclic tensile strain varying between 0 and 7%, simulating normal shear stress/tensile strain conditions in a healthy coronary artery; (2) pulsatile shear stress varying between 0 and 3.7 Pa42,66 and cyclic tensile strain between 0 and 3%, simulating pathological shear stress/tensile strain conditions occurring at the stenosis throat (70%) in a diseased coronary artery; (3) pulsatile shear stress varying between 0 and 0.7 Pa66 and cyclic tensile strain varying between 0 and 5%, simulating pathological shear stress/tensile strain conditions in the recirculation zones developed downstream of a stenosis. One cardiac cycle lasted for 0.9 s. Two shear stress peaks occurred in one cardiac cycle. The first peak was due to the increased blood flow rate during diastole, and the second peak was due to myocardial contraction during systole.52,77 The frequency of the cyclic tensile strain was 1.1 Hz (i.e., one tensile strain peak per cardiac cycle).
Figure 1.

The schematic drawing of the shearing and stretching device. Endothelial cells grow to confluence in the in-plane area of the flexible membrane. Vacuum pressure causes the flexible membrane to stretch, generating tensile strain in the endothelial cells. A cone is placed in the well above the membrane. The rotational motion of the cone generates shear stress on the endothelial cell monolayer.
Figure 2.
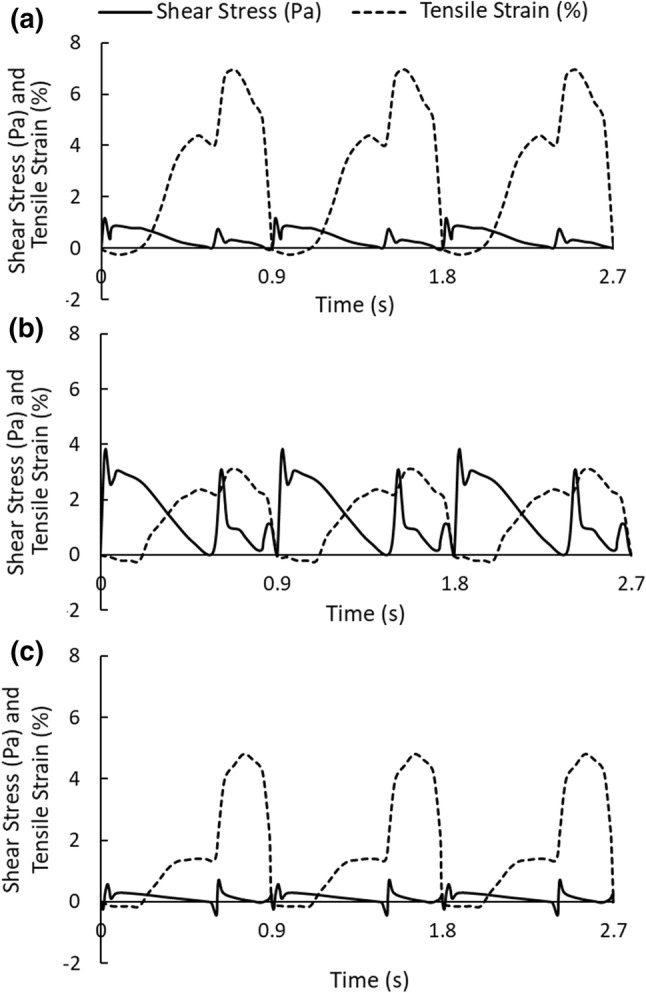
Pulsatile fluid shear stress and cyclic tensile strain waveforms generated using the shearing–stretching device. (a) Normal condition: pulsatile shear stress varied between 0 and 1 Pa and cyclic tensile strain varied between 0 and 7%; (b) Pathological condition occurring at a 70%-stenosis throat: pulsatile shear stress varied between 0 and 3.7 Pa and cyclic tensile strain varied between 0 and 3%; (c) Pathological condition occurring in the recirculation zone downstream of a stenosis: pulsatile shear stress varied between 0 and 0.7 Pa and cyclic tensile strain varied between 0 and 5%. Two shear stress peaks occurred in one cardiac cycle (0.9 s). The first peak was due to the increased blood flow rate during diastole, and the second peak was due to myocardial contraction during systole. The frequency of the cyclic tensile strain was 1.1 Hz.
EC were exposed to concurrent shear stress and/or tensile strain for 1 h53,85 at 37°C. Cells that were not treated (resting cells) were used as the experimental control. Before and after mechanical treatment (shearing, stretching or both), EC within the Flexcell plates were examined under a microscope to ensure that only confluent cells were used, and that mechanical stimulation did not cause any cell loss, i.e., the total number of cells (in a confluent monolayer) remained the same for the follow-up measurements.
EC Morphology
Following shear stress and/or tensile strain treatment, EC were washed with warm phosphate buffered saline (i.e., aspirate, wash, and repeat for three times) and fixed with 0.5% glutaraldehyde (Sigma Aldrich, 15 min at 37 °C). Cell cytoskeleton F-actin filaments were detected by Alexa Fluor 488-conjugated phalloidin using immunofluorescence microscopy (Nikon Ti inverted microscope). Cell area and cell aspect ratio (the ratio between the cell long axis and the short axis) were quantified using the Image J software. Using the polygon selection tool, a cell was selected. The program automatically measured the area (in pixels) surrounded by the polygon, as well as the lengths of the major axis and the minor axis. This information was then used to calculate the cell area (converted from pixels to µm2) and the aspect ratio of the cell (major axis/minor axis). Multiple single cells were selected randomly for each image, and the measured results from multiple cells were averaged for that particular image. To avoid bias, image analysis was conducted by multiple individuals independently.
EC Surface ICAM-1 Expression
Following fluid shear stress and/or tensile strain treatment, HCAEC were washed and fixed. Cell surface ICAM-1 expression was measured using a solid phase ELISA approach. A murine monoclonal anti-human ICAM-1 antibody (Abcam, Cambridge, MA) was used to detect endothelial cell surface ICAM-1 expression. Primary antibody binding was detected using an alkaline phosphatase-conjugated goat anti-mouse secondary antibody (Sigma-Aldrich, St. Louis, MO) and a P-nitrophenyl phosphate substrate. Absorbance was read at 405 nm using a SpectraMax i3 microplate reader (Molecular Devices LLC, Sunnyvale, CA).
EC Surface PECAM-1 Phosphorylation
In a similar manner, following shear stress/tensile strain treatment, EC were washed and fixed. Cell surface PECAM-1 activation (i.e., phosphorylation) was measured using solid phase ELISA. The primary antibody used was a polyclonal rabbit anti human phosphorylated (at tyrosine713) PECAM-1 antibody (Abcam). Primary antibody binding was detected using an alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (Sigma-Aldrich, St. Louis, MO) and a P-nitrophenyl phosphate substrate. Absorbance was read at 405 nm using a microplate reader.
ERK 1/2 (Extracellular Signal-Regulated Kinases 1/2) Expression
After fluid shear stress and/or tensile strain treatment, EC were washed and removed from the culture plate using Trypsin/EDTA (Sigma Aldrich). Each well was checked under a microscope to ensure that all cells were collected. Collected cell solution was centrifuged at 1000 × g for 5 min. Cell pellet was then re-suspended in lysis buffer containing Triton X-100, protease and phosphatase inhibitors (Life Technologies, Carlsbad, CA), and incubated at 4 °C for 1 h. Total protein concentration of the lysate was quantified using a standard BCA (bicinchoninic acid) assay (Thermo Scientific, Waltham, MA). EC ERK 1/2 activation was measured by Western blot, using a monoclonal rabbit anti human phosphor-p44/42(ERK1/2) antibody (Cell Signaling Technology Inc., Danvers, Massachusetts). Primary antibody binding was detected using an EU-conjugated goat anti-rabbit secondary antibody (Molecular Devices). β-actin was used as the loading control. Expressed protein bands were visualized and quantified using the SpectraMax®i3 ScanLater™ Western blot detection system/software (Molecular Devices). For statistical analysis, the raw band intensity data obtained from the scanner was normalized to that of untreated control cells.
NF-κB Activation
To determine if transcriptional changes would occur in shear stress- and/or tensile strain-treated EC, NF-κB activation was measured using a NF-κB pathway activation ELISA kit (eBioscience). It measured IκB proteins (phosphor-IκBa, IKKa and NFκBa) present in the cell lysate, using a sandwich approach. When NF-κB is inactive, IκB proteins bind to NF-κB transcription factor complex; upon extracellular stimulation such as shear stress and tensile strain, IκB phosphorylates and dissociates from the complex, resulting in NF-κB activation and translocation to the nucleus.
Statistical Analysis
Statistical analysis was conducted using Microsoft Excel and SAS (9.3) Statistical Software. Two-way ANOVA was used to detect significant differences (p < 0.05) between treatment groups. Least square means post hoc test was conducted for further comparison when significant difference was detected.
Results
Shear stress and tensile strain conditions used to treat EC were validated experimentally (using tracers) before any experiments were conducted.51 Following shear stress and/or tensile strain treatment, EC were examined under a microscope to ensure that EC remained attached to the bottom of the Flexcell plate as a confluent monolayer.
EC Morphology
Following shear stress/tensile strain treatment, EC cytoskeleton F-actin filaments were detected by Alexa Fluor 488-conjugated phalloidin using immunofluorescence microscopy. Figure 3 depicts representative EC images under various shear stress and tensile strain conditions. Untreated EC (control, panel d) appeared relatively small and randomly oriented, without distinct cell boundaries. EC that were exposed to pathological low (panel i, 0.7 Pa) or high (panel f, 3.7 Pa) shear stress, alone or combined with pathological (i.e., reduced) tensile strain (panel g, with 3% strain and panel j, with 5% strain), looked enlarged and elongated, with highlighted cell boundaries. Changes in EC area and aspect ratio (ratio between long axis and short axis) induced by shear stress and/or tensile strain were quantified using the Image J software. As depicted in Fig. 4a, untreated cells (control) had an average size of 1979 ± 113 µm2 (mean ± standard error, n = 197). Compared to untreated cells, elevated shear stress alone (3.7 Pa, stenosis condition) significantly increased cell area to 2633 ± 110 µm2 (p < 0.0001, n = 221). Concurrent shear stress and tensile strain also increased cell area compared to untreated control cells (normal: 2418 ± 125 µm2, n = 84, p = 0.0185; stenosis: 3430 ± 176 µm2, n = 128, p < 0.0001; recirculation: 2559 ± 169 µm2, n = 49, p = 0.0628). Tensile stretch alone did not cause any significant changes in cell area when compared to untreated control cells. Changes in cell aspect ratio are depicted in Fig. 4b. Compared to untreated control cells (1.89 ± 0.03, n = 261, mean ± standard error), shear stress alone under all conditions caused significant increase in cell aspect ratio (normal: 2.23 ± 0.08, n = 97, p < 0.0001; stenosis: 2.07 ± 0.05, n = 221, p = 0.0016; recirculation: 2.06 ± 0.06, n = 120, p = 0.009). Tensile strain alone did not cause any significant changes in cell aspect ratio compared to untreated cells. Under concurrent shear stress and tensile strain, cell aspect ratio remained unchanged (normal condition) or decreased, especially under recirculation condition (1.56 ± 0.04, n = 55, p = 0.0004). Significant P values for individual comparisons (within each treatment group) are marked in Fig. 4. P values obtained from two-way ANOVA are listed in Tables 1 and 2. These results indicated that mechanical loading conditions (i.e., normal, stenosis, or recirculation, p < 0.0001 for cell area and p = 0.0426 for cell aspect ratio), type of mechanical stimulation (i.e., fluid shear stress, tensile stretch, or combined, p < 0.0001 for cell area and p < 0.0001 for cell aspect ratio), and the interaction between the two (p < 0.0001 for cell area and p = 0.0405 for cell aspect ratio), all had significant effects on EC area and aspect ratio. The concurrent stimulation from shear stress and tensile strain was significantly different from each single stimulation alone. However, changes in cell morphology (cell area and cell aspect ratio) induced by the concurrent stimulation was less than the sum of that induced by each individual stimulation.
Figure 3.

Representative images of EC following shear stress and tensile strain treatment. Untreated EC (panel d) were used as the experimental control. Scale bars represent 50 μm. (a) EC under normal tensile strain (7%); (b) EC under normal shear stress (1 Pa); (c) EC under normal/physiological tensile strain (7%) and shear stress (1 Pa); (d) untreated EC; (e) EC under pathological low tensile strain (3%); (f) EC under elevated shear stress (3.7 Pa); (g) EC under low tensile strain (3%) and elevated shear stress (3.7 Pa; stenosis condition); (h) EC under slightly reduced tensile strain (5%); (i) EC under pathological low shear stress (0.7 Pa); (j) EC under low tensile strain (5%) and low shear stress (0.7 Pa; recirculation condition). Cell cytoskeleton F-actin filaments were detected by Alexa Fluor 488-conjugated phalloidin.
Figure 4.
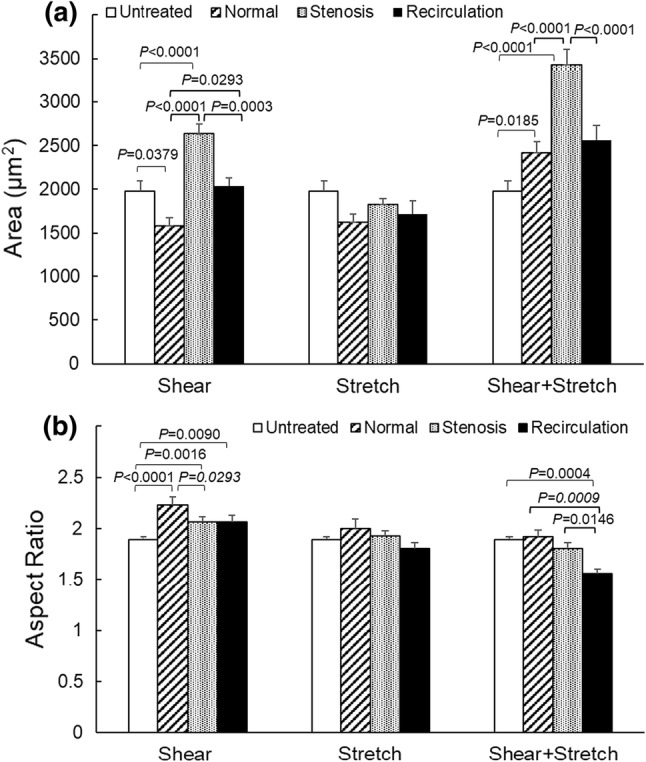
EC area (A) and aspect ratio (B) following shear stress and/or tensile strain treatment. Data is presented as mean + standard error (n = 49–221). Significant differences (p < 0.05, by two-way ANOVA) within each stress/strain treatment group are marked.
Table 1.
Two-way ANOVA was used for statistical analysis of cell area data.
| Normal vs. stenosis | Normal vs. recirculation | Stenosis vs. recirculation |
|---|---|---|
| p<0.0001 | p = 0.2347 | p<0.0001 |
| Shear vs. stretch | Shear vs. shear + stretch | Shear vs. control | Shear vs. shear + stretch | Shear vs. control | Shear + stretch vs. control |
|---|---|---|---|---|---|
| p<0.0031 | p<0.0001 | p = 0.2748 | p<0.0001 | p = 0.0228 | p<0.0001 |
Significant p values (< 0.05) are bolded
Table 2.
Two-way ANOVA was used for statistical analysis of cell aspect ratio data (ratio between long axis and short axis).
| Normal vs. stenosis | Normal vs. recirculation | Stenosis vs. recirculation |
|---|---|---|
| p = 0.0253 | p<0.0001 | p = 0.0253 |
| Shear vs. stretch | Shear vs. shear + stretch | Shear vs. control | Shear vs. shear + stretch | Shear vs. control | Shear + stretch vs. control |
|---|---|---|---|---|---|
| p<0.0001 | p<0.0001 | p<0.0001 | p = 0.0095 | p = 0.5927 | p = 0.0070 |
Significant p values (< 0.05) are bolded
EC Surface ICAM-1 Expression
EC surface ICAM-1 expression following shear stress and/or tensile strain treatment was measured using solid phase ELISA. As depicted in Fig. 5, mechanical loading conditions (normal, stenosis or recirculation, p < 0.0001), type of mechanical stimulation (shear stress, tensile stretch, or combined, p = 0.0144) and the interaction between the two (p < 0.0001) had significant effects on EC surface ICAM-1 expression. Compared to normal shear stress (1 Pa, 0.40 ± 0.05, n = 6, mean ± standard error), both stenosis shear stress (3.7 Pa, 0.54 ± 0.02, n = 5, p = 0.0373) and recirculation shear stress (0.7 Pa, 0.58 ± 0.06, n = 6, p = 0.0047) significantly increased EC surface ICAM-1 expression. Compared to normal tensile strain (7%, 0.29 ± 0.01, n = 7), both stenosis strain (3%, 0.72 ± 0.03, n = 6, p < 0.0001) and recirculation strain (5%, 0.59 ± 0.03, n = 6, p < 0.0001) caused significant increase in EC surface ICAM-1 expression. When shear stress and tensile strain were applied to EC simultaneously, stenosis condition (0.78 ± 0.03, n = 5, p < 0.0001) and recirculation condition (0.64 ± 0.03, n = 5, p < 0.0001) both caused significant increases in EC surface ICAM-1 expression compared to that under normal condition (0.36 ± 0.04, n = 6). Overall, altered shear stress (increased for stenosis condition and decreased for recirculation condition) and/or tensile strain (reduced for both stenosis and recirculation conditions) significantly enhanced EC surface ICAM-1 expression, suggesting cell activation. P values obtained from two-way ANOVA are listed in Table 3. The concurrent stimulation from shear stress and tensile strain caused significant changes in EC surface ICAM-1 expression compared to shear stress alone (p = 0.0275), but not to tensile strain alone (p = 0.0956). Changes in cell surface ICAM-1 expression induced by the concurrent stimulation was less than the sum of that induced by each individual stimulation.
Figure 5.
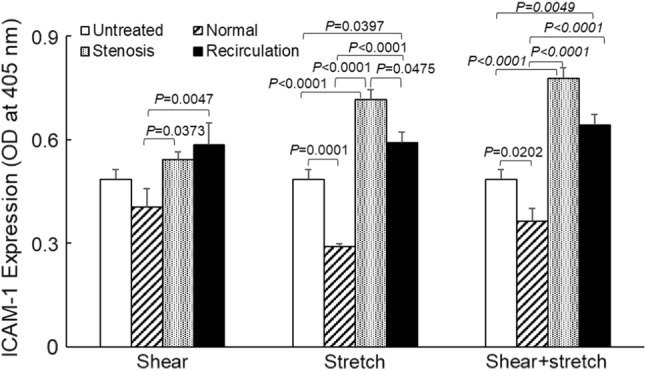
EC surface ICAM-1 expression following shear stress and/or tensile strain treatment. Data is presented as mean + standard error (n = 5–7). Significant differences within each treatment group (p < 0.05, by two-way ANOVA) are marked.
Table 3.
Two-way ANOVA was used for statistical analysis of EC surface ICAM-1 expression data.
| Normal vs. stenosis | Normal vs. recirculation | Stenosis vs. recirculation |
|---|---|---|
| p<0.0001 | p<0.0001 | p = 0.0725 |
| Shear vs. stretch | Shear vs. shear + stretch | Shear vs. control | Shear vs. shear + stretch | Shear vs. control | Shear + stretch vs. control |
|---|---|---|---|---|---|
| p = 0.5320 | p = 0.0275 | p< 0.4072 | p = 0.0956 | p = 0.1027 | p = 0.0006 |
Significant p values (< 0.05) are bolded
EC Surface PECAM-1 Phosphorylation
EC surface PECAM-1 phosphorylation (at tyrosine713) was measured using a solid phase ELISA approach, and the results are depicted in Fig. 6. As indicated by two-way ANOVA, mechanical loading conditions (normal, stenosis or recirculation, p < 0.0001), type of mechanical stimulation (shear stress, tensile stretch, or combined, p < 0.0001), and the interaction between the two (p < 0.0001) had significant effects on EC surface PECAM-1 phosphorylation. Shear stress alone (physiological or pathological) did not induce any significant change in PECAM-1 activation (phosphorylation) compared to untreated control cells. However, under stenosis conditions, tensile stretch alone (0.77 ± 0.05, n = 6, mean ± standard error) reduced PECAM-1 phosphorylation significantly compared to that under normal tensile stretch (1.60 ± 0.19, n = 6, p < 0.0001) or recirculation tensile stretch (1.63 ± 0.24, n = 6, p < 0.0001). When shear stress and tensile strain were applied to EC concurrently, stenosis condition (0.80 ± 0.03, n = 6) also caused significant decrease in PECAM-1 phosphorylation (normal, 2.10 ± 0.12, n = 6, p < 0.0001; recirculation, 1.80 ± 0.34, n = 4, p < 0.0001). While under normal or recirculation conditions, tensile strain, alone or together with shear stress, significantly enhanced EC surface PECAM-1 phosphorylation, compared to untreated EC (1.13 ± 0.05, p = 0.0006 for normal stretch vs. control, p < 0.0001 for combined normal shear and stretch vs. control; p = 0.0003 for recirculation stretch vs. control, p < 0.0001 for combined recirculation shear and stretch vs. control). P values obtained from two-way ANOVA are shown in Table 4. Concurrent shear stress and tensile strain caused significant changes in EC PECAM-1 phosphorylation compared to shear stress (p = 0.0005) or tensile strain (p = 0.0246) alone. However, changes in EC PECAM-1 phosphorylation induced by the concurrent stimulation was less than the sum of that induced by each individual stimulation.
Figure 6.
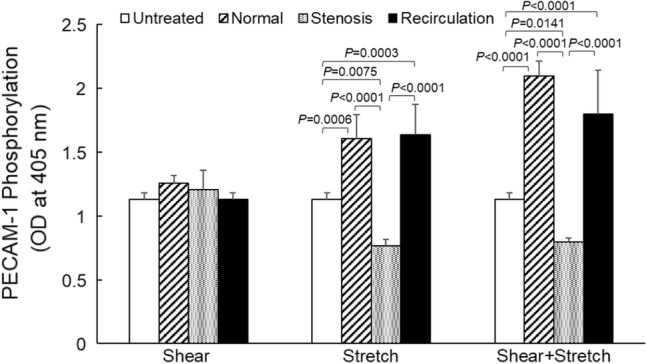
EC surface PECAM-1 phosphorylation following shear stress and/or tensile strain treatment. Data is presented as mean + standard error (n = 4–7). Significant differences within each treatment (p < 0.05, by two-way ANOVA) are marked.
Table 4.
Two-way ANOVA was used for statistical analysis of EC surface PECAM-1 phosphorylation data.
| Normal vs. stenosis | Normal vs. recirculation | Stenosis vs. recirculation |
|---|---|---|
| p<0.0001 | p = 0.2187 | p<0.0001 |
| Shear vs. stretch | Shear vs. shear + stretch | Shear vs. control | Shear vs. shear + stretch | Shear vs. control | Shear + stretch vs. control |
|---|---|---|---|---|---|
| p = 0.1608 | p = 0.0005 | p = 0.4087 | p = 0.0248 | p = 0.0095 | p = 0.0001 |
Significant p values (< 0.05) are bolded
ERK1/2 Activation
For Western blot, following shear stress/tensile strain treatment, cell lysate was prepared using lysis buffer as described previously, and total protein concentration was measured using a standard BCA assay (Supplement Fig. 1). Phosphorylated ERK1/2 expression was measured using Western blot (Fig. 7). Detected band intensity (representative bands are shown in Supplement Fig. 1) was normalized to that of untreated EC. Due to large variances, no significant changes were detected in phosphorylated ERK1/2 expression in EC following shear stress and/or tensile strain treatment, by two-way ANOVA.
Figure 7.
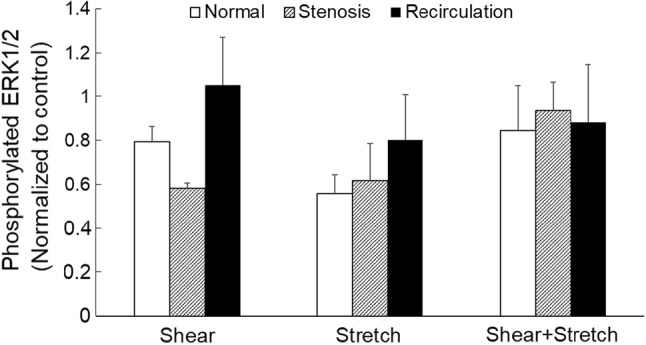
EC phosphorylated ERK1/2 expression following shear stress and/or tensile strain treatment. Data is presented as mean + standard error (n = 4–7). All data was normalized to the matching control.
NF-κB Activation
NF-κB activation was measured using a NF-κB pathway activation ELISA kit. As shown in Fig. 8, different treatment conditions caused significant changes in NF-κB activation (p = 0.0323 by two-way ANOVA). But individually, neither mechanical loading conditions (normal, stenosis or recirculation, p = 0.0551) nor the type of mechanical stimulation (shear, stretch or both, p = 0.1736) had significant effects. Reduced tensile strain alone under recirculation conditions (1.37 ± 0.15, n = 3, mean ± standard error) caused a significant increase in NF-κB activation (p = 0.0325), compared to normal conditions (0.86 ± 0.12, n = 6). When pathological (stenosis and recirculation) shear stress and tensile strain were applied to EC simultaneously, NF-κB activation was significantly enhanced (1.67 ± 0.10, n = 4, p = 0.0026 and 1.40 ± 0.08, n = 5, p = 0.0426 respectively) from that under normal conditions (0.97 ± 0.08, n = 5). When compared to untreated EC (1.13 ± 0.12, n = 11), concurrent shear stress and tensile strain under stenosis conditions caused a significant increase in NF-κB activation (p = 0.0069). P values obtained from two-way ANOVA are shown in Table 5. Concurrent shear stress and tensile strain caused significant changes in NF-κB activation compared to untreated cells (p = 0.0427), which was not seen under shear stress or tensile strain alone. However, EC NF-κB activation induced by the concurrent stimulation was less than the sum of that induced by each individual stimulation.
Figure 8.
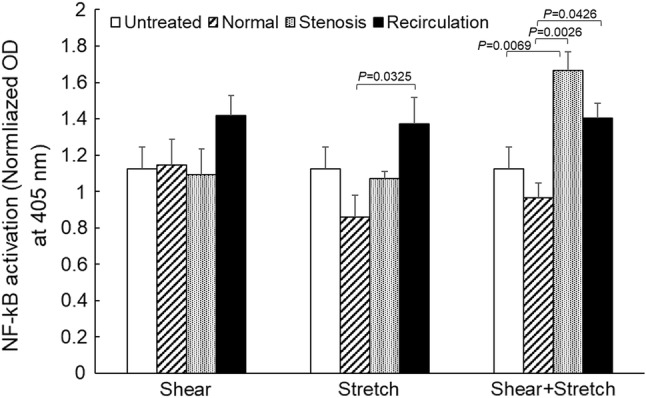
Transcription factor NF-κB activation following shear stress and/or tensile strain treatment. Data is presented as mean + standard error (n = 3–11). All O.D. values were normalized by the total protein concentration. Significant differences (p < 0.05, by two-way ANOVA) within each treatment group are marked.
Table 5.
Two-way ANOVA was used for statistical analysis of EC NF-κB activation data.
| Normal vs. stenosis | Normal vs. recirculation | Stenosis vs. recirculation |
|---|---|---|
| p<0.0285 | p = 0.0038 | p = 0.3916 |
| Shear vs. stretch | Shear vs. shear + stretch | Shear vs. control | Shear vs. shear + stretch | Shear vs. control | Shear + stretch vs. control |
|---|---|---|---|---|---|
| p = 0.3512 | p = 0.3065 | p = 0.3668 | p = 0.0618 | p = 0.8219 | p = 0.0427 |
Significant p values (< 0.05) are bolded
Discussion
EC can respond to altered mechanical conditions by morphology change. The morphology data obtained from this study (Fig. 4 and Tables 1, 2) demonstrated that under all mechanical loading conditions (normal, stenosis and recirculation), cell area increased significantly in response to concurrent shear stress and tensile strain, compared to each single stimulation. However, such changes in cell area were not synergistic. EC can elongate in response to fluid shear stress to decrease resistance to flow.15 In the present study, untreated EC (resting EC without shear stress or tensile strain treatment) were used as the experimental control. The mean aspect ratio of control cells was 1.89 ± 0.0.03, about 10–20% less than that under shear stress (physiological and pathological, in Fig. 4b), matching observations from our previous study (28) and by others.40,44,60 Tensile stretch also caused noticeable changes in cell aspect ratio compared to control cells (Table 1). This trend matches that reported by Russo et al.65 In their study, rabbit aorta endothelial cells were exposed to 5% or 15% tensile strain in a Flexcell tension system; cell length significantly increased under 5% stretch (4 h), and further increased when tensile strain increased to 15%. However, the concurrent stimulation from shear stress and tensile strain did not have a synergistic effect on EC aspect ratio. EC did not become longer when shear stress and tensile strain were applied simultaneously, especially under reduced shear and strain (i.e., recirculation condition).64 These results demonstrated that both shear stress and tensile strain are very important in affecting EC morphology. Even though they are not synergistic, various combinations of shear stress and tensile strain at physiological and pathological levels can significantly alter the size and shape of EC, which are likely to cause changes in EC mechanotransduction and subsequent physiological responses.
Besides morphology change, changes in proteins associated with EC biomechanics12,39,53 and MAPK pathway23,36 (mitogen activated protein kinase) activation were observed in many studies following EC mechanical stimulation, especially by shear stress. PECAM-1 is one of the well-studied mechanotransducers on EC surface. It is highly localized at EC junctions and plays important roles in endothelial mechanotransduction and many inflammatory responses.81 PECAM-1 phosphorylates upon mechanical stimulation.11 As shown in Fig. 6, normal tensile strain (7%) induced a 41.5% increase in EC surface PECAM-1 phosphorylation (compared to untreated EC), which further increased by another 30% when normal pulsatile shear stress was applied simultaneously. However, this trend was not observed under other conditions. Recirculation tensile strain (5%) caused 42% increase in PECAM-1 phosphorylation, which was not further enhanced when recirculation shear stress (0.7 Pa) was applied simultaneously. Under stenosis conditions, neither shear stress (3.7 Pa) nor tensile strain (3%) increased EC surface PECAM-1 phosphorylation (stenosis tensile strain, alone or with stenosis shear stress, significantly reduced PECAM-1 phosphorylation). These results differed from our previous observation that in a cone and plate shearing device, low pulsatile shear stress (0.3 Pa, 1.1 Hz) induced a significant increase in EC PECAM-1 phosphorylation.53 This inconsistency could have resulted from the different shear stress conditions used in the two studies. In the present study, low shear stress used to simulate recirculation conditions had a magnitude of 0.7 Pa and frequency of 2 Hz.52 Figure 6 also suggests that tensile strain near physiological level (7%) had a more dominant effect on EC surface PECAM-1 phosphorylation, compared to shear stress. This could be explained by the fact that stretch applied to EC is transmitted through cell junctions.48,76 PECAM-1 localized at cell junctions is thus directly pulled by forces resulted from tensile stretch. Furthermore, reduced tensile strain at 3%, applied alone or together with pathological high shear stress (3.7 Pa), caused significant decreases in PECAM-1 phosphorylation. This may indicate that pathological low tensile strain could decrease the sensitivity of PECAM-1 as a mechanotransducer to elevated pathological shear stress.
PECAM phosphorylation can lead to MAPK pathway activation,19,72 which can regulate many cellular processes such as growth, proliferation, differentiation, motility, survival, apoptosis, stress response and inflammation.59 The members of the MAPK family, i.e., extracellular signal-regulated kinase (ERK1/2), N-terminal Jun kinase (JNK1/2) and p38 MAPK, are often involved in shear- and cyclic stretch-induced EC inflammation.34,43,46,47,82 As depicted in Fig. 7, EC that were subject to shear stress and/or tensile strain did not have any significant changes in phosphor-ERK1/2 expression compared to control cells. This might be due to the transient nature of ERK 1/2 phosphorylation.35,45,74 It has been reported that under larger cyclic stretch (20–30%), ERK activation peaks between 10 and 15 min13,79; and under shear stress, it takes about 30 min.3 In the present study, ERK1/2 activation was measured 1 h following shear stress/tensile strain treatment, at which point, we suspect EC might have adapted to the mechanical loading conditions. In other words, we might have missed the transient change in ERK1/2 activation by conducting the measurements at 1 h. In order to capture the transient changes in ERK1/2 activation, measured time-response will be needed.
Nevertheless, significant changes were detected in downstream transcription factor NF-κB activation. As demonstrated in Fig. 8, the concurrent application of pathological shear stress and tensile strain caused significant increases in NF-κB activation (compared to normal conditions). This is in agreement with the in vivo study reported by Hejira et al. that NF-κB signal transduction pathway in coronary artery endothelial cells was activated in regions predisposed to atherosclerotic lesion formation.24 These results also highlight the importance of considering both shear stress and tensile strain regarding NF-κB activation. One discrepancy we noticed was that pathological shear stress (stenosis or recirculation) alone did not cause any significant changes in NF-κB activation compared to normal shear stress. This is different from literature that shear stress can cause marked changes in NF-κB activation.28,38,57 This could be due to the relatively short duration of mechanical stimulation in the present study (1 h vs. 8, 12 or 24 h in other studies). In our future studies, we will need to increase the duration of mechanical stimulation to address this limitation.
NF-κB activation can induce cytokine and adhesion molecule expression, including ICAM-1.63 As shown in Fig. 5, pathological shear stress and tensile strain, alone or combined, significantly increased EC surface ICAM-1 expression compared to normal stress/strain conditions, in agreement with that has been reported.7,10,22,71,78 Many studies have suggested that enhanced PECAM-1 phosphorylation can increase ICAM-1 expression.25,75 However, in the present study, no direct correlation between PECAM-1 phosphorylation and ICAM-1 expression was observed. Under normal shear stress and tensile strain, PECAM-1 phosphorylation increased but ICAM-1 expression did not; under stenosis shear and strain, however, PECAM-1 phosphorylation was low while ICAM-1 expression increased significantly. Trend observed with NF-κB activation did not match that of either PECAM-1 or ICAM-1, indicating that NF-κB activation and EC activation (enhanced ICAM-1 expression) are not likely resulted from PECAM-1 phosphorylation. Other mechanotransducers are likely involved in EC response to shear stress and/or cyclic strain signals.72
It is also interesting to notice that in most of the measurements conducted in this study (EC morphology, PECAM-1 phosphorylation, NF-κB activation and ICAM-1 expression), two-way ANOVA suggested both the type of mechanical stimulation (shear stress, tensile strain, or combined) and the measure of the mechanical stimulation (1 Pa and 7% for normal condition, 3.7 Pa and 3% for stenosis condition, and 0.7 Pa and 5% for recirculation condition) had significant effects. Reduced tensile strain, low shear stress or elevated shear stress can all trigger changes in EC morphology, mechanotransduction and inflammatory protein expression. Shear stress and tensile strain are equally important in the above-mentioned responses. However, their effects are not synergistic, different from what we have hypothesized.
This study has a few limitations. Under physiological conditions, most blood vessels are only exposed to circumferential (uniaxial) strain. Coronary arteries undergo large bending motion due to contraction/relaxation of the heart. Therefore, coronary arteries are exposed to not only circumferential strain but also axial strain. As reported previously,52 the magnitudes of coronary artery axial strain and circumferential strain could be quite different, especially during systole. The shearing–stretching device used in this study can only generate uniform biaxial strain, which was not sufficient to accurately replicate the strain conditions (axial and circumferential) within the left coronary artery. Due to the limitation in our shearing–stretching device, only circumferential tensile strain was applied to EC in the present study.
Endothelial cells were treated with shear stress and/or tensile strain for 1 h, which may not be the best treatment duration to catch all the changes that can occur. There is no agreement on how long endothelial cells should be treated with shear stress or tensile strain to cause detectable biomechanical responses. A study by Yamamoto and Ando reported that endothelial cell membrane can respond to shear stress and tensile stretch within minutes.84 Chatterjee et al. reported that when bovine endothelial cells were exposed to laminar flow, cell boundaries altered within 5 min.9 Seko et al. reported that pulsatile stretch at 15% could cause MAPK pathway protein activation within 5 min in cultured rat cardiac myocytes.68 Hurley et al. reported that 20% cyclic stretch caused a significant increase in c-Myc gene expression in cultured HUVEC within 1–2 h.32 Previously, we reported morphological changes in human coronary artery endothelial cells induced by shear stress and/or tensile strain within 30 min of mechanical stimulation.51 However, these may all be transient responses. Steady-state morphological responses to mechanical stimulation could take 24 h or longer to occur.44,62 In the present study, EC morphological changes (size and aspect ratio), ICAM-1 expression, PECAM-1 phosphorylation, and NF-κB activation were observed after 1 h of shear stress/tensile strain application, suggesting that human coronary artery endothelial cells can sense the mechanical stimulation and respond accordingly. However, as mentioned previously, the 1-h shearing–stretching time may be too long to catch the transient changes in ERK1/2 phosphorylation, but too short to detect changes in NF-κB activation. Therefore, time response studies (15 min, 30 min, 2 h, 4 h, and up to 24 h) will be needed to evaluate short-term and long-term changes induced by shear stress and/or tensile strain. To measure ERK1/2 activation, EC were taken off from the 6-well plates and lysed in lysis buffer before Western blot. Taking adherent cells from the plate using Trypsin-ETDA could affect ERK1/2 phosphorylation state. Therefore, directly lysing EC from the 6-well plate without trypsinizing could be a helpful alternative approach.
In the present study, shear stress and tensile strain with different magnitudes were used to simulate physiological and pathological mechanical conditions. How variations in other factors, such as stimulation frequency, duration and shape of the waveform, on cellular response were not investigated. It has been reported that oscillatory low shear stress at a higher frequency is more likely to induce endothelial cell inflammatory responses.67 Frequency of tensile strain can also significantly affect stress fiber (F-actin) distribution in endothelial cells.30 Flow velocity acceleration (which affects the shape of shear stress) has been reported to have a significant impact on endothelial cell morphology and mechanotransduction.29,70,80 Therefore, frequency and perturbation of mechanical stimulation may need to be investigated in the future studies.
In the present study, only confluent EC were used. However, in each Flexcell well, confluent EC density could still vary (EC were seeded at 1000 cells/mm2; when they reached confluence, the average density increased to 2685 ± 180 cells/mm2). In literature, different seeding densities were reported for Flexcell culture. Morrow et al. grew HUVEC in 6-well Flexcell plates and reported a seeding density of 200,000 cells/well (~ 208 cells/mm2).56 Juan el al also used HUVEC, but they seeded cells at 20,000/well (~ 21 cells/mm2).37 When bovine aortic endothelial cells were used, both Azuma et al. and Fujoka et al. seeded cells at approximately 400 cells/mm2.4,18 However, the exact cell density at confluence in those studies was not reported. It is important to be aware that confluent cell density could vary due to cell type, culture substrate, and cell passage, etc., and the variation in total cell numbers could have had an impact on the measured experimental results. To rule out cell number-induced effects, in the future studies, the total numbers of cells before and after each experiment will need to be counted and taken into consideration when results are analyzed.
Overall, our results demonstrated that EC respond to combined shear stress and tensile strain differently than to each single simulation. Concurrent shear stress and tensile strain signal at physiological and pathological levels can be relayed from cell membrane to nucleus, and cause significant changes in EC morphology (cell area and aspect ratio) and biomechanical responses (ICAM-1 expression, PECAM-1 phosphorylation, and NF-κB activation). However, shear stress and tensile strain did not have a synergistic effect on EC biomechanical responses, as we hypothesized originally. To investigate how altered mechanical conditions affect cardiovascular disease initiation and development, both shear stress and tensile strain need to be considered.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
Funding
This study was in part supported by an American Heart Association Grant in Aid Award (16GRNT30440002).
Conflict of Interest
None for Daphne Meza, Bryan Musmacker, Elisabeth Steadman, Thomas Stransky, David A. Rubenstein, and Wei Yin.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Amaya R, Cancel LM, Tarbell JM. Interaction between the stress phase angle (SPA) and the oscillatory shear index (OSI) affects endothelial cell gene expression. PLoS ONE. 2016;11(11):e0166569. doi: 10.1371/journal.pone.0166569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaya R, Pierides A, Tarbell JM. The interaction between fluid wall shear stress and solid circumferential strain affects endothelial gene expression. PLoS ONE. 2015;10(7):e0129952. doi: 10.1371/journal.pone.0129952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azuma N, Duzgun SA, Ikeda M, Kito H, Akasaka N, Sasajima T, Sumpio BE. Endothelial cell response to different mechanical forces. J. Vasc. Surg. 2000;32(4):789–794. doi: 10.1067/mva.2000.107989. [DOI] [PubMed] [Google Scholar]
- 4.Azuma N, Duzgun SA, Ikeda M, Kito H, Akasaka N, Sasajima T, Sumpio BE. Endothelial cell response to different mechanical forces. J. Vasc. Surg. 2000;32(4):789–794. doi: 10.1067/mva.2000.107989. [DOI] [PubMed] [Google Scholar]
- 5.Birukova AA, Chatchavalvanich S, Rios A, Kawkitinarong K, Garcia JG, Birukov KG. Differential regulation of pulmonary endothelial monolayer integrity by varying degrees of cyclic stretch. Am. J. Pathol. 2006;168(5):1749–1761. doi: 10.2353/ajpath.2006.050431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Breen LT, McHugh PE, Murphy BP. Multi-axial mechanical stimulation of HUVECs demonstrates that combined loading is not equivalent to the superposition of individual wall shear stress and tensile hoop stress components. J. Biomech. Eng. 2009;131(8):081001. doi: 10.1115/1.3127248. [DOI] [PubMed] [Google Scholar]
- 7.Breen LT, McHugh PE, Murphy BP. HUVEC ICAM-1 and VCAM-1 synthesis in response to potentially athero-prone and athero-protective mechanical and nicotine chemical stimuli. Ann. Biomed. Eng. 2010;38(5):1880–1892. doi: 10.1007/s10439-010-9959-8. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB. Membrane depolarization is the trigger for PI3 K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012;302(1):H105–114. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chatterjee S, Fujiwara K, Perez NG, Ushio-Fukai M, Fisher AB. Mechanosignaling in the vasculature: emerging concepts in sensing, transduction and physiological responses. Am. J. Physiol. Heart Circ. Physiol. 2015;308(12):H1451–1462. doi: 10.1152/ajpheart.00105.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain enhances adhesion of monocytes to endothelial cells by increasing intercellular adhesion molecule-1 expression. Hypertension. 1996;28(3):386–391. doi: 10.1161/01.HYP.28.3.386. [DOI] [PubMed] [Google Scholar]
- 11.Chiu, Y.-J. Identification of the kinase for flow-and stretch-elicited phosphorylation of platelet endothelial cell adhesion molecule-1 in endothelial cells. Thesis (Ph.D.), University of Rochester. School of Medicine and Dentistry. Dept. of Biochemistry and Biophysics, 2008. http://hdl.handle.net/1802/6626.
- 12.Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr. Biol. 2013;23(11):1024–1030. doi: 10.1016/j.cub.2013.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Correa-Meyer E, Pesce L, Guerrero C, Sznajder JI. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;282(5):L883–891. doi: 10.1152/ajplung.00203.2001. [DOI] [PubMed] [Google Scholar]
- 14.Dancu MB, Tarbell JM. Large Negative Stress Phase Angle (SPA) attenuates nitric oxide production in bovine aortic endothelial cells. J. Biomech. Eng. 2006;128(3):329–334. doi: 10.1115/1.1824120. [DOI] [PubMed] [Google Scholar]
- 15.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J. Biomech. Eng. 1981;103(3):177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 16.Doriot PA, Dorsaz PA, Dorsaz L, De Benedetti E, Chatelain P, Delafontaine P. In-vivo measurements of wall shear stress in human coronary arteries. Coron. Artery Dis. 2000;11(6):495–502. doi: 10.1097/00019501-200009000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Dorland YL, Huveneers S. Cell–cell junctional mechanotransduction in endothelial remodeling. Cell. Mol. Life Sci. 2017;74(2):279–292. doi: 10.1007/s00018-016-2325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujioka K, Azuma N, Kito H, Gahtan V, Esato K, Sumpio BE. Role of caveolin in hemodynamic force-mediated endothelial changes. J. Surg. Res. 2000;92(1):7–10. doi: 10.1006/jsre.2000.5838. [DOI] [PubMed] [Google Scholar]
- 19.Fujiwara K. Platelet endothelial cell adhesion molecule-1 and mechanotransduction in vascular endothelial cells. J. Intern. Med. 2006;259(4):373–380. doi: 10.1111/j.1365-2796.2006.01623.x. [DOI] [PubMed] [Google Scholar]
- 20.Gambillara V, Chambaz C, Montorzi G, Roy S, Stergiopulos N, Silacci P. Plaque-prone hemodynamics impair endothelial function in pig carotid arteries. Am. J. Physiol. Heart Circ. Physiol. 2006;290(6):H2320–2328. doi: 10.1152/ajpheart.00486.2005. [DOI] [PubMed] [Google Scholar]
- 21.Gawlak G, Son S, Tian Y, O’Donnell JJ, 3rd, Birukov KG, Birukova AA. Chronic high-magnitude cyclic stretch stimulates EC inflammatory response via VEGF receptor 2-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016;310(11):L1062–1070. doi: 10.1152/ajplung.00317.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golledge J, Turner RJ, Harley SL, Springall DR, Powell JT. Circumferential deformation and shear stress induce differential responses in saphenous vein endothelium exposed to arterial flow. J. Clin. Invest. 1997;99(11):2719. doi: 10.1172/JCI119461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gulino-Debrac D. Mechanotransduction at the basis of endothelial barrier function. Tissue Barriers. 2013;1(2):e24180. doi: 10.4161/tisb.24180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. 2000;97(16):9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hashimoto K, Kataoka N, Nakamura E, Tsujioka K, Kajiya F. Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis. 2007;194(2):e9–e17. doi: 10.1016/j.atherosclerosis.2006.11.029. [DOI] [PubMed] [Google Scholar]
- 26.Hay DC, Beers C, Cameron V, Thomson L, Flitney FW, Hay RT. Activation of NF-kappaB nuclear transcription factor by flow in human endothelial cells. Biochim. Biophys. Acta. 2003;1642(1–2):33–44. doi: 10.1016/S0167-4889(03)00084-3. [DOI] [PubMed] [Google Scholar]
- 27.Heart Disease Facts American Heart Association. Heart Disease and Stroke Update 2015. Cited 2 February 2016. http://www.theheartfoundation.org/heart-disease-facts/heart-disease-statistics/.
- 28.Heuslein JL, Meisner JK, Li X, Song J, Vincentelli H, Leiphart RJ, Ames EG, Blackman BR, Blackman BR, Price RJ. Mechanisms of amplified arteriogenesis in collateral artery segments exposed to reversed flow direction. Arterioscler. Thromb. Vasc. Biol. 2015;35(11):2354–2365. doi: 10.1161/ATVBAHA.115.305775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiai TK, Cho SK, Honda HM, Hama S, Navab M, Demer LL, Ho CM. Endothelial cell dynamics under pulsating flows: significance of high versus low shear stress slew rates (d(tau)/dt) Ann. Biomed. Eng. 2002;30(5):646–656. doi: 10.1114/1.1484222. [DOI] [PubMed] [Google Scholar]
- 30.Hsu HJ, Lee CF, Kaunas R. A dynamic stochastic model of frequency-dependent stress fiber alignment induced by cyclic stretch. PLoS ONE. 2009;4(3):e4853. doi: 10.1371/journal.pone.0004853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hunt BJ, Jurd KM. Endothelial cell activation. A central pathophysiological process. BMJ. 1998;316(7141):1328–1329. doi: 10.1136/bmj.316.7141.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurley NE, Schildmeyer LA, Bosworth KA, Sakurai Y, Eskin SG, Hurley LH, McIntire LV. Modulating the functional contributions of c-Myc to the human endothelial cell cyclic strain response. J. Vasc. Res. 2010;47(1):80–90. doi: 10.1159/000235928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, Hsiai TK. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ. Res. 2003;93(12):1225–1232. doi: 10.1161/01.RES.0000104087.29395.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jalali S, Li Y-S, Sotoudeh M, Yuan S, Li S, Chien S, Shyy JY. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998;18(2):227–234. doi: 10.1161/01.ATV.18.2.227. [DOI] [PubMed] [Google Scholar]
- 35.Jo H, Sipos K, Go YM, Law R, Rong J, McDonald JM. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal Jun kinase in endothelial cells. Gi2- and Gbeta/gamma-dependent signaling pathways. J. Biol. Chem. 1997;272(2):1395–1401. doi: 10.1074/jbc.272.2.1395. [DOI] [PubMed] [Google Scholar]
- 36.Johnson BD, Mather KJ, Wallace JP. Mechanotransduction of shear in the endothelium: basic studies and clinical implications. Vasc. Med. 2011;16(5):365–377. doi: 10.1177/1358863X11422109. [DOI] [PubMed] [Google Scholar]
- 37.Juan SH, Chen JJ, Chen CH, Lin H, Cheng CF, Liu JC, Hsieh MH, Chen YL, Chao HH, Chen TH, Chan P, Cheng TH. 17beta-estradiol inhibits cyclic strain-induced endothelin-1 gene expression within vascular endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004;287(3):H1254–1261. doi: 10.1152/ajpheart.00723.2003. [DOI] [PubMed] [Google Scholar]
- 38.Keegan PM, Anbazhakan S, Kang B, Pace BS, Platt MO. Biomechanical and biochemical regulation of cathepsin K expression in endothelial cells converge at AP-1 and NF-kappaB. Biol. Chem. 2016;397(5):459–468. doi: 10.1515/hsz-2015-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kevil CG, Orr AW, Langston W, Mickett K, Murphy-Ullrich J, Patel RP, Kucik DF, Bullard DC. Intercellular adhesion molecule-1 (ICAM-1) regulates endothelial cell motility through a nitric oxide-dependent pathway. J. Biol. Chem. 2004;279(18):19230–19238. doi: 10.1074/jbc.M312025200. [DOI] [PubMed] [Google Scholar]
- 40.Kohn JC, Zhou DW, Bordeleau F, Zhou AL, Mason BN, Mitchell MJ, King MR, Reinhart-King CA. Cooperative effects of matrix stiffness and fluid shear stress on endothelial cell behavior. Biophys. J . 2015;108(3):471–478. doi: 10.1016/j.bpj.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kou B, Zhang J, Singer DR. Effects of cyclic strain on endothelial cell apoptosis and tubulogenesis are dependent on ROS production via NAD(P)H subunit p22phox. Microvasc. Res. 2009;77(2):125–133. doi: 10.1016/j.mvr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 42.Kumar A, Thompson EW, Lefieux A, Molony DS, Davis EL, Chand N, Fournier S, Lee HS, Suh J, Sato K, Ko Y-A, Molloy D, Chandran K, Hosseini H, Gupta S, Milkas A, Gogas B, Chang H-J, Min JK, Fearon WF, Veneziani A, Giddens DP, King SB, De Bruyne B, Samady H. High coronary shear stress in patients with coronary artery disease predicts myocardial infarction. J. Am. Coll. Cardiol. 2018;72(16):1926–1935. doi: 10.1016/j.jacc.2018.07.075. [DOI] [PubMed] [Google Scholar]
- 43.Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 2012;92(2):689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- 44.Levesque MJ, Nerem RM. The elongation and orientation of cultured endothelial cells in response to shear stress. J. Biomech. Eng. 1985;107(4):341–347. doi: 10.1115/1.3138567. [DOI] [PubMed] [Google Scholar]
- 45.Li Y-SJ, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2005;38(10):1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 46.Li LF, Ouyang B, Choukroun G, Matyal R, Mascarenhas M, Jafari B, Bonventre JV, Force T, Quinn DA. Stretch-induced IL-8 depends on c-Jun NH2-terminal and nuclear factor-kappaB-inducing kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003;285(2):L464–475. doi: 10.1152/ajplung.00031.2003. [DOI] [PubMed] [Google Scholar]
- 47.Li Y-S, Shyy J, Li S, Lee J, Su B, Karin M, Chien S. The Ras-JNK pathway is involved in shear-induced gene expression. Mol. Cell. Biol. 1996;16(11):5947–5954. doi: 10.1128/MCB.16.11.5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z, Tan JL, Cohen DM, Yang MT, Sniadecki NJ, Ruiz SA, Nelson CM, Chen CS. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl Acad. Sci. U.S.A. 2010;107(22):9944–9949. doi: 10.1073/pnas.0914547107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu D, Kassab GS. Role of shear stress and stretch in vascular mechanobiology. J. R. Soc. Interface. 2011;8(63):1379–1385. doi: 10.1098/rsif.2011.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maniotis AJ, Chen CS, Ingber DE. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl Acad. Sci. U.S.A. 1997;94(3):849–854. doi: 10.1073/pnas.94.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meza D, Abejar L, Rubenstein DA, Yin W. A shearing-stretching device that can apply physiological fluid shear stress and cyclic stretch concurrently to endothelial cells. J. Biomech. Eng. 2016;138(3):4032550. doi: 10.1115/1.4032550. [DOI] [PubMed] [Google Scholar]
- 52.Meza D, Rubenstein DA, Yin W. A comprehensive fluid–structure interaction model of the left coronary artery. J. Biomech. Eng. 2018;140:121006. doi: 10.1115/1.4040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meza D, Shanmugavelayudam SK, Mendoza A, Sanchez C, Rubenstein DA, Yin W. Platelets modulate endothelial cell response to dynamic shear stress through PECAM-1. Thromb. Res. 2017;150:44–50. doi: 10.1016/j.thromres.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 54.Mohan S, Mohan N, Sprague EA. Differential activation of NF-kappa B in human aortic endothelial cells conditioned to specific flow environments. Am. J. Physiol. Cell Physiol. 1997;273(2):C572–C578. doi: 10.1152/ajpcell.1997.273.2.C572. [DOI] [PubMed] [Google Scholar]
- 55.Moore JE, Jr, Burki E, Suciu A, Zhao S, Burnier M, Brunner HR, Meister JJ. A device for subjecting vascular endothelial cells to both fluid shear stress and circumferential cyclic stretch. Ann. Biomed. Eng. 1994;22(4):416–422. doi: 10.1007/BF02368248. [DOI] [PubMed] [Google Scholar]
- 56.Morrow D, Cullen-John P, Cahill Paul A, Redmond Eileen M. Cyclic strain regulates the notch/CBF-1 signaling pathway in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007;27(6):1289–1296. doi: 10.1161/ATVBAHA.107.142778. [DOI] [PubMed] [Google Scholar]
- 57.Nagel T, Resnick N, Dewey CF, Jr, Gimbrone MA., Jr Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler. Thromb. Vasc. Biol. 1999;19(8):1825–1834. doi: 10.1161/01.ATV.19.8.1825. [DOI] [PubMed] [Google Scholar]
- 58.Pedrigi RM, Papadimitriou KI, Kondiboyina A, Sidhu S, Chau J, Patel MB, Baeriswyl DC, Drakakis EM, Krams R. Disturbed cyclical stretch of endothelial cells promotes nuclear expression of the pro-atherogenic transcription factor NF-kappaB. Ann. Biomed. Eng. 2017;45(4):898–909. doi: 10.1007/s10439-016-1750-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Plotnikov A, Zehorai E, Procaccia S, Seger R. The MAPK cascades: signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta. 2011;1813(9):1619–1633. doi: 10.1016/j.bbamcr.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 60.Potter CMF, Schobesberger S, Lundberg MH, Weinberg PD, Mitchell JA, Gorelik J. Shape and compliance of endothelial cells after shear stress in vitro or from different aortic regions: scanning ion conductance microscopy study. PLoS ONE. 2012;7(2):e31228. doi: 10.1371/journal.pone.0031228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Raaz U, Kuhn H, Wirtz H, Hammerschmidt S. Rapamycin reduces high-amplitude, mechanical stretch-induced apoptosis in pulmonary microvascular endothelial cells. Microvasc. Res. 2009;77(3):297–303. doi: 10.1016/j.mvr.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 62.Remuzzi A, Dewey CF, Jr, Davies PF, Gimbrone MA., Jr Orientation of endothelial cells in shear fields in vitro. Biorheology. 1984;21(4):617–630. doi: 10.3233/BIR-1984-21419. [DOI] [PubMed] [Google Scholar]
- 63.Riou S, Mees B, Esposito B, Merval R, Vilar J, Stengel D, Ninio E, van Haperen R, de Crom R, Tedgui A, Lehoux S. High pressure promotes monocyte adhesion to the vascular wall. Circ. Res. 2007;100(8):1226–1233. doi: 10.1161/01.RES.0000265231.59354.2c. [DOI] [PubMed] [Google Scholar]
- 64.Rouleau L, Farcas M, Tardif JC, Mongrain R, Leask RL. Endothelial cell morphologic response to asymmetric stenosis hemodynamics: effects of spatial wall shear stress gradients. J. Biomech. Eng. 2010;132(8):081013. doi: 10.1115/1.4001891. [DOI] [PubMed] [Google Scholar]
- 65.Russo TA, Stoll D, Nader HB, Dreyfuss JL. Mechanical stretch implications for vascular endothelial cells: altered extracellular matrix synthesis and remodeling in pathological conditions. Life Sci. 2018;213:214–225. doi: 10.1016/j.lfs.2018.10.030. [DOI] [PubMed] [Google Scholar]
- 66.Samady H, Eshtehardi P, McDaniel MC, Suo J, Dhawan SS, Maynard C, Timmins LH, Quyyumi AA, Giddens DP. coronary artery wall shear stress is associated with progression and transformation of atherosclerotic plaque and arterial remodeling in patients with coronary artery disease. Circulation. 2011;124(7):779–788. doi: 10.1161/CIRCULATIONAHA.111.021824. [DOI] [PubMed] [Google Scholar]
- 67.Sei YJ, Ahn SI, Virtue T, Kim T, Kim Y. Detection of frequency-dependent endothelial response to oscillatory shear stress using a microfluidic transcellular monitor. Sci. Rep. 2017;7(1):10019. doi: 10.1038/s41598-017-10636-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seko Y, Takahashi N, Tobe K, Kadowaki T, Yazaki Y. Pulsatile stretch activates mitogen-activated protein kinase (MAPK) family members and focal adhesion kinase (p125(FAK)) in cultured rat cardiac myocytes. Biochem. Biophys. Res. Commun. 1999;259(1):8–14. doi: 10.1006/bbrc.1999.0720. [DOI] [PubMed] [Google Scholar]
- 69.Shanmugavelayudam SK, Rubenstein DA, Yin W. Effects of physiologically relevant dynamic shear stress on platelet complement activation. Platelets. 2011;22(8):602–610. doi: 10.3109/09537104.2011.585257. [DOI] [PubMed] [Google Scholar]
- 70.Stoner L, Young JM, Fryer S, Sabatier MJ. The importance of velocity acceleration to flow-mediated dilation. Int. J. Vasc. Med. 2012;2012:589213. doi: 10.1155/2012/589213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4–and TGF-β1-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2009;29(2):254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sumpio BE, Yun S, Cordova AC, Haga M, Zhang J, Koh Y, Madri JA. MAPKs (ERK½, p38) and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J. Biol. Chem. 2005;280(12):11185–11191. doi: 10.1074/jbc.M414631200. [DOI] [PubMed] [Google Scholar]
- 73.Thacher T, Gambillara V, da Silva RF, Silacci P, Stergiopulos N. Reduced cyclic stretch, endothelial dysfunction, and oxidative stress: an ex vivo model. Cardiovasc. Pathol. 2010;19(4):e91–98. doi: 10.1016/j.carpath.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 74.Tseng H, Peterson TE, Berk BC. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ. Res. 1995;77(5):869–878. doi: 10.1161/01.RES.77.5.869. [DOI] [PubMed] [Google Scholar]
- 75.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 76.Ueki YA, Sakamoto N, Ohashi T, Sato M. Morphological responses of vascular endothelial cells induced by local stretch transmitted through intercellular junctions. Exp. Mech. 2009;49(1):125. doi: 10.1007/s11340-008-9143-3. [DOI] [Google Scholar]
- 77.van Wolferen SA, Marcus JT, Westerhof N, Spreeuwenberg MD, Marques KM, Bronzwaer JG, Henkens IR, Gan CT, Boonstra A, Postmus PE, Vonk-Noordegraaf A. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur. Heart J. 2008;29(1):120–127. doi: 10.1093/eurheartj/ehm567. [DOI] [PubMed] [Google Scholar]
- 78.Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arterioscler. Thromb. Vasc. Biol. 1995;15(1):2–10. doi: 10.1161/01.ATV.15.1.2. [DOI] [PubMed] [Google Scholar]
- 79.Wang JG, Miyazu M, Matsushita E, Sokabe M, Naruse K. Uniaxial cyclic stretch induces focal adhesion kinase (FAK) tyrosine phosphorylation followed by mitogen-activated protein kinase (MAPK) activation. Biochem. Biophys. Res. Commun. 2001;288(2):356–361. doi: 10.1006/bbrc.2001.5775. [DOI] [PubMed] [Google Scholar]
- 80.White CR, Stevens HY, Haidekker M, Frangos JA. Temporal gradients in shear, but not spatial gradients, stimulate ERK1/2 activation in human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2005;289(6):H2350–2355. doi: 10.1152/ajpheart.01229.2004. [DOI] [PubMed] [Google Scholar]
- 81.Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler. Thromb. Vasc. Biol. 2007;27(12):2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 82.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ. Res. 2014;114(4):616–625. doi: 10.1161/CIRCRESAHA.114.302157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wung BS, Cheng JJ, Hsieh HJ, Shyy YJ, Wang DL. Cyclic strain-induced monocyte chemotactic protein-1 gene expression in endothelial cells involves reactive oxygen species activation of activator protein 1. Circ. Res. 1997;81(1):1–7. doi: 10.1161/01.RES.81.1.1. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto K, Ando J. Emerging role of plasma membranes in vascular endothelial mechanosensing. Circ. J. 2018;82(11):2691–2698. doi: 10.1253/circj.CJ-18-0052. [DOI] [PubMed] [Google Scholar]
- 85.Yin W, Shanmugavelayudam SK, Rubenstein DA. The effect of physiologically relevant dynamic shear stress on platelet and endothelial cell activation. Thromb. Res. 2011;127(3):235–241. doi: 10.1016/j.thromres.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 86.Yoshizumi M, Abe J, Tsuchiya K, Berk BC, Tamaki T. Stress and vascular responses: atheroprotective effect of laminar fluid shear stress in endothelial cells: possible role of mitogen-activated protein kinases. J. Pharmacol. Sci. 2003;91(3):172–176. doi: 10.1254/jphs.91.172. [DOI] [PubMed] [Google Scholar]
- 87.Zhao S, Suciu A, Ziegler T, Moore JE, Jr, Burki E, Meister JJ, Brunner HR. Synergistic effects of fluid shear stress and cyclic circumferential stretch on vascular endothelial cell morphology and cytoskeleton. Arterioscler. Thromb. Vasc. Biol. 1995;15(10):1781–1786. doi: 10.1161/01.ATV.15.10.1781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.