

Abstract
Reactions between Mo(N-t-Bu)2(CH2-t-Bu)2 or Mo(NAdamantyl)2(CH2CMe2Ph)2 and 3 equiv of HCl in the presence of 1 equiv of PPh2Me yield Mo(NR)(CHR’)(PPh2Me)Cl2 complexes, from which Mo(NR)(CHR’)(PPh2Me)(OAr)Cl complexes (OAr = a 2,6-terphenoxide) can be prepared. The Mo(NR)(CHR’)(PPh2Me)(OAr)Cl complexes were evaluated as cross-metathesis catalysts between cyclooctene and Z-1,2-dichloroethylene. The efficiencies of the test reaction for complexes in which OAr = OTPP, OHMT, OHIPT, or OHTBT (where OTPP is 2,3,5,6-tetraphenylphenoxide, OHMT is hexamethylterphenoxide, OHIPT is hexaisopropylterphenoxide, and OHTBT is hexa-t-butylterphenoxide) maximize when OAr is OHMT or OHIPT. Mo(N-t-Bu)(CH-t-Bu)(PPh2Me)Cl2 is essentially inactive for the reaction between cyclooctene and Z-1,2-dichloroethylene. X-ray structural studies were carried out on Mo(NAd)(CHCMe2Ph)(PPh2Me)Cl2, Mo(N-t-Bu)(CH-t-Bu)(PPh2Me)(OHMT)Cl, Mo(NAd)(CHCMe2Ph)(Cl)(OHTBT)(PMe3), and [Mo(NAd)(CHCMe2Ph)(PMe3)(Cl)]2(μ-O), the product of the reaction between Mo(NAd)(CHCMe2Ph)(Cl)(OHTBT)(PMe3) and 0.5 equiv of water.
Graphical Abstract
INTRODUCTION
A key step in the synthesis of molybdenum and tungsten imido alkylidene complexes traditionally has been the addition of 3 equiv of triflic acid to a M(NR)2(CH2R’)2 complex (R = aryl, t-butyl, or 1-adamantyl; R’ = t-Bu or CMe2Ph) in the presence of dimethoxyethane to give RNH3OTf, CH3R’, and a M(NR)(CHR’)(OTf)2(DME) complex.1 The triflate ligands can be replaced with a variety of sterically demanding alkoxide (or aryloxide; X) or pyrrolide (Y) ligands to give 14e M(NR)(CHR’)(X)2, M(NR)(CHR’)(Y)2, or M(NR)(CHR’)(X)(Y) complexes. Monoaryloxide pyrrolide M(NR)(CHR’)(X)(Y) complexes have proven useful for E-selective or Z-selective reactions,2 including stereoselective ROMP reactions,3 when the aryloxide is a sterically demanding 2,6-terphenoxide such as 2,6-dimesitylphenoxide (OHMT = hexamethylterphenoxide). In the process of exploring cross-metathesis reactions in which one of the olefins is Z-ClCH=CHCl,4 we discovered that M(NR)(CHR’)(OAr)(L)Cl complexes are especially reactive if the donor ligand (L = a 2e donor such as a pyridine or a nitrile) is labile enough to expose the 14e M(NR)(CHR’)(OAr)Cl core. Although we devised methods of preparing some M(NR)(CHR’)(OAr)(L)Cl complexes,5 the monochloride complexes would be more directly accessible from M(NR)(CHR’)(L)Cl2 or M(NR)(CHR’)(L)2Cl2 complexes simply through substitution of one chloride with a sterically demanding aryloxide. They should be useful catalysts if L readily dissociates from M(NR)(CHR’)(OAr)(L)Cl to a significant degree.
M(NR)(CHR’)(L)Cl2 or M(NR)(CHR’)(L)2Cl2 complexes (M = Mo or W) are relatively rare. The first M(NR)(CHR’)(L)2Cl2 complexes were prepared through addition of 3 equiv of HCl (as pyridinium chloride) to M(NR)2(CH2R’)2, where NR is N-2,6-(mesityl)2C6H3 (NAr*).6 Molybdenum and tungsten complexes that contain a N-2,6-(2,4,6-i-Pr3C6H2)2C6H3 (NAr**) ligand were prepared in a similar manner.7 The steric demand of the NAr** ligand even allowed a W(NAr**)(CHCMe2Ph)Cl2 complex to be prepared, the only isolable monomeric 14e dichloride complex of this general type. W(NR)(CHR’)Cl2(pyridine)2 complexes (R = adamantylimido or t-butylimido) were reported soon thereafter,8 as were similar dichlorides in reactions between W(N-t-Bu)2(CH2-2-OMeC6H4)2 and pyridine hydrochloride.9 Unfortunately, in most cases pyridine is bound too strongly to the metal in the resulting M(NR)(CHR’)(OAr)(L)Cl complexes for them to be highly active in olefin metathesis reactions. In some cases the pyridine is labile enough to be removed from solution through addition of B(C6F5)3 to yield (py)B(C6F5)3, in which case the 14e complexes can be generated in situ.
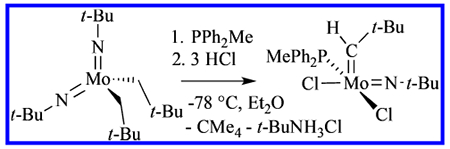
Phosphine adducts of imido alkylidene complexes are relatively well-known.1 Therefore, we explored the possibility of using phosphonium chlorides as the means of delivering HCl and chloride to generate imido alkylidene dichloride complexes as phosphine adducts. We discovered that addition of 2 equiv of PhMe2PHCl to Mo(NC6F5)2(CH2CMe2Ph)2 led to formation of Mo(NC6F5)(CHCMe2Ph)(PhMe2P)2Cl2 complexes in good yield.5 In that paper, we reported that “treatment of Mo(N-t-Bu)2(CH2-t-Bu)2 with phosphonium halides generated a complex mixture of alkylidene-containing compounds along with other unidentified products.” Upon optimization of conditions and choice of phosphine, we have now found that reactions between Mo(N-t-Bu)2(CH2-t-Bu)2 or Mo(NAdamantyl)2(CH2CMe2Ph)2 and 3 equiv of HCl in the presence of 1 equiv of PPh2Me produce Mo(NR)(CHR’)(PPh2Me)Cl2 complexes, from which Mo(NR)(CHR’)(PPh2Me)(OAr)Cl complexes can be prepared. In this paper we report the syntheses of Mo(NR)(CHR’)(PPh2Me)Cl2 and Mo(NR)(CHR’)(PPh2Me)(OAr)Cl complexes, along with a side-by-side comparison of all catalysts for a ring-opening cross-metathesis between cyclooctene and Z-ClCH=CHCl.
RESULTS
The molybdenum imido alkylidene dichloride complexes, Mo(NAd)(CHCMe2Ph)(PPh2Me)Cl2 (1a(PPh2Me)) and Mo(N-t-Bu)(CH-t-Bu)(PPh2Me)Cl2 (1b(PPh2Me)) are obtained in approximately 50% yield from the corresponding Mo(NR)2(CH2R’)2 complexes upon addition of 1 equiv of PPh2Me, followed by 3 equiv of HCl in diethyl ether, or alternatively, 1 equiv of PPh2Me•HCl, followed by 2 equiv of HCl in ether (eq 1). Three equivalents of HCl are required to complete the reaction because the t-butylamine or adamantylamine that is formed is readily protonated to give the ammonium salt, which is not a strong enough acid to allow the reaction to be completed. The reactions typically are carried out at −78 °C initially, followed by warming the reaction mixtures slowly to 22 °C, although reactions that start at ~−30 °C have been equally successful. Exploratory reactions in the presence of 1 equiv of PPh3 or PPhMe2 so far have not led to acceptable yields of complexes analogous to 1a(PPh2Me) and 1b(PPh2Me).
 |
All NMR data are consistent with 1a(PPh2Me) and 1b(PPh2Me) being monophosphine complexes that do not contain a plane of symmetry. Two alkylidene doublet resonances are found in the proton NMR spectra of each. The major alkylidene α proton resonances found near 13 ppm (in CD2Cl2) have 1JCH values (123 Hz for 1a(PPh2Me) and 118 Hz for 1b(PPh2Me)) that are consistent with the alkylidene being in the syn orientation. In the minor isomer (5–10 % of the mixture) the value for 1JCH (148 Hz for 1a(PPh2Me)) is characteristic of an anti isomer. The doublet resonance for the syn alkylidene α carbon in 1a(PPh2Me) is found at 309.8 ppm (2JCP = 18.9 Hz; CD2Cl2) and in 1b(PPh2Me) is found at 314.5 ppm (2JCP = 18.7 Hz; CD2Cl2). Recrystallization of mixtures of syn and anti isomers leads to samples that are essentially pure syn isomers, which suggests that syn and anti isomers do not interconvert readily in solution, most likely because alkylidenes can interconvert only as 14e phosphine-free species10,11 and PPh2Me is bound too strongly in 1a(PPh2Me) and 1b(PPh2Me).
An X-ray structural study of 1a(PPh2Me) (Figure 1) shows the overall structure to be approximately halfway between a trigonal bipyramid and a square pyramid (τ11 = 0.54) with the alkylidene in the syn orientation. Relevant bond angles at the metal are N1–Mo–Cl1 = 132.38(7)°, Cl1–Mo–C1 = 120.92(8)°, C1–Mo–N1 = 106.24(10)°, Cl2–Mo–P1 = 164.85(2)°, C1–Mo–P1 = 89.15(8)°, and C1–Mo–Cl2 = 96.40(7)°. The Mo=C1 (1.887 (2) Å) and Mo=N1 (1.717 (2) Å) distances and Mo=C–C (147.39(19)°) and Mo=N–C (163.15(17)°) angles are normal for five-coordinate complexes of this general type.
 |
Figure 1.
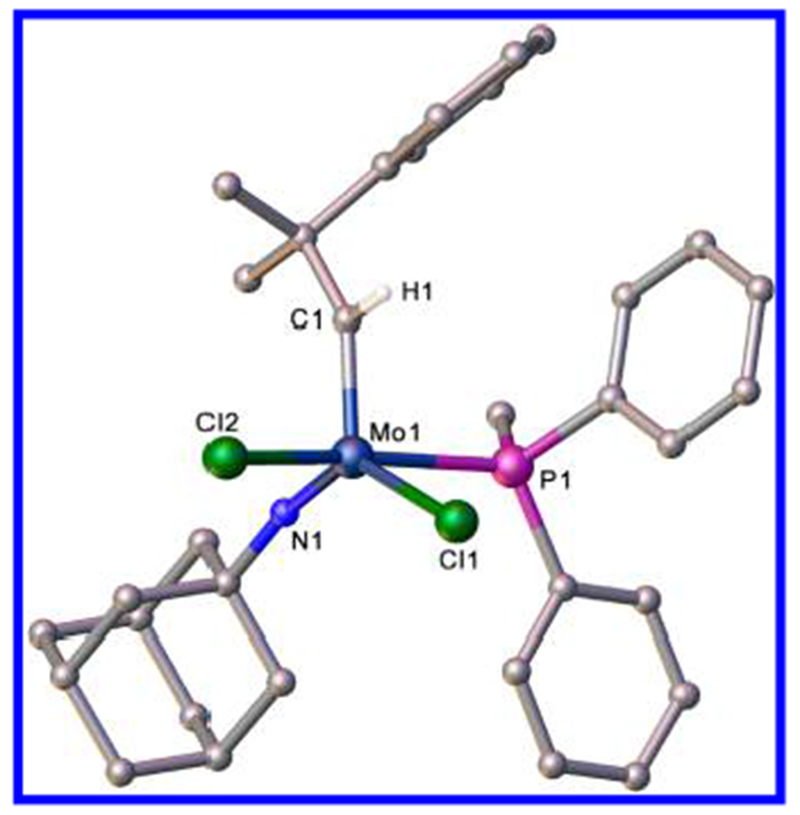
Structure of Mo(NAd)(CHCMe2Ph)(PPh2Me)Cl2 (1a(PPh2Me)). Hydrogen atoms, except on C1, have been omitted for clarity.
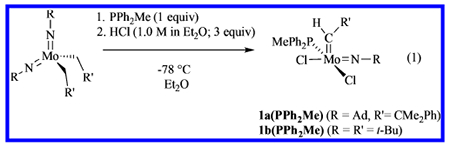
Monoaryloxide monochloride complexes were prepared in reactions between 1a(PPh2Me) or 1b(PPh2Me) and 1 equiv of LiOAr (OAr = OTPP, OHMT, OHIPT, OHTBT) in benzene, where OTPP is 2,3,5,6-tetraphenylphenoxide, OHMT is hexamethylterphenoxide (2,6-dimesitylphenoxide), OHIPT is hexaisopropylterphenoxide, and OHTBT is hexa-t-butylterphenoxide.12 We chose OTPP, OHMT, OHIPT, and OHTBT because of the dramatic degree to which their steric properties vary in the order OTPP<OHMT<OHIPT<OHTBT. Compound 5a(PMe3) is the first alkylidene complex to be prepared that contains the OHTBT ligand.12
Proton NMR spectra of complexes 2a(PPh2Me) and 2b(PPh2Me) reveal largely a doublet resonance for a syn alkylidene isomer, and under some conditions a small doublet for an anti alkylidene isomer, as found for 1a(PPh2Me) and 1b(PPh2Me). However, a few percent of each sample (~5%, depending upon the concentration) are the phosphine-free 14e complexes, 2a and 2b, respectively, as syn isomers, according to 1H NMR spectra. Free phosphine also can be observed in the 31P NMR spectra (see the SI). The amount of free phosphine increases for 3a(PPh2Me) and 3b(PPh2Me), and again for 4b(PPh2Me). As expected, addition of PPh2Me shifts the equilibrium toward the PPh2Me adduct (as a syn isomer), while addition of 1 equiv of B(C6F5)3 results in scavenging of the PPh2Me to produce spectra consistent with formation of the phosphine-free species. It is clear that the phosphine-free complexes are extremely soluble, even in pentane, and none has been isolated in crystalline form. Mo(NR)(CHR’)(OAr)Cl complexes also appear to be exceedingly sensitive to water (vide infra).
An X-ray crystallographic study of 3b(PPh2Me) reveals that the crystals contain two molecules that are enantiomers,13 with equal occupancy over the entire unit cell (see the SI for details). The disorder could be resolved and both structures of 3b(PPh2Me) solved. They are best described as approximately square pyramids (τ = 0.24 and 0.36) with the apical alkylidene in a syn conformation and the PPh2Me bound trans to the chloride. Solvents are commonly found in the crystal lattices of the compounds reported here (sometimes partial occupation; see the SI), a complication that has compromised the precision of elemental analyses in some cases.
The structure of the isomer of 3b(PPh2Me) with τ = 0.24 is shown in Figure 2. Relevant bond angles around the metal are Cl1–Mo1–P1 = 167.39(4)°, N1–Mo1–O1 = 153.12(9)°, C1–Mo1–N1 = 101.32(11)°, C1–Mo1–O1 = 105.51(9)°, C1–Mo1–Cl1 = 100.33(8)° and C1–Mo1–P1 = 89.01(8)°. The Mo=C1 (1.921(2) Å), Mo=N1 (1.722(2) Å), Mo–Cl1 (2.4457(7) Å), Mo–P1 (2.5587(13) Å), and Mo–O1 (1.9895(14) Å) distances and Mo=C1–C2 (142.4(2)°), Mo=N1–C6 (160.15 (19)°), and Mo–O1–C10 (146.90(12)°) angles are typical of other five-coordinate complexes of this general type.5
Figure 2.
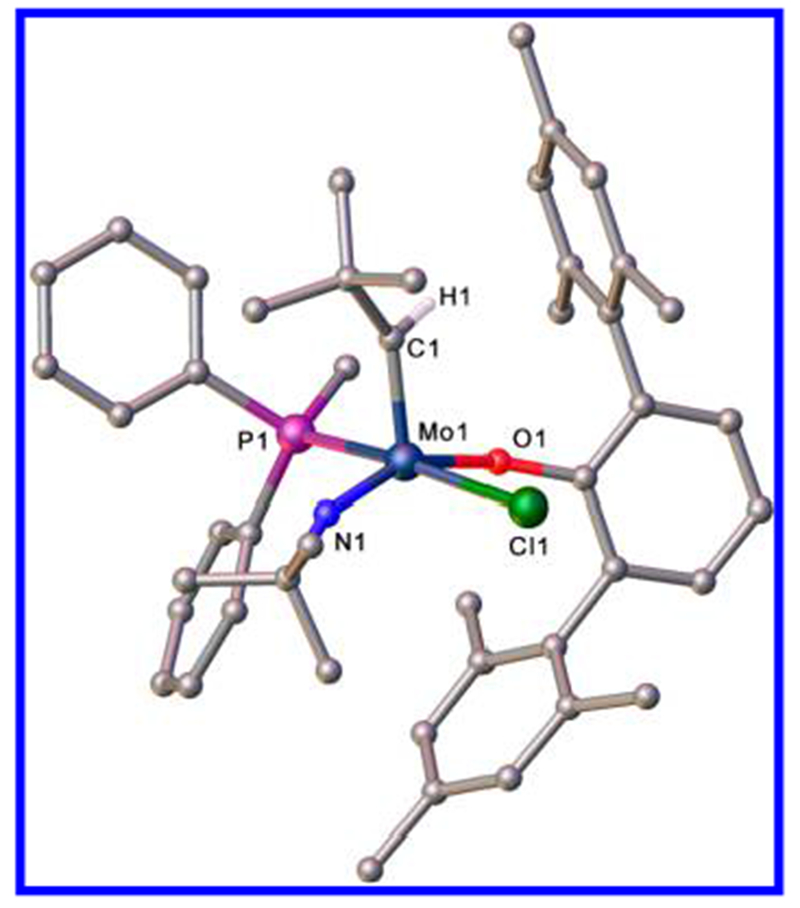
Structure of one of the molecules of Mo(N-t-Bu)(CH-t-Bu)(PPh2Me)(OHMT)Cl (3b(PPh2Me)); τ = 0.24. Hydrogen atoms (except on C1), disordered atoms, and lattice solvent toluene have been omitted for clarity.
The reaction of 1a(PPh2Me) with KOHTBT (OHTBT = hexa-t-butylterphenoxide) gives essentially phosphine-free 4-coordinate 5a, according to NMR studies; it is too soluble, even in pentane, to be crystallized (so far). However, a PMe3 complex, 5a(PMe3), could be isolated in crystalline form in 47% yield upon addition of 5 equiv of PMe3 to 5a in dichloromethane. Compound 5a(PMe3) is an off-white powder in the solid state, but its solutions are yellow because PMe3 dissociates to a significant degree to give yellow 5a, as can be shown upon scavenging PMe3 in solutions of 5a(PMe3) with B(C6F5)3. The proton NMR spectrum of 5a(PMe3) in Figure 3 (top) shows that the ratio of 5a(PMe3) to 5a is 73:27 (20 mg of 5a(PMe3) in 0.6 mL of C6D6, 31 mM initial concentration); therefore Keq is 3.1 mM in C6D6 at 20 °C. The rate of PMe3 exchange in 5a(PMe3) is such that the alkylidene proton is essentially decoupled from phosphorus. Addition of 2 equiv of PMe3 to the solution of 5a(PMe3) results in a mixture that is ~97% 5a(PMe3) (Figure 3 bottom). The small doublet alkylidene resonance at 13.55 ppm in the spectra in Figure 3 is the result of a reaction between 5a(PMe3) and traces of water (vide infra).
Figure 3.
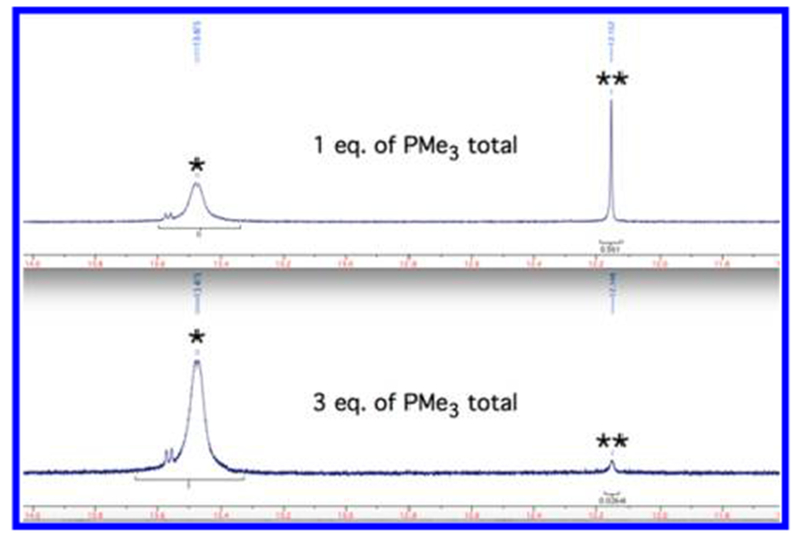
Alkylidene resonances in proton NMR spectra of Mo(NAd)(CHCMe2Ph)(Cl)(OHTBT)(PMe3) 5a(PMe3) in C6D6 at 20 °C; *5a(PMe3); **5a.
An X-ray study of 5a(PMe3) (Figure 4) shows its structure to be closer to a square pyramid (τ = 0.27) with the alkylidene in the syn orientation, analogous to that of 3b(PPh2Me) (Figure 2). Relevant bond angles at the metal are Cl1–Mo–P1 = 168.122 (13)°, N1–Mo–O1 = 151.86(5)°, C1–Mo–O1 = 106.44(5)°, C1–Mo–N1 = 101.36(6)°, C1–Mo1–Cl1 = 97.67(4)°, and C1–Mo1–P1 = 94.21(4)°. The Mo=C1 (1.8973(14) Å), Mo=N1 (1.7345(12) Å), Mo–Cl1 (2.4480(5) Å), and Mo–P (2.5168(5) Å) distances are similar to what they are in 3b(PPh2Me). The Mo-O1 (2.0243(10) Å) distance is slightly longer than what it is in 3b(PPh2Me), presumably as a consequence of the extreme steric bulk of the OHTBT ligand. The steric bulk of the OHTBT ligand must also be the origin of the relatively large Mo–O1–C21 (159.86(9)°) angle. For comparison, in Mo(N-t-Bu)(CH-t-Bu)Cl(OHIPT)(3-Bromopyridine) the Mo–O–Caryl angle is 151.4(3)°.4 The Mo=C-C (142.48(11)°) and Mo=N–C (166.59(11)°) angles within 5a(PMe3) are consistent with complexes of this type.
Figure 4.
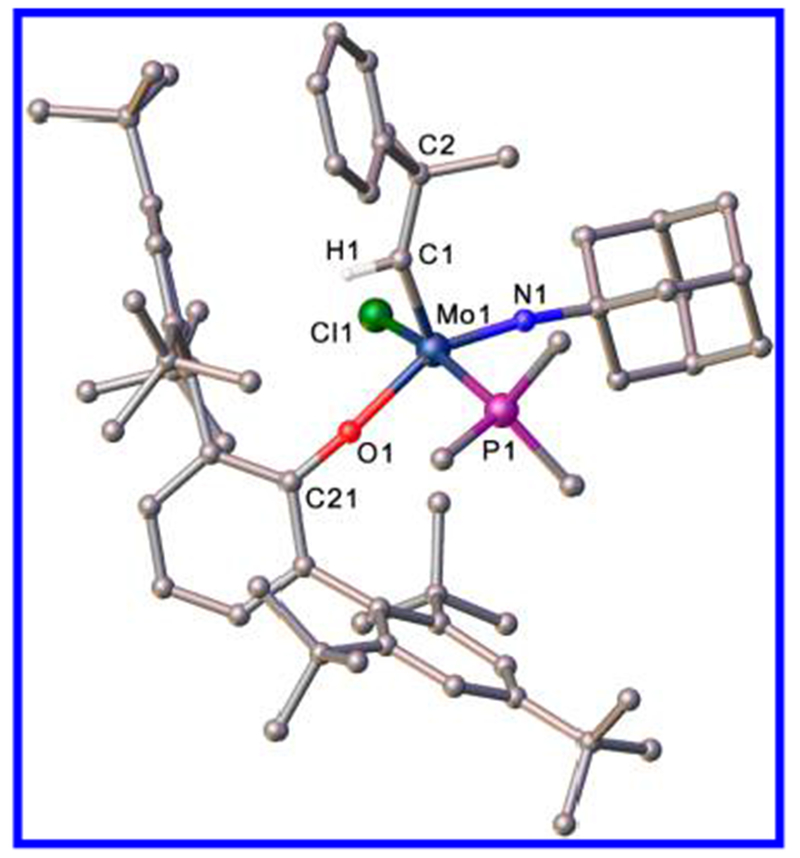
Structure of Mo(NAd)(CHCMe2Ph)(Cl)(OHTBT)(PMe3) 5a(PMe3); τ = 0.27. Hydrogen atoms (except on C1) and lattice solvent dichloromethane have been omitted for clarity.
In an attempt to remove PMe3 from 5a(PMe3) in the solid state to give 5a, a sample of 50 mg of 5a(PMe3) in a Pyrex tube was exposed to a vacuum (25 mTorr) for 24 h. We were surprised to find that 5a(PMe3) decomposed under these conditions to give HTBTOH and a single alkylidene complex that is relatively insoluble in pentane and that contains PMe3, but not OHTBT. HTBTOH can be separated from the decomposition product (6a(PMe3)) through addition of a small quantity of pentane to the mixture of 6a(PMe3) and HTBTOH. A 1H NMR spectrum of 6a(PMe3) shows that the syn-alkylidene proton resonance is a doublet at 13.55 ppm (3JPH = 6.4 Hz; 1JCH = 127 Hz). Because 6a(PMe3) also could be obtained in 67% yield in a reaction between 5a(PMe3) and 0.5 equiv of water in ether, we suspect that 6a(PMe3) is formed from a selective hydrolysis of the OHTBT ligand in 5a(PMe3), even in vacuo in the particular experiment described above.
An X-ray study of 6a(PMe3) (Figure 5) showed it to be [Mo(NAd)(CHCMe2Ph)(PMe3)(Cl)]2(μ-O). The overall structure of each Mo center is close to a square pyramid (τ = 0.21 at Mo1 and τ = 0.16 at Mo2) with each alkylidene in the syn orientation. Relevant bond angles at the metal are Mo1–O–Mo2 = 120.37(7)°, N1–Mo1–O1 = 149.41(7)°, Cl1–Mo1–P1 = 161.730(19)°, N2–Mo2–O1 = 152.02 (7)°, Cl2–Mo2–P2 = 161.34(2)°. The Mo1–O1 (1.9423(13) Å), Mo2–O1 (1.9774(13) Å), Mo1=C101 (1.907(2) Å), Mo2=C201 (1.907(2) Å), Mo1=N1 (1.7507(17) Å), Mo2=N2 (1.7492(17) Å), Mo1–Cl1 (2.4871(6) Å), Mo2–Cl2 (2.4870(6) Å), Mo1–P1 (2.4980(6) Å), Mo2–P2 (2.4894(7) Å) distances and Mo1=C–C (141.95(6)°), Mo2=C–C (143.61(16)°), Mo1=N–C (158.81(14)°), and Mo2=N–C (160.82(15)°). In view of the steric crowding within 6a(PMe3), we were surprised to find that the Mo1–O1–Mo2 angle is only 120°.
Figure 5.
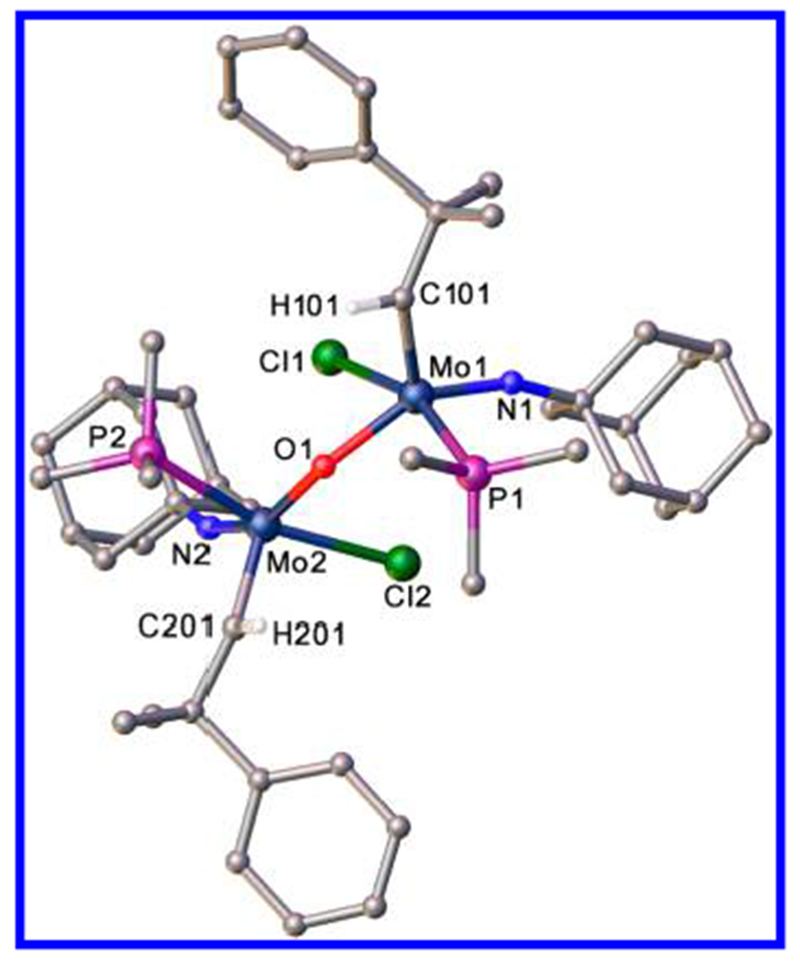
Structure of [Mo(NAd)(CHCMe2Ph)(PMe3)(Cl)]2(μ-O) (6a(PMe3)).
We evaluated the efficacy of 1b, 2b, 3b, 4b, 5a, and 6a, and their phosphine adducts), for the ring-opening cross-metathesis of cyclooctene and Z-dichloroethylene (equation 3), as has been described in an earlier paper for closely related monomeric complexes.5 The results are shown in Table 1.
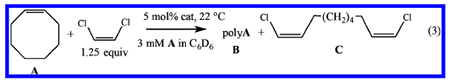 |
Table 1.
Results (A/B/C Percentages) of Reactions between Cyclooctene (A) and Z-1,2-Dichloroethylene (1.25 equiv) in C6D6 to Give Polycyclooctene (B) and/or the Cross-metathesis Product (C)
| initiator | 20 min | 1 h | 6 h | % Z,Z of C |
|---|---|---|---|---|
| 1b(PPh2Me) | 100/0/0 | 100/0/0 | 100/0/0 | n/a |
| 2b(PPh2Me) | 100/0/0 | 92/8/0 | 45/55/0 | |
| 3b(PPh2Me) | 3/51/46 | 0/9/91 | 0/1/99 | 86.4 |
| 4b(PPh2Me) | 4/32/64 | 0/24/76 | 0/4/96 | 100 |
| 5a(PMe3) | 100/0/0 | 100/0/0 | 100/0/0 | n/a |
| 6a(PMe3) | 100/0/0 | 100/0/0 | 100/0/0 | n/a |
| 1ba | 100/0/0 | 100/0/0 | 100/0/0 | n/a |
| 2ba | 0/2/98 | 0/1/99 | 89.6 | |
| 3ba | 0/3/97 | 0/3/97 | 99.3 | |
| 4ba | 0/0/100 | 99.7 | ||
| 5aa | 100/0/0 | 100/0/0 | 100/0/0 | n/a |
| 6aa | 78/22/0 | 61/39/0 | 5/95/0 | n/a |
Generated in situ through addition of 1 equiv of B(C6F5)3 to the phosphine adduct.
We found that product C is formed readily when 3b and 4b, in which the PPh2Me is dissociated to the greatest extent, are the initiators. Compound C is essentially 100% Z,Z when 4b(PPh2Me) is the initiator and 86.4% Z,Z when 3b(PPh2Me) is the initiator, as a consequence of the greater steric demand of the OHIPT versus the OHMT terphenoxide ligand.5 Some poly(cyclooctene) is formed when 2b(PPh2Me) is the initiator, but the cross-metathesis reaction is comparatively sluggish unless B(C6F5)3 is added, in which case the activity is high, but the stereoselectivity is only ~90%. Compound 1b(PPh2Me) is inactive for the cross-metathesis reaction, most likely because PPh2Me is too tightly bound to the metal. 1b(PPh2Me), cyclooctene, and ClCH=CHCl are still present after 10 h and no initial metathesis product (t-BuCH=CHCl) is formed. We were surprised to find that 5a(PMe3) is inactive, even though PMe3 is dissociated to a significant degree from Mo in 5a(PMe3) (Figure 3, top) and even when B(C6F5)3 has been added to 5a(PMe3) to give 5a. No C is formed using 6a(PMe3) or 6a.
DISCUSSION
It has long been known through experiments,1,14 and has now been confirmed through extensive calculations,15 that the 14e core of a high oxidation state Mo or W complex must be accessible in order to be an efficient olefin metathesis catalyst, and the metallacyclobutane intermediate must be a trigonal bipyramid with the MC3 ring in two equatorial positions.15 As we have shown here and elsewhere,4,5 monoaryloxide monochloride complexes are unusually reactive and capable of cross-metathesizing electron-poor Z-ClCH=CHCl with an “ordinary” olefin such as cyclooctene. Fortunately, the adamantylimido and t-butylimido alkylidene complexes now can be prepared relatively directly as diphenylmethylphosphine adducts using HCl to trigger formation of the alkylidene through protonation and removal of one imido ligand and to provide the chloride anion, all in one event. Phosphine is required in order to isolate the dichloride or monochloride complexes, but in the right circumstances the phosphine is labile enough for the compound to initiate and sustain a metathesis reaction that involves Z-ClCH=CHCl. The degree of dissociation of PPh2Me depends dramatically upon the size of the aryloxide (OAr) ligand in the order OTPP < OHMT < OHIPT < OHTBT, but the reactivity of the resulting 14e complex is likely to follow the opposite trend (OTPP > OHMT > OHIPT > OHTBT). These opposing trends result in 3b(PPh2Me) and 4b(PPh2Me) being the “best compromise” for the reaction shown in equation 3. Sterically less demanding and more strongly bound phosphines (PMe3) or other ligands (e.g., pyridine) are more likely to hinder or block reaction with an olefin through formation of a 16e or 18e complex. More reactive, smaller olefins (e.g., ethylene) may be able to compete successfully for a 14e complex, whereas Z-ClCH=CHCl, or even cyclooctene, cannot.
We have to ascribe the low activity of phosphine-free 5a entirely to the fact that OHTBT is simply too sterically demanding for the reaction shown in equation 3, or even polymerization of cyclooctene, under the conditions employed. Low reactivity has been observed, largely for steric reasons, for other 14e imido alkylidene complexes. One example is Mo(NAd)(CHCMe2Ph)[OSi(t-Bu)3]2, which does not react readily with ethylene at 5 atm and 120 °C.16 The low reactivity of phosphine-free 6a also can be ascribed to the steric demand of one of the Mo entities with respect to reaction of substrate at the other Mo center.
Detailed studies that document the consequence of reaction of water with high oxidation state Mo or W alkylidene complexes17 (or alkylidyne complexes18) are rare. What we have found here in the case of 6a(PMe3) is that the alkylidene ligand is not necessarily the first ligand to be protonated upon addition of water. The protonation process is likely to consist of water first binding to the metal followed by a proton being transferred (intramolecularly or intermolecularly) to the aryloxide oxygen. It also cannot be assumed that, if an alkylidene ligand survives partial hydrolysis, the resulting complex will be a more active catalyst than the original alkylidene complex, especially if a μ-oxo dimer or larger cluster is formed, as is found for 6a(PMe3). In fact, a lower reactivity may be more likely in view of the propensity of an oxo ligand to bridge between metals in many circumstances.
CONCLUSIONS
We have shown that adamantylimido and t-butylimido alkylidene dichloride complexes can be prepared in the presence of diphenylmethylphosphine and 3 equiv of HCl. These serve as starting materials for the synthesis of monoaryloxide chloride complexes in which the aryloxide is a terphenoxide (OTPP, OHMT, OHIPT, or OHTBT). A 16e monoaryloxide monochloride complex is active in metathesis reactions when the terphenoxide is large enough to labilize the phosphine, but small enough to allow reactions at the 14e metal center to proceed. The metathesis reaction is most stereoselective when the terphenoxide is OHIPT.
Experimental Section
General Procedures.
All air- and moisture-sensitive materials were manipulated under a nitrogen atmosphere in a Vacuum Atmospheres glovebox or on a dual-manifold Schlenk line. Glassware was either oven-dried or flame-dried prior to use. Acetonitrile, benzene, methylene chloride, diethyl ether, 1,2-dimethoxyethane, and toluene were degassed, passed through activated alumina columns, and stored over 4 Å Linde-type molecular sieves. Pentane was washed with H2SO4, followed by water and a saturated solution of aqueous NaHCO3, and dried over CaCl2 pellets for at least 2 weeks prior to use in the solvent purification system. Deuterated solvents were dried over 4 Å Linde-type molecular sieves prior to use. 1H and 31P{1H} NMR spectra were acquired on a Varian Mercury 300 MHz or a Varian Inova-500 MHz NMR spectrometer. 13C{1H} NMR spectra were acquired on a Varian Inova-500 MHz NMR spectrometer. 2D NMR spectra were acquired on a Varian Inova-500 MHz NMR spectrometer. Chemical shifts for 1H, 31P, and 13C NMR spectra are reported as parts per million relative to tetramethylsilane and referenced to the residual 1H or 13C resonances of the deuterated solvent (1H δ: benzene 7.16 ppm, methylene chloride 5.32 ppm; 13C δ: benzene 128.06 ppm, methylene chloride 53.84 ppm). Gas Chromatography was performed on an Agilent system equipped with an HP-5 column (ID 320 μm, 0.25 μm, and length 30 m). TMSCl was purchased from Alfa Aesar and degassed by a free-pump-thaw method prior to use. B(C6F5)3 was purchased from Strem and used as received. LiOAr was prepared by the addition of 1 equiv of n-butyllithium to a cold pentane or Et2O solution of ArOH, and the solid was collected on a glass frit, washed with pentane, and dried in vacuo. PMe3, PPhMe2, and PPh2Me were purchased from Strem chemicals and used as received. The hydrochloride salts of the phosphines were prepared through addition of ethereal HCl (1.0 M; Sigma Aldrich) to the phosphine in diethyl ether while stirring the mixture; the phosphine hydrochloride salt was collected on a glass frit, washed with minimal ether, and dried in vacuo. Mo(NAd)2Cl2(DME),19 Mo(NAd)2(CH2CMe2Ph)2,9 and Mo(N-t-Bu)2Cl2(DME)20 were prepared as described in the literature. Syntheses of Mo(N-t-Bu)2(CH2-t-Bu)2 from Mo(N-t-Bu)2Cl2 and neopentyllithium21 and from Mo(N-t-Bu)2Cl2(DME) and neopentyl Grignard22 have been reported in the literature, but no detailed procedures were provided; we provide a procedure here. Elemental analyses were performed at the CENTC Elemental Analysis Facility at the University of Rochester, New York.
Mo(N-t-Bu)2(CH2-t-Bu)2.
Mo(N-t-Bu)2Cl2(DME) (4.01 g, 10.03 mmol) was dissolved in ~30 mL of diethyl ether in a glovebox under nitrogen. The reaction vessel was connected to a Schlenk manifold and cooled to −78 °C with a dry ice/acetone bath. Neopentylmagnesium chloride (20.1 mL, 20.09 mmol, 2 equiv., 1.0 M in THF) was syringed into the reaction vessel over a few minutes. The reaction mixture was stirred overnight to give a turbid brown-orange solution. All volatiles were removed in vacuo. In a glovebox ~30 mL of pentane was added and the mixture was filtered through Celite. All volatiles were removed from the filtrate in vacuo to yield Mo(N-t-Bu)2(CH2-t-Bu)2 as a brown oil that was used without purification; crude yield 3.77 g (99%): 1H NMR (500 MHz, C6D6, 22 °C) 1.76 (s, 4H), 1.41 (s, 18 H), 1.19 (s, 18 H) ppm.
Mo(NAd)(CHCMe2Ph)(PPh2Me)Cl2 (1a(PPh2Me)).
Mo(NAd)2(CH2CMe2Ph)2 (1.05 g, 1.59 mmol, 1.00 equiv) was dissolved in 30 mL of diethyl ether. The solution was stirred rapidly and solid PPh2Me·HCl (0.375 g, 1.59 mmol, 1.00 equiv) was added. The reaction mixture was stirred for 30 min at room temperature and then chilled to – 78 °C. HCl (1.0 M in diethyl ether; 3.17 mL, 2.00 equiv) was added to the mixture with a syringe. The cooling bath was removed and the mixture was stirred overnight. The volatiles were removed from the reaction in vacuo, the crude residue was triturated with 1–2 mL of diethyl ether, and the off-white solid was collected on a glass frit. The crude product was dissolved in ~20 mL of a mixture of benzene and toluene (1:1 v/v) and the solution was filtered through Celite to remove the t-butylammonium chloride. The solvents were removed from the extract in vacuo to afford the product; yield 0.540 g (53 %). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 13.10 (d, 3JHP = 4.9 Hz, 1JCH = 123 Hz, 1H, Mo=CHsyn); 13C{1H} NMR (126 MHz, CD2Cl2) δ 309.8 (d, 2JCP = 22.4 Hz, Mo=C). Anal. Calcd for C33H40Cl2MoNP (648.52 g/mol): C, 61.12%; H, 6.22%; N, 2.16%. Found: C, 61.21%; H, 6.19%; N, 2.51%.
Slow evaporation of benzene from a saturated solution of 1a(PPh2Me) at room temperature gave crystals suitable for X-ray data collection.
Mo(N-t-Bu)(CH-t-Bu)Cl2(PPh2Me) (1b(PPh2Me)).
The procedure was virtually the same as that for 1a(PPh2Me) starting with Mo(N-t-Bu)2(CH2-t-Bu)2 (2.00 g, 5.26 mmol, 1.00 equiv) in 30 mL of diethyl ether, PPh2Me·HCl (1.24 g, 5.26 mmol, 1.00 equiv), and HCl (1.0 M in diethyl ether; 10.5 mL, 2.00 equiv); yield 1.31 g (49 %). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 13.01 (d, 3JHP = 4.9 Hz, 1JCH = 118 Hz, 1H, Mo=CHsyn); 13C{1H} NMR (126 MHz, CD2Cl2) δ 314.5 (d, 2JCP = 22.7 Hz, Mo=C). Anal. Calcd for C22H32Cl2MoNP (508.34 g/mol): C, 51.98%; H, 6.35%; N, 2.76%. Found: C, 52.04%; H, 6.29%; N, 2.61%.
Mo(NAd)(CHCMe2Ph)(Cl)(OTPP)(PPh2Me) (2a(PPh2Me)).
A solution of LiOTPP (0.218 g, 0.216 mmol, 1 equiv) in benzene solution was added dropwise to 1a(PPh2Me) (0.140 g, 0.216 mmol, 1 equiv) in benzene. The reaction was stirred overnight and filtered through Celite. The solvents were removed in vacuo and the tacky yellow solid was triturated in 5 mL of pentane to afford the product as an off-white solid; yield 0.187 g (86%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 11.55 (d, 3JHP = 4.4 Hz, 1H, Mo=CH); 13C{1H} NMR (126 MHz, CD2Cl2) δ 311.9 (d, 2JCP = 16.7 Hz, Mo=C). Anal. Calcd for C63H61ClMoNOP (1010.57 g/mol): C, 74.88%; H, 6.08%; N, 1.39%. So far elemental analyses that agree with the calculated figures has not been successful.
Mo(N-t-Bu)(CHCMe2Ph)(Cl)(OTPP)(PPh2Me) (2b(PPh2Me)).
A solution of LiOTPP (0.114 g, 0.238 mmol, 1 equiv) in benzene was added dropwise to 1b(PPh2Me) (0.121 g, 0.238 mmol, 1 equiv) in benzene. The isolation procedure was the same as that for 2a(PPh2Me); yield 0.151 g (72.9%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 11.58 (d, 3JHP = 4.5 Hz, 1H, Mo=CH); 13C{1H} NMR (126 MHz, CD2Cl2) δ 316.5 (d, 2JCP = 16.9 Hz, Mo=C). Anal. Calcd for C52H53ClMoNOP (870.39 g/mol): C, 71.76%; H, 6.14%; N, 1.61%. Found: C, 72.05%; H, 6.25%; N, 1.19%.
Mo(NAd)(CHCMe2Ph)(Cl)(OHMT)(PPh2Me) (3a(PPh2Me)).
A solution of LiOHMT (0.063 g, 0.154 mmol, 1 equiv) was added dropwise to 1a(PPh2Me) (0.100 g, 0.154 mmol, 1 equiv) in benzene. The isolation procedure was the same as that for 2a(PPh2Me); yield 0.111 g (76.4%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 12.45 (d, 3JHP = 4.6 Hz, 1H, Mo=CH); 13C{1H} NMR (126 MHz, CD2Cl2) δ 313.6 (d, 2JCP = 17.9 Hz, Mo=C). Anal. Calcd for C57H65ClMoNOP (942.54 g/mol): C, 72.64%; H, 6.95%; N, 1.49%. Found: C, 72.13%; H, 6.88%; N, 1.31%.
Mo(N-t-Bu)(CH-t-Bu)(Cl)(OHMT)(PPh2Me) (3b(PPh2Me)).
A solution of LiOHMT (0.067 g, 0.163 mmol, 1 equiv) in benzene solution was added dropwise to 1b(PPh2Me) (0.083 g, 0.163 mmol, 1 equiv) in benzene. The isolation procedure was the same as that for 2a(PPh2Me); yield 0.072 g (55%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, CD2Cl2) δ 12.27 (d, 3JHP = 4.7 Hz, 1H, Mo=CH); 13C{1H} NMR (126 MHz, CD2Cl2) δ 318.5 (d, 2JCP = 17.7 Hz, Mo=C). Anal. Calcd for C49.5H61ClMoNOP (848.42 g/mol): C, 70.07%; H, 7.25%; N, 1.65%. Found: C, 70.05%; H, 7.27%; N, 1.51%.
X-ray quality crystals were grown from a saturated toluene solution at –25 °C. The lattice contains 0.5 molecules of toluene per 3b(PPh2Me).
Mo(NAd)(CHCMe2Ph)(Cl)(OHIPT)(PPh2Me) (4a(PMe3)).
A solution of LiOHIPT (0.089 g, 0.177 mmol, 1 equiv) was added dropwise to 1a(PPh2Me) (0.115 g, 0.177 mmol, 1 equiv) in benzene. The solution was stirred for 30 h and filtered through Celite. The solvents were removed from the filtrate in vacuo and the tacky residue was extracted with pentane to give an orange solution. PMe3 (0.100 g, 1.31 mmol, 7.4 equiv) was added to the orange pentane solution. The crystals that grew when the solution was cooled to – 25 °C were collected by filtration; yield 0.040 g (23%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, C6D6) δ 12.74 (d, 3JHP = 4.8 Hz, 1H, Mo=CH); 13C{1H} NMR (126 MHz, C6D6) δ 300.9 (d, 2JCP = 18.4 Hz, Mo=C). Anal. Calcd for C59H85ClMoNOP (986.72 g/mol): C, 71.82%; H, 8.68%; N, 1.42%. Found: C, 71.49%; 8.55%; N, 1.20%.
Mo(N-t-Bu)(CH-t-Bu)(Cl)(OHIPT)(PPh2Me) (4b(PPh2Me)).
A solution of LiOHIPT (0.139 g, 0.275 mmol, 1 equiv) in benzene solution was added drop wise to a solution of Mo(N-t-Bu)(CH-t-Bu)(PPh2Me)Cl2, 1b(PPh2Me) (0.140 g, 0.275 mmol, 1 equiv) in benzene. The solution was stirred rapidly overnight. The mixture was filtered through Celite, and the volatiles were removed from the filtrate in vacuo. The tacky yellow solid was stirred in 5 mL pentane to afford 4b(PPh2Me) as an off-white solid; yield 0.168 g (62.8%). Only alkylidene NMR resonances are listed here: 1H NMR (500 MHz, C6D6) δ 12.99 (br d, 1H, 5-coordinate Mo=CH), 11.62 (s, 1H, 4-coordinate Mo=CH); 13C NMR (obtained from 1H-13C deHSQC spectrum 500 MHz, C6D6) δ 292.9 (4-coordinate Mo=C). Anal. Calcd for C58H81ClMoNOP (970.68 g/mol): C, 71.77%; H, 8.41%; N, 1.44%. Found: C, 71.39%; H, 8.27%; N, 1.22%.
Mo(NAd)(CHCMe2Ph)(Cl)(OHTBT)(PMe3) (5a(PMe3)).
KOHTBT (300 mg, 0.482 mmol, 1 equiv) was added to 20 mL of benzene containing Mo(NAd)(CHCMe2Ph)(Cl)2(PPh2Me) 1a(PPh2Me) (313 mg, 0.482 mmol, 1 equiv), and the mixture was heated at 60 °C for 18 h. The benzene was evaporated in vacuo, 5 mL of dichloromethane was added, and the mixture filtered through Celite to give a red solution. PMe3 (0.245 mL, 184 mg, 2.41 mmol, 5 equiv) was added, and the resulting solution was kept at −20 °C overnight. During this time, pale off-white crystals formed, which were filtered off and washed with 1 mL of cold dichloromethane. The product was dried in vacuo to give Mo(NAd)(CHCMe2Ph)(OHTBT)(Cl)(PMe3).2CH2Cl2 as off-white crystals; yield 280 mg (47%). The product was stored in the glovebox refrigerator at – 20 °C in order to avoid slow decomposition. Only alkylidene NMR resonances are listed here: 1H NMR (400 MHz, C6D6) δ 13.47 (br s, 1H, Mo=CH); 13C{1H} NMR (151 MHz, C6D6) δ 311.1 (d, 2JCP = 15.1 Hz, Mo=C). Anal. Calcd for C65H97ClMoNOP (1070.88 g/mol): C, 72.90%; H, 9.13%; N, 1.31%. Found: C, 73.49%; H, 9.40%; N, 1.05%.
Crystals suitable for X-ray data collection were obtained by crystallizing 5a(PMe3) from dichloromethane in the presence of 2 equiv of PMe3 at room temperature.
[Mo(NAd)(CHCMe2Ph)(Cl)(PMe3)]2(μ-O) (6a(PMe3)).
Mo(NAd)(CHCMe2Ph)(OHTBT)(Cl)(PMe3).2CH2Cl2 (5a(PMe3)) (100 mg, 0.08 mmol, 1 equiv) was suspended in 2 mL of ether and the mixture was cooled to – 78 °C. A stock solution of wet ether was prepared through addition of 0.005 mL of water to 1 mL ether. A 0.145 mL (0.726 mg, 0.04 mmol, 0.5 equiv of water) of this solution was added, and the resulting suspension was stirred at room temperature overnight. The solid was filtered and washed by 1 mL of ether. All solvent was removed in vacuo to give [Mo(NAd)(CHCMe2Ph)(Cl)(PMe3)]2(μ-O) (6a(PMe3) as an off-white powder; yield 27 mg (67%). Only alkylidene NMR resonances are listed here: 1H NMR (400 MHz, C6D6) δ 13.57 (d, 3JHP = 6.3 Hz, 2H, Mo=CH); 13C{1H} NMR (151 MHz, C6D6) δ 307.7 (d, 2JCP = 19.8 Hz, Mo=C). Anal. Calcd for C46H72Cl2Mo2N2OP2 (993.86 g/mol): C, 55.59%; H, 7.30%; N, 2.82%. Found: C, 55.60%; H, 7.11%; N, 2.24%.
Crystals of 6a(PMe3) suitable for X-ray data collection were obtained through crystallization from a mixture of pentane and toluene – 20 °C.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful for financial support from the National Institutes of Health (GM-59426) and from the National Science Foundation under the CCI Center for Enabling New Technologies through Catalysis (CENTC; CHE-1205189). We thank Jonathan Becker for one of the structural solutions and the group of Professor T. F. Jamison for GC studies.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACSPublicationswebsite at DOI: 10.1021/acs.organomet.7b00647.
NMR data and spectra for all compounds, details of the cross-metathesis experiments, and X-ray data for the four structures (PDF)
Accession Codes
CCDC 1572637–1572640 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Schrock RR; Murdzek JS; Bazan GC; Robbins J; DiMare M; O’Regan M J. Am. Chem. Soc 1990, 112, 3875–3886. [Google Scholar]; (b) Schrock RR Chem. Rev 2002, 102, 145–180. [DOI] [PubMed] [Google Scholar]; (c) Schrock RR Chem. Rev 2009, 109, 3211–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Schrock RR Handbook of Metathesis, Vol 1, 2nd Ed., Wiley-VCH; Weinheim, Grubbs RH and Wenzel AG, Ed., 2015, pp. 1–32. [Google Scholar]
- (2).(a) Wang C; Yu M; Kyle AF; Jacubec P; Dixon DJ; Schrock RR; Hoveyda AH Chem. Eur. J 2013, 19, 2726–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang C; Haeffner F; Schrock RR; Hoveyda AH Angew. Chem. Int. Ed 2013, 52, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kiesewetter ET; O’Brien RV; Yu EC; Meek SJ; Schrock RR; Hoveyda AH J. Am. Chem. Soc 2013, 135, 6026–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mann TJ; Speed AWH; Schrock RR; Hoveyda AH Angew. Chem. Int. Ed 2013, 52, 8395–8400. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Speed AWH; Mann TJ; O’Brien RV; Schrock RR; Hoveyda AH J. Am. Chem. Soc 2014, 136, 16136–16139. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Zhang H; Yu EC; Torker T; Schrock RR; Hoveyda AH J. Am. Chem. Soc 2014, 136, 16493–16496. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nguyen TT; Koh M-J; Shen X; Romiti F; Schrock RR; Hoveyda AH Science 2016, 352, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Shen X; Xu D; Nguyen TT; Koh MJ; Speed AWH; Schrock RR; Hoveyda AH Nature 2017, 541, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Jeong H; John JM; Schrock RR Organometallics 2015, 34, 5136–5145. [Google Scholar]; (b) Flook MM; Ng VWL; Schrock RR J. Am. Chem. Soc 2011, 133, 1784–1786. [DOI] [PubMed] [Google Scholar]; (c) Flook MM; Börner J; Kilyanek S; Gerber LCH; Schrock RR Organometallics 2012, 31, 6231–6243. [Google Scholar]; (d) Jeong H; Ng VWL; Börner J; Schrock RR Macromolecules 2015, 48, 2006–2012. [Google Scholar]; (e) Autenrieth B; Schrock RR Macromolecules 2015, 48, 2493–2503. [Google Scholar]; (f) Hyvl J; Autenrieth B; Schrock RR Macromolecules 2015, 48, 3148–3152. [Google Scholar]; (g) Schrock RR Acc. Chem. Res 2014, 47, 2457–2466. [DOI] [PubMed] [Google Scholar]; (h) Jang ES; John JM; Schrock RR ACS Cent. Sci 2016, 2, 631–636. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Jang ES; John JM; Schrock RR J. Am. Chem. Soc 2017, 139, 5043–5046. [DOI] [PubMed] [Google Scholar]
- (4).(a) Koh MJ; Nguyen TT; Lam J; Torker S; Hyvl J; Schrock RR; Hoveyda AH Nature 2017, 542, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Koh M-J; Nguyen TT; Zhang H; Schrock RR; Hoveyda AH Nature, 2016, 531, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lam JK; Zhu C; Bukhryakov KV; Schrock RR; Müller PM; Hoveyda AH J. Am. Chem. Soc 2016, 138, 15774–15783. [DOI] [PubMed] [Google Scholar]
- (6).(a) Gerber LCH; Schrock RR; Müller P; Takase MK J. Am. Chem. Soc 2011, 133, 18142–18144. [DOI] [PubMed] [Google Scholar]; (b) Gerber LCH; Schrock RR; Müller P Organometallics 2013, 32, 2373–2378. [Google Scholar]
- (7).Axtell JC; Schrock RR; Müller P; Hoveyda AH Organometallics 2015, 34, 2110–2113. [Google Scholar]
- (8).Jeong H; Axtell J; Török B; Schrock RR; Müller P Organometallics 2012, 31, 6522–6525. [Google Scholar]
- (9).Jeong H; Schrock RR; Müller P Organometallics 2015, 34, 4408–4418. [Google Scholar]
- (10).Oskam JH; Schrock RR J. Am. Chem. Soc 1993, 115, 11831–11845. [Google Scholar]
- (11).Addison AW; Rao TN; Van Rijn J; Veschoor GC; Reedijk JJ Chem. Soc., Dalton Trans 1984, 1349–1356. [Google Scholar]
- (12).Bukhryakov KV; Schrock RR; Hoveyda AH; Müller P; Becker J Organic Letters 2017, 19, 2607–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Schrock RR; Crowe WE; Bazan GC; DiMare M; O’Regan MB; Schofield MH Organometallics 1991, 10, 1832–1843. [Google Scholar]
- (14).Sues PE; John JM; Schrock RR; Müller P Organometallics 2016, 35, 758–761. [Google Scholar]
- (15).Gordon C; Yamamoto K; Liao W-C; Allouche F; Andersen RA; Copéret C; Raynaud C; Eisenstein O ACS Cent. Sci 2017, 3, 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wampler KM; Hock AS; Schrock RR Organometallics 2007, 26, 6674–6680. [Google Scholar]
- (17).Schoettel T; Kress J; Fischer J; Osborn JAJ Chem. Soc., Chem. Commun 1988, 914–915. [Google Scholar]
- (18).(a) Chen P; Zhang L; Xue Z-L; Wu Y-D; Zhang X Inorg. Chem 2017, 56, 7111–7119. [DOI] [PubMed] [Google Scholar]; (b) O’Reilly ME; Ghiviriga I; Abboud KA; Veige AS J. Am. Chem. Soc 2012, 134, 11185–11195. [DOI] [PubMed] [Google Scholar]; (c) Feinstein-Jaffe I; Dewan JC; Schrock RR Organometallics 1985, 4, 1189. [Google Scholar]; (d) Feinstein-Jaffe I; Pedersen SF; Schrock RR J. Am. Chem. Soc 1983, 105, 7176. [Google Scholar]; (e) Feinstein-Jaffe I; Gibson D; Lippard SJ; Schrock RR; Spool AJ Am. Chem. Soc 1984, 106, 6305. [Google Scholar]; (f) Morton LA; Miao M; Callaway TM; Chen T; Chen S-J; Tuinman AA; Yu X; Lu Z; Xue Z-L Chem. Commun 2013, 49, 9555. [DOI] [PubMed] [Google Scholar]
- (19).Oskam JH; Fox HH; Yap KB; McConville DH; O’Dell R; Lichtenstein BJ Schrock RRJ Organometal. Chem 1993, 459, 185–198. [Google Scholar]
- (20).(a) Fox HH; Yap KB; Robbins J; Cai S; Schrock RR Inorg. Chem 1992, 31, 2287–2289. [Google Scholar]; (b) Dyer PW; Gibson VC; Howard JAK; Whittle B; Wilson CJ Chem. Soc., Chem. Commun 1992, 1666–1668. [Google Scholar]
- (21).Schoettel G; Kress J; Osborn JAJ Chem. Soc., Chem. Commun 1989, 1062–1063. [Google Scholar]
- (22).Jeong H; Kozera DJ; Schrock RR; Smith SJ; Zhang J; Ren N; Hillmyer MA Organometallics 2013, 32, 4843–4850. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.