

Abstract
Background
Information about the association with alpha thalassemia in sickle cell patients is unknown in the Democratic Republic of Congo. There is very little data on the alpha thalassemia in patients suffering from sickle cell anemia in Central Africa, and their consequences on the clinical expression of the disease.
Methods
A cross‐sectional study was conducted in 106 sickle cell patients living in the country's capital Kinshasa. The diagnosis of sickle cell anemia was confirmed with a molecular test using PCR‐RFLP (restriction fragment length polymorphism) technique. The diagnosis of thalassemia was performed by the technique of multiplex ligation dependent probe amplification.
Results
The mean age of our patients was 22.4±13.6 years. The α3.7 heterozygous deletion, the α3.7 homozygous deletion and the α3.7 triplication were respectively encountered in 23.6%, 25.5% , and 11.3% of patients. Patients with normal αα/αα genotype represented 39.6% of the study population. The average of severe vaso‐occlusive crises, the rates of blood transfusions per year, the rate of osteonecrosis, cholelithiasis and leg ulcers were significantly lower in the group of patients with α3.7 homozygous deletion and α3.7 triplication.
Conclusion
The prevalence of α3.7 triplication was higher in sickle cell patients in the Democratic Republic of Congo than in worldwide series. The α3.7 triplication and α3.7 homozygous deletion were associated with less severe forms of the Sickle cell anemia in Congolese patients. These results showed the need to investigate systematically the alpha‐globin gene mutations in sickle cell population in Central Africa.
Keywords: alpha thalassemia; Kinshasa; multiplex ligation dependent probe amplification; sickle cell anemia; the Democratic Republic of Congo, Africa; α3.7 triplication
1. Introduction
Sickle cell anemia (SCA) is a qualitative hemoglobinopathy characterized by the substitution of glutamic acid for valine at the sixth codon of the beta chain S globin.1 This substitution results in the production of an abnormal protein called hemoglobin S (HbS) .
The disease is characterized by its extreme clinical heterogeneity. This clinical polymorphism is influenced by environmental factors and genetic factors.2, 3, 4 Genetic factors modulators of sickle cell phenotype have been associated with asymptomatic phenotype. These genetic factors include the influence of fetal hemoglobin (HbF) (α2γ2) and the association between SCA and alpha‐thalassemia.5, 6, 7
The modulator and protective effect of alpha‐thalassemia on clinical manifestations of sickle cell anemia is due to the fact that the decrease of the mean corpuscular hemoglobin concentration. The hypochromic anemia resulting from this situation reduces the risk of the polymerization of the HbS.2
Thalassemic syndromes are very common in malaria‐endemic areas.8 Silent alpha thalassemia form (−α/αα) and cis or trans alpha‐thalassemia minor forms are most encountered in sub‐Saharan Africa.9 These forms are often clinically asymptomatic.10 The 3.7 kb alpha‐globin chain deletion (α3.7 kb deletion) is a common silent form encountered.11, 12, 13 In previous studies, the α‐triplication was reported with low frequency in sub‐Saharan populations.14
The Democratic Republic of Congo (DRC) has the second highest incidence of SCA patients in Africa after Nigeria. The prevalence of the disease is estimated from 0.97% to 1.4% in newborns and an incidence of SCA is estimated to be 30 000 to 40 000 neonates per year.15, 16, 17 In Congolese population, the disease is characterized by a low level of fetal hemoglobin and F‐Cells, and the risk of developing severe clinical symptomatology.
Despite high prevalence of this hemoglobinopathy in the DRC, information about the association between SCA and alpha thalassemia in population suffering from SCA are unknown. We therefore conducted a cross‐sectional study in sickle cell patients living in Kinshasa, the DRC. The objectives of this study were to estimate the prevalence of this association and to assess its influence on clinical and hematological profile in our midst.
2. Methods
2.1. Study design and population
The present cross‐sectional study is the first part of a larger ongoing study of genetic overview in sickle cell anemia in the DRC. The study was conducted in Sickle cell center of Yolo/Kalamu in Kinshasa February 2013 and April 2014, the capital of the DRC. This health facility provides most of the public beds in the DRC for sickle cell patients.
The samples were collected from patients in steady state regularly followed up at the outpatient clinic of Sickle cell center of Yolo. All patients were free of pain for at least 1 month and had not been hospitalized for at least 100 days before the study.19
We excluded subjects with (i) initiated antibiotics treatment prior to seeking medical care; (ii) previous blood transfusion in the 3 months prior to the study (iii) under hydroxyurea (iv) under chronic transfusion program.
2.2. Data collection procedure and blood analysis
Five milliliters (mL) of venous blood sample were drawn from each study participant into an Ethylenediaminetetraacetic acid (EDTA) tube, used to determine hematologic parameters. These were determined using an automated Hematology Analyzer Sysmex XS‐1000i (Lincolnshire, IL, USA). Hematological and biochemical analyzes were performed in the laboratory of the Institut National de Recherche Biomédicale de of Kinshasa (INRB). Molecular analyzes were performed at Center for Human Genetics, University hospital, Katholieke Universiteit de Leuven, Belgium.
2.3. DNA extraction
Genomic DNA was extracted from 5 mL of venous blood collected in a tube with EDTA, using the method of “Salting out”.
2.4. Diagnosis of sickle cell anemia
Sickle cell screening was performed using semi‐automated electrophoresis technique with the Hydrasis II apparatus (Sebia, Lysse, France). The electrophoresis technique separates hemoglobin in acid and alkaline agarose gel. SCA was diagnosed in the presence of production of HbS with no Hb ‐A. The concentrations were measured by an integrated densitometer.
2.5. Confirmation of the diagnosis of sickle cell anemia
The DNA was concentrated to 50 ηg/μL with a robot dropsence (Trinean NV, Gand, Belgium).
The diagnosis of sickle cell anemia was confirmed with a molecular test with a molecular test using PCR‐RFLP technique. This test sought E6V mutation in the mutated HBB gene. A fragment of 440 bp HBB gene was amplified with the following primers: For: TGTGGAGCCACACCCTAGGGTTG, Rev: CATCAGGAGTGGACAGATCC. The primers were supplied by Integrated DNA Technologies. The amplified fragment contains two sites of restriction for the enzyme DdeI. The first restriction site is eliminated in the presence of the mutation E6V. After enzymatic digestion, normal individuals (AA) had three fragments of 72 bp, 167 bp and 201 bp, respectively. The heterozygous individuals (AS) had four fragments of 72 bp, 167 bp, 201 bp and 368 bp, respectively. Homozygous subject (SS) had two fragments of 72 bp and 368 bp, respectively. The amplification after digestion was performed with a thermal cycler ABI GenAmp 2700 (Foster City, CA, USA).
2.6. The diagnosis of thalassemia
The diagnosis of thalassemia was performed by the technique of multiplex ligation dependent probe amplification (MLPA). The analysis was performed using the SALSA MLPA kit P140 probemix HBA provided by MRC‐Holland (Amsterdam, the Netherlands).
The technique used for the diagnosis comprises five steps: (a) denaturation: in a mixture of 4 μL of water with 1 μL of genomic DNA concentration to 50 ηg/μL at 98°C for 5 min.(b) hybridization: In each microtube, 3 μL of mix consisting of 1.5 μL of probe mix and 1.5 μL of MLPA buffer were added at room temperature. The hybridization was performed at 60°C for 16 hours (c) ligation: In each microtube, 32 μL of mix consisting of 3 μl of buffer A, 3 μL of buffer B, 25 μL of water and 1 μL of ligase were added at 54°C. The ligation of the program comprises a phase at 54°C for 15 min and another phase at 98°C for 5 min. (d) PCR MLPA: 10 μL of a mixture consisting of 2 μL of primers, 7.5 μL of water and 0.5 μL of polymerase were added at room temperature. Amplification was performed for 35 cycles and included an initiation phase at 95°C for 30 s, followed by hybridization at 60°C for 30 s, an elongation at 72 ° C for 1 minute and a final elongation at 72°C for 20 min. (e) The analysis of the fragments was carried out after denaturation at 96°C for 3 min with a mixture made of 1 μL of the PCR mixture and 20 μL HiDi Formamide/Rox (Applied Biosystems, Foster City, CA, USA). The fragments of MLPA were analyzed with the software GeneMarker v1.91.
The various alpha‐thalassemia deletions were categorized according to the ratios of the probes of the deletion area (Table 1). The normal ratio is between 0.75 and 1.25. Deletion was defined for a ratio lower than 0.75 and triplication for a ratio higher than 1.25. Details are shown in Figures 1, 2 and 3.
Table 1.
The ratio of the probes in the different alpha‐thalassemia polymorphisms
| Probes | Heterozygous α3.7 deletion (Figure 1) ratio | Homozygous α3.7 deletion (Figure 2) ratio | α3.7 Triplication (Figure 3) ratio |
|---|---|---|---|
| 08498‐L08422 | 0.5 | 0 | +1 copy |
| 04633‐L23748 | 0.5 | 0 | +1 copy |
| 18096‐L22520 | 0.5 | 0 | +1 copy |
| 18880‐L24428 | 0.5 | 0 | +1 copy |
| 08494‐L08417 | 0.5 | 0 | +1 copy |
| 14855‐L23604 | 0.5 | 0 | + 1 copy |
| 18093‐L22517 | 0.5 | 0 | +1 copy |
Figure 1.
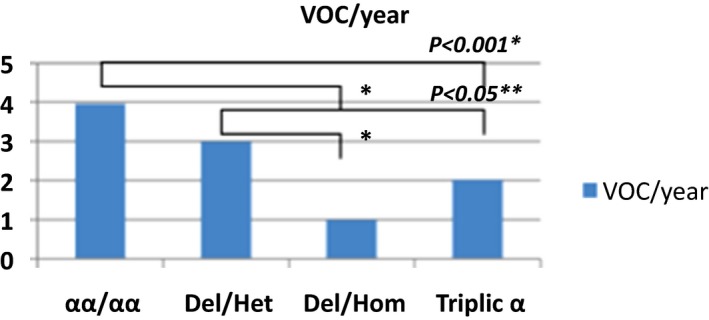
Severe Vaso‐occlusives crises per year
Figure 2.
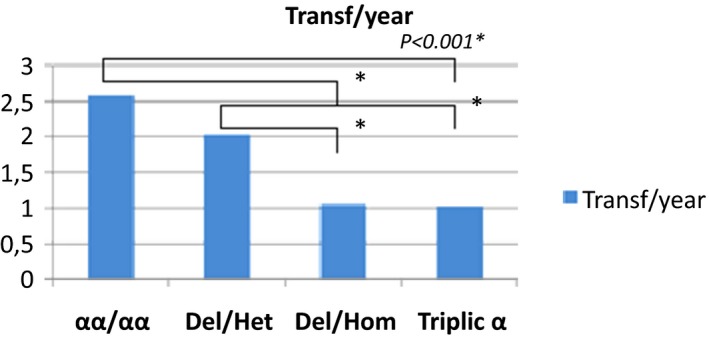
Mean of blood transfusion per year in the study population
Figure 3.
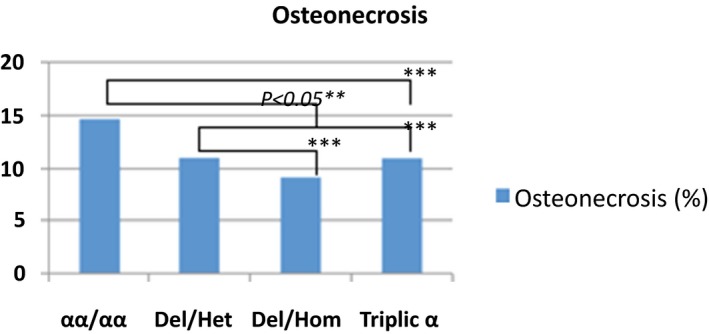
Frequency of osteonecrosis in the study population
2.7. Ethical considerations
All major participants provided written consent for study participation. Since some participants were minors, their legal guardians provided written consent for study participation. This consent procedure and the study were reviewed and approved by the National Ethical Committee of the Public Health School of the University of Kinshasa, Kinshasa, the DRC (ESP/CE/027B/2011), in compliance with the principles of the Helsinki Declaration II. The aim and the procedures of the study were explained to the participants. The participants were informed that they could withdraw anytime without further obligation. None of the authors collected samples. Samples were collected and sent to the authors by the Research unit of Sickle cell Centre of Yolo. Anonymity of the participants was guaranteed and no personal details were recorded.
2.8. Data management and analysis
Data are represented as means±standard deviation (SD) when the distribution was normal or median with range when the distribution was not normal. Frequency of various clinical and laboratory findings are expressed as percentages. The confidence interval at 95% was calculated. Associations between variables and subtypes of polymorphisms were evaluated using chi‐square and fisher exact test. A P value <.05 was considered significant.
2.9. Role of the funding source
None of the funders had involvement in the study design, data collection, data analysis, data interpretation, writing of the report, or decision to submit for publication. The corresponding author had full access to all the data in the study and had the final responsibility for the decision to submit for publication.
3. Results
The mean age of our patients was 22.4±13.6 years. The sex ratio female/male was 3.2.
3.1. Alpha thalassemia polymorphism
The α3.7 heterozygous deletion (silent thalassemia) was encountered in 25 (23.6%) patients, α3.7 homozygous deletion (thalassemia minor) in 27 (25.5%) patients, the α3.7 triplication was encountered in 12 (11.3%) patients. Forty two (39.6%) patients with normal genotype αα/αα were identified.
3.2. Hematologic parameters in the study population
The Hb and HbF were significantly lower in the group of patients with triplication. Leukocytosis and reticulocytosis were similar in all groups. However, the group of patients with α3.7 triplication had the lowest rate of platelets with a statistically significant difference. Mean corpuscular volume (MCV) and mean concentration hemoglobin (MCHC) were significantly lower in the group of patients with homozygous α3.7 deletion and α3.7 triplication. Other details are shown in Table 2.
Table 2.
Hematologic characteristics of the study population according to alpha‐thalassemia polymorphisms
| Variables | αα/αα (n=42) | Heterozygous α3.7 deletion (n=25) | Homozygous α3.7 deletion (n=27) | α3.7 Triplication (n=12) | P (ANOVA) |
|---|---|---|---|---|---|
| Age (years) | 22.5±5.1 | 20.1±3.6 | 23.8±4.5 | 23.8±3.3 | .017 |
| Hb (g/dL) | 8.6±1.4 | 9.0±1.2 | 8.6±1.9 | 7.2±0.4 | .009 |
| HbF (%) | 9.1±5.1 | 13.6±4.6 | 9.7±6.0 | 5.2±2.9 | <.001 |
| HbA2 (%) | 2.0±0.5 | 1.7±0.7 | 2.0±0.6 | 2.9±0.5 | <.001 |
| GB (×103/μL) | 10.3±4.0 | 9.0±3.1 | 8.2±2.6 | 8.4±3.0 | .07 |
| Reticul (%) | 12.8±7.9 | 12.2±6.7 | 10.2±6.8 | 9.9±4.9 | .37 |
| Platelets (×103/μL) | 360.4±246.9 | 272.4±145.6 | 250.4±95.0 | 249.4±96.9 | .047 |
| MCV (ƒL) | 77.6±9.0 | 80.5±7.9 | 72.2±6.8 | 78.6±9.4 | .003 |
| MCHC (g/dL) | 34.0±1.5 | 33.3±1.2 | 32.8±1.0 | 32.6±1.1 | <.001 |
MCH, mean corpuscular volume; MCHC, mean corpuscular hemoglobin concentration.
3.3. Frequency of acute and chronic complications of SCA
The average of severe vasoocclusive crisis (VOC), the rates of blood transfusions per year, the rate of osteonecrosis, cholelithiasis and the rate of leg ulcers were significantly lower in the group of patients with homozygous α3.7 deletion and α3.7 triplication (Figures 1, 2, 3, 4, 5).
Figure 4.
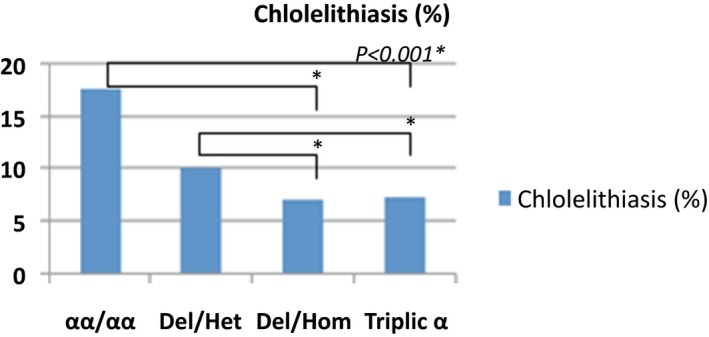
Frequency of cholelithiasis in the study population
Figure 5.
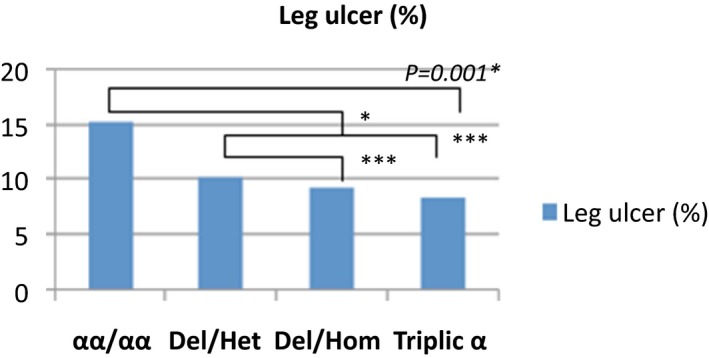
Frequency of leg ulcers in the study population
3.4. Relationship between alpha‐thalassemia polymorphism and microcytosis
Microcytosis was more frequent in sickle cell patients with homozygous deletion (thalassemia minor) and was found in 17 (16.03%) cases. Microcytosis was a rare event in sickle cell patients with heterozygous α3.7 heterozygous deletion (1.9%), α3.7 triplication (0.9%) and also in those with normal α genotype (2.8%).
4. Discussion
Alpha‐thalassemia associated with SCA has been described as a modulating factor of sickle cell phenotype.6, 20, 21 Despite the high prevalence and incidence of the disease in Central Africa, there is still very little information on alpha‐thalassemia in patients suffering from SCA living in this part of the world.9, 21, 22, 23 This study was the first to be performed specifically in Central Africa where sickle cell anemia is most frequent with a predominance of Bantu haplotype.
Our study showed that association between alpha‐thalassemia and SCA is frequent in our midst. Indeed, nearly 49% of sickle cell patients are carriers of a deletional thalassemia. Among them, 25.5% had a homozygous α3.7 deletion and 23.6% had a heterozygous α3.7 deletion. This frequent association between alpha‐thalassemia and sickle cell anemia has been reported in several African and Arab‐Indian studies.9, 24, 25, 26 On the other hand, the frequency of α3.7 triplication observed in this Congolese series is the highest reported compared to worldwide series where the frequencies range from 1.3% to 5%.24, 27, 28, 29 This difference may be explain by the fact that the frequency of α‐triplication varies according to population origin.30 However, the frequency in non SCA Congolese population is unknown.
We report a lower level of Hb, leukocytes, platelets, HbF, MCV and MCHC in patients with sickle cell anemia with co‐existent α3.7 triplication. Low MCHC and low MCV are described as factors that could reduce morbidity in SCA.2, 31 The decrease of MCHC is a major factor which reduces the risk of polymerization of HbS, while the low MCV is a factor which improves rheology in SCA patients.2 Furthermore, it is well known that leukocytosis, thrombocytosis and increased reticulocyte production are major factors of acute sickle cell crises.31, 32, 33, 34 Thus, the low expression of these disease markers could explain the protective effect of homozygous deletion and triplication on the severity of the SCA.
Our study showed that the annual average of severe VOC and lower blood transfusions were low in patients with α3.7 triplication and homozygous α3.7 deletion compared to patients with normal genotype αα/αα and heterozygous α3.7 deletion. The reduction of VOC and lower blood transfusions may be associated with the modification of hematological parameters changes observed in these two groups. Indeed, the risk of VOC risk in SCA is increased by the cytoadherence of granulocytes and reticulocytes to endothelium. Additionally, the risk of VOC in SCA is associated with increased cytoadherence of polynuclear and reticulocytes to endothelium, while platelets are involved in VOC by the secretion of thrombospondin.35, 36, 37
Hemolysis in sickle cell disease is often secondary to the process of polymerization and the vaso‐occlusion. Chronic complications of hyperhemolysis are gallstone cholecystitis and leg ulcers.38, 39 In our series, cholelithiasis and leg ulcers were less frequent in groups of patients with α3.7 triplication and homozygous α3.7 deletion. Alpha thalassemia was identified as a protective factor against cholelithiasis and leg ulcers.40
In this report, patients with homozygous α3.7 deletion and α3.7 triplication were less likely to develop osteonecrosis of the femoral head compared to other groups. The protective effect of alpha‐thalassemia of osteonecrosis is not well established. In a previous study, SCA patients with alpha‐gene deletion were at the greatest risk for osteonecrosis.41, 42, 43 In this series, the protective effect of the homozygous α3.7 deletion and α3.7 triplication observed could be associated with the observed hematological changes in these two groups.
These results are in contrast with those of India in which clinical symptoms occurred at higher frequency and were more severe. This difference may be due to the fact that in their series, the authors included patients with α3.7 triplication in the without α‐deletion group of patients.24 Furthermore, in sickle cell β‐thalassemia patients, the presence of an alpha triplication worsens the clinical state.44
5. Conclusion
The prevalence of the association between SCA and alpha thalassemia encountered has been described and the influence on clinical and hematological profile assessed in our midst. In our study, the prevalence of α3.7 triplication was higher than those reported in worldwide series. The homozygous deletion and triplication were associated with less severe forms of the SCA in Congolese patients. These results showed the need to investigate systematically the alpha‐globin gene mutations in sickle cell population in our midst. Providing data that can properly be used for advocacy as well as for developing further strategies for improving the care of the SCA patients is very important for region with high prevalence of the disease such as the Central Africa.
Authors' contribution
TMM, PLT, and JMMM conceived, designed, deployed and directed the case–control study at the Sickle cell center of Yolo and at the Faculty of Medicine of University of Kinshasa. TMM carried out patient recruitment and follow‐up, sample collection, storage and transport. TMM, VR, AL PLT, and JMMM analyzed data. TMM and MNA wrote the manuscript. PLT, JMMM, KD, GM, AL and VR brought some precious corrections. All authors read and approved the final manuscript.
Acknowledgments
The authors thank all the patients who participated in this study. We thank all our colleagues involved in the collection of samples, all physicians and the nurses of the Sickle Cell Center of Yolo for the support given for the present study. This research project was supported by grant from the ALUMNI/KU Leuven, Belgium.
Mikobi TM, Lukusa PT, Aloni MN, et al. Association between sickle cell anemia and alpha thalassemia reveals a high prevalence of the α3.7 triplication in congolese patients than in worldwide series. J Clin Lab Anal. 2018;32:e22186 10.1002/jcla.22186
References
- 1. Ingram VM. A specific chemical difference between the globins of normal human and sickle‐cell anaemia haemoglobin. Nature. 1956;178:792‐794. [DOI] [PubMed] [Google Scholar]
- 2. Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129:465‐481. [DOI] [PubMed] [Google Scholar]
- 3. Thein SL. Genetic modifiers of the beta‐haemoglobinopathies. Br J Haematol. 2008;141:357‐366. [DOI] [PubMed] [Google Scholar]
- 4. Sebastiani P, Solovieff N, Hartley SW, et al. Genetic modifiers of the severity of sickle cell anemia identified through a genome‐wide association study. Am J Hematol. 2010;85:29‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87:795‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thein SL. Genetic association studies in β‐hemoglobinopathies. Hematology Am Soc Hematol Educ Program. 2013;2013:354‐361. [DOI] [PubMed] [Google Scholar]
- 8. Hedrick PW. Selection and mutation for α Thalassemia in nonmalarial and malarial environments. Ann Hum Genet. 2011;75:468‐474. doi: 10.1111/j.1469-1809.2011.00653.x. [DOI] [PubMed] [Google Scholar]
- 9. Rumaney MB, Ngo Bitoungui VJ, Vorster AA, et al. The co‐Inheritance of alpha‐thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rates in Cameroonian patients and could improve their survival. PLoS ONE. 2014;9:e100516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harteveld CL, Higgs DR. Alpha‐thalassaemia. Orphanet J Rare Dis. 2010;5:13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodgers GP, Steinberg MH. Pharmacologic treatment of sickle cell disease and thalassemia: the augmentation of fetal hemoglobin In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, eds. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management, 1st edn Cambridge, United Kingdom: Cambridge University Press; 2001:1028‐1051. [Google Scholar]
- 12. Suchdev PS, Ruth LJ, Earley M, et al. The burden and consequences of inherited blood disorders among young children in western Kenya. Matern Child Nutr. 2014;10:135‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Veenemans J, Andang'o PE, Mbugi EV, et al. Alpha+‐thalassemia protects against anemia associated with asymptomatic malaria: evidence from community‐based surveys in Tanzania and Kenya. J Infect Dis. 2008;198:401‐8. [DOI] [PubMed] [Google Scholar]
- 14. Bernini LF, Harteveld CL. Alpha‐thalassaemia, In: Rodgers GP, ed. Bailliere's Clinical Haematology International Practice and Research: Sickle Cell Disease and Thalassaemia, Vol. 11 London: Bailliere Tindall; 1998:53‐90. [DOI] [PubMed] [Google Scholar]
- 15. Piel FB, Hay SI, Gupta S, et al. Global burden of sickle cell anaemia in children under five, 2010‐2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10:e1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tshilolo L, Aissi LM, Lukusa D, et al. Neonatal screening for sickle cell anaemia in the Democratic Republic of the Congo: experience from a pioneer project on 31 204 newborns. J Clin Pathol. 2009;62:35‐38. [DOI] [PubMed] [Google Scholar]
- 17. Agasa B, Bosunga K, Opara A, et al. Prevalence of sickle cell disease in a northeastern region of the Democratic Republic of Congo: what impact on transfusion policy? Transfus Med. 2010;20:62‐65. [DOI] [PubMed] [Google Scholar]
- 18. Tshilolo L, Summa V, Gregorj C, et al. Foetal haemoglobin, erythrocytes containing foetal haemoglobin, and hematological features in congolese patients with sickle cell anaemia. Anemia. 2012;2012:105349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ballas SK. More definitions in sickle cell disease: steady state v base line data. Am J Hematol. 2012;87:338. [DOI] [PubMed] [Google Scholar]
- 20. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mouélé R, Pambou O, Feingold J, et al. Alpha‐thalassemia in Bantu population from Congo‐Brazzaville: its interaction with sickle cell anemia. Hum Hered. 2000;50:118‐125. [DOI] [PubMed] [Google Scholar]
- 22. Lubega I, Ndugwa CM, Mworozi EA, et al. Alpha thalassemia among sickle cell anaemia patients in Kampala. Uganda. Afr Health Sci. 2015;15:682‐689. doi: 10.4314/ahs.v15i2.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox SE, Makani J, Soka D, et al. Haptoglobin, alpha‐thalassaemia and glucose‐6‐phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol. 2014;165:699‐706. doi: 10.1111/bjh.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pandey S, Pandey S, Mishra RM, et al. Genotypic influence of α‐deletions on the phenotype of Indian sickle cell anemia patients. Korean J Hematol. 2011;46:192‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nouraie M, Lee JS, Zhang Y, et al. Walk‐PHASST Investigators and Patients. The relationship between the serverity of hemolysis clinical manifestation and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013;98:464‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wali YA, Al‐Lamki Z, Hussein SS, et al. Splenic function in Omani children with sickle cell disease: correlation with severity index, hemoglobin phenotype, irion status, and alpha‐thalassemia trait. Pediatr Hematol Oncol. 2002;19:491‐500. [DOI] [PubMed] [Google Scholar]
- 27. Kumar R, Panigrahi I, Dalal A, et al. Sickle cell anemia–molecular diagnosis and prenatal counseling: SGPGI experience. Indian J Pediatr. 2012;79:68‐74. doi: 10.1007/s12098-011-0510-1. [DOI] [PubMed] [Google Scholar]
- 28. Ataulfo González F, Blázquez C, Ropero P Grupo de Eritropatología , et al. Association of hemoglobinopathy and alpha thalassemia. Study of 45 patients. Med Clin (Barc). 2005;124:726‐9. [DOI] [PubMed] [Google Scholar]
- 29. Kéclard L, Ollendorf V, Berchel C, et al. Beta S haplotypes, alpha‐globin gene status, and hematological data of sickle cell disease patients in Guadeloupe (F.W.I.). Hemoglobin. 1996;20:63‐74. [DOI] [PubMed] [Google Scholar]
- 30. Goossens M, Dozy AM, Embury SH, et al. Triplicated α‐globin loci in humans. Proc Natl Acad Sci USA. 1980;77:518‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El‐Hazmi MAF. Clinical manifestation and laboratory findings of sickle cell anaemia in association with alpha‐thalassemia in Saudi Arabia. Acta Haematol. 1985;74:155‐160. [DOI] [PubMed] [Google Scholar]
- 32. Conran N, Costa FF. Hemoglobin disorders and endothelial cell interactions. Clin Biochem. 2009;42:1824‐1838. [DOI] [PubMed] [Google Scholar]
- 33. Hebbel RP, Schwartz RS, Mohandas N. The adhesive sickle erythrocyte: cause and consequence of abnormal interactions with endothelium, monocytes/macrophages and model membranes. Clin. Haematol. 1985;191:141‐161. [PubMed] [Google Scholar]
- 34. Meier ER, Byrnes C, Weissman M, et al. Absolute reticulocyte count acts as a surrogate for fetal hemoglobin in infants and children with sickle cell anemia. PLoS ONE. 2015;10:e0136672. doi: 10.1371/journal.pone.0136672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hebbel RP. Blockade of adhesion of sickle cells to endothelium by monoclonal antibodies. N Engl J Med. 2000;342:1910‐1912. [DOI] [PubMed] [Google Scholar]
- 36. Morris CR. Vascular risk assessment in patients with sickle cell disease. Haematologica. 2011;96:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mikobi TM, Lukusa Tshilobo P, Aloni MN, et al. Correlation between the lactate dehydrogenase levels with laboratory variables in the clinical severity of sickle cell anemia in congolese patients. PLoS ONE. 2015;10:e0123568. doi: 10.1371/journal.pone.0123568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Longo‐Mbenza B, Ngiyulu R, Kizunda P, et al. Gallbladder disease in young Congolese with sickle cell anemia: an ultrasound survey. J Trop Pediatr. 2004;50:73‐77. [DOI] [PubMed] [Google Scholar]
- 40. Steinberg MH. Genetic etiologies for phenotypic diversity in sickle cell anemia. ScientificWorldJournal. 2009;9:46‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ballas SK, Talacki CA, Rao VM, et al. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha‐thalassemia. Hemoglobin. 1989;13:649‐655. [DOI] [PubMed] [Google Scholar]
- 42. Adekile AD, Gupta R, Yacoub F, et al. Avascular necrosis of the hip in children with sickle cell disease and high Hb F: magnetic resonance imaging findings and influence of alpha‐thalassemia trait. Acta Haematol. 2001;105:27‐31. [DOI] [PubMed] [Google Scholar]
- 43. Milner PF, Kraus AP, Sebes JI, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med. 1991;325:1476‐1481. [DOI] [PubMed] [Google Scholar]
- 44. Pandey SK, Pandey S, Ranjan R, et al. Phenotypic effect of α‐globin gene numbers on Indian sickle β‐thalassemia patients. J Clin Lab Anal. 2014;28:110‐113. doi: 10.1002/jcla.21652. [DOI] [PMC free article] [PubMed] [Google Scholar]