

Abstract
Aim
To select an optimal whole‐genome amplification (WGA) method to improve the efficiency of the preimplantation genetic diagnosis and screening (PGD/PGS) of beta‐thalassaemia disorders.
Methods
Fifty‐seven fibroblast samples with defined beta‐thalassaemia variations and forty‐eight single‐blastomere samples were amplified from single‐, two‐, and five‐cell samples by multiple annealing and looping‐based amplification cycles (MALBAC) and the multiple displacement amplification (MDA) method. Low‐depth, high‐throughput sequencing was performed to evaluate and compare the coefficiencies of the chromosomal copy number variation (CNV) detection rate and the allele dropout (ADO) rate between these two methods.
Results
At the single‐cell level, the success rates of the CNV detection in the fibroblast samples were 100% in the MALBAC group and 91.67% in the MDA group; the coefficient of variation in the CNV detection in the MALBAC group was significantly superior to that in the MDA group (0.15 vs 0.37). The total ADO rate in the HBB allele detection was 4.55% in the MALBAC group, which was significantly lower than the 22.5% rate observed in the MDA group. However, when five or more cells were used as the starting template, the ADO rate significantly decreased, and these two methods did not differ significantly.
Conclusions
For the genetic diagnosis of HBB gene variation at the single‐cell level, MALBAC is a more suitable method due to its higher level of uniformity and specificity. When five or more cells are used as the starting template, both methods exhibit similar efficiency, increased accuracy, and a similar success rate in PGD/PGS.
Keywords: beta‐thalassaemia, multiple annealing and looping‐based amplification cycles, multiple displacement amplification, whole‐genome amplification efficiency
1. INTRODUCTION
Beta‐thalassaemia is the most common anemia disorder and is caused by mutations in the beta‐globin gene.1 In Southeast China, beta‐thalassaemia accounts for most of the monogenic‐inherited diseases, with an average carrier rate as high as 2.54%.2 Although a prenatal diagnosis of beta‐thalassaemia using fetal genetic material provides a method to prevent the birth of a genetically affected child,3 the invasive amniocentesis or chorionic villus sampling procedure may expose pregnant women to the risk of miscarriage,4 and the selective termination of pregnancy often causes severe psychological and physiological damage to the patients and their families. Therefore, preimplantation genetic diagnosis (PGD) is a preferred technique that may be used to select embryos that are unaffected by monogenic disease for uterine transfer.5 Moreover, preimplantation genetic screening (PGS) is considered to significantly improve the pregnancy success rate because a euploid embryo may be selected for the transfer.6 Currently, PGD/PGS is utilized in many in vitro fertilization (IVF) centers worldwide.7
A routine PGD/PGS procedure involves an embryo biopsy using oocyte polar bodies, one blastomere cell from a cleavage‐stage embryo or 3‐5 trophectoderm cells from a blastocyst embryo.3, 5, 8, 9 Because the genetic material used for diagnosis is highly limited, a strong demand exists for a whole‐genome amplification (WGA) method with high uniformity and fidelity. The development of WGA methods and the advancement of next‐generation sequencing (NGS) techniques are auspicious for detecting mutated alleles and concomitant screening for aneuploidy.7, 10
WGA plays a key role in the PGD/PGS process. The following three main categories of WGA methods are currently available: degenerate oligonucleotide primed‐polymerase chain reaction (DOP‐PCR),11, 12 the multiple displacement amplification (MDA) method,13, 14, 15, 16 and very recently, multiple annealing and looping‐based amplification cycles (MALBAC).17, 18, 19 By introducing a quasilinear amplification step, the MALBAC method has been reported to significantly reduce the amplification bias and has the advantages of accurately detecting copy number variations (CNV).17, 20 In addition, compared to the DOP‐PCR and MDA methods, MALBAC has a lower allele dropout (ADO) rate.21 However, MALBAC is reported to have a high false‐positive rate in the detection of single‐nucleotide variations (SNV) and a bias toward high GC‐content regions.22, 23
Because no WGA method is superior in all aspects, the selection of the optimal WGA method based on its specific applications is critical. To date, the efficiencies of the various WGA methods in detecting the HBB gene and diagnosing beta‐thalassaemia have not been evaluated. As performed in the PGD/PGS procedure, embryos are often biopsied in the cleavage stage (one blastomere cell) or in the blastocyst stage (3‐5 trophectoderm cells). Therefore, in this study we compared the WGA‐sequencing efficiencies of the two most commonly used WGA techniques, MDA and MALBAC, using one to five cells. We then compared the key performance parameters, such as the detection of an ADO in the HBB gene mutation and the detection of variations in CNV and amplification success rates. To the best of our knowledge, this comparison has not been performed previously, and the results might increase the understanding and selection of WGA techniques in the contexts of single‐gene PGD and chromosomal PGS.
2. MATERIALS AND METHODS
2.1. Ethics approval
This study was approved by the Ethics Committee of the Third Affiliated Hospital of Guangzhou Medical University (No. 071). A pregnant woman who was diagnosed with beta‐thalassaemia, had a compound heterozygous mutation at codon 17 (CD17) and had the IVSII654 locus in her amniotic fluid sample along with couples who donated discarded poor‐quality embryos were recruited from the IVF centre; all provided written informed consent for this study.
2.2. Fibroblast cell preparation
An amniotic fluid fibroblast cell line that had the compound heterozygous beta‐thalassaemia mutation at CD17 and the IVSII654 locus was derived for the WGA efficiency comparison. The two heterozygous mutations were diagnosed using a conditional reverse dot‐blot probe method (Yanengbio, Shenzhen, Guangdong, China). After thawing the cell line and culturing for 3 days, 11 single‐cell fibroblast samples were randomly selected using a mouth pipette under a dissection microscope, and each sample was washed three times with phosphate‐buffered saline (PBS) and then transferred into separate 200‐μL PCR tubes. In addition, 11 two‐cell and 11 five‐cell fibroblast samples were prepared for WGA with the MALBAC method. For comparison, eight replicates of each of the three cell type samples were prepared to evaluate the efficiency of the MDA method. All samples were clearly marked, immediately frozen, and stored at −20°C for subsequent testing.
2.3. Single‐blastomere preparation
Twenty‐four discarded poor‐quality embryos, which were developmentally arrested at the 8‐cell stage on day 3, were donated and collected for single‐blastomere biopsy. All these embryos were derived from insemination by intracytoplasmic sperm injection. Single blastomeres were biopsied twice from each embryo following procedures used in a previous study,24 and the individual blastomeres were washed three times with freshly prepared droplets of PBS. Therefore, a total of 48 single‐blastomere samples were prepared. The parallel samples were clearly marked, immediately frozen, and stored at −20°C until further use.
2.4. WGA
In this study, the efficiencies of the MALBAC and MDA WGA methods in the diagnosis of beta‐thalassaemia disorders were evaluated. The amplification of the collected fibroblast cells and biopsied blastomeres using the MALBAC method was performed using standard protocols provided by the manufacturer (Yikon Genomics, Shanghai, China). For the comparison, parallel samples were amplified using the MDA method (Qiagen, Hilden, Germany). The amplified DNA products were purified using a High Pure PCR Product Purification Kit (Roche, Mannheim, Germany). The quantity and quality of amplified DNA were determined using a NanoDrop 2000 instrument (ThermoFisher, Wilmington, DE, USA) and gel electrophoresis. The remaining DNA was stored at −20°C until further processing.
2.5. Library construction and aneuploidy screening
Approximately 500 ng of the WGA product, which was amplified using either MALBAC or MDA, was used to prepare the libraries for the low‐depth whole‐genome sequencing on an Illumina HiSeq 2500 platform (Illumina, Inc., San Diego, CA, USA). The read numbers were counted along the whole genome with a bin size of 1 Mb to calculate copy number states. The chromosomal CNVs were determined using local Perl scripts; unique mapped reads were normalized to the relative read number (RRN) after the GC correction in 1000‐kb bins. The CNV visualization was performed using the R programming language.
2.6. Library construction of the HBB allele and SNP sites
In this study, two primers were designed to amplify the target HBB allele mutations (CD17 and IVSII654) using the WGA products. The primers are listed in Supplementary Table 1. In addition to the allelic detection, 60 pairs of primers were used to amplify the SNPs for linkage analysis using two multiple‐PCR systems. All SNPs were located 1 Mb downstream or upstream of the target HBB region. These linkage SNPs may be used to confirm the mutations and to avoid false‐negative or false‐positive occurrences. The primers used for individual SNP amplification reactions are available upon request.
After amplification, the PCR products of the HBB allele and the SNP sites were mixed together and fragmented using a Covaris M220 system (Covaris, Woburn, MA, USA). After the end repair and the ligated “A” adaptor ligation in each fragment, 10 cycles of PCR were performed using eight‐base barcode primers. The PCR products were purified with Agencourt AMPure XP beads (Agencourt Bioscience, Beverly, MA, USA). Libraries with product sizes of approximately 200 bp were chosen and sequenced by NGS on the Illumina HiSeq 2500 platform.
2.7. NGS data analysis and validation
After trimming the raw data using Trimmomatic software (version 0.33, Illumina, Inc., San Diego, CA, USA) to remove the adaptors and the low‐quality data, the remaining high‐quality read sequences were aligned to a human reference genome (Hg19; NCBI Build 37) using BWA software (version 0.7.12, Illumina, Inc.). Samtools (version 1.2, Illumina, Inc.) was used to count the total number of mutations. In this study, the heterozygous mutated allele was identified in more than 10% of the total reads. The HBB allele mutation detected by NGS was validated using Sanger sequencing.
2.8. Statistical analysis
The t‐test and the Chi‐square test were performed using SPSS software (Version 20.0, Chicago, IL, USA) to assess the concentration of the DNA yield after the WGA and the ADO rates. Significance was set at P<.05.
3. RESULTS
3.1. HBB gene mutation validation in cultured fibroblast cells
Genomic DNA derived from the day 3 cultured fibroblast cells was used to validate the HBB variations using the conditional reverse dot‐blot probe method.25 Compound heterozygous beta‐thalassaemia mutations at CD17 and the IVSII654 locus were identified in the fibroblast cells (Supplementary Figure 1).
3.2. WGA efficiencies in fibroblast cells and single blastomeres
In this study, in total, 57 fibroblast cells and 48 single‐blastomere embryo samples were prepared for WGA (Table 1). Two WGA methods, MALBAC and MDA, were used to evaluate the efficiency of WGA at the specific HBB gene region and at the whole‐genome level. After WGA, all three types (single‐, two‐, and five‐cell) of amplified fibroblast samples showed clear bands after electrophoresis (Figure 1), demonstrating the high WGA efficiency of both methods. However, the yield after WGA with the MDA method was significantly higher than the yield after MALBAC amplification (Table 1).
Table 1.
Evaluation of WGA efficiency by the detection of CNV at the whole‐genome level
| WGA method | Cell types | Number of tested samples | Number of detected samples | CNV detection rate (%) | Mean concentration (ng/μL) |
|---|---|---|---|---|---|
| MALBAC | 1 cell (fibroblast) | 11 | 11 | 100.00a | 48.35 |
| 1 cell (blastomere) | 24 | 21 | 87.50b | 44.66 | |
| 2 cells (fibroblasts) | 11 | 11 | 100.00 | 52.49 | |
| 5 cells (fibroblasts) | 11 | 11 | 100.00 | 54.89 | |
| MDA | 1 cell (fibroblast) | 8 | 6 | 75.00a | 333.13 |
| 1 cell (blastomere) | 24 | 19 | 79.17b | 340.90 | |
| 2 cells (fibroblasts) | 8 | 8 | 100.00 | 345.00 | |
| 5 cells (fibroblasts) | 8 | 8 | 100.00 | 357.38 |
Significance (P value) in the CNV detection rate. aFibroblast single‐cell group; bSingle‐blastomere group. Chi‐square testing (SPSS software, Chicago, IL, USA) showed that the CNV detection rates in these two groups did not differ significantly. a χ 2=3.074, P=.164; b χ 2=0.600, P=.701.
Figure 1.
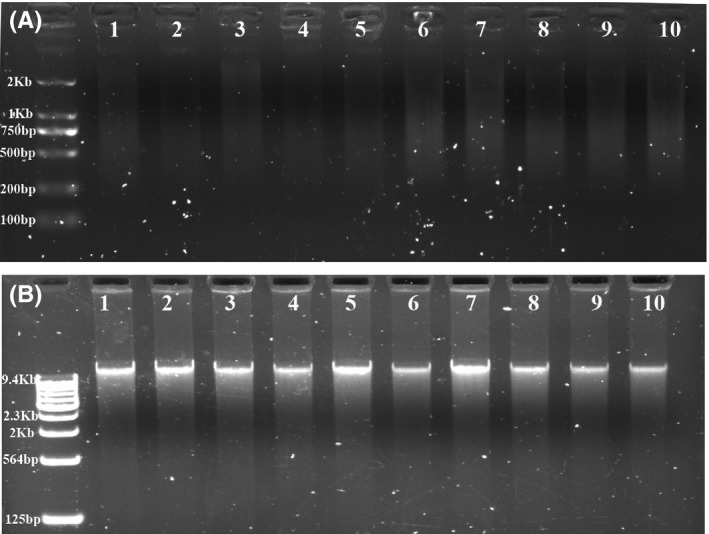
Efficiency evaluation of the whole‐genome amplification methods using single‐cell fibroblasts. Single‐cell fibroblast samples were collected for WGA efficiency evaluations. Paralleled single‐cell samples were amplified using the MALBAC and MDA methods, respectively. (A) The upper column shows the amplified single‐cell samples (samples 1‐4), two‐cell samples (samples 5‐7), and five‐cell samples (samples 8‐10) using MALBAC; all samples produced DNA bands with sizes ranging from 400 to 2000 bp, as shown by gel imaging. (B) The lower column shows that all parallel samples amplified with the MDA method had clear DNA bands with sizes larger than 10 kb
While evaluating the WGA efficiency using single‐blastomere samples, we observed that the amplification of 3 of the 24 MALBAC samples failed, as indicated by their low concentrations and the absence of specific bands after gel electrophoresis (Supplementary Figure 2). In contrast, all amplified samples showed clear bands using the MDA method.
3.3. Evaluation of the chromosomal CNV status at the whole‐genome level
Approximately 1.5 million sequencing reads were obtained from each fibroblast sample. The chromosomal CNV status was analyzed to evaluate the efficiency of the WGA methods at the whole‐genome level. After sequencing, read mapping, and alignment, we demonstrated the high efficiencies of the chromosomal CNV detection rates in both the MALBAC and the MDA groups. In the MALBAC group, the chromosomal CNV detection rates were 100% for the single‐, two‐, and five‐cell samples (Figure 2). All two‐ and five‐cell samples were successfully used to detect CNV in the MDA group. However, the two single‐cell samples failed in CNV detection (Figure 2); therefore, the total CNV detection rate in the MDA group was 91.67% (Table 1). Although the CNV detection rate at the single‐cell level using MDA was lower than that in the MALBAC group, the detection rates of these two methods did not differ significantly (Table 1).
Figure 2.
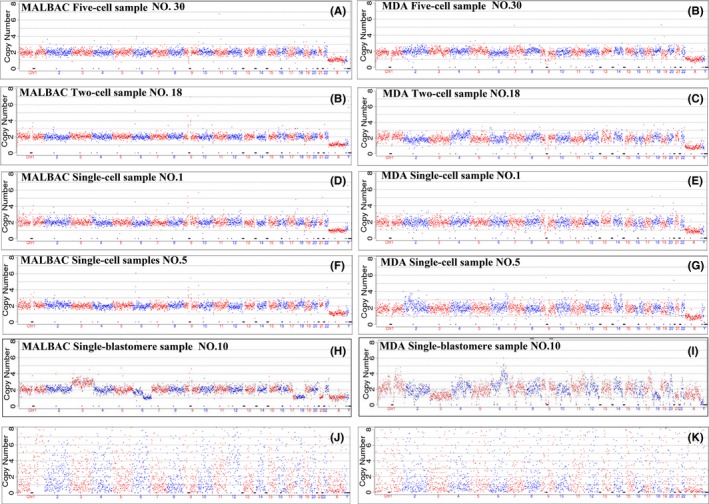
Chromosomal copy number variation analysis of amplified samples. Chromosomal copy number variation (CNV) analysis was performed in parallel samples that were amplified using either the MALBAC or MDA method. (A, B) CNV was successfully analyzed using five‐cell fibroblast samples. (C, D) A normal XY karyotype was identified using two‐cell fibroblast samples. (E‐H) Normal diploid results were observed in single‐cell fibroblast samples. The signal derived from the MALBAC group was smoother than that from the MDA group. (I, J) CNV was analyzed using single blastomeres, which were biopsied from discarded poor‐quality embryos. Multiple chromosomal aneuploidy was detected after data analysis, demonstrating many chromosomal abnormalities in those discarded embryos. (K, L) Two single‐cell fibroblast samples failed CNV analysis. After sequencing and data mapping, the scatter plot demonstrates the failed CNV results
For single‐blastomere samples, the successful CNV detection rate was slightly lower than that in the fibroblast samples, with CNV detection rates of 87.50% (21/24) and 79.17% (19/24) in the MALBAC and MDA groups, respectively. The detection rates of these two methods did not differ significantly (Table 1).
We then analyzed the coefficient of variation (cv) of the CNV in the fibroblast cell samples to evaluate the stability of the detected CNV. The mean cv of the 33 tested fibroblasts in the MALBAC group was 0.15, whereas in the 22 MDA fibroblast samples, it was 0.37 (t=−3.885, P=.007), indicating that the CNV detection using the MALBAC method was more accurate and reproducible than that using the MDA method.
3.4. The efficiency of amplification of the target HBB gene
To compare the amplification efficiency of the MALBAC and MDA methods at the specific HBB gene region, two allelic sites in the HBB gene, CD17 and IVSII654, were analyzed in this study. All successful CNV‐detected fibroblast samples were positive for HBB allele amplification, with a 100% allele detection rate (Supplementary Figure 3). In the two samples that failed CNV analysis, HBB allele amplification also failed, which confirmed the WGA failure of these two samples. Therefore, the total HBB allele amplification rates in the fibroblast samples were 100% (33/33) and 91.67% (22/24) in the MALBAC and MDA groups, respectively.
3.5. Evaluation of ADO in the two identified HBB alleles
The NGS data of each fibroblast sample obtained for HBB allele and SNP site sequencing reached approximately 1.0 M reads. In this experiment, all HBB alleles were successfully amplified from the fibroblast samples, including 33 fibroblast samples amplified by MALBAC and 22 fibroblast samples amplified by MDA, and then they were sequenced on the Illumina HiSeq 2500 platform to evaluate the ADO rates of the two identified HBB alleles (Figure 3). Therefore, a total of 66 and 44 allele sites were included for the investigation of the ADO rates in the MALBAC and MDA groups, respectively.
Figure 3.
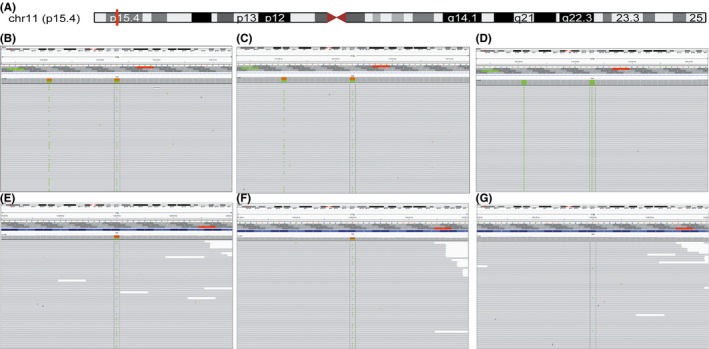
High‐throughput sequencing of the HBB gene IVSII654 mutation. (A) The genomic location of the HBB gene on chromosome 11 (red grid). (B, C) A heterozygous genotype at the c.316‐197 site (IVSII654) was detected in the single‐cell fibroblast samples, which were amplified using either the MALBAC or MDA method. (D) The ADO was observed in a single‐cell fibroblast sample amplified using the MDA method, indicating that the wild‐type IVSII654 allele was missing. (E, F) A heterozygous genotype at the c.52A>AT (CD17 allele) was detected in a single‐cell fibroblast sample, which was amplified using the MALBAC and MDA methods. (G) A homozygous wild‐type CD17 allele was detected in a single‐cell fibroblast sample that was amplified using the MALBAC method. The dashed black lines indicate the positions of IVSII654 and CD17, respectively
In this study, 3 and 9 ADOs in HBB alleles were observed in the MALBAC group and MDA group, respectively. Thus, the total defined allele ADO rate in the MALBAC group (4.55%, 3/66) was significantly lower than that observed in the MDA group (20.45%, 9/44) (Table 2).
Table 2.
ADO rates of the CD17 and IVSII654 alleles using the two WGA methods
| WGA method | Cell types | Number of tested alleles | ADO | ADO rate (%) |
|---|---|---|---|---|
| MALBACd | 1 cell (fibroblast) | 22 | 3 | 13.64a |
| 2 cells (fibroblasts) | 22 | 0 | 0.00b | |
| 5 cells (fibroblasts) | 22 | 0 | 0.00c | |
| MDAd | 1 cell (fibroblast) | 12 | 4 | 33.33a |
| 2 cells (fibroblasts) | 16 | 3 | 18.75b | |
| 5 cells (fibroblasts) | 16 | 2 | 12.50c |
Significance (P value) in the defined HBB allele ADO rate detection. aFibroblast single‐cell group; bFibroblast two‐cell group; cFibroblast five‐cell group; dThe total ADO rate in the two method groups. Chi‐square test (SPSS software, Chicago, IL, USA) data of the HBB allele ADO rate in each group: a χ 2=1.843, P=.175; b χ 2=4.479, P=.034; c χ 2=2.903, P=.088; and d χ 2=6.875, P=.009.
In addition, we used Sanger sequencing to validate the CD17 and IVSII654 locus mutations. Although the allele data of most samples were concordant between the Sanger sequencing and the NGS, higher allele ADO rates were detected in the Sanger sequencing data than in the NGS results (data not shown), indicating that NGS has advantages when applied in mutation identification (Supplementary Figure 4).
3.6. Evaluation of ADO in SNP sites downstream or upstream of the HBB gene
In this study, 28 well‐amplified SNP sites were selected to evaluate the amplification efficiencies and ADO rates of the two WGA methods. The information from all 28 SNP sites was validated using genomic DNA derived from a fibroblast cell line. All of the four parallel samples in the MALBAC and MDA groups were analyzed. Therefore, in total, 112 SNP sites were analyzed in each of the three tested cell‐type groups. Using genomic DNA derived from the fibroblast cell line as a reference, we found that the SNP site amplification efficiencies using the MDA method were significantly superior to those using the MALBAC method. The ADO rate detected using the MALBAC method was lower than that derived using the MDA method (Table 3). In this study, we found that compared to the single‐ and two‐cell groups, when five cells were used as the starting template, the ADO rates of both methods decreased significantly (Table 3).
Table 3.
Comparison of the ADO rates between the two WGA methods using SNP alleles
| WGA method | Cell types | Number of tested SNP sites | Number of detected SNP sitesa | ADO in detected SNP sites | ADO rateb (%) |
|---|---|---|---|---|---|
| MALBAC | 1 cell (fibroblast) | 112 | 96 (85.71%) | 6 | 6.25 |
| 2 cells (fibroblasts) | 112 | 91 (81.25%) | 3 | 3.30 | |
| 5 cells (fibroblasts) | 112 | 109 (97.32%) | 1 | 0.92 | |
| MDA | 1 cell (fibroblast) | 112 | 108 (97.62%) | 10 | 9.26 |
| 2 cells (fibroblasts) | 112 | 104 (92.86%) | 9 | 8.65 | |
| 5 cells (fibroblasts) | 112 | 112 (100.00%) | 0 | 0.00 |
SNP amplification efficiencies between the MALBAC and MDA methods for single‐cell groups: χ 2=7.906, P=.004; two‐cell groups; χ 2=6.694, P=.010; and five‐cell groups: χ 2=3.041, P=.081. The ADO rates in each cell‐type group did not differ significantly.
Significance (P value) evaluation between the MALBAC and MDA groups: aTotal SNP amplification rate; bThe total ADO rate. Chi‐square test (SPSS software, Chicago, IL, USA) data: a χ 2=16.341, P=.000; b χ 2=2.144, P=.143.
4. DISCUSSION
The PGD/PGS of embryos depends on an efficient WGA strategy that provides a reliable and sufficient DNA template from a single‐cell sample or a small amount of starting material for the downstream genetic analysis.6 However, the WGA method may also introduce artifacts and lead to a disproportionate amplification of genomic regions,26 and different WGA methods may yield different performances and biases when applied in procedures used for different purposes.21 Therefore, for the amplification of a target genomic region or the diagnosis of a specific disorder with target variations, the selection of an appropriate WGA method is critical to the success and accuracy of the diagnostic approach.
Currently, three commercially available WGA kits exist that are widely applied in PGD/PGS and single‐cell genetic analysis, and comparisons of the detection performance variations among the different WGA methods have been conducted.14, 16, 21, 23, 27 Previous studies focused on the different efficiencies in terms of genome coverage, uniformity, reproducibility, ADOs, and the ability to identify CNVs among these different WGA kits. The different WGA methods have specific advantages in one or several parts of the aforementioned key parameters; however, none performs satisfactorily for all evaluated parameters.21 Moreover, information regarding the performance efficiencies of the WGA methods when applied to the detection of a specific disorder or targeted genome regions, such as for the diagnosis of beta‐thalassaemia disease, is lacking. Therefore, to select the most appropriate method, an assessment of the efficiencies of the WGA methods as applied to the analysis of specific disorders at the single‐cell level is necessary.
MDA and MALBAC are the two most widely used WGA methods. Previously, researchers have indicated that at the whole‐genome level, the genome coverage of the MDA method was superior to that of the MALBAC method (84% vs 72%), whereas the ADO rate of the MALBAC method was lower than that of the MDA method (21% vs 33%).21 In addition, compared to the MDA method, MALBAC was able to significantly reduce the amplification bias.28 However, comparisons of the efficiencies of these two methods on the HBB target region have not been performed thus far. Therefore, in this study, we provided a comparison of the performance in terms of variation detection in the PGD/PGS of beta‐thalassaemia disease (via the HBB gene) using these two methods. The WGA efficiency, CNV detection, HBB gene amplification, HBB gene target variation detection efficiencies, and ADO rates were evaluated at the single‐cell level. Because trophectoderm tissue cells are commonly used as the material for embryonic genetic diagnosis,10, 29, 30, 31 we also investigated the WGA efficiencies of these two methods using two‐ and five‐cell samples.
Our results demonstrate that the MALBAC and MDA methods have comparable amplification efficiencies at the single‐cell level. Although the DNA yield with MDA was significantly higher than that with MALBAC, the WGA efficiencies of these two methods did not differ significantly when evaluated at the whole‐genome level. When two or five cells were used as a starting template, the WGA efficiencies of both methods increased to 100%, indicating that the reproducibility of these two methods is more stable when more cells are used as the starting amplification material.
In addition, we observed that two samples were unsuccessful for WGA in the MDA single‐cell group. Although the two failed WGA samples showed clear bands and detectable DNA concentrations using the MDA method, the sequencing data from these two samples did not map to the human genome reference and failed in the aneuploidy screening. Our data demonstrated a higher false amplification rate using the MDA method in the WGA procedure. The false amplification results in the MDA group may be attributed to the isothermal amplification conditions, including the use of random hexamers as primers and phi29 DNA polymerase, which may cause a nonspecific amplification.24
In contrast to a previous study,32 our data indicated that MDA and MALBAC had similar favorable detection accuracies and efficiencies in CNV detection using single‐, two‐, and five‐cell samples. However, the lower cv in the MALBAC group demonstrated greater uniformity and reproducibility of that method than the MDA method, which further indicated MALBAC as a superior method for CNV analysis. Our results are consistent with the data obtained by Huang et al.,21 who found that MDA creates variations throughout the genome that are not reproducible from cell to cell and cannot be smoothed via normalization, whereas MALBAC yields the most uniform CNV after normalization, along with reproducible results from cell to cell.
To achieve the goal of improving the success rate of PGD for a hereditary single‐gene disorder, such as beta‐thalassaemia, it is critical for a WGA method to have a high target amplification rate and a low ADO rate, and low false‐positive and false‐negative detection rates are crucial for the accurate diagnosis of pathogenic SNV. Therefore, in addition to CNV evaluation, we evaluated the ADO rates using two identically compound heterozygous variations and sixty SNPs between these two WGA methods. Although the HBB target region amplification efficiency using MDA was higher than that using MALBAC at the single‐ and two‐cell levels, the amplification efficiencies were no different when five cells were used as a template. The total ADO rates of the two target variations and the SNP markers within 1 Mb downstream or upstream of the HBB gene region in the MALBAC group were lower than those of the MDA group; however, the two WGA methods did not show a significant difference in ADO rates at the single‐cell level. In addition, the ADO rate significantly decreased when five cells were used as the starting material, indicating that trophectoderm cells are highly superior to single blastomeres for PGD/PGS.
In this study, although we evaluated the efficiencies of two WGA methods for the genetic diagnosis of beta‐thalassaemia disorders using single‐, two‐, and multiple‐cell fibroblasts samples and using discarded single‐blastomere samples, a limitation of this study is that we did not perform the comparison of these two methods using biopsied day 5 or day 3 high scoring and grading blastocysts or single‐blastomere samples, and the evaluated locus is also limited. Therefore, a preclinical evaluation of these two methods is further suggested before its application in clinical PGD/PGS procedures.
In summary, for the genetic diagnosis and aneuploidy screening of a target HBB gene variation at the single‐cell level, MALBAC is a more suitable method due to its higher levels of uniformity and specificity. To increase the accuracy and success rate of the PGD/PGS procedures, five or more cells are recommended as the starting template, by which both methods performed with a similar efficiency.
Supporting information
ACKNOWLEDGMENTS
This study was supported by grants from theScience and Technology Program of Guangdong Province (2014A020212354, 2013B051000087), from the Guangdong Medicine‐Science Research Program (A2015327), and from the Science and Technology Program of Guangzhou City (201400000004‐4, 201400000003‐4). We gratefully acknowledge the support of Prof. JianQiao Liu and Hongzi Du from the Department of Reproductive Medicine of The Third Affiliated Hospital of Guangzhou Medical University for single‐blastomere samples preparation.
Liu W, Zhang H, Hu D, Lu S, Sun X. The performance of MALBAC and MDA methods in the identification of concurrent mutations and aneuploidy screening to diagnose beta‐thalassaemia disorders at the single‐ and multiple‐cell levels. J Clin Lab Anal. 2018;32:e22267 10.1002/jcla.22267
REFERENCES
- 1. Olivieri NF. The beta‐thalassemias. N Engl J Med. 1999;341:99‐109. [DOI] [PubMed] [Google Scholar]
- 2. Xu XM, Zhou YQ, Luo GX, et al. The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province: implications for the future health burden and population screening. J Clin Pathol. 2004;57:517‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ray PF, Kaeda JS, Bingham J, Roberts I, Handyside AH. Preimplantation genetic diagnosis of beta‐thalassaemia major. Lancet. 1996;347:1696. [DOI] [PubMed] [Google Scholar]
- 4. Tabor A, Vestergaard CH, Lidegaard O. Fetal loss rate after chorionic villus sampling and amniocentesis: an 11‐year national registry study. Ultrasound Obstet Gynecol. 2009;34:19‐24. [DOI] [PubMed] [Google Scholar]
- 5. Kokkali G, Vrettou C, Traeger‐Synodinos J, et al. Birth of a healthy infant following trophectoderm biopsy from blastocysts for PGD of beta‐thalassaemia major. Hum Reprod. 2005;20:1855‐1859. [DOI] [PubMed] [Google Scholar]
- 6. Van der Aa N, Zamani Esteki M, Vermeesch JR, Voet T. Preimplantation genetic diagnosis guided by single‐cell genomics. Genome Med. 2013;5:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gui B, Yang P, Yao Z, et al. A new next‐generation sequencing‐based assay for concurrent preimplantation genetic diagnosis of Charcot‐Marie‐tooth disease type 1A and aneuploidy screening. J Genet Genomics. 2016;43:155‐159. [DOI] [PubMed] [Google Scholar]
- 8. Milachich T. New advances of preimplantation and prenatal genetic screening and noninvasive testing as a potential predictor of health status of babies. Biomed Res Int. 2014;2014:306505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Rycke M, Belva F, Goossens V, et al. ESHRE PGD Consortium data collection XIII: cycles from January to December 2010 with pregnancy follow‐up to October 2011. Hum Reprod. 2015;30:1763‐1789. [DOI] [PubMed] [Google Scholar]
- 10. Yan L, Huang L, Xu L, et al. Live births after simultaneous avoidance of monogenic diseases and chromosome abnormality by next‐generation sequencing with linkage analyses. Proc Natl Acad Sci USA. 2015;112:15964‐15969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheung VG, Nelson SF. Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci USA. 1996;93:14676‐14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide‐primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718‐725. [DOI] [PubMed] [Google Scholar]
- 13. Dean FB, Hosono S, Fang L, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA. 2002;99:5261‐5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deleye L, De Coninck D, Christodoulou C, et al. Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method. Sci Rep. 2015;5:11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mairinger FD, Walter RF, Vollbrecht C, et al. Isothermal multiple displacement amplification: a methodical approach enhancing molecular routine diagnostics of microcarcinomas and small biopsies. Onco Targets Ther. 2014;7:1441‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hou Y, Wu K, Shi X, et al. Comparison of variations detection between whole‐genome amplification methods used in single‐cell resequencing. Gigascience. 2015;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zong C, Lu S, Chapman AR, Xie XS. Genome‐wide detection of single‐nucleotide and copy‐number variations of a single human cell. Science. 2012;338:1622‐1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang MY, Chen HM, Jenq CC, et al. Novel PKD1 and PKD2 mutations in Taiwanese patients with autosomal dominant polycystic kidney disease. J Hum Genet. 2013;58:720‐727. [DOI] [PubMed] [Google Scholar]
- 19. Lu S, Zong C, Fan W, et al. Probing meiotic recombination and aneuploidy of single sperm cells by whole‐genome sequencing. Science. 2012;338:1627‐1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen M, Song P, Zou D, et al. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping‐based amplification cycles (MALBAC) in single‐cell sequencing. PLoS ONE. 2014;9:e114520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang L, Ma F, Chapman A, Lu S, Xie XS. Single‐cell whole‐genome amplification and sequencing: methodology and applications. Annu Rev Genomics Hum Genet. 2015;16:79‐102. [DOI] [PubMed] [Google Scholar]
- 22. Lasken RS. Single‐cell sequencing in its prime. Nat Biotechnol. 2013;31:211‐212. [DOI] [PubMed] [Google Scholar]
- 23. Ning L, Li Z, Wang G, et al. Quantitative assessment of single‐cell whole genome amplification methods for detecting copy number variation using hippocampal neurons. Sci Rep. 2015;5:11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Voet T, Kumar P, Van Loo P, et al. Single‐cell paired‐end genome sequencing reveals structural variation per cell cycle. Nucleic Acids Res. 2013;41:6119‐6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai SP, Wall J, Kan YW, Chehab FF. Reverse dot blot probes for the screening of beta‐thalassemia mutations in Asians and American blacks. Hum Mutat. 1994;3:59‐63. [DOI] [PubMed] [Google Scholar]
- 26. Macaulay IC, Voet T. Single cell genomics: advances and future perspectives. PLoS Genet. 2014;10:e1004126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Bourcy CF, De Vlaminck I, Kanbar JN, Wang J, Gawad C, Quake SR. A quantitative comparison of single‐cell whole genome amplification methods. PLoS ONE. 2014;9:e105585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply‐primed rolling circle amplification. Genome Res. 2001;11:1095‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liss J, Chromik I, Szczyglinska J, Jagiello M, Lukaszuk A, Lukaszuk K. Current methods for preimplantation genetic diagnosis. Ginekol Pol. 2016;87:522‐526. [DOI] [PubMed] [Google Scholar]
- 30. Yang Z, Liu J, Collins GS, et al. Selection of single blastocysts for fresh transfer via standard morphology assessment alone and with array CGH for good prognosis IVF patients: results from a randomized pilot study. Mol Cytogenet. 2012;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Capalbo A, Ubaldi FM, Cimadomo D, et al. Consistent and reproducible outcomes of blastocyst biopsy and aneuploidy screening across different biopsy practitioners: a multicentre study involving 2586 embryo biopsies. Hum Reprod. 2016;31:199‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Evrony GD, Cai X, Lee E, et al. Single‐neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151:483‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials