

Abstract
Background
Severe hypertriglyceridemia usually results from a combination of genetic and environmental factors and is most often attributable to mutations in the lipoprotein lipase (LPL) gene.
Objectives
The aim of this study was to identify rare mutations in the LPL gene causing severe hypertriglyceridemia.
Methods
A Chinese infant who presented classical features of severe hypertriglyceridemia recruited for DNA sequencing of the LPL gene. The pathogenicity grade of the variants was defined based on the prediction of pathogenicity using in silico prediction tools. Review some studies to understand the molecular mechanisms underlying the severe hypertriglyceridemia.
Results
We identified a rare mutation in the LPL gene causing severe hypertriglyceridemia: a nucleotide substitution (c.836T>G) resulting in a leucine to arginine substitution at position 279 of the protein (p.Leu279Arg).The pathogenicity of the variant was predicted by in silico analysis using PolyPhen2 and SIFT prediction programs, which indicated that mutation p.Leu279Arg is probably harmful. We have also reviewed published studies concerning the molecular mechanisms underlying severe hypertriglyceridemia. A missense mutation in the 6 exon of the LPL gene is reportedly associated with LPL deficiency.
Conclusions
We have here identified a rare pathogenic mutation in the LPL gene in a Chinese infant with severe hypertriglyceridemia.
Keywords: heterozygous, Hypertriglyceridemia, LPL gene, missense
Abbreviations
- apoA I
apolipoproein A I
- apoB
apolipoprotein B
- HDL
high‐density lipoprotein
- LDL
low‐density lipoprotein
- Lp(a)
lipoprotein(a)
- LPL
Lipoprotein lipase
- TC
total cholesterol
- TG
triglyceride
1. INTRODUCTION
Lipoprotein lipase (LPL), a member of the serine esterase family, was discovered in 1943 and is mainly produced and secreted by myocardial, skeletal muscle, and adipose tissue.1After synthesis, it is secreted and transported to the luminal surfaces of vascular endothelial cells and embedded in the inner walls of vessels by ion reaction with heparan sulfate proteoglycan or glycosylphosphatidylinositol.2 The main physiological function of LPL in humans is to hydrolyze chitos an particles in plasma and triglycerides in very low‐density lipoproteins, resulting in free fatty acids entering the myocardium and skeletal muscle to undergo oxygenolysis, release energy, or re‐synthesize triglycerides stored in adipose tissue.3 LPL is a rate‐limiting enzyme in triglyceride degradation, which plays important roles in lipid metabolism, insulin resistance, and adipocyte differentiation.1
The human LPL gene is located on chromosome 8 short arm 8p22 and comprises ten exons and nine introns, including 30 kp bases, that encode a 475 amino acid lipoprotein.4, 5 Thus far, 114 mutations have been reported in the LPL gene, most of which are missense mutations in the highly conserved 4, 5, and 6 exons,6 throughout the regulatory region, introns, and exons. This protein is among those with the most abundant gene mutations.
Mutations in lipoprotein lipase may affect its catalytic function and even cause chylomicronemia. Chylomicronemia syndrome, which presents in childhood, is a rare recessive genetic disorder characterized by severe hypertriglyceridemia (sHTG, >12 mmol/L), fasting chylomicronemia, eruptive xanthomas, and lipemia retinalis and occasionally also manifesting hepatosplenomegaly, abdominal pain, and/or recurrent acute pancreatitis.7 In newborns and very young children (1 year of age), chylomicronemia is often diagnosed after the incidental finding of milky plasma during routine blood tests or laboratory investigations performed for unrelated conditions.8In most cases, it is attributable to mutations in the lipoprotein lipase (LPL) gene.
In this article, we report a case of heterozygous LPL mutation in an infant with severe hypertriglyceridemia and convulsions.
2. MATERIALS AND METHODS
2.1. Subjects
A 5‐month‐old baby boy of Chinese Zhuang ethnicity was admitted to the First Affiliated Hospital of Guangxi Medical University, China, for repeated convulsions and dyslipidemia. On admission, no abnormalities were detected on routine clinical examination. His laboratory tests were as follows. (i) Routine blood analysis: white blood cell (WBC) count, 14.06 × 109/L; platelet count, 617.2 × 109/L; hemoglobin (Hb), 111.90 g/L; neutrophil‐to‐lymphocyte ratio, 0.423; and platelet‐to‐lymphocyte ratio, 0.501.(ii) Biochemical tests showed no abnormalities in liver or kidney function or electrolytes. Total cholesterol (TC) concentration was 4.69 mmol/L, triglycerides (TG) 24.9 mmol/L, high‐density lipoprotein cholesterol (HDL‐C) 0.44 mmol/L, low‐density lipoprotein cholesterol (LDL‐C) 2.44 mmol/L, apolipoprotein AI (apoAI) 0.63 g/L, apolipoprotein B (apoB) 0.93 g/L, apolipoprotein AI/B (apoAI/B) 0.7, and lipoprotein (a) (Lp[a]) 0.020 g/L. Abdominal color Doppler ultrasonography showed hepatosplenomegaly. Cranial magnetic resonance and electroencephalogram were normal. Considering these manifestations and laboratory test results, the infant was diagnosed as having chylomicronemia. The proband's sister was also found to have hypertriglyceridemia but without any clinical signs. The child's parents gave informed consent for the tests on the proband and family members, which were approved by the Ethics Committee of our institution.
2.2. DNA isolation and sequencing
With informed consent, 2‐mL blood samples were extracted from the proband and his family members and ethylenediaminetetraacetic acid added. A Qiagen FlexiGene DNA Kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from these samples according to the manufacturer's instructions. All LPL exons and their flanking intronic regions were amplified by a primer set design based on the sequence of the LPL gene (NC_000008.11) in GenBank. The primers were diluted, after which ABI PCR amplification was performed according to primer conditions. The PCR products were electrophoresed by 1% agarose gel electrophoresis to identify, purify, and recover the products, which were then sequenced. The sequencing products were run on a Genetic Analyzer 3730 (Applied Biosystems).
2.3. Plasma lipid analysis
A morning fasting blood sample was taken, and serum concentrations of TC (cholesterol oxidase method), TG (glycerol phosphate oxidase method), HDL‐C (phosphotungstic acid‐magnesium precipitation method), LDL‐C (polyethylene sulfuric acid precipitation method), and apoA I, apoB, and Lp (a) (immunoturbidimetry method) were measured using a 7600 Automatic Biochemical Analyzer (Hitachi High‐Technologies, Tokyo, Japan).
2.4. Bioinformatic analysis
The conserved amino acid sequences of LPL were partially analyzed by multiple sequence alignment. The pathogenicity of the variants was predicted using in silico prediction tools, namely PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT (http://sift.jcvi.org).
3. RESULT
3.1. Clinical data analysis
As shown in Table 1, the lipid profiles of the proband and his family were compared. Triglycerides were very high and high‐density lipoprotein was low in the proband and his sister showed moderate hypertriglyceridemia, whereas his parents had normal lipid profiles (Figure 1). When he first diagnosed chylomicronemia, the doctor suggested weaning. The TG concentration after weaning was 22.07 mmol/L. The proband's parents were of Zhuang ethnicity from Nanning, Guangxi Zhuang Autonomous Region in China, and denied both in‐law marriage and any relevant family history.
Table 1.
Lipid concentrations in the proband and his family
| Name | Age (years) | Lipid | |||||||
|---|---|---|---|---|---|---|---|---|---|
| TC (mmol/L) | TG (mmol/L) | HDL‐C (mmol/L) | LDL‐C (mmol/L) | apoA I (g/L) | apoB (g/L) | apoA I/B | Lp(a) (g/L) | ||
| Proband | 0.42 | 4.69 | 24.9 | 0.44 | 2.44 | 0.63 | 0.93 | 0.7 | 0.020 |
| Father | 29 | 4.84 | 1.54 | 0.90 | 2.99 | 0.91 | 1.11 | 0.8 | 0.983 |
| Mother | 22 | 3.8 | 0.55 | 1.96 | 1.62 | 1.49 | 0.6 | 2.5 | 0.067 |
| Sister | 2 | 4.55 | 3.47 | 0.86 | 2.41 | 0.85 | 1.01 | 0.8 | 1.018 |
apoA I, apolipoproein A I; apoB, apolipoprotein B; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; Lp(a), lipoprotein(a); TC, total cholesterol; TG, triglyceride.
Figure 1.
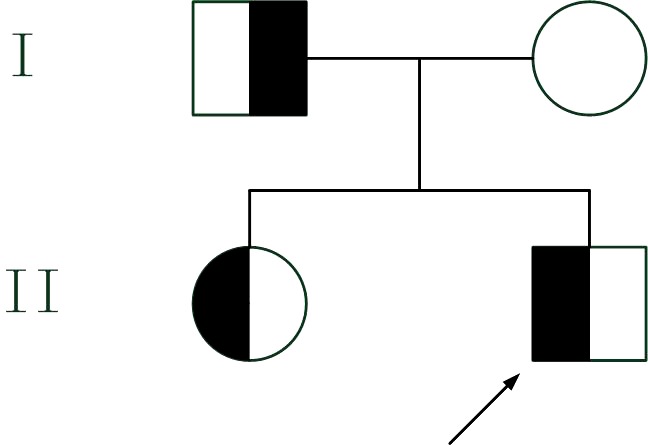
Pedigree of the reported family. The proband is indicated by an arrow. Half‐filled symbols denote affected individuals, carriers of the mutation p.Leu279Arg of the LPL gene, and empty symbols denote noncarriers
3.2. Analysis of mutations in the LPL gene
PCR sequencing revealed a c.836T>G heterozygous missense mutation in exon 6 of the LPL gene in the proband, and this mutation resulted in the conversion of amino acid 279 from Leu to Arg (p.Leu279Arg). His father and sister were found to be heterozygous carriers of c.836T>G missense mutations, as shown in Figure 2.
Figure 2.
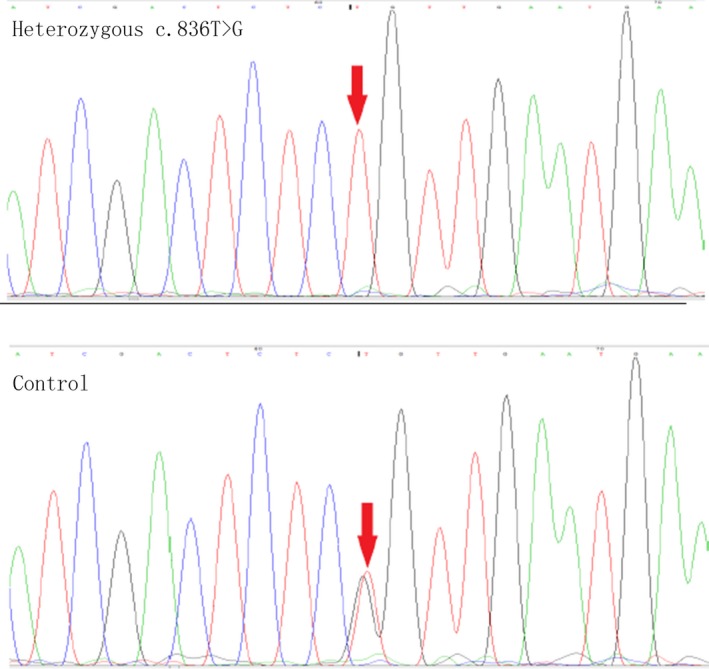
Sequence analysis of the LPL mutations. Upper panel: segments of genomic DNA sequences of the proband showing a nucleotide substitution (c.836T>G) in the region indicated by the red arrows. Lower panel: segments of genomic DNA sequences of the control individual (the proband's mother); his father and sister were found to be heterozygous carriers of c.836T>G missense mutations
3.3. Bioinformatic analysis
Partial amino acid sequences of LPL in 11 species were obtained and found to have highly conserved p.L279 leucine sequences (Figure 3). In silico analysis using PolyPhen2 and SIFT prediction programs indicated that mutation p.Leu279Arg is probably harmful.
Figure 3.
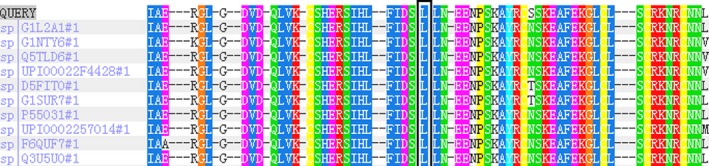
Partial amino acid sequences of LPL in 11 species were compared. A highly conserved p.L279 leucine sequence is present in these 11 species (black)
4. DISCUSSION
Familial lipoprotein lipase deficiency is rare and caused by mutations in the LPL gene that have an autosomal‐recessive pattern of inheritance.9 In this study, we report a Chinese infant with hypertriglyceridemia and convulsions who we found to have a heterozygous missense mutation in the LPL gene.
Recent studies have shown that missense mutations in the LPL gene are associated with hypertriglyceridemia. LPL is a key enzyme in lipid metabolism and, when abnormal, the underlying cause of hyperlipidemia.10 Missense mutations in the LPL gene play an important role in patients with severe hypertriglyceridemia.11 Chan et al12 reported a patient with hypertriglyceridemia, two mutations in the LPL gene(Leu252Val and Leu252Arg), and decreased LPL activity and quantity in their postheparin plasma. Extracellular culture showed that the Leu252Val and Leu252Arg mutations affected the release of LPL and damaged the catalytic activity of LPL. Hoffmann et al13 showed that a C266W mutation in the 6 exon of the LPL gene prevents formation of the second disulfide bridge of LPL, which is an essential part of the lid covering the catalytic center. Consequently, misfolded LPL is rapidly degraded within the cells, resulting in the absence of LPL immunoreactive protein in this patient's plasma. Saika et al14 reported a patient with L303F mutation in the 6 exon of the LPL gene who had approximately 6% of normal LPL activity and 40% of LPL quantity in their postheparin plasma. An in vitro expression study demonstrated that COS7 cells transfected with L303F mutant cDNA produced 40% of LPL protein in cell lysates compared with normal cDNA. No protein was detected in the media and lipoprotein lipase activity was completely absent in both lysates and media of the cells transfected with the mutant cDNA, suggesting that this mutation in the LPL gene results in production of a functionally inactive protein. Takagi et al15 have systematically investigated an F297L mutation in exon 6 resulting in a primary lipoprotein lipase (LPL) deficiency in a Japanese male infant. They failed to detect LPL activity or any immunoreactive LPL in thus infant's pre‐ or postheparin plasma. In vitro expression experiments on the mutant F297L LPL cDNA in COS‐1 cells demonstrated that the mutant LPL protein was synthesized as a catalytically inactive form and its total amount being almost equal to that of normal LPL. Moreover, the synthesized mutant LPL was impaired because the structure of the mutant LPL protein was abnormal. Most studies have shown that a missense mutation in the 6 exon of the LPL gene is associated with LPL deficiency and consequently hypertriglyceridemia (Table 2).
Table 2.
Missense mutations of LPL gene in exon 6 identified in individuals with hypertriglyceridemia
| Reference | Effect on LPL Protein | Description |
|---|---|---|
| 13 | C266W | In LPL deficiency; prevents the formation of the second disulfide bridge of LPL; the absence of LPL immunoreactive protein in the plasma of this patient |
| 23, 24 | R270C and S271C | In LPL deficiency |
| 25 | D277N | In LPL deficiency; 5% of full activity |
| 26 | L279R and L279V | In LPL deficiency; decreases the specific activity of the enzyme; the total LPL mass is reduced compared to that of the wild‐type construct. |
| 27 | S286G | No detectable LpL mass or activity in postheparin plasma |
| 28 | S286R | A clear 50% reduction in LPL activity |
| 29 | A288T | In LPL deficiency; the LPL mass level is approximately 67% of the normal; the activity is 32% of the normal |
| 30 | Y289H | Undetectable pre‐ and postheparin plasma LPL mass and activity |
| 15 | F297L | In LPL deficiency and hyperlipidemia; synthesized as a catalytically inactive form; total amount is almost equal to that of the normal enzyme; nonreleasable by heparin due to the abnormal structure of the mutant protein. |
| 14 | L303F | In LPL deficiency; approximately 6% of normal LPL activity and 40% of LPL mass are detected in the patient's postheparin plasma; results in the production of a functionally inactive enzyme, produced a 40% amount of LPL protein in cell lysates compared with normal cDNA, but no protein was detected in the media |
| 31 | N318S | In LPL deficiency; loss of activity |
| 26 | S325R | In LPL deficiency; has no effect on the specific activity of the enzyme; has a mild effect on the secretion of the mutant enzyme; the total LPL mass is reduced compared to that of the wild‐type construct |
| 32 | M328R and M328T | In LPL deficiency |
| 29 | L 330F and L330P | In LPL deficiency |
Recently, Weerapan et al16 resequenced the LPL gene in 101 individuals with hypertriglyceridemia and 111 normotriglyceridemic controls and found six rare variants in the coding region of LPL. All these known variants, p.Ala98Thr, p.Leu279Val, and p.Leu279Arg, have been associated with severe hypertriglyceridemia. Three different in silico prediction programs consistently predicted that the novel variant p.Arg270Gly and the known variant p.Arg432Thr would impair the relevant protein (p.Asp308Glyfs*3). The rare variant (p.Arg270Gly) was predicted to result in a truncated LPL protein lacking the C‐terminus, which is important for interaction with lipoproteins and lipoprotein receptors. None of these rare variants were found in 111 control subjects with normal triglyceride concentrations. Unfortunately, the severity of the hypertriglyceridemia‐associated phenotype was not reported. The patient we here describe has a heterozygous nonsense mutation (c.836T>G–p.Leu279Arg) of LPL that is associated with a severe hypertriglyceridemia phenotype. In silico PolyPhen2 and SIFT prediction programs have indicated that mutation p.Leu279Arg is probably harmful.
Leucine 279, located in the loop structure covering the catalytic triad, is conserved in the LPL gene in various mammalian species(Figure 3) and may play a significant role in the catalytic function of LPL.12 Ma et al17 assessed postheparin and in vitro expression of LPL activity and mass and showed that Leu279Arg abolishes both the catalytic function and secretion of LPL. Replacement of the nonpolar leucine by the positively charged arginine at codon 279 may result in loss of catalytic activity by disrupting the folding of LPL. Arginine substitution may lead to unstable dimers and thus rapid dissociation resulting in the absence of secretion of the mutant protein.12 Chan et al12 reported that after transfection with Leu279Arg cDNA in COS‐1 cells, LPL activity was undetectable in both the cell medium and lysate. It has become evident that normal LPL is required to bind to heparin in order for it to separate from parenchymal cells and be released into the extracellular fluid from which LPL is transported across the capillary endothelial cells in the presence of GPIHBP1.18 Heparin‐binding sites for LPL are located at residues 279–282 and 292–304 in the N‐terminal region.19 The present newly identified mutation Leu279Arg is in the heparin‐binding sites in the N‐terminal region. Taken together, the evidence points to the possibility that electrical charge transfer in the vicinity of the heparin‐binding sites compromises the conformational stability of LPL, resulting in a marked reduction in the affinity of LPL to heparin.20
This finding is interesting in that our proband has severe hypertriglyceridemia, whereas his sister, a carrier of the mutation, has mild‐to‐moderate hypertriglyceridemia, and the father's triglyceride concentrations are normal. These findings raise the question of why the presence of LPL mutations does not consistently correlate with total triglyceride concentrations in individuals with identical mutations in the same family. There are only a few published data that partially answer this question. Apolipoprotein E2 isoform is another factor that may influence plasma triglyceride concentrations. Bennet et al21 reported that carriers of the APOE2 allele have higher triglyceride concentrations than carriers of the more common APOE3 allele, which may partially explain why family members carrying the same heterozygous missense mutation, and have differing triglyceride concentrations. We did not assess this possibility, which is a limitation of our study. Other factors likely also contribute to these discrepancies in triglyceride concentrations: phenotypic expression is a result of interactions between LPL deficiency caused by LPL gene mutations and secondary environmental factors such as age, lifestyle, diet, and drugs.22
In conclusion, we here identified a rare missense mutation of LPL gene, c.836T>G–p.Leu279Arg, that is associated with a severe hypertriglyceridemia phenotype and has variable penetrance. Our findings and those reported by others indicate that searching for LPL gene mutations should be part of the genetic work‐up in individuals with primary hypertriglyceridemia.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Yes.
AVAILABILITY OF DATA AND MATERIALS
Data sharing not applicable to this article as no dataset is generated or analysed during the current study.
AUTHOR'S CONTRIBUTIONS
QYY and LFQ designed study. QYY, WAQ, SQW, XXY, WYY, LL, YJ, LZF, and LFQ analyzed and discussed data, wrote or revised the manuscript, and have primary responsibility for final content. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
We would like to thank the Department of Clinical Laboratory, First Affiliated Hospital of Guangxi Medical University, China.
Qin Y‐y, Wei A‐q, Shan Q‐w, et al. Rare LPL gene missense mutation in an infant with hypertriglyceridemia. J Clin Lab Anal. 2018;32:e22414 10.1002/jcla.22414
REFERENCES
- 1. Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271‐E288. [DOI] [PubMed] [Google Scholar]
- 2. Pingitore P, Lepore SM, Pirazzi C, et al. Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia. J Clin Lipidol. 2016;10:816‐823. [DOI] [PubMed] [Google Scholar]
- 3. Young SG, Zechner R. Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev. 2013;27:459‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hegele RA, Pollex RL. Hypertriglyceridemia: phenomics and genomics. Mol Cell Biochem. 2009;326:35‐43. [DOI] [PubMed] [Google Scholar]
- 5. Deeb SS, Peng RL. Structure of the human lipoprotein lipase gene. Biochemistry. 1989;28:4131‐4135. [DOI] [PubMed] [Google Scholar]
- 6. Brahm AJ, Hegele RA. Chylomicronaemia–current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352‐362. [DOI] [PubMed] [Google Scholar]
- 7. Stefanutti C, Gozzer M, Pisciotta L, et al. A three month‐old infant with severe hyperchylomicronemia: molecular diagnosis and extracorporeal treatment. Atheroscler Suppl. 2013;14:73‐76. [DOI] [PubMed] [Google Scholar]
- 8. Buonuomo PS, Bartuli A, Rabacchi C, Bertolini S, Calandra S. A 3‐day‐old neonate with severe hypertriglyceridemia from novel mutations of the GPIHBP1 gene. J Clin Lipidol. 2015;9:265‐270. [DOI] [PubMed] [Google Scholar]
- 9. Kassner U, Salewsky B, Wuhle‐Demuth M, et al. Severe hypertriglyceridemia in a patient heterozygous for a lipoprotein lipase gene allele with two novel missense variants. Eur J Hum Genet. 2015;23:1259‐1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagasaka H, Kikuta H, Chiba H, et al. Two cases with transient lipoprotein lipase (LPL) activity impairment: evidence for the possible involvement of an LPL inhibitor. Eur J Pediatr. 2003;162:132‐138. [DOI] [PubMed] [Google Scholar]
- 11. Evans D, Arzer J, Aberle J, Beil FU. Rare variants in the lipoprotein lipase (LPL) gene are common in hypertriglyceridemia but rare in Type III hyperlipidemia. Atherosclerosis. 2011;214:386‐390. [DOI] [PubMed] [Google Scholar]
- 12. Chan L, Mak Y, Tomlinson B, et al. Compound heterozygosity of Leu252Val and Leu252Arg causing lipoprotein lipase deficiency in a chinese patient with hypertriglyceridemia. Eur J Clin Invest. 2000;30:33‐40. [DOI] [PubMed] [Google Scholar]
- 13. Hoffmann MM, Jacob S, Luft D, et al. Type I hyperlipoproteinemia due to a novel loss of function mutation of lipoprotein lipase, Cys(239)–>Trp, associated with recurrent severe pancreatitis. J Clin Endocrinol Metab. 2000;85:4795‐4798. [DOI] [PubMed] [Google Scholar]
- 14. Saika Y, Sakai N, Takahashi M, et al. Novel LPL mutation (L303F) found in a patient associated with coronary artery disease and severe systemic atherosclerosis. Eur J Clin Invest. 2015;33:216‐222. [DOI] [PubMed] [Google Scholar]
- 15. Takagi A, Ikeda Y, Takeda E, Yamamoto A. A newly identified lipoprotein lipase (LPL) gene mutation (F270L) in a Japanese patient with familial LPL deficiency. Biochem Biophys Acta. 2000;1502:433‐446. [DOI] [PubMed] [Google Scholar]
- 16. Khovidhunkit W, Charoen S, Kiateprungvej A, Chartyingcharoen P, Muanpetch S, Plengpanich W. Rare and common variants in LPL and APOA5 in Thai subjects with severe hypertriglyceridemia: a resequencing approach. J Clin Lipidol. 2016;10:505‐511 e1. [DOI] [PubMed] [Google Scholar]
- 17. Ma Y, Ooi TC, Liu MS, et al. High frequency of mutations in the human lipoprotein lipase gene in pregnancy‐induced chylomicronemia: possible association with apolipoprotein E2 isoform. J Lipid Res. 1994;35:1066‐1075. [PubMed] [Google Scholar]
- 18. Tian GP, Chen WJ, He PP, Yin WD, Tnag CK. [Current progress in lipoprotein lipase and atherosclerosis]. Prog Physiol Sci. 2012;43:345‐350. [PubMed] [Google Scholar]
- 19. Lookene A, Nielsen MS, Gliemann J, Olivecrona G. Contribution of the Carboxy‐Terminal Domain of Lipoprotein Lipase to Interaction with Heparin and Lipoproteins ☆. Biochem Biophys Res Comm. 2000;271:15‐21. [DOI] [PubMed] [Google Scholar]
- 20. Lun Y, Sun X, Wang P, Chi J, Hou X, Wang Y. Severe hypertriglyceridemia due to two novel loss‐of‐function lipoprotein lipase gene mutations (C310R/E396V) in a Chinese family associated with recurrent acute pancreatitis. Oncotarget. 2017;8:47741‐47754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bennet AM, Di AE, Ye Z, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. 2007;298:1300‐1311. [DOI] [PubMed] [Google Scholar]
- 22. Fisher RM, Humphries SE, Talmud PJ. Common variation in the lipoprotein lipase gene: effects on plasma lipids and risk of atherosclerosis. Atherosclerosis. 1997;135:145. [DOI] [PubMed] [Google Scholar]
- 23. Allain CC. Premature atherosclerosis in patients with familial chylomicronemia caused by mutations in the lipoprotein lipase gene &mdash. N Engl J Med. 1996;335:848. [DOI] [PubMed] [Google Scholar]
- 24. Foubert L, Gennes JLD, Lagarde JP, et al. Assessment of French patients with LPL deficiency for French Canadian mutations. J Med Genet. 1997;34:672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishimura‐Oka K, Semenkovich CF, Faustinella F, et al. A missense (Asp250—Asn) mutation in the lipoprotein lipase gene in two unrelated families with familial lipoprotein lipase deficiency. J Lipid Res. 1992;33:745‐754. [PubMed] [Google Scholar]
- 26. Chan LYS, Lam CW, Mak YT, et al. Genotype‐phenotype studies of six novel LPL mutations in Chinese patients with hypertriglyceridemia. Hum Mutat. 2002;20:232‐233. [DOI] [PubMed] [Google Scholar]
- 27. Evans D, Wendt D, Ahle S, Guerra A, Beisiegel U. Compound heterozygosity for a new (S259G) and a previously described (G188E) mutation in lipoprotein lipase (LpL) as a cause of chylomicronemia. Mutations in brief no. 183. Online. Hum Mutat. 1998;12:217. [PubMed] [Google Scholar]
- 28. Pimstone SN, Clee SM, Gagné SE, et al. A frequently occurring mutation in the lipoprotein lipase gene (Asn291Ser) results in altered postprandial chylomicron triglyceride and retinyl palmitate response in normolipidemic carriers. J Lipid Res. 1996;37:1675‐1684. [PubMed] [Google Scholar]
- 29. Maruyama T, Yamashita S, Matsuzawa Y, et al. Mutations in Japanese subjects with primary hyperlipidemia–results from the Research Committee of the Ministry of Health and Welfare of Japan since 1996. J Atheroscler Thromb. 2004;11:131. [DOI] [PubMed] [Google Scholar]
- 30. Gehrisch S. Common mutations of the lipoprotein lipase gene and their clinical significance. Curr Atheroscler Rep. 1999;1:70‐78. [DOI] [PubMed] [Google Scholar]
- 31. Gerhard DS, Wagner L, Feingold EA, et al. The status, quality, and expansion of the NIH full‐length cDNA project: the Mammalian Gene Collection (MGC). Genome Res. 2004;14:2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kavazarakis E, Stabouli S, Gourgiotis D, et al. Severe hypertriglyceridaemia in a Greek infant: a clinical, biochemical and genetic study. Eur J Pediatr. 2005;163:462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no dataset is generated or analysed during the current study.