

Abstract
Background
To offer 4‐year clinical prenatal diagnosis experience of Duchenne muscular dystrophy (DMD).
Methods
Denaturing high‐performance liquid chromatography (DHPLC) and Sanger sequencing were used for molecular diagnosis of 237 DMD families.
Results
In the study, deletions, duplications, complex rearrangement and small mutations accounted for 47.3%, 8.4%, 1.7% and 42.6% of 237 families, respectively. Sixty‐six different deletion patterns were identified in 112 families. Fourteen different duplication patterns were identified in 20 families and 4 complex rearrangements were identified. About 87.1% different small mutation patterns were identified, including 37.6% different nonsense mutation patterns, 24.8% different frameshift mutation patterns, 7.9% different missense mutation patterns, and 16.8% different splice site mutation patterns. There was no significant difference in the age of onset and mutation patterns (P > .05). The follow‐up examinations revealed that the pregnancies of 14 cases were interrupted. Two cases were preterm births, 151 cases were delivered at term, 63 cases continued to pregnancy, and 7 cases were lost to follow‐up.
Conclusion
DHPLC and Sanger sequencing technique are efficient, sensitive, and specific in screening for DMD gene mutations. And pre‐pregnancy DMD gene examination is an important step to assess mutation type of family with suspected DMD and guides exactly prenatal diagnosis in high‐risk families.
Keywords: denaturing high‐performance liquid chromatography, dystrophin gene, prenatal diagnosis, Sanger sequencing
1. INTRODUCTION
Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy and presents in early childhood with proximal muscle weakness. Most patients cannot walk by the age of 12 and die because of cardiorespiratory complications at their late teens. DMD is inherited in an X‐linked recessive fashion, which affects approximately 1/5000 boys. Most of females are asymptomatic due to one single copy of the defective gene, however, 20% also show mild symptoms.1, 2 DMD is related to the mutation of DMD gene mutation, including deletions, duplications, small mutations, or other smaller gene rearrangements.3, 4 DMD severely affects the survival and life quality of the patients. Currently, comprehensive management including, corticosteroid therapy, surveillance of the respiratory, cardiac, orthopedic, nutritional, and general medical issues, greatly improve the life expectancy and quality of life.2 Gene therapy and stem cell therapy also bring great promises for the disease, however, there is still a long way.5, 6, 7 So the disease prevention is still the key, especially screening of female carrier and prenatal diagnosis.
The diagnosis of DMD relies on postnatal genetic testing using MLPA analysis,8 DHPLC9, 10 or next‐generation sequencing technology11, 12 or direct sequencing methods.13 Performing prenatal diagnosis can offer sufficient and valuable genetic information for DMD families, to make appropriate reproductive choices. Prenatal molecular diagnosis for DMD was firstly introduced by the Netherlands in 1985.14 At present, multiplex PCR, MLPA, and other techniques were tested to make prenatal diagnosis.15, 16, 17 However, there are also barriers associated with prenatal diagnosis of DMD, which include probands and carries screening, pre and post‐test counseling, and early completion of fetal diagnosis. In this study, we retrospectively analyzed clinical data on 237 DMD cases and the clinical application of DHPLC and direct Sanger sequencing in DMD families and assessed the validity of the strategy.
2. MATERIALS AND METHODS
2.1. Subjects
The study was approved by the Chinese People Liberation Army General Hospital medical ethics committee (Approval no. S2016‐120‐02). This retrospective cohort study included 237 DMD families, who underwent prenatal diagnostic testing in the prenatal diagnostic center of Shaanxi Xi Jing Hospital from November. 2011 to May 2016. Each family signed informed consent. Inclusion criteria were as follows: (i) a history of Duchenne muscular dystrophy or (ii) diagnosed as DMD by physical and biochemical examination finding or muscle biopsy (iii) from Nov. 2011 to May 2016. The exclusion criteria were as follows: (i) indefinite diagnosis (ii) wheelchair‐bound after age 13 years.
2.2. Karyotype analysis
Amniocentesis was performed under ultrasonographic guidance. Amniotic fluid samples (20 mL) were collected and centrifuged at 395 g for 8 minutes. After discarding the supernatant, the cells were suspended in 6 mL culture medium (GIBCO AmnioMAX‐II complete, Grand Island, NY, USA) and cultured for 9 days at 37°C and 5% CO2.18 Karyotype analysis was performed to exclude fetal abnormal karyotype.
2.3. Gene mutation examination
Genomic DNA was extracted from peripheral blood specimens or fetal samples using the NP986‐S nucleic acid extraction system (Tian Long, Xi'an, China). The major rearrangement of DMD gene was examined by denaturing high‐performance liquid chromatography (DHPLC) and multiplex PCR (mPCR). The copy number variation of samples was performed according to Zou et al.10 If a large deletion/duplication was not detected by DHPLC and multiplex PCR, PCR amplification, and Sanger sequencing using primer sets were used to confirm the diagnosis. The mutations were described according to the Leiden Muscular Dystrophy database (http://www.dmd.nl/). The nomenclature system was in 2000 on human mutation (reference sequence GenBank fileNM_004006.1).19
2.4. Gene mutation analysis
For prenatal diagnosis of monogenic disorders, thirteen STR markers within the dystrophin gene were applied to rule out maternal contamination (markers AMEL, D3S1358, D5S818, D8S1179, D13S317, D16S539, D18S51, D21S11, FGA, CSF1PO, TPOX, VWA, and TH01).
These sequences were analyzed by preliminary comparison with sequences in the GenBank database and software Vector NTI. The nucleotide position was assessed using the DMD reference sequence (RefSeq NM_004006.2) and mutation nomenclature followed the guidelines of the Human Genome Variation Society (HGVS). Small mutations can be determined according to LOVD database (Leiden Open Variation Database: www.dmd.nl), dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/), and clinVar database (http://www.ncbi.nlm.nih.gov/clinvar/?term=DMD%5Bgene%5D)20 as references. Unreported missense mutation or splice site mutation was evaluated using the Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/), SIFT algorithms (http://sift.jcvi.org/), and Alamut 2.7.2 to analyze pathogenicity. All substitutions with a Polyphen‐2 score below 1 and a SIFT score below 0.05 were considered damaging substitutions. In addition, when an amino acid substitution was detected in any of the 100 healthy controls, pathogenicity was rejected.
2.5. Genetic counseling and follow‐up
All pregnant women received counseling by genetic counselor according to a standardized protocol consistent with national guidelines.21 The pregnancy outcomes were acquired from delivery records if the pregnant women were delivered at our hospital; otherwise, they were gained by telephone follow‐up.
2.6. Statistical analysis
Statistical analyses for clinical data were performed using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). Between‐group comparisons were calculated using a chi‐square test. All values are described as mean ± SD. For all tests, P‐value less than .05 was considered as significant difference.22
3. RESULTS
3.1. Case characteristics
During the 4 years, we identified a total of 237 DMD families (214 with alive patients and 23 not alive patients) in our center. A total of 23 cases were primarily diagnosed as DMD by their relatives' description and case history. The mean gestational age at prenatal diagnostic testing was 26 ± 2 weeks (range of 18‐32 weeks). All fetuses had normal karyotype results.
3.2. Mutation spectrum
The diagnostic process and spectrum of mutations of 237 DMD families were summarized as a flowchart in Figure 1. In the 237 DMD families detected by DHPLC, deletions and duplications accounted for 47.3% (112/237) and 8.4% (20/237) of 237 families, respectively. We detected 1.7% (4/237) complex rearrangements in DMD families. The remaining 42.6% (101/237) were small mutations. 172 DMD different mutations were found in 237 cases.
Figure 1.
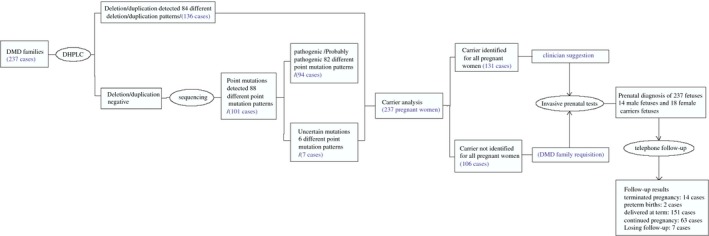
Stepwise duchenne muscular dystrophy patients mutation analysis and fetuses prenatal gene diagnosis results
3.3. Detection and duplication mutations
About 48.5% (66/136) different deletion patterns were detected in this study. The commonly observed deletion patterns were deletion of exons 49‐50 (6.3%, n = 7), deletion of exons 45‐50 (3.6%, n = 4), deletion of exons 46‐55(3.6%, n = 4), and deletion of exon 51 (3.6%, n = 4). 10.3% (14/136) different duplication patterns were identified in 20 families. The most common duplication patterns were duplication of exon 2 (20.0%, n = 4), followed by duplication of exon 66 (15%, n = 3), and duplication of exons 3‐4 (10%, n = 2). 2.9% (4/136) different complex rearrangements (Del 45‐47 and Dup 72; Del 49‐50 and Dup 66; Del 51 and Dup 48; Del 3‐5 and Dup 66) were identified in this study. Both deletions and duplications showed a great heterogeneity, because 52 of the 136 identified deletions and duplications were tested only once (Figure 2).
Figure 2.
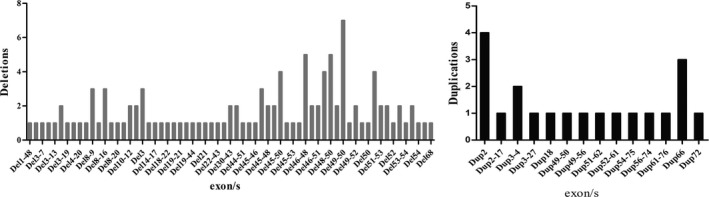
Duchenne muscular dystrophy mutations detected using DHPLC
3.4. Small mutations
We identified 87.1% (88/101) different small mutation patterns including 37.6% (38/101) different nonsense mutation patterns, 24.8% (25/101) different frameshift mutation patterns, 7.9% (8/101) different missense mutation patterns, and 16.8% (17/101) different splice site mutation patterns in 101 families. Eighty‐two different small mutations, which were reported to affect the DMD gene function, were identified in 101 families. However, six new small mutations (p.Asp556Asn, p.Gly882Asp, p.Gln2366Lys, p.Gln2806Arg, c.5586 + 18A>G, and c.9649 + 15T>C were firstly identified in this study. (Table 1).
Table 1.
Summary of different point mutation patterns mutations in 101 DMD patients
| Family No. | Disease onset | Mutation site | Protein level | Mutation type | Pathogenicity analysis |
|---|---|---|---|---|---|
| F132 | 4 | c.4178T>A | p.Leu1393X | nonsense | (++) |
| F52 | 6 | c.10033C>T | p.Arg3345X | nonsense | (+++) |
| F24 | 5 | c.10108C>T | p.Arg3370X | nonsense | (+++) |
| F29 | 6 | c.10141C>T | p.Arg3381X | nonsense | (+++) |
| F214 | 4 | c.1033C>T | p.Gln345X | nonsense | (++) |
| F235 | 5 | c.10546G>T | p.Glu3516X | nonsense | (++) |
| F79, F197 | 3,4 | c.1594C>T | p.Gln532X | nonsense | (++) |
| F164 | 5 | c.1886C>G | p.Ser629X | nonsense | (+++) |
| F152 | 6 | c.2137C>T | p.Thr715X | nonsense | (+++) |
| F149 | 6 | c.2302C>T | p.Arg768X | nonsense | (+++) |
| F189 | 3 | c.2308A>T | p.Lys770X | nonsense | (+++) |
| F62 | 6 | c.2665C>T | p.Arg889X | nonsense | (++) |
| F158 | 3 | c.2816T>A | p.Leu939X | nonsense | (+++) |
| F44 | 10 | c.2833C>T | p.Gln945X | nonsense | (++) |
| F147 | 4 | c.3388G>T | p.Glu1130X | nonsense | (++) |
| F184 | 4 | c.3940C>T | p.Arg1314X | nonsense | (+++) |
| F22, F45 | 2,2 | c.4174C>T | p.Gln1392X | nonsense | (++) |
| F15 | 6 | c.4232C>T | p.Gln1411X | nonsense | (++) |
| F206 | 5 | c.4996C>T | p.Arg1666X | nonsense | (+++) |
| F25, F104 | 2 | c.5287C>T | p.Arg1763X | nonsense | (+++) |
| F43 | 6 | c.5633C>T | p.Gln1878X | nonsense | (++) |
| F30 | 4 | c.615T>A | p.Tyr205X | nonsense | (+++) |
| F190 | 3 | c.6292C>T | p.Arg2098X | nonsense | (+++) |
| F78, F103 | 6,4 | c.6547G>T | p.Glu2183X | nonsense | (++) |
| F35 | 1 | c.7105G>T | p.Glu2369X | nonsense | (+++) |
| F2, F148 | 2,4 | c.7657C>T | p.Arg2553X | nonsense | (+++) |
| F4 | 6 | c.7705C>T | p.Gln2569X | nonsense | (++) |
| F185 | 4 | c.8038C>T | p.Arg2680X | nonsense | (+++) |
| F28 | 2 | c.829C>T | p.Gln277X | nonsense | (++) |
| F100 | 4 | c.8713C>T | p.Arg2905X | nonsense | (+++) |
| F156 | 2 | c.8740G>T | p.Glu2914X | nonsense | (++) |
| F151 | 2 | c.8944C>T | p.Arg2982X | nonsense | (+++) |
| F47 | 9 | c.9072G>A | p.Trp3024X | nonsense | (+++) |
| F150 | 4 | c.9337C>T | p.Arg3113X | nonsense | (+++) |
| F153 | 4 | c.9568C>T | p.Arg3190X | nonsense | (+++) |
| F171 | 3 | c.10171C>T | p.Arg3391X | nonsense | (+++) |
| F130 | 4 | c.1615C>T | p.Arg539X | nonsense | (+++) |
| F124 | 3 | c.3295C>T | p.Gln1099X | nonsense | (+++) |
| F11 | 5 | c.10231dupT | p.Thr3411 fs | frameshift | (+++) |
| F229 | 1 | c.1365_1366delGA | p.Gln455 fs | frameshift | (++) |
| F180 | 2 | c.1713insT | p.Phe571 fs | frameshift | (++) |
| F216 | 4 | c.1758delC | p.His586 fs | frameshift | (++) |
| F188 | 4 | c.2191delC | p.Leu731 fs | frameshift | (++) |
| F159 | 3 | c.2300delA | p.Glu767 fs | frameshift | (++) |
| F208 | 4 | c.2571delC | p.Pro857 fs | frameshift | (++) |
| F163 | 2 | c.264_264 + 4delTGTAA | p.Asn88 fs | frameshift | (++) |
| F26 | 5 | c.3257delA | p.Lys1086 fs | frameshift | (++) |
| F19 | 9 | c.4584inA | p.Gln1528 fs | frameshift | (++) |
| F186 | 6 | c.4630delA | p.Arg1544 fs | frameshift | (++) |
| F154 | 5 | c.4746_4747delCT | P. Val582 fs | frameshift | (++) |
| F155 | 3 | c.4808_4809insGGAA | p.Met1603 fs | frameshift | (++) |
| F146 | 1 | c.6033insTTAA | p.Leu2011 fs | frameshift | (++) |
| F187 | 3 | c.6045delA | p.Glu2015 fs | frameshift | (++) |
| F82 | 3 | c.6804_6807delACAA | p.Lys2268 fs | frameshift | (++) |
| F194 | 3 | c.7327_7328insA | p.Thr2443 fs | frameshift | (++) |
| F40 | 4 | c.7433inG | p.Ala2478 fs | frameshift | (++) |
| F32 | 4 | c.7885delAG | p.Asp2629 fs | frameshift | (++) |
| F94 | 6 | c.8104insT | p.Leu2702 fs | frameshift | (++) |
| F192 | 1 | c.8147‐8149insCAGAAGCTGAAACAACTGCCAATGTCCTACA | p.Gln2716 fs | frameshift | (++) |
| F105 | 4 | c.841_844delAGTC | p.Ser281 fs | frameshift | (++) |
| F215, F217 | 7,4 | c.8877delC | p.Ser2959 fs | frameshift | (++) |
| F96 | 3 | c.9461delT | p.Lys3154 fs | frameshift | (++) |
| F160 | 3 | c.9722_9723delCT | p.Ser456 fs | frameshift | (++) |
| F18 | 3 | c.1666G>A | p.Asp556Asn | missense | (+) |
| F57 | 3 | c.2645G>A | p.Gly882Asp | missense | (+) |
| F195 | 8 | c.3432G>T | p.Gln1144His | missense | (++) |
| F97 | 7 | c.5163G>C | p.Lys1721Asn | missense | (++) |
| F34, F39, F107 | 3,7,7 | c.5234G>A | p.Arg1745His | missense | (++) |
| F143, F173 | 5,3 | c.5234G>A, c.7096C>A | p.Arg1745His;p.Gln2366Lys | missense | (++) |
| F1, F141 | 4,6 | c.7096C>A | p.Gln2366Lys | missense | (+) |
| F136 | 6 | c.8417A>G | p.Gln2806Arg | missense | (+) |
| F9, F51 | 7,3 | c.10797 + 1G>A | p.spl | Splice site mutation | (++) |
| F12 | 5 | c.1149 + 2T>G | p.spl | Splice site mutation | (++) |
| F232 | 6 | c.1704 + 1G>C | p.spl | Splice site mutation | (+++) |
| F157 | 3 | c.2169‐1G>T | p.spl | Splice site mutation | (++) |
| F162 | 8 | c.2803 + 1G>A | p.spl | Splice site mutation | (+++) |
| F167, F177 | 7,3 | c.358‐2A>G | p.spl | Splice site mutation | (+++) |
| F50 | 4 | c.3602‐2G>C | p.spl | Splice site mutation | (++) |
| F46 | 3 | c.3603 + 2dupT | p.spl | Splice site mutation | (++) |
| F127, F139 | 2,3 | c.4518 + 3A>T | p.spl | Splice site mutation | (++) |
| F101 | 6 | c.5449‐2A>G | p.spl | Splice site mutation | (++) |
| F118 | 1 | c.5586 + 18A>G | p.spl | Splice site mutation | (+) |
| F102 | 4 | c.8027 + 1G>A | p.spl | Splice site mutation | (++) |
| F193 | 2 | c.8028‐1G>C | p.spl | Splice site mutation | (++) |
| F92 | 2 | c.8217 + 3delAAGT | p.spl | Splice site mutation | (++) |
| F93 | 9 | c.8668 + 1G>A | p.spl | Splice site mutation | (++) |
| F69 | 3 | c.9649 + 15T>C | p.spl | Splice site mutation | (+) |
| F161 | 4 | c.9807 + 5G>A | p.spl | Splice site mutation | (+++) |
Pathogenicity Analysis was evaluated using the LOVD (www.dmd.nl), ClinVar database, Polyphen‐2 (http://genetics.bwh.harvard.edu/pph2/), SIFT algorithms (http://sift.jcvi.org/), Alamut 2.7.2. and ExAc, 1000 Genomes. (+++), pathogenic; (++), probably pathogenic and (+), uncertainly.
3.5. Analysis the association between the age of onset and mutation patterns
According to clinical data, the mean age at loss of ambulation among deletion, duplication, and small mutation were 4.7 ± 2.2, 4.3 ± 1.9, and 4.2 ± 1.9 years, respectively. No relationships were found between the age of onset and mutation patterns (P > .05).
3.6. Prenatal diagnosis of 237 fetuses
For 237 families, we analyzed the carrier status of all pregnant women, among whom 131 had the same mutation as their children or patients. Fourteen pathogenic mutations and 18 female carriers were detected in fetal samples (Table 2). Two cases (F174 and F162) with deletion asked for repeat analytical tests. DHPLC analysis was performed to confirm. Fetal echocardiography showed ventricular septal defect and congenital heart disease in family F23 and F87, respectively.
Table 2.
Prenatal diagnosis and clinical risk in 14 cases
| Family | Proband | Potential Pregnant Carrier | Fetus | Clinical Risk | Pregnancy Outcome |
|---|---|---|---|---|---|
| F14 | Del 45 | C | + | Affected | Top |
| F23 | Del 45‐49 | C | + | Affected | Top |
| F33 | Del 5‐44 | C | C | Low Risk | − |
| F37 | Del 45‐47 | C | + | Affected | Top |
| F56 | Del 45‐46 | C | C | Low Risk | − |
| F61 | Del 46‐48 | C | C | Low Risk | − |
| F68 | Del 3‐9 | C | C | Low Risk | − |
| F73 | Dup 2 | C | + | Affected | Top |
| F87 | Del 21 | C | C | Low Risk | − |
| F98 | Del 46‐48 | C | + | Affected | Top |
| F117 | Del 8‐16 | C | + | Affected | Top |
| F128 | Del 22‐43 | C | C | Low Risk | − |
| F134 | Del 51‐53 | C | C | Low Risk | − |
| F140 | Del 3‐19 | C | C | Low Risk | − |
| F170 | Dup 2‐17 | C | + | Affected | Top |
| F174 | Del 3‐13 | C | + | Affected | Top |
| F200 | Del 49‐51 | C | C | Low Risk | − |
| F204 | Dup 51‐62 | C | + | Affected | Top |
| F212 | Del 45‐50 | C | C | Low Risk | − |
| F226 | Del 46‐48 | C | C | Low Risk | − |
| F228 | Del 45‐53 | C | C | Low Risk | − |
| F24 | C.10108C>T | C | C | Low Risk | − |
| F29 | C.10141C>T | C | + | Affected | Top |
| F162 | C.2803 + 1G>A | C | C | Low Risk | − |
| F171 | C.10171C>T | C | + | Affected | Top |
| F28 | C.829C>T | C | + | Affected | Top |
| F40 | C.7433ing | C | C | Low Risk | − |
| F96 | C.9461delt | C | + | Affected | Top |
| F34 | C.5234G>A | C | + | Affected | Top |
| F57 | C.2645G>A | C | C | Low Risk | − |
| F97 | C.5163G>C | C | C | Low Risk | − |
| F157 | C.2169‐1G>T | C | C | Low Risk | − |
+, positive for DMD mutation; −, negative for DMD mutation; C, carrier for DMD; Top, termination of pregnancy.
3.7. Follow‐up and genetic counseling
Among the total cases, 14 pregnant women chose the termination of pregnancy. The pedigree diagnosed as negative and carriers continued the pregnancy. In total, the pregnancies of 14 cases were interrupted, 2 were preterm births (>34 gestational weeks), 151 were delivered at term, 63 continued the pregnancy and 7 were lost to follow‐up. For 18 female carrier fetuses, long time monitoring is necessary.
4. DISCUSSION
Lacking effective therapies, DMD brings great harm to many patients and their families. Families with DMD history showed anxiety and depression and strongly requested prenatal diagnosis. It has been known that 1/3 sporadic mutations of DMD gene occurs in male patients.23, 24 Germinal mosaicism cases were detected in some DMD families.25, 26, 27, 28 So, the prenatal DMD diagnosis has been recommended as the most effective way to avoid congenital malformed children. In this retrospective cohort study, genetic analysis and prenatal diagnosis results of the 237 Chinese families with DMD history were presented. Fourteen pathogenic mutations (5.9%) and 18 female carriers (7.6%) were detected in fetal samples. The size of the samples was good for a rare disorder.
Molecular genetic analysis of DMD patients is essential for establishing a definitive diagnosis, directing appropriate clinical management, and providing for further prenatal diagnosis, and genetic counseling.29 Cho A et al reported that the rates of deletions, duplications, and sequence variations were reported as 65.4%, 13.3%, and 12.3%, respectively.22 Chen C et al reported that they were 79%, 19.8%, and 9.2%, respectively.9 In our study, we found the rate of deletions, duplications, and sequence variations as 47.3%, 8.4%, 1.7%, and 42.6%, respectively. We believed that these differences between the rates were due to sample size for inspection patients and regional differences.
In recent years, the prenatal diagnosis techniques acquired great improvements. The advancement in ultrasound‐monitored amniocentesis enhanced the capability for diagnosis of DMD in utero. TA et al30 reported combination of STR and MLPA could be a rapid, reliable, and affordable detection protocol for determination of the carrier's status and prenatal diagnosis of DMD.30 Zhang et al31 reported that real‐time PCR assay was successfully used to make prenatal diagnosis of DMD families in thirty Chinese families.31 Yoo et al reported the single targeted sequencing platform was applied to detect carriers and make non‐invasive prenatal diagnosis.17 Parks et al reported that haplotype dosage analysis for X‐linked disorders was one non‐invasive prenatal diagnosis for DMD.32 Therefore, we identified DMD mutations in a cohort of 237 Chinese patients using denaturing high‐performance liquid chromatography (DHPLC) followed by Sanger sequencing according to local conditions and platforms in our prenatal diagnosis center. Chen et al (2014)29 also reported DHPLC analysis exhibited high efficiency and specificity for identifying any small alterations in the DMD gene.9
In this study, 82 different small mutations were previously reported or possibly affect the DMD gene function (Table 1). However, the pathogenicity of six different small mutations (p.Asp556Asn, p.Gly882Asp, p.Gln2366Lys, p.Gln2806Arg, c.5586 + 18A>G, and c.9649 + 15T>C) was not clear.
This study offered 4‐year clinical prenatal diagnosis experience of Duchenne muscular dystrophy, which was one relatively large sample size to perform mutational analysis. The limitation of this study was that some novel mutations had not been fully proved to be related to the DMD.
In conclusion, this study offers informative data for proper prenatal genetic counseling of pregnant women and their partner. DHPLC technology has high sensitivity and specificity.
CONFLICT OF INTERESTS
All authors declare no conflict of interests. The authors alone are responsible for the content and writing of the paper.
ETHICAL APPROVAL
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
INFORMED CONSENT
Informed consent was obtained from all individual participants included in the study.
ACKNOWLEDGMENTS
The authors are grateful for the valuable discussion with Xu Zhang, Beijing GH Epigen Medical Institute, Beijing 100089, China.
Xu Y, Li Y, Song T, et al. A retrospective analysis of 237 Chinese families with Duchenne muscular dystrophy history and strategies of prenatal diagnosis. J Clin Lab Anal. 2018;32:e22445 10.1002/jcla.22445
Funding information
This study was funded by Research and the development of non‐invasive prenatal screening and diagnostic technology platform for single gene and genomic diseases and its standardization application system establishment. National Key R&D Program (Grant No. 2016YFC1000700).
Contributor Information
Biliang Chen, Email: cblxjh@fmmu.edu.cn.
Jianfang Zhang, Email: zjf516@yeah.net.
REFERENCES
- 1. Hoogerwaard EM, Bakker E, Ippel PF, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: a cohort study. Lancet. 1999;353:2116‐2119. [DOI] [PubMed] [Google Scholar]
- 2. Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health. 2015;51:759‐764. [DOI] [PubMed] [Google Scholar]
- 3. Bladen CL, Salgado D, Monges S, et al. The TREAT‐NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Juan‐Mateu J, Gonzalez‐Quereda L, Rodriguez MJ, et al. DMD Mutations in 576 Dystrophinopathy Families: a step forward in genotype‐phenotype correlations. PLoS ONE. 2015;10:e0135189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chun JL, O'Brien R, Song MH, Wondrasch BF, Berry SE. Injection of vessel‐derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn‐/‐ but not aged mdx mouse models for duchenne muscular dystrophy. Stem Cells Transl Med. 2013;2:68‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev. 2015;26:57‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nance ME, Duan D. Perspective on Adeno‐Associated Virus Capsid Modification for Duchenne Muscular Dystrophy Gene Therapy. Hum Gene Ther. 2015;26:786‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lalic T, Vossen RH, Coffa J, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231‐1234. [DOI] [PubMed] [Google Scholar]
- 9. Chen C, Ma H, Zhang F, et al. Screening of Duchenne muscular dystrophy (DMD) mutations and investigating its mutational mechanism in Chinese patients. PLoS ONE. 2014;9:e108038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Muscarella LA, Piemontese MR, Barbano R, et al. Novel mutations of dystrophin gene in DMD patients detected by rapid scanning in biplex exons DHPLC analysis. Biomol Eng. 2007;24:231‐236. [DOI] [PubMed] [Google Scholar]
- 11. Okubo M, Minami N, Goto K, et al. Genetic diagnosis of Duchenne/Becker muscular dystrophy using next‐generation sequencing: validation analysis of DMD mutations. J Hum Genet. 2016;61:483‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang Y, Yang Y, Liu J, et al. Whole dystrophin gene analysis by next‐generation sequencing: a comprehensive genetic diagnosis of Duchenne and Becker muscular dystrophy. Mol Genet Genomics. 2014;289:1013‐1021. [DOI] [PubMed] [Google Scholar]
- 13. Magri F, Del Bo R, D'Angelo MG, et al. Clinical and molecular characterization of a cohort of patients with novel nucleotide alterations of the Dystrophin gene detected by direct sequencing. BMC Med Genet. 2011;12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bakker E, Hofker MH, Goor N, et al. Prenatal diagnosis and carrier detection of Duchenne muscular dystrophy with closely linked RFLPs. Lancet. 1985;1:655‐658. [DOI] [PubMed] [Google Scholar]
- 15. Carsana A, Frisso G, Intrieri M, et al. Molecular analysis of Duchenne/Becker muscular dystrophy. Front Biosci (Elite Ed). 2010;2:547‐558. [DOI] [PubMed] [Google Scholar]
- 16. Esposito G, Ruggiero R, Savarese M, et al. Prenatal molecular diagnosis of inherited neuromuscular diseases: Duchenne/Becker muscular dystrophy, myotonic dystrophy type 1 and spinal muscular atrophy. Clin Chem Lab Med. 2013;51:2239‐2245. [DOI] [PubMed] [Google Scholar]
- 17. Yoo SK, Lim BC, Byeun J, et al. Noninvasive prenatal diagnosis of duchenne muscular dystrophy: comprehensive genetic diagnosis in carrier, proband, and fetus. Clin Chem. 2015;61:829‐837. [DOI] [PubMed] [Google Scholar]
- 18. Tao H, Xiao J, Yang C, Wang J, Tang Y, Guo C. Retrospective analysis of 4761 cases who underwent amniocentesis in southeast China. J Obstet Gynaecol 2017;38:1‐4. [DOI] [PubMed] [Google Scholar]
- 19. Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15:7‐12. [DOI] [PubMed] [Google Scholar]
- 20. Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862‐D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kwon JM, Abdel‐Hamid HZ, Al‐Zaidy SA, et al. Clinical Follow‐Up For Duchenne Muscular Dystrophy Newborn Screening: A Proposal. Muscle Nerve. 2016;54:186‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cho A, Seong MW, Lim BC, et al. Consecutive analysis of mutation spectrum in the dystrophin gene of 507 Korean boys with Duchenne/Becker muscular dystrophy in a single center. Muscle Nerve. 2017;55:727‐734. [DOI] [PubMed] [Google Scholar]
- 23. Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 2016;12:1795‐1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mukherjee M, Chaturvedi LS, Srivastava S, Mittal RD, Mittal B. De novo mutations in sporadic deletional Duchenne muscular dystrophy (DMD) cases. Exp Mol Med. 2003;35:113‐117. [DOI] [PubMed] [Google Scholar]
- 25. Bermudez‐Lopez C, Garcia‐de Teresa B, Gonzalez‐del Angel A, Alcantara‐Ortigoza MA. Germinal mosaicism in a sample of families with Duchenne/Becker muscular dystrophy with partial deletions in the DMD gene. Genet Test Mol Biomarkers. 2014;18:93‐97. [DOI] [PubMed] [Google Scholar]
- 26. Boileau C, Junien C. Misdiagnosed normal fetus owing to undetected germinal mosaicism for DMD deletion. J Med Genet. 1989;26:790‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bullock S, Felix C, Iskander‐Gabra S, Davison V. Detection of germinal mosaicism in a DMD family. Biochem Soc Trans. 1996;24:273S. [DOI] [PubMed] [Google Scholar]
- 28. Covone AE, Caroli F, Cereseto A, Lerone M, Romeo G. [Identification of the dystrophin gene deletions in DMD/BMD patients. Analysis of the reading frame shift and germinal mosaicism]. Minerva Pediatr. 1991;43:65‐66. [PubMed] [Google Scholar]
- 29. Aartsma‐Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53:145‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ta MH, Tran TH, Do NH, et al. Rapid method for targeted prenatal diagnosis of Duchenne muscular dystrophy in Vietnam. Taiwan J Obstet Gynecol. 2013;52:534‐539. [DOI] [PubMed] [Google Scholar]
- 31. Zhang T, Liu S, Wei T, et al. Development of a comprehensive real‐time PCR assay for dystrophin gene analysis and prenatal diagnosis of Chinese families. Clin Chim Acta. 2013;424:33‐38. [DOI] [PubMed] [Google Scholar]
- 32. Parks M, Court S, Cleary S, et al. Non‐invasive prenatal diagnosis of Duchenne and Becker muscular dystrophies by relative haplotype dosage. Prenat Diagn. 2016;36:312‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]