

Abstract
Background
Wilson's disease (WD) is a rare autosomal recessive disorder characterized by the deposition of copper mainly in the liver or nerve system that leads to their dysfunction. Mutations in the gene encoding ATPase, Cu+ transporting, beta polypeptide (ATP7B) are causative for WD. The aim of this study was to develop a rapid and convenient assay for detection of the three most common causative ATP7B mutations, p.R778L, p.P992L, and p.V1106I.
Methods
Plasmids containing DNA fragments harboring each of the three ATP7B mutations were constructed. High‐resolution melting (HRM) analysis was conducted by asymmetric polymerase chain reaction (PCR) amplification with paired primer and unlabeled probe, performed in a 96‐well plate formatted LightCycler 480 Real‐Time PCR System. The assay was evaluated for accuracy and reproducibility by genotyping of 41 WD cases.
Results
The unlabeled probe HRM assays performed on constructs with the p.R778L, p.P992L, and p.V1106I mutations in the ATP7B gene resulted in additional melting peaks. According to the unlabeled probe HRM molecular signature, we could differentiate homozygous mutations from wild‐type with the ΔTm (difference between melting temperatures) >4°C, and the coefficient of variation in repeatability tests was <5%. In the validation assay using our method to examine clinical samples, a 100% accuracy rate was achieved.
Conclusions
The newly developed assay to rapidly genotype the ATP7B mutations is convenient, accurate, and reproducible, and represents a favorable alternative to Sanger sequencing in the identification of specific ATP7B mutations.
Keywords: ATP7B, genotype, mutation detection, unlabeled probe HRM, Wilson's disease
1. INTRODUCTION
Wilson's disease (WD), or hepatolenticular degeneration, is a rare autosomal recessive disorder characterized by the decrease of biliary copper excretion and the reduction of copper into apoceruloplasmin. WD is a treatable disorder, and treatment involves removing excess copper by chelating agents, such as D‐penicillamine and zinc sulfate. However, because of iron accumulation in various organs, the symptoms of WD can include chronic liver disease (mild abnormalities, acute or chronic hepatitis, cirrhosis, or even fulminant hepatitis), neurological impairment (Parkinson‐like or neuropsychiatric symptoms), and Kayser‐Fleischer rings.1, 2, 3 Therefore, early diagnosis and treatment can decrease the rates of disability or mortality and lead to better prognoses.
The gene responsible for WD is located in the 13q14.3 chromosome and encodes a copper‐specific transporting P‐type ATPase (ATP7B).4 Mutations in the ATP7B gene change the biosynthetic and transporting role of ATPase in cells, leading to impaired biliary excretion of copper. To date, more than 500 ATP7B mutations have been identified, and more than 70% of WD cases are associated with an ATP7B mutation [WD Mutation Database (http://www.wilsondisease.med.ualberta.ca/database.asp) and Human GeneMutation Database (http://www.hgmd.org/)]. Reported mutations in ATP7B include missense mutations, frameshift mutations, nonsense mutations, and splicing mutations, and missense mutations are the most common ATP7B mutation in WD. Among these mutations, p.Arg778Leu (c.2333G>T) has been identified as the most common. Both p.Pro992Leu (c.2975C>T) and p.Val1106Ile (c.3317G>A) are also relatively frequent mutations in WD, and also play important roles in the detection of WD.5, 6, 7
Various approaches have been described for the detection of ATP7B gene mutations, such as multi‐allele genotyping, genotyping microarray, and Sanger sequencing, but the clinical uses of these approaches are limited.8, 9 Recently, high‐resolution melting (HRM) analysis was developed as a closed‐tube technology for genotyping DNA variants and mutation screening, with advantages over other techniques such as its high‐throughput, rapid, and non‐destructive nature.10 However, amplicon melting analysis of HRM cannot always differentiate homozygous mutations from wild‐type, particularly in cases where the mutation results in the insertion of a complementary base and the bases closest to the mutation are identical on both strands (nearest‐neighbor thermodynamics symmetry).11 The use of an unlabeled probe confers an added degree of sequence specificity desirable for clinical application. The Tm shift induced by a mutation depends on the binding strength between the mutated base and its counterpart on the opposite strand, as well as nearest‐neighbor effects. Incorporation of locked nucleic acids (modified bases) into the probe may increase the ability to discriminate different sequence variants.12, 13, 14 Thus, these modifications could satisfy the clinical demand of developing a simple and accurate method for the detection of ATP7B gene mutations with high specificity and sensitivity.
In this study, we reported a novel method based on the use of HRM analysis and unlabeled probes to rapidly genotype the three most common variants of the ATP7B gene in a clinical laboratory setting.
2. MATERIALS AND METHODS
2.1. Plasmid construction
Part of the ATP7B gene was amplified by polymerase chain reaction (PCR) using clinical DNA samples and primers. The PCR reaction (50 μL) was composed of 5 μL of 10× reaction buffer, 1 μL of 25 mmol L−1 MgCl2, 2 μL of 10 mmol L−1 dNTP, 0.5 μL of the Pfu s15TMDNA polymerase (TransGen Biotech Co., Ltd, Beijing, China), 2 μL of forward primer (5 mmol L−1), 2 μL of reverse primer (5 mmol L−1), and 2 μL of template DNA. The PCR condition was 95°C for 5 minutes followed by 50 cycles of 95°C for 30 seconds, 46°C for 40 seconds, and 72°C for 30 seconds. An additional 3‐minute extension at 72°C was performed after the last PCR cycle to replenish PCR product followed by cooling at 4°C. The PCR product was then cloned into the pMD19‐T‐simple cloning vector as described by the manufacturer's instructions (Takara, Dalian, China), and the construct was confirmed by DNA sequencing.
2.2. Primers and probes
Primers and probes were designed with Primer 5 software (Table 1). Unlabeled nucleotide probes bind to the negative strand of wild allele at the three loci, respectively, with ~40 GC content (except the ATP7B 992 probe). A 3′ amino modifier (AmM) was incorporated to prevent extension during the PCR reaction.
Table 1.
Primers and unlabeled probe sequences
| Primer/probe type | Primer/probe name | Sequences | PCR product size |
|---|---|---|---|
| Forward primer | p.R778L‐F | 5′‐GAGGAGCCCTGTGACATTCT‐3′ | 159 bp |
| Reverse primer | p.R778L‐R | 5′‐AACATGGTGTTCAGAGGAAGTG‐3′ | |
| Unlabeled probe | p.R778L‐P | 5′‐TGGGCCGGTGGCTGGAACACTTGGCAA/AmMo‐3′ | |
| Forward primer | p.P992L‐F | 5′‐TTCCAGACGTCCATCACGGT‐3′ | 181 bp |
| Reverse primer | p.P992L‐R | 5′‐TCTCAGGATGGGGAAAGCCG‐3′ | |
| Unlabeled probe | p.P992L‐P | 5′‐CTGGGGCTGGCCACGCCCACGGCTGTCATG/AmMo‐3′ | |
| Forward primer | p.V1106I‐F | 5′‐ACCTTGGGATACTGCACGGA‐3′ | 175 bp |
| Reverse primer | p.V1106I‐R | 5′‐GCAGAGACAAAAGCCAGCAAT‐3′ | |
| Unlabeled probe | p.V1106I‐P | 5′‐GAATTGGGTGCAAAGTCAGCAACGTGGAA/AmMo‐3′ |
2.3. Unlabeled probe HRM analysis
Asymmetric PCR amplification was performed in a 96‐well plate formatted LightCycler 480 Real‐Time PCR System (Roche Applied Science, Mannheim, Germany). The PCR reaction (20 μL) was composed of 10 μL of 2×HRM master mix, 2 μL of 25 mmol L−1 MgCl2, 0.8 μL of 1.25 mmol L−1 forward primer, 4 μL of 2.5 mmol L−1 reverse primer, 1 μL of 10 mmol L−1 unlabeled probe for different ATP7B gene variants (Table 1), and 2 μL of template DNA (concentration ranged from 20 to 60 ng/μL). The amplification condition was optimized by incubating the reaction mixtures at 95°C for 15 minutes, followed by 65 cycles of 95°C for 15 seconds, 60°C for 20 seconds, and 72°C for 30 seconds with the ramp to 95°C at 4.8°C/s, to 60°C at 2.5°C/s, and to 72°C at 4.8°C/s. An additional 1 minute at 72°C was added to replenish the PCR product. For HRM analysis, the PCR product was denatured by rising temperature to 95°C at 4.8°C/s and was then cooled down to 55°C at 2.5°C/s for hybridization. The melting curve was acquired by increasing the temperature from 55°C to 95°C at a ramp rate of 4.8°C/s with 25 acquisitions per degree of temperature.
2.4. Samples
Blood samples were collected from 41 patients at the Beijing Friendship Hospital and with informed consent. Genomic DNA was extracted using the QIAamp DNA mini kit (Hilden, Germany) and eluted in 50 μL elution buffer. Quantification of all DNA samples was performed using the NanoDrop™ 8000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and checked by conventional PCR using the housekeeping gene glyceraldehyde 3‐phosphate dehydrogenase as a reference.
2.5. Statistical analysis
Melting data were analyzed using the LightCycler 480 Software SW1.5 (Roche Applied Science) and the MeltingWizard High‐Resolution Melting Analysis vMW4.1 software (University of Utah).15 For reproducibility studies, intra‐run reproducibility was performed using six samples representing three genotypes. These samples were assessed in triplicate within a common run. Inter‐run reproducibility was performed with six samples representing three genotypes. These samples were analyzed in five runs on different days. Precision was measured by the standard deviation (SD) of the melting temperature (Tm) and the SD of the Tm differences (ΔTm) between alleles. All statistical analyses were performed using SPSS 16.0 software (SPSS Inc., Chicago, IL, USA).
3. RESULTS
3.1. Construction plasmids of different genotypes of ATP7B
To assess the use of unlabeled probe HRM analysis for genotyping the three most common variants of the ATP7B gene in WD (R778L, P992L, and V1106I), we amplified fragments corresponding to these different genotypes and subcloned the fragments into T&A cloning vectors. These plasmids were used as templates for HRM assays using the corresponding primers in Table 1. Sequences of the wild‐type ATP7B gene and the three variants, as well as the location of the primers and probes, are shown in Fig. 1. All of the plasmids were confirmed by sequencing.
Figure 1.

Locations of the primers and probes. (a) The amplified region of ATP7B 778 loci is shown and the primers and probe sequences are indicated. (b) The amplified region of ATP7B 992 loci is shown and the primers and probe sequences are indicated. (c) The amplified region of ATP7B 1106 loci is shown and the primers and probe sequences are indicated
3.2. Detection of different genotypes by unlabeled probe HRM assay
We next performed unlabeled probe HRM assays for all three variants. As shown in Fig. 2, the melting peaks include both the probe region (left of the vertical line) and the PCR product region (right of the vertical line). The melting peaks obtained directly using the PCR products did not allow us to distinguish wild‐type from mutant, whereas the sensitivity and accuracy of HRM were dramatically improved when the unlabeled probe was used. The unlabeled probes resulted in additional melting peaks when analyzing p.R778L, p.P992L, and p.V1106I mutations and were sufficient for differentiating mutants from wild‐type sequences. The melting peak at 72.10°C represents the R778L mutation, while the melting peak at 76.40°C represents the wild‐type (Fig. 2a). The melting peak at 77.20°C represents the P992L mutation, while the melting peak at 82.00°C represents the wild‐type (Fig. 2b). The melting peak at 66.50°C represents the V1106I mutation, while the melting peak at 71.10°C represents the wild‐type (Fig. 2c).
Figure 2.
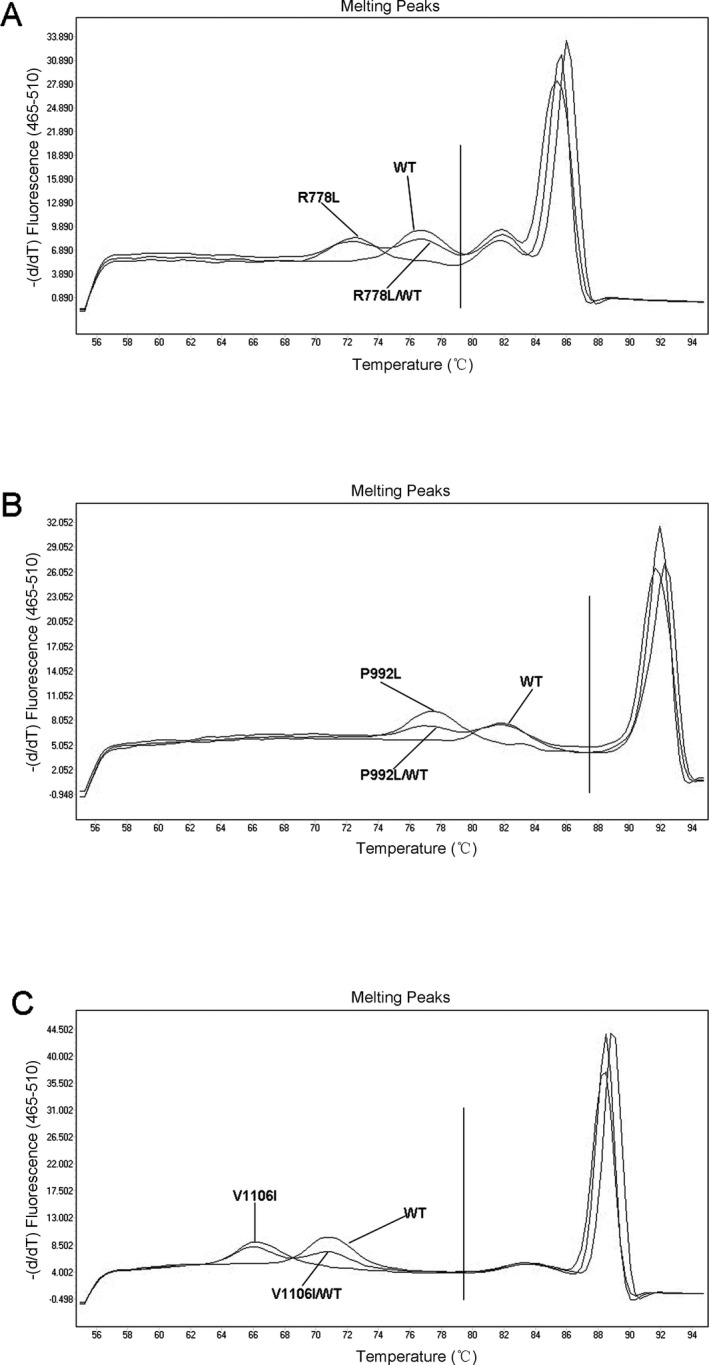
The results of unlabeled probe HRM assays for different ATP7B genotypes using the plasmids of different ATP7B genotypes. (a) The results of unlabeled probe HRM assays for detection of different genotypes of ATP7B 778 loci. (b) The results of unlabeled probe HRM assays for detection of different genotypes of ATP7B 992 loci. (c) The results of unlabeled probe HRM assays for detection of different genotypes of ATP7B 1106 loci
The controls with no templates showed no peaks, and the amplicon melting peaks did not interfere with the probe melting peaks.
3.3. Repeatability of the unlabeled probe HRM assay
To determine within‐run reproducibility, wild‐type plasmids, homozygous plasmids, and heterozygous plasmids (mixed wild‐type and homozygous plasmids) of different variants were tested in triplicate in one run. The averaged Tm and delta Tm for different genotypes were described in Table 2.
Table 2.
Repeatability of unlabeled probe HRM
| Genotype | Within‐run | Between‐run | ||
|---|---|---|---|---|
| Homozygotes | Tm (SD), °C | CV, % | Tm (SD), °C | CV, % |
| 778WT | 76.36 (0.11) | 0.04 | 76.44 (0.06) | 0.07 |
| p.R778L | 72.14 (0.05) | 0.06 | 72.19 (0.12) | 0.17 |
| 992WT | 82.06 (0.09) | 0.11 | 81.98 (0.21) | 0.26 |
| p.P992L | 77.25 (0.09) | 0.12 | 77.34 (0.23) | 0.17 |
| 1106WT | 71.09 (0.06) | 0.08 | 71.21 (0.21) | 0.29 |
| p.V1106I | 66.47 (0.05) | 0.08 | 66.60 (0.18) | 0.27 |
| Heterozygotes | ΔTm (SD), °C | CV, % | ΔTm (SD), °C | CV, % |
|---|---|---|---|---|
| 778WT/p.R778L | 4.47 (0.29) | 1.96 | 4.57 (0.15) | 3.28 |
| 992WT/p.P992L | 4.57 (0.08) | 1.75 | 4.70 (0.17) | 3.61 |
| 1106WT/p.V1106I | 4.73 (0.04) | 0.85 | 4.86 (0.13) | 4.73 |
The probe melting temperatures (Tm) from within‐run and between‐run showing averaged Tms, difference between melting temperatures (ΔTm) for heterozygotes, standard deviations (SD, in parentheses), and coefficient of variation (CV) are listed.
The between‐run reproducibility was tested using the same plasmids as in the within‐run reproducibility test. The assays were run five consecutive times and the Tm values from each run were averaged (Table 2).
3.4. Clinical evaluation for accuracy of the unlabeled probe HRM assay
We further assessed our method in clinical specimens containing different variants. When clinical samples with the indicated genotypes were subject to the unlabeled probe HRM analyses, the melting patterns were consistent with those obtained from plasmid DNA templates (Fig. 3). The averaged Tm and delta Tm for different genotypes are described in Table 3.
Figure 3.
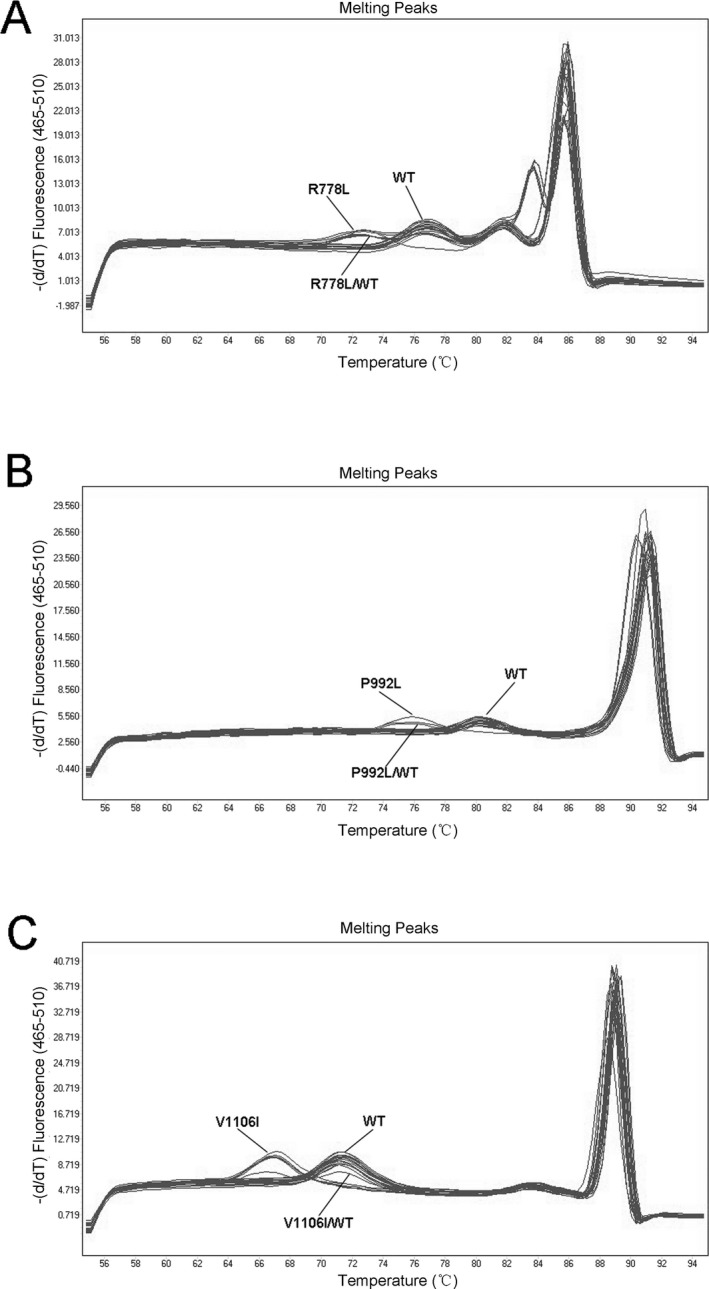
The results of unlabeled probe HRM assays for clinical patient samples. (a) The results of unlabeled probe HRM assays for detection of ATP7B 778 loci of clinical patient samples. (b) The results of unlabeled probe HRM assays for detection of ATP7B 992 loci of clinical patient samples. (c) The results of unlabeled probe HRM assays for detection of ATP7B 1106 loci of clinical patient samples
Table 3.
Validation assay for 41 clinical WD patient samples
| Genotype | No. of samples | Tm (SD), °C | CV, % | Correct genotyping rate (%) |
|---|---|---|---|---|
| Homozygotes | ||||
| 778WT | 29 | 76.75 (0.17) | 0.22 | 100 |
| p.R778L | 1 | 72.73 (N/A) | N/A | 100 |
| 992WT | 31 | 81.43 (0.37) | 0.46 | 100 |
| p.P992L | 6 | 76.96 (0.39) | 0.51 | 100 |
| 1106WT | 29 | 71.17 (0.22) | 0.31 | 100 |
| p.V1106I | 7 | 66.90 (0.11) | 0.16 | 100 |
| Heterozygotes | No. of samples | ΔTm (SD), °C | CV, % | Correct genotyping rate (%) |
|---|---|---|---|---|
| 778WT/p.R778L | 11 | 4.27 (0.16) | 3.74 | 100 |
| 992WT/p.P992L | 4 | 4.31 (0.54) | 12.53 | 100 |
| 1106WT/p.V1106I | 5 | 4.63 (0.05) | 1.07 | 100 |
The probe melting temperatures (Tm) from within‐run and between‐run showing averaged Tms, difference between melting temperatures (ΔTm) for heterozygotes, standard deviations (SD, in parentheses), and coefficient of variation (CV) are listed. N/A, not available (only one sample with this genotype was run, thus SD and CV could not be calculated).
The results were further verified by sequencing and were found to be consistent with those of the unlabeled probe HRM assay (correct genotyping rates were 100%).
4. DISCUSSION
Wilson's disease is an autosomal recessive inheritance disorder of copper metabolism. Early diagnosis and treatment are associated with better outcome. However, current diagnostic criteria are based on symptoms and copper detection. Patients with mild symptoms may be misdiagnosed, resulting in delayed treatment. Genetic testing of the ATP7B gene mutation may lead to reliable early diagnosis and treatment, which can prevent copper accumulation and tissue damage.16
The methods currently used to detect ATP7B mutation include multi‐allele genotyping, genotyping microarray, and Sanger sequencing, but the clinical use of these approaches are limited. HRM analysis has emerged as a rapid and sensitive method for mutation scanning as well as targeted single‐nucleotide polymorphism genotyping.17 In HRM, samples can be genotyped by Tm as well as curve shape, and the amplification products from different genotypes can be resolved by melt analysis. However, HRM can be affected by such factors as sequence length and mutation type, and sometimes HRM cannot detect homozygous mutations with a small difference in Tm.18 Therefore, HRM is also characterized by poor sensitivity and specificity. To overcome this disadvantage, a method based on fluorescently labeled probe assays was developed. However, this approach requires two sets of fluorescently labeled probes and employs fluorescent resonance energy transfer in the LightCycler platform, which is costly and royalty bearing.19 The unlabeled probe HRM technique was developed as a novel approach by integrating HRM with asymmetric PCR using an unlabeled modified base probe, which facilitated detection of mutation sites and added the sequence specificity desirable for clinical applications.20, 21, 22
The unlabeled probe HRM assays use ~30 bp C3 blocked probes to target the site of interest during the melting process. A single base pair difference in such a short probe can result in a significant Tm shift (e.g., 3–5°C). Genotyping with unlabeled probes has been successfully applied for the detection of disease‐related singlenucleotide polymorphisms such as factor V Leiden, and the detection and genotyping of herpes simplex virus.15, 22 Therefore, unlabeled probe HRM is a high‐throughput, inexpensive, closed‐tube genotyping method that may substantially reduce the need for sequencing. Using unlabeled probe HRM, we developed a sensitive and clinically useful method for the high‐throughput detection of ATP7B mutations: p.Arg778Leu, p.Pro992Leu, and p.Val1106Ile. The difference of the wild‐type peak and mutation peak Tm values was more than 4°C, and thus it was easy to distinguish mutant peaks from wild‐type peaks via the probe HRM curve, suggesting its reliability for clinical gene mutation detection.
We further assessed the application of our method in clinical specimens. A total of 41 clinical samples from patients who were suspected of WD were blind tested. All tested samples displayed distinguishable HRM patterns that can be assigned to the correct genotype with the rate equivalent to 100% compared with sequencing.
In conclusion, our study demonstrates that the unlabeled probe HRM is useful for the detection of ATP7B mutations and is suitable for clinical application for WD diagnosis.
ACKNOWLEDGMENTS
This study was supported by a grant from the National Natural Science Foundation of China (no. 81071973), and a grant from the Wang Bao‐En Liver Fibrosis Foundation (grant no. 20100013).
REFERENCES
- 1. Coffey AJ, Durkie M, Hague S, et al. A genetic study of Wilson's disease in the United Kingdom. Brain. 2013;136:1476–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ferenci P, Czlonkowska A, Stremmel W, et al. EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56:671–685. [DOI] [PubMed] [Google Scholar]
- 3. Bacon BR, Adams PC, Kowdley KV, et al. American Association for the Study of Liver D. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bull PC, Thomas GR, Rommens JM, et al. The Wilson disease gene is a putative copper transporting P‐type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. [DOI] [PubMed] [Google Scholar]
- 5. Mak CM, Lam CW, Tam S, et al. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: Identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet. 2008;53:55–63. [DOI] [PubMed] [Google Scholar]
- 6. Lee BH, Kim JH, Lee SY, et al. Distinct clinical courses according to presenting phenotypes and their correlations to ATP7B mutations in a large Wilson's disease cohort. Liver Int. 2011;31:831–839. [DOI] [PubMed] [Google Scholar]
- 7. Wei Z, Huang Y, Liu A, et al. Mutational characterization of ATP7B gene in 103 Wilson's disease patients from Southern China: Identification of three novel mutations. NeuroReport. 2014;25:1075–1080. [DOI] [PubMed] [Google Scholar]
- 8. Gojova L, Jansova E, Kulm M, et al. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease. Clin Genet. 2008;73:441–452. [DOI] [PubMed] [Google Scholar]
- 9. Amvrosiadou M, Petropoulou M, Poulou M, et al. Multi‐allele genotyping platform for the simultaneous detection of mutations in the Wilson disease related ATP7B gene. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1006:201–208. [DOI] [PubMed] [Google Scholar]
- 10. Fox M, Hebbard G, Brasseur J, et al. The advantages of high resolution oesophageal manometry (HRM) in clinical practice. Gut. 2003;52(Suppl 1):A42. [Google Scholar]
- 11. Palais RA, Liew MA, Wittwer CT. Quantitative heteroduplex analysis for single nucleotide polymorphism genotyping. Anal Biochem. 2005;346:167–175. [DOI] [PubMed] [Google Scholar]
- 12. Chou LS, Meadows C, Wittwer CT, et al. Unlabeled oligonucleotide probes modified with locked nucleic acids for improved mismatch discrimination in genotyping by melting analysis. Biotechniques. 2005;39:644–650. [DOI] [PubMed] [Google Scholar]
- 13. Lyon E. Discovering rare variants by use of melting temperature shifts seen in melting curve analysis. Clin Chem. 2005;51:1331–1332. [DOI] [PubMed] [Google Scholar]
- 14. Margraf R, Mao R, Highsmith WE, et al. Mutation scanning of the RET protooncogene using high‐resolution melting analysis. Clin Chem. 2006;52:138–141. [DOI] [PubMed] [Google Scholar]
- 15. Svensson AM, Chou LS, Mesdows C, et al. Implementation of a cost‐effective unlabeled probe high‐resolution melt assay for genotyping of Factor V Leiden. Genet Test Mol Biomarkers. 2011;15:207–213. [DOI] [PubMed] [Google Scholar]
- 16. Li XH, Lu Y, Ling Y, et al. Clinical and molecular characterization of Wilson's disease in China: Identification of 14 novel mutations. BMC Med Genet. 2011;12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Erali M, Voelkerding KV, Wittwer CT. High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol. 2008;9:290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liew M, Pryor R, Palais R, et al. Genotyping of single mucleotide polymorphisms by high‐resolution melting of small amplicons. Clin Chem. 2004;50:1156–1164. [DOI] [PubMed] [Google Scholar]
- 19. Phillips M, Meadows CA, Huang MY, et al. Simultaneous detection of C282Y and H63D hemochromatosis mutation by dual‐color probes. Mol Diagn. 2000;5:107–116. [DOI] [PubMed] [Google Scholar]
- 20. Guo W, Zhang C, Wu J, et al. Unlabeled‐probe high‐resolution melting to detect KRAS codon 12 and 13 mutations in pancreatic adenocarcinoma tissues. Clin Chem Lab Med. 2012;50:1035–1040. [DOI] [PubMed] [Google Scholar]
- 21. Sumner K, Hubley L, Pont‐Kingdon G, et al. Validation of an unlabeled probe melting analysis assay combined with high‐throughput extractions for genotyping of the most common variants in HFE‐associated hereditary hemochromatosis, C282Y, H63D, and S65C. Genet Test Mol Biomarkers. 2012;16:656–660. [DOI] [PubMed] [Google Scholar]
- 22. Lee TH, Wu TS, Tseng CP, et al. High‐resolution melting molecular signatures for rapid identification of human papillomavirus genotypes. PLoS ONE. 2012;7:e42051. [DOI] [PMC free article] [PubMed] [Google Scholar]