

Abstract
Background
The current transfusion policy recommended for individuals with serologic weak‐D phenotype is based on data derived from European‐descent populations. Data referring to the distribution of RH alleles underlying weak‐D phenotype among people of mixed origin are yet incomplete, and the applicability of European‐based transfusion guidelines to this specific population is questionable.
Goal
To evaluate the distribution of RHD variant genotype among individuals with serologic weak‐D phenotype of both African and European descent.
Methods
Donors and patients of mixed origin and with serologic weak‐D phenotype were selected for the study. They were investigated using conventional RHD‐PCR assays and RHD whole‐coding region direct sequencing.
Results
One hundred and six donors and 58 patients were included. There were 47 donors and 29 patients with partial‐D genotype (47/106, 44.3%, and 29/58, 50%, respectively). RHD*DAR and RHD*weak D type 38 represented the most common altered RHD alleles among donors (joint frequency of 39.6%), while weak D types 1‐3 accounted for 10.4% of the total D variant samples. RHD*DAR was the most common allele identified in the patient group (frequency of 31%), and weak D types 1‐3 represented 29.3% of the total.
Conclusion
The frequency of partial D among mixed individuals with serologic weak‐D phenotype is high. They should be managed as D‐negative patients until molecular tests are complete.
Keywords: alloimmunization, donors, mixed population, partial D, weak D
1. INTRODUCTION
D antigen is highly immunogenic and relevant to transfusion practice, as the development of anti‐D antibody is associated with delayed hemolytic transfusion reactions and potentially severe hemolytic disease of the fetus/newborn.1 Several D variants have been described in the literature, which are classically organized in three groups: partial D, lacking immunogenic D epitopes and prone to anti‐D development; weak D, with reduced antigen density and at no risk of alloimmunization and DEL, where the D antigen can only be identified on RBC membrane after adsorption with anti‐D followed by elution.2 It is well established that the distinction between these three groups is in fact imprecise, as some D variants lacking immunogenic epitopes present with reduced antigen density on erythroid membrane and, consequently, can develop anti‐D.2, 3
A serologic weak‐D phenotype is defined as weak reactivity of RBCs with anti‐D reagent in initial testing, less than expected for the applied serologic method.4 In the immunohematological routine of blood donors, identifying individuals with very weak‐D phenotype is encouraged, as the correspondent RBC unit must be labeled as D‐positive, minimizing the risks of anti‐D development by recipients.5 The recommendation here is to confirm D‐negative results through sensitive indirect antiglobulin test (IAT) methods and, preferably, RHD‐PCR.6, 7 Weak results obtained on the D typing routine of patients may uncover a D‐partial phenotype and, as a consequence, classifying the weak‐D recipients as D‐positive can potentially cause alloimmunization.3, 5 On the other hand, the stocks of D‐negative units are commonly short and the decision to indiscriminately classify all weak‐D blood recipients as D‐negative may be non‐attractive, as many of them would not be at risk of making anti‐D and could be transfused with D‐positive units.
Most studies exploring the distribution of altered RHD genotypes encoding a weak‐D phenotype are from European cohorts of donors and patients, in which nearly 95% of the individuals type as weak D types 1‐3.2, 8 These RH variants are not at risk of developing alloanti‐D and can be served with D‐positive units to spare the stocks of D‐negative RBC units.8 This finding justifies why UK guidelines determine that patients exhibiting weak results in the D typing routine should be treated as D+ unless they are a female of childbearing age or a patient under chronic transfusion support.9 However, studies evaluating patients of mixed origin with discrepancies in D typing have shown that the distribution of RH variant alleles significantly differ from that observed among European‐descent individuals, mainly referring to the partial‐D phenotype, but also concerning the weak‐D phenotype.10, 11 Transposing the recommendation of European transfusion guidelines to this different scenario may cause anti‐D alloimmunization, especially in services with no access to RH genotyping.
Based on the exposed, our aim was to evaluate the distribution of RHD variant genotype among blood donors and recipients with serologic weak‐D phenotype of both African and European descent.
2. METHODS
2.1. Donor and patient recruiting
Blood donors were sequentially recruited as they donated whole‐blood units at Fundação Pró‐Sangue Hemocentro de São Paulo (São Paulo, Brazil) between October 2014 and September 2017. Patients were recruited in the same period as donors. The study was conducted following Helsinki principles and approved by our local ethics committee.
The genetic ancestry of the studied donor population has been previously characterized using the FYB*‐67T/C polymorphism (rs 2814778) as an informative marker of African ancestry.12 The mutated c.‐67T>C allele is present in 99.9% of the HAPMAP‐LWK (Luhya in Webuye, Kenya) population and in 2.27% of the HAPMAP‐TSI (Toscans in Italy) population. In the donor population selected for this study, 36.7% of the individuals exhibited the c.‐67T>C allele either in homozygosis or in heterozygosis.12
2.2. Immunohematological tests
From August 2014 to July 2016, donors were typed for D antigen using direct hemagglutination in microplates and, in case of negative results, solid‐phase methodology was performed for confirmation (Capture‐R technology). All these tests were held under secure automation (NEO, Immucor, Norcross, GA, USA). From August 2016 to September 2017, donor D typing was performed using gel methodology (anti‐D clones ESD‐1M + 175‐2) in the IH‐1000 equipment (Bio‐Rad, Cressier, FR, Switzerland). For patients, D typing was also performed using gel methodology, but using different clones (IgM: P3x61; IgM and IgG: P3x290, P3x61, P3x35, and P3x21223 B10) and equipment (Erytra, Grifols, Barcelona, Spain).
All samples with weak‐D phenotype in solid phase (confirmatory phase) and gel method (initial testing) were selected for the study. Samples with weak‐D phenotype detected by direct hemagglutination using microplate were not selected for the study. A serologic weak‐D phenotype was defined as reactivity of RBCs with an anti‐D reagent giving weak (≤2+) reactivity in initial testing. The threshold was established in line with the manufacturers’ recommendation.
2.3. Nucleic acid purification
DNA was individually extracted from all selected samples using the PureLink Genomic Kit (Invitrogen, Carlsbad, CA, USA), following the manufacturer's instructions. Purity and concentration of the material were evaluated by spectrophotometry (Nanodrop1000 Spectrophotometer, Wilmington, DE, USA). DNA samples were diluted to a final concentration of 100 ng/mL for RHD genotyping.
2.4. RHD genotyping
Initial molecular analyses aiming to identify possible partial and weak RHD alleles were performed by amplifying RHD exons 3‐7 and 9, using gene‐specific primers, as described elsewhere.13 All samples were genotyped for RHD*Ψ, RHC, and RHc 14 and also had the RHD zygosity confirmed using two different conventional PCR‐based methods designed to detect the RHD deletion 15, 16 and also a multiplex real‐time quantitative PCR approach.15 When the sample was homozygous for RHD and genotyped RHCE*ce/RHCE*ce, the multiplex PCR RHC/RHc/hex3 was performed aiming to detect the presence of r'S haplotype.17
PCRs using sequence‐specific primers (PCR‐SSP) designed to detect some weak D types (D weak types 2, 3, 4, and 5) were applied for all included samples.18 In situations where the altered RHD genotype could not be confirmed based on the conventional molecular methods, the direct sequencing of RHD whole‐coding regions was performed.
2.5. RHD direct sequencing
All RHD exons and short flanking intron sequences were amplified using gene‐specific primers.19 Direct sequencing was performed by standard Sanger methodology using the cycle sequencing kit (ABI BigDye Terminator v3.1, Applied Biosystems) and run on automated sequencer equipment (ABI 310, Applied Biosystems), as previously described.19
3. RESULTS
3.1. RHD genotyping of blood donors with weak‐D phenotype
One hundred and six donors were included in the study, all typing as D‐positive with low antigen expression (≤2+ reactivity with anti‐D in initial testing using gel method or in confirmatory testing using solid‐phase method). All participants were genotyped for RHD/RHCE and classified as R1r (CcDdee) (47 donors, 44.3%); R0r (ccDdee) (25 donors, 23.6%); R1R0 (CcDDee) (10 donors, 9.4%); R0R0 (ccDDee) (8 donors, 7.5%); R2r (ccDdEe) (7 donors, 6.6%); R2R0 (ccDDEe) (3 donors, 2.8%); R1R1 (CCDDee) (2 donors, 1.9%); R1R2 (CcDDEe) (2 donors, 1.9%); R1r′ (CCDdee) (1 donor, 1%); and R2r″ (ccDdEE) (1 donor, 1%).
The following alleles were identified among the studied individuals, with their respective frequencies: RHD*01N.01 (deleted D) (39.61%); RHD*weak D type 38 (19.81%); RHD*DAR (18.4%); RHD*weak D type 2 (6.13%); RHD*weak D type 1 (3.8%); RHD*weak partial 11 (3.8%); RHD*Ѱ (1.4%); RHD*IIIc (0.94%); RHD*D‐CE 4‐7 ‐D (0.94%); RHD*weak D type 4 (0.94%); RHD*weak partial 15 (0.94%); RHD*DFR (0.47%); RHD*VI type 1 (0.47%); RHD*1157A;IVS5‐41delCTCT (0.47%); RHD*weak D type 3 (0.47%); RHD*weak D type 5 (0.47%); RHD*weak D type 104 (0.47%); and RHD*weak D type 112 (0.47%). The final RHD genotype of all studied individuals is shown in Table 1.
Table 1.
RHD genotype of the Brazilian donors with serologic weak‐D phenotype
| ID | RH genotype | Phenotype | N (%) | |
|---|---|---|---|---|
| Allele 1 | Allele 2 | |||
| 1 | RHD*DAR (RHD*09.01) | RHD*DAR (RHD*09.01) | R0R0 | 3 (2.8) |
| 2 | RHD*DAR (RHD*09.01) | RHD*DAR (RHD*09.01) | R1R0 | 3 (2.8) |
| 3 | RHD*DAR (RHD*09.01) | Deleted D (RHD*01N.01) | R0r | 24 (22.6) |
| 4 | RHD*DAR (RHD*09.01) | Deleted D (RHD*01N.01) | R1r | 1 (0.9) |
| 5 | RHD*DAR (RHD*09.01) | RHD*Pseudogene (RHD*08N.01) | R0R0 | 2 (1.9) |
| 6 | RHD*DFR1 (RHD*17.01) | Deleted D (RHD*01N.01) | R0r | 1 (0.9) |
| 7 | RHD*DIIIc (RHD*03.03) | Deleted D (RHD*01N.01) | R1r | 2 (1.9) |
| 8 | RHD*DVI.1 (RHD*06.01) | RHD*DIIIa‐CE(4‐7)‐D (RHD*03N.01) | R1R2 | 1 (0.9) |
| 9 | RHD*1157A; IVS5‐41delCTCT (RHD*01N.58) | Deleted D (RHD*01N.01) | R1r | 1 (0.9) |
| 10 | RHD*weak D type 1 (RHD*01W.1) | Deleted D (RHD*01N.01) | R1r | 3 (2.8) |
| 11 | RHD*weak D type 1 (RHD*01W.1) | Deleted D (RHD*01N.01) | R1r″ | 1 (0.9) |
| 12 | RHD*weak D type 1 (RHD*01W.1) | RHD*weak D type 1 (RHD*01W.1) | R1R0 | 2 (1.9) |
| 13 | RHD*weak D type 2 (RHD*01.W2) | Deleted D (RHD*01N.01) | R2r | 7 (6.6) |
| 14 | RHD*weak D type 2 (RHD*01.W2) | Deleted D (RHD*01N.01) | R2r″ | 1 (0.9) |
| 15 | RHD*weak D type 2 (RHD*01.W2) | Deleted D (RHD*01N.01) | R0r | 1 (0.9) |
| 16 | RHD*weak D type 2 (RHD*01.W2) | RHD*weak D type 2 (RHD*01.W2) | R2R0 | 2 (1.9) |
| 17 | RHD*weak D type 3 (RHD*01W.3) | Deleted D (RHD*01N.01) | R1r | 1 (0.9) |
| 18 | RHD*weak D type 4.0 (RHD*09.03.01) | RHD*weak D type 4.0 (RHD*09.03.01) | R0R0 | 1 (0.9) |
| 19 | RHD*weak D type 5 (RHD01W.5) | RHD*DIIIa‐CE(4‐7)‐D (RHD*03N.01) | R1R2 | 1 (0.9) |
| 20 | RHD*weak partial 11 (RHD*11) | Deleted D (RHD*01N.01) | R1r | 8 (7.5) |
| 21 | RHD*weak partial 15 (RHD*15) | RHD*weak partial 15 (RHD*15) | R2R0 | 1 (0.9) |
| 22 | RHD*weak D type 38 (RHD*01W.38) | Deleted D (RHD*01N.01) | R1r | 31 (29.2) |
| 23 | RHD*weak D type 38 (RHD*01W.38) | RHD*weak D type 38 (RHD*01W.38) | R1R1 | 2 (1.9) |
| 24 | RHD*weak D type 38 (RHD*01W.38) | RHD*weak D type 38 (RHD*01W.38) | R1R0 | 3 (2.8) |
| 25 | RHD*weak D type 38 (RHD*01W.38) | RHD*Pseudogene (RHD*08N.01) | R1R0 | 1 (0.9) |
| 26 | RHD*weak D type 104 (RHD*01W.104) | Deleted D (RHD*01N.01) | R0r | 1 (0.9) |
| 27 | RHD*weak D type 112 (RHD*01W.112) | Deleted D (RHD*01N.01) | R0r | 1 (0.9) |
Among the identified RHD genotypes, 44.3% (47 of 106) encoded partial‐D antigens with weak expression and, as a consequence, could lead to anti‐D alloimmunization following incompatible transfusions or pregnancies.
3.2. Serological data of the donors with altered RHD genotype
All studied samples were tested either by Capture‐R solid‐phase technology (NEO, Immucor, Norcross, GA) or gel methodology (IH‐1000; Bio‐Rad, Cressier, Switzerland). The intensity of reactions is shown in Table 2. Both methodologies exhibited similar accuracy in identifying the weak‐D variants, as no discrepant results were observed among the samples tested in parallel by the two techniques (95 of 106 samples).
Table 2.
Serologic reactivity of different RHD genotypes encoding serologic weak‐D
| ID | RHD allele | Reactivity intensitya | Samples tested (%) | ||
|---|---|---|---|---|---|
| Neo (Immucor) | IH‐1000 (Bio‐Rad) | Adsorption/elution | |||
| 1 | RHD*DAR (RHD*09.01) | 1+ | 1+ or 2+ | nt | 32 (30.18) |
| 2 | RHD*DFR (RHD*17.01) | (+) | nt | 4+ | 1 (0.94) |
| 3 | RHD*DIIIc (RHD*03.03) | (+) | nt | 4+ | 2 (1.88) |
| 4 | RHD*DVI type 1(RHD*06.01) | nt | 2+ | nt | 1 (0.94) |
| 5 | 1157T>A; IVS5‐41delCTCT (RHD*01N.58) | (+) | nt | nt | 1 (0.94) |
| 6 | RHD*weak D type 1 (RHD*01W.1) | 1+ | 1+ | nt | 6 (5.66) |
| 7 | RHD*weak D type 2 (RHD*01W.2) | 1+ | 2+ | nt | 11 (10.37) |
| 8 | RHD*weak D type 3 (RHD*01W.3) | 1+ | nt | nt | 1 (0.94) |
| 9 | RHD*weak D type 4.0 (RHD*09.03.01) | nt | 2+ | nt | 1 (0.94) |
| 10 | RHD*weak D type 5 (RHD*01W.5) | (+) | nt | 4+ | 1 (0.94) |
| 11 | RHD*weak partial 11 (RHD*11) | (+) | 1+ | 4+ | 8 (7.54) |
| 12 | RHD*weak partial 15 (RHD*15) | (+) | nt | 4+ | 1 (0.94) |
| 13 | RHD*weak D type 38 (RHD*01W.38) | (+) | 1 or 2+ | 4+ | 38 (35.84) |
| 14 | RHD*weak D type 104 (RHD*01W.114) | nt | 2+ | nt | 1 (0.94) |
| 15 | RHD*weak D type 112 (RHD*01W.112) | (+) | 2+ | 4+ | 1 (0.94) |
Nt, not tested.
The reactions considered for Neo (Immucor) refer to the confirmatory testing using solid‐phase method. In the case of IH‐1000 (Bio‐Rad), the reactions refer to the initial testing using gel method.
The lower reaction intensity of the samples classified as RHD*weak D type 1 was remarkable, at both solid phase and gel methodology (Table 2). In one sample, the RHD*weak D type 1 allele was in trans to the dCe haplotype, further reducing D antigen expression (Table 1). Sample number 5 exhibited the RHD*1157T>A;IVS5‐41delCTCT/RHD*01N.01 genotype and weakly reacted with anti‐D at solid phase (Table 2). This allele has been previously described by Garcia F, 2015.20 It was named RHD*01N.58, and the corresponding phenotype was D‐negative by adsorption and elution test. And so, this donor was summoned for result confirmation.
3.3. RHD genotyping of patients with weak‐D phenotype
Fifty‐eight patient samples which were weakly reactive with anti‐D at gel method were genotyped for RHD and RHCE. The following alleles were identified among recipients, with the respective frequencies: RHD*01N.01 (deleted D) (42.4%); RHD*DAR (27.4%); RHD*weak D type 2 (18.9%); RHD*weak D type 1 (5.2%); RHD*weak D type 3 (5.2%); and RHD*Ѱ (0.9%). The final genotype of the patients is shown in Table 3. The overall frequency of patients exhibiting partial‐D phenotype was 50% (29 of 58 samples).
Table 3.
RHD genotype of Brazilian patients with serologic weak‐D phenotype
| ID | RHD genotype | Phenotypes (alternative) | Anti‐D | Grifols | N (%) | |
|---|---|---|---|---|---|---|
| 1 | RHD*DAR (RHD*09.01) | RHD*DAR (RHD*09.01) | R0R0 | 2+ | 2+ | 2 (3.4) |
| 2 | RHD*DAR (RHD*09.01) | RHD*DAR (RHD*09.01) | R2R0 | 2+ | 2+ | 1 (1.7) |
| 3 | RHD*DAR (RHD*09.01) | Deleted D (RHD*01N.01) | R0r | 2+ | 2+ | 26 (44.7) |
| 4 | RHD*weak D type 1 (RHD*01W.1) | Deleted D (RHD*01N.01) | R1r | 2+ | 2+ | 5 (8.6) |
| 5 | RHD*weak D type 1 (RHD*01W.1) | RHD*Pseudogene(RHD*08N.01) | R1R0 | 1+ | Negative | 1 (1.7) |
| 6 | RHD*weak D type 2 (RHD*01W.2) | Deleted D (RHD*01N.01) | R2r | 2+ | 1+ or 2+ | 14 (24.1) |
| 7 | RHD*weak D type 2 (RHD*01W.2) | RHD*weak D type 2 (RHD*01W.2) | R2R0 | 1+ or 2+ | Negative or 1+ or 2+ | 4 (6.8) |
| 8 | RHD*weak D type 3 (RHD*01W.3) | Deleted D (RHD*01N.01) | R1r | 2+ | 2+ | 3 (5.1) |
| 9 | RHD*weak D type 3 (RHD*01W.3) | Deleted D (RHD*01N.01) | R1r′ | 2+ | 2+ | 1 (1.7) |
| 10 | RHD*weak D type 3 (RHD*01W.3) | RHD*weak D type 3 (RHD*01W.3) | R1R0 | 2+ | 2+ | 1 (1.7) |
The variant alleles RHD*weak D type 38 and RHD*weak partial 11 were relatively prevalent among donors, but could not be identified in the patient group. This may reflect the different anti‐D clones used in this routine or the very low intensity of agglutination of both variants, which lead to their classification as D‐negative in the analysis of results.
4. DISCUSSION
This study describes the distribution of altered RHD alleles encoding weak‐D expression among blood donors and patients of mixed origin, mainly between African and European descent. It was shown that 44% (47/106) of the donors and 50% (29/58) of the blood recipients whose samples were weakly reactive (≤2+ agglutination intensity) with anti‐D (solid phase in confirmatory testing or gel method in initial testing) had a partial‐D phenotype and, as a consequence, were at risk of alloimmunization if transfused with D‐positive units. RHD*DAR was the most prevalent variant allele in blood donors, together with RHD*weak D type 38, and in patients. The distribution of the identified RHD variant alleles reflects the high degree of admixture of the Brazilian population. The recommendation is that patients with low expression of D antigen be transfused with D‐negative units and receive RhIG in case of pregnancy.
Brazil represents one of the most diverse populations in the world, result of centuries of inter‐ethnical crossing of individuals from several continents.21 This ancestry diversity is a major factor underlying the peculiar distribution of RBC antigen phenotype among Brazilians, especially referring to the RH system, in which both African and European genomes are represented.10, 22 This miscegenation can be clearly demonstrated by the allele distribution found in the present study, which comprised individuals with weak‐D phenotype. RHD*DAR, typically encountered in D variant patients of African descent, represented the most prevalent altered RHD allele followed by RHD*weak D type 38, which is found in Portuguese and whose presence among Brazilian donors probably originates in the colonization period.5 Even though RHD*weak D type 1, RHD*weak D type 2 and RHD*weak D type 3 correspond to 95% of D variants in Europeans,2 they accounted for less than 30% of the D variant genotypes in the present cohort. This peculiar distribution of altered RHD alleles reinforces the need for precisely classifying the RH variants when suspected and, until the molecular investigation is complete, follow guidelines specifically designed for populations of mixed origin, as the one proposed in the present study.
Our results have shown a high frequency of RHD partial genotype among individuals of mixed origin with weak‐D expression. This information is very relevant to the routine of patients, as the standard practice in some transfusion services is to detect weaker forms of D variant phenotype in order to save D‐negative units.5, 9 In European samples, weak D type 1, weak D type 2, and weak D type 3 underlie the vast majority of the weak‐D phenotype and, in these situations, patients are not at risk of alloimmunization.2 This scenario cannot be transposed to non‐European patient populations, because, as demonstrated by our data, only 29.3% of the individuals with weak‐D expression are classified as weak D type 1, 2 or 3. Here, the transfusion of D‐negative units should be preferred for all recipients with suspected weak‐D phenotype until molecular investigation is complete. This significantly differs from the recommendation proposed by the UK guidelines on 2015, which states that the recipients with weak‐D phenotype should be treated as D‐positive until molecular clarification.9
The present study also shed light into some important information for the immunohematological routine of mixed blood donors. The most important one refers to the need for applying ultra‐sensitivity methods for D typing or RHD‐PCR in the donor routine in order to identify the weak‐D phenotypes. Our data showed a high prevalence of weak‐D variants with very low antigen density among blood donors, such as weak D type 38 and weak D type 11, which could have been classified as D‐negative if methodologies other than gel was used for direct D typing or if solid phase was not indicated to confirm D‐negative results obtained through tube or microplate hemagglutination. If the chosen method for direct D typing is tube or microplate hemagglutination and sensitive IAT‐methods, such as solid phase, are unavailable to confirm negative results, RHD‐PCR should be applied to exclude D variant donors which could potentially sensitize D‐negative recipients.6, 7 Another important observation drawn from our results is that performing the molecular investigation of donors with weak‐D phenotype is an alternative strategy for identifying donors exhibiting RHD*DAR/RHD*DAR and RHD*DAR/RHD*01N.01 genotype. RHD*DAR in cis to RHCE*ceAR is encountered among sickle cell disease patients requiring RH variant‐identical transfusions, and in this situation, the selection of compatible donors is mandatory.23, 24
Every transfusion service should establish the workflow for investigating cases of weak‐D phenotype, as well as determining the criteria for transfusing D‐units or administering RhIG. The present study describes a strategy for the molecular investigation of patients with weak‐D phenotype based on the RHD genotypes identified in the Brazilian patient and donor population (shown in Figure 1). The first step of the proposed workflow is the accomplishment of a multiplex PCR for identification of RHD variants, which involves the amplification of RHD exons 3, 4, 5, 6, 7, and 9.13 Depending on the results of this multiplex PCR and the patient's Rh phenotype, tests are directed for the identification of weak D weak types 2, 3, 4, and 5 using allele specific primers 18, 19 followed by the direct sequencing of specific RHD exons.19 RHD exons 6 and 7 were included in the workflow because they contained key mutations for RHD*weak D type 1, RHD*weak D type 11, RHD*weak D type 15, RHD*weak D type 38, and RHD*DAR, respectively.
Figure 1.
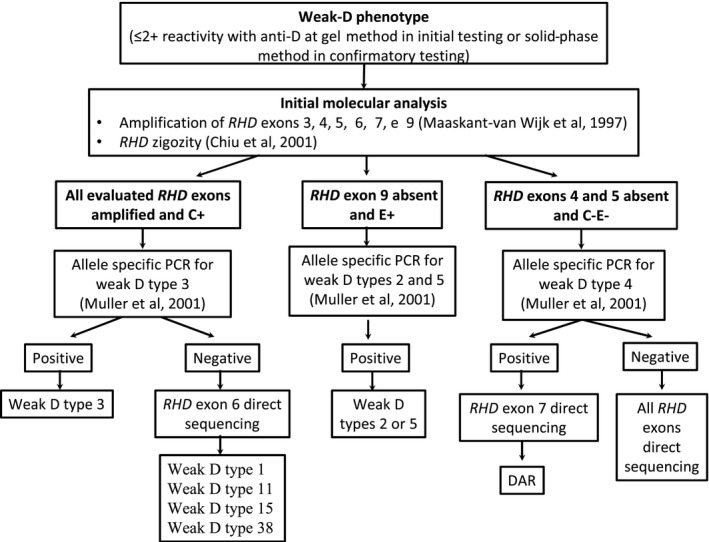
Workflow for the molecular investigation of donors and patients with serologic weak‐D phenotype originated from mixed population
Arnoni et al25, 26 have previously published an interesting strategy to investigate D variants among Brazilians using the same multiplex PCR which was also the cornerstone of the molecular investigation workflow proposed by the present study. The main differences between the two protocols are that our focused on investigating only individuals with serologic weak‐D phenotype, aiming to determine the best transfusion protocol in a relatively short period of time, while the other protocol is more extensive and focusing on the evaluation of all samples with suspected variant D phenotype.
Previous studies have also evaluated the frequency of D variants among Brazilian donors and patients, but with different criteria for performing the molecular investigation.11, 25 All studies were concordant with respect to the high prevalence of RHD*DAR and to the relatively lower representability of RHD*weak D types 1, 2, and 3 among Brazilian donors with weak‐D phenotype. As most studies included samples with agglutination discrepancies between different anti‐D clones or <3+ agglutination intensity using tube or gel method, a high frequency of RHD*weak D type 4.0 was detected. In our study, the inclusion criteria were more stringent, as only samples with <2+ agglutination in solid phase or gel method were included, and, as a consequence, D variants with not so low antigen density were not detected.
This study has some limitations. The most important one refers to the fact that there is a wide variation in the contribution of African ancestry in different mixed populations. And so, the conclusions drawn from the present study are applicable to populations where the racial ancestry is similar to Brazilian. On the other hand, the greatest strength of this study is that it sheds to light the need for having specific transfusion guidelines to meet the needs of mixed patients with serologic weak‐D phenotype. One proposed transfusion policy, based on our results, is shown in Figure 2. Similar to what have been published by other groups, the recommendation to transfuse D‐positive units to some weak‐D genotypes was based on the lack of adverse clinical reports, rather than on the presence of studies ensuring the impossibility of anti‐D formation.8, 26
Figure 2.
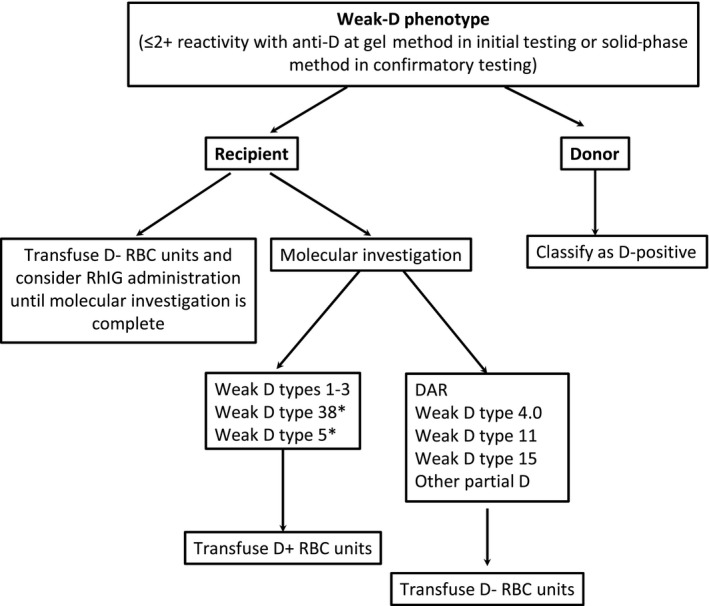
Suggested transfusion policy designed for patients of mixed origin with serologic weak‐D phenotype. *The reason for recommending the transfusion of D‐positive units for individuals bearing these variants was based on the lack of alloimmunization reports rather than on definite evidences that they are not prone to anti‐D development
In conclusion, nearly half of the Brazilian patients and donors with serologic weak‐D phenotype were eventually classified as partial D and, consequently, at risk of alloimmunization. The recommendation is that blood recipients of mixed origin be transfused with D‐negative units and receive RhIG in case of pregnancy until the molecular investigation is complete, differing from current guidelines directed to the European population.
ACKNOWLEDGMENTS
We thank Dr. Mayra Altobelli de Brito, Dr. Rita Fontão Wendel, and Dr. Silvano Wendel of Sírio Libanês Hospital (São Paulo, Brazil) for gently providing the primers used for confirmation of RHD zygosity in this study.
Dezan MR, Oliveira VB, Gomes CN, et al. High frequency of variant RHD genotypes among donors and patients of mixed origin with serologic weak‐D phenotype. J Clin Lab Anal. 2018;32:e22596 10.1002/jcla.22596
Funding information
This study was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPQ)—Grant 420168/2016‐8.
REFERENCES
- 1. Flegel WA. Molecular genetics and clinical applications for RH. Transfus Apher Sci. 2011;44:81‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wagner FF, Gassner C, Muller TH, Schonitzer D, Schunter F, Flegel WA. Molecular basis of weak D phenotypes. Blood. 1999;93:385‐393. [PubMed] [Google Scholar]
- 3. Flegel WA, Denomme GA, Yazer MH. On the complexity of D antigen typing: a handy decision tree in the age of molecular blood group diagnostics. J Obstet Gynaecol Can. 2007;29:746‐752. [DOI] [PubMed] [Google Scholar]
- 4. Sandler SG, Flegel WA, Westhoff CM, et al. It's time to phase in RHD genotyping for patients with a serologic weak D phenotype. College of American Pathologists Transfusion Medicine Resource Committee Work Group. Transfusion. 2015;55:680‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daniels G. Variants of RhD–current testing and clinical consequences. Br J Haematol. 2013;161:461‐470. [DOI] [PubMed] [Google Scholar]
- 6. Flegel WA, von Zabern I, Wagner FF. Six years’ experience performing RHD genotyping to confirm D‐ red blood cell units in Germany for preventing anti‐D immunizations. Transfusion. 2009;49:465‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dezan MR, Guardalini LGO, Pessoa E, et al. Evaluation of the applicability and effectiveness of a molecular strategy for identifying weak D and DEL phenotype among D‐ blood donors of mixed origin exhibiting high frequency of RHD*Psi. Transfusion. 2018;58:317‐322. [DOI] [PubMed] [Google Scholar]
- 8. Wagner FF, Frohmajer A, Ladewig B, et al. Weak D alleles express distinct phenotypes. Blood. 2000;95:2699‐2708. [PubMed] [Google Scholar]
- 9. Milkins C, Berryman J, Cantwell C, et al. Guidelines for pre‐transfusion compatibility procedures in blood transfusion laboratories. British Committee for Standards in Haematology. Transfus Med. 2013;23:3‐35. [DOI] [PubMed] [Google Scholar]
- 10. Campos FC, Mota MA, Aravechia MG, et al. Variant RHD types in Brazilians with discrepancies in RhD typing. J Clin Lab Anal. 2016;30:845‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bub CB, Aravechia MG, Costa TH, Kutner JM, Castilho L. RHD alleles among pregnant women with serologic discrepant weak D phenotypes from a multiethnic population and risk of alloimmunization. J Clin Lab Anal. 2018;32:e22221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dezan MR, Oliveira VB, Bianchi JVS, et al. Effectiveness of a red cell antigen‐matching transfusion protocol in sickle cell disease patients. ISBT Sci Ser. 2016;11:132‐139. [Google Scholar]
- 13. Maaskant‐van Wijk PA, Faas BH, de Ruijter JA, et al. Genotyping of RHD by multiplex polymerase chain reaction analysis of six RHD‐specific exons. Transfusion. 1998;38:1015‐1021. [DOI] [PubMed] [Google Scholar]
- 14. Singleton BK, Green CA, Avent ND, et al. The presence of an RHD pseudogene containing a 37 base pair duplication and a nonsense mutation in africans with the Rh D‐negative blood group phenotype. Blood. 2000;95:12. [PubMed] [Google Scholar]
- 15. Chiu RW, Murphy MF, Fidler C, Zee BC, Wainscoat JS, Lo YM. Determination of RhD zygosity: comparison of a double amplification refractory mutation system approach and a multiplex real‐time quantitative PCR approach. Clin Chem. 2001;47:667‐672. [PubMed] [Google Scholar]
- 16. Perco P, Shao CP, Mayr WR, Panzer S, Legler TJ. Testing for the D zygosity with three different methods revealed altered Rhesus boxes and a new weak D type. Transfusion. 2003;43:335‐339. [DOI] [PubMed] [Google Scholar]
- 17. Tax MG, van der Schoot CE, van Doorn R, Douglas‐Berger L, van Rhenen DJ, Maaskant‐vanWijk PA. RHC and RHc genotyping in different ethnic groups. Transfusion. 2002;42:634‐644. [DOI] [PubMed] [Google Scholar]
- 18. Muller TH, Wagner FF, Trockenbacher A, et al. PCR screening for common weak D types shows different distributions in three Central European populations. Transfusion. 2001;41:45‐52. [DOI] [PubMed] [Google Scholar]
- 19. Wagner FF, Gassner C, Muller TH, Schonitzer D, Schunter F, Flegel WA. Three molecular structures cause rhesus D category VI phenotypes with distinct immunohematologic features. Blood. 1998;91:2157‐2168. [PubMed] [Google Scholar]
- 20. Garcia F, Rodriguez MA, Goldman M, et al. New RHD variant alleles. Transfusion. 2015;55:427‐429. [DOI] [PubMed] [Google Scholar]
- 21. Parra FC, Amado RC, Lambertucci JR, Rocha J, Antunes CM, Pena SD. Color and genomic ancestry in Brazilians. Proc Natl Acad Sci U S A. 2003;100:177‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dezan MR, Ribeiro IH, Oliveira VB, et al. RHD and RHCE genotyping by next‐generation sequencing is an effective strategy to identify molecular variants within sickle cell disease patients. Blood Cells Mol Dis. 2017;65:8‐15. [DOI] [PubMed] [Google Scholar]
- 23. Reid ME, Halter Hipsky C, Hue‐Roye K, Hoppe C. Genomic analyses of RH alleles to improve transfusion therapy in patients with sickle cell disease. Blood Cells Mol Dis. 2014;52:195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chou ST, Jackson T, Vege S, Smith‐Whitley K, Friedman DF, Westhoff CM. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh‐matched minority donors. Blood. 2013;122:1062‐1071. [DOI] [PubMed] [Google Scholar]
- 25. Arnoni CP, Latini FR, Muniz JG, et al. How do we identify RHD variants using a practical molecular approach? Transfusion. 2014;54:962‐969. [DOI] [PubMed] [Google Scholar]
- 26. Ouchari M, Srivastava K, Romdhane H, Jemni Yacoub S, Flegel WA. Transfusion strategy for weak D Type 4.0 based on RHD alleles and RH haplotypes in Tunisia. Transfusion. 2018;58:306‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]