

Abstract
Background
Lipoprotein(a) [Lp(a)] level is a novel risk factor for atherosclerotic cardiovascular disease in patients with familial hypercholesterolemia (FH), while its impact on the different sites of arteries remains undetermined. We aim to examine the associations of Lp(a) levels with coronary and carotid atherosclerosis in patients with heterozygous FH (HeFH).
Methods
A total of 148 patients with HeFH who have received carotid ultrasonography and coronary angiography due to chest pain were enrolled. Plasma Lp(a) was measured using immunoturbidimetric method. Finally, the associations between Lp(a) and coronary as well as carotid lesions were evaluated.
Results
Patients with Lp(a) ≥ 300 mg/L had similar carotid intima‐media thickness (IMT, 0.782 ± 0.16 mm vs 0.798 ± 0.18 mm, P = .579) and plaque prevalence (66.7% vs 65%, P = .833) compared to those with Lp(a) < 300 mg/L, but had a higher prevalence of coronary artery disease (CAD, 69.7% vs 50.0%, P = .016) and higher Gensini score (GS, median 27 vs 3, P = .006). Moreover, no correlations were found between carotid mean IMT with either Lp(a) level or Lp(a) year score, while positive relation of Lp(a) with GS did. Multivariate regression analysis revealed that Lp(a), Lp(a) year score, and Lp(a) ≥ 300 g/L were all independent predictors for the presence of CAD (OR = 4.99, P = .007; OR = 4.73, P = .009; OR = 4.46, P = .006, respectively) but not for carotid plaques.
Conclusions
This study suggested that Lp(a) level was associated with the presence and severity of CAD but not with carotid atherosclerosis in patients with HeFH.
Keywords: carotid intima‐media thickness, carotid plaque, coronary artery disease, familial hypercholesterolemia, lipoprotein(a)
1. INTRODUCTION
Familial hypercholesterolemia (FH), characterized by highly elevated level of low‐density lipoprotein cholesterol (LDL‐C) and premature coronary artery disease (CAD), is an autosomal dominant disease caused mainly by the mutations in low‐density lipoprotein receptor (LDLR), apolipoprotein B (APOB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes.1 In spite of early and aggressive lipid‐lowering therapy, patients with FH still have a high risk for atherosclerotic cardiovascular disease (ASCVD). It has been demonstrated that ASCVD is a multifactorial disease and other risk factors also play important roles in the development of atherosclerosis besides elevating LDL‐C level.2
Lipoprotein(a) [Lp(a)] is a kind of lipoprotein particles with a LDL‐like structure linked to apolipoprotein(a) [apo(a)] and is mainly genetically determined.3 Epidemiological and genetic studies have demonstrated that Lp(a) is an important risk factor for the development of ASCVD.4, 5, 6 Recent evidences have showed that concentration of Lp(a) is higher in patients with FH compared with general population and has the same hazard effect on CAD, which may explain the residue risk and heterogeneity of CAD in patients with FH.7, 8 Interestingly, the previous study suggested that high level of Lp(a) was an independent risk factor for aortic valve calcification but not for coronary artery calcification in asymptomatic patients with FH,9 indicating the involvement of Lp(a) in the pathological mechanism of lesions in various sites may be different. It is necessary to compare the effect of Lp(a) on coronary and carotid arteries. Apart from close associations with CAD, carotid plaque and the resulting carotid stenosis also lead to stroke, which also has high morbidity and mortality as CAD in Chinese population.
As far as we know, studies concerning the association between Lp(a) and carotid lesions in patients with FH are scarce. Moreover, whether Lp(a) exerts similar effects on coronary and carotid arteries of the same individual is unknown. Thus, we aim to illustrate the associations between Lp(a) and coronary as well as carotid manifestations.
2. MATERIALS AND METHODS
2.1. Study population
In this study, patients referring for coronary angiography (CAG) due to chest pain were included from the patient group with clinical definite/probable FH consecutively recruited from 2011 to 2017 in the division of dyslipidemia of Fuwai Hospital. The exclusion criteria included (i) age <18 years old; (ii) clinical homozygous FH; (iii) without complete clinical data and blood samples; and (iv) without carotid ultrasound examination. Finally, a total of 148 patients with clinical heterozygous FH (HeFH) were enrolled into this study.
We made the diagnosis of clinical FH using the Dutch Lipid Clinic Network (DLCN) criteria as previously reported. Of note, this study did not include the physical examination of corneal arcus. During the diagnosis, if the untreated lipid profiles were unavailable, we got the untreated LDL‐C levels adjusted by a correction factor depending on the type and potency of lipid‐lowering drugs for the individuals receiving lipid‐lowering medications.10 Patients were screened for mutations in LDLR, APOB, and PCSK9 genes as previously reported.11 After admission, clinical data of each participant were collected at length, including medical history of hypertension (HT), diabetes mellitus (DM), smoking, family history of dyslipidemia and CAD of first‐degree relatives, body mass index (BMI), and, of particular, the presence of tendon xanthoma.
Our study complied with the Declaration of Helsinki and was approved by the hospital's ethical review board (Fu Wai Hospital & National Center for Cardiovascular Diseases, Beijing, China). Informed written consents were obtained from each participant.
2.2. Laboratory analyses
Upon admission, fasting blood samples of patients were collected from cubital veins into EDTA‐containing tubes. The concentrations of serum total cholesterol (TC), triglyceride (TG), high‐density lipoprotein cholesterol (HDL‐C), apolipoprotein B (apoB), apolipoprotein A (apoA), and LDL‐C were measured using automatic biochemistry analyzer (Hitachi 7150, Tokyo, Japan). Lp(a) level was determined using an immunoturbidimetric method [LASAY Lp(a) auto; SHIMA Laboratories] as previously described,12 with its normal reference value less than 300 mg/L. Lp(a) year score was calculated as Lp(a) level multiplying the patient's age.7
2.3. Severity of CAD
The diagnosis of CAD was defined as the presence of coronary lesions ≥50% in at least one major coronary artery assessed using CAG analyzed by at least two experienced interventional physicians. The severity of coronary atherosclerosis was quantified using Gensini score (GS), which assigning the severity score based on the degree and location of stenosis. In detail, the narrowing was scored as 1 point for 1%‐25%, 2 points for 26%‐50%, 4 points for 51%‐75%, 8 points for 76%‐90%, 16 points for 91%‐99%, and 32 points for 100% occluded artery. Then, the score was multiplied by a coefficient, indicating the functional significance of each segment. The coefficient was as follows: 5 for left main coronary artery (LM), 2.5 for proximal left anterior descending artery (LAD) and proximal left circumflex artery (LCX), 1.5 for middle LAD, 1.0 for distal LAD, mid‐distal LCX and right coronary artery (RCA), and 0.5 for residual major segments.13
2.4. Carotid ultrasound examination
Bilateral carotid arteries of each patient were examined by two experienced operators who were blind to the clinical characteristics of the patients using 128 System (Acuson, Mountain View, CA, USA) with a high‐resolution 7.5‐10.0 MHz transducer. Intima‐media thickness (IMT) was measured on the proximal and distal common carotid artery as well as the bifurcation in both left and right carotid arteries. The mean cIMT represented the average value of IMT in bilateral arteries. Carotid plaque was defined as a local enlargement of the IMT of more than 50% of the surrounding IMT, or the IMT was above 1.5 mm, and was documented as present or absent.
2.5. Statistical analysis
The statistical analysis was performed with SPSS version 21.0 software (SPSS Inc., Chicago Illinois, USA). Continuous variables were presented as mean ± standard deviation (SD) if normally distributed and analyzed with analysis of variance (ANOVA). Otherwise, median (Q1‐Q3 quartiles) represented abnormal variables, and differences among groups were assessed using Mann‐Whitney U test. Categorical variable, shown as number (percentage), were analyzed using chi‐square test. The correlation of Lp(a) with GS and cIMT was assessed by Spearman's or Pearson's correlation analysis. Multivariable logistic regression analysis was performed to determine the odds ratios (OR) and 95% confidential interval (CI) of Lp(a) and CAD as well as carotid plaque, adjusted by potential confounding factors. A P‐value <.05 was considered as significantly different.
3. RESULTS
3.1. Baseline characteristics
The clinical characteristics of the study population are shown in Table 1. The average age was 49 years old, and 52% were males. Of the 148 subjects, 61 patients (41.2%) were identified as CAD. Most patients (94, 63.5%) were undergoing statin treatment upon admission, and patients diagnosed with CAD were taking significantly more statin treatment. Thus, the mean level of LDL‐C was not significantly different between patients with and without CAD (5.04 mmol/L vs 5.32 mmol/L, P = .314). Carotid plaques were present in 97 patients (65.5%), and the mean cIMT of whole population was 0.79 ± 0.17 mm. Lp(a) level ranged from 17.2 to 1180.2 mg/L with the media value of 263.1 mg/L. A total of 66 patients (44.6%) had Lp(a) level ≥300 mg/L.
Table 1.
Baseline characteristic of study population with FH
| Variables | Overall N = 148 | Non‐CAD N = 61 | CAD N = 87 | P‐value |
|---|---|---|---|---|
| Age, year | 49 ± 13 | 45 ± 14 | 52 ± 12 | <.001 |
| Male, n (%) | 77 (52%) | 27 (44.3%) | 50 (57.5%) | .113 |
| Female, n (%) | 71 (48%) | 34 (55.7%) | 37 (42.5%) | .113 |
| BMI, kg/(m2) | 24.68 ± 3.25 | 23.77 ± 3.11 | 25.3 ± 3.22 | .01 |
| Family history of CAD, n (%) | 57 (40.1%) | 18 (30%) | 39 (47.6%) | .035 |
| Xanthoma, n (%) | 11 (7.5%) | 5 (8.3%) | 6 (6.9%) | .745 |
| Statin, n (%) | 94 (63.5%) | 28 (45.9%) | 66 (75.9%) | <.001 |
| HT, n (%) | 53 (36.8%) | 12 (20%) | 41 (48.8%) | <.001 |
| DM, n (%) | 22 (15.3%) | 4 (6.7%) | 18 (21.4%) | .015 |
| Current smoking, n (%) | 28 (19.4%) | 2 (3.3%) | 26 (31%) | <.001 |
| Alcohol drinker, n (%) | 20 (13.9%) | 6 (10%) | 14 (16.7%) | .254 |
| Glucose, mmol/L | 5.34 ± 1.2 | 5.04 ± 0.69 | 5.56 ± 1.42 | .012 |
| HbA1C (%) | 6 ± 0.9 | 5.6 ± 0.6 | 6.2 ± 1 | <.001 |
| TC, mmol/L | 6.98 ± 1.75 | 7.33 ± 1.38 | 6.73 ± 1.94 | .03 |
| HDL‐C, mmol/L | 1.14 ± 0.39 | 1.22 ± 0.44 | 1.08 ± 0.34 | .033 |
| LDL‐C, mmol/L | 5.15 ± 1.7 | 5.32 ± 1.68 | 5.04 ± 1.72 | .314 |
| ApoA, g/L | 1.3 ± 0.33 | 1.33 ± 0.35 | 1.27 ± 0.33 | .319 |
| ApoB, g/L | 1.5 ± 0.44 | 1.52 ± 0.43 | 1.48 ± 0.45 | .625 |
| Non‐HDL‐C, mmol/L | 5.84 ± 1.76 | 6.11 ± 1.51 | 5.65 ± 1.9 | .116 |
| TG, mmol/L | 1.6 (1.1‐2.12) | 1.61 (1.06‐2.11) | 1.59 (1.14‐2.13) | .713 |
| Lp(a), mg/L | 263.1 (124.3‐562.6) | 214.58 (79.45‐460.98) | 347.3 (162‐681.4) | .007 |
| Lp(a) year score, (g y/L) | 12.57 (5.3‐30.81) | 7.89 (3.36‐19.14) | 17.71 (7.94‐35.51) | <.001 |
| Lp(a) ≥ 300 mg/L, (%) | 66 (44.6%) | 20 (32.8%) | 46 (52.9%) | .016 |
Data are expressed as mean ± SD, median (25th‐75th percentile) or n (%). Bold values indicate statistical significance.
BMI: body mass index; CAD:coronary artery disease; DM: diabetes mellitus; FH: familial hypercholesterolemia; HbA1C: glycated hemoglobin; HDL‐C: HDL cholesterol; HT: hypertension; LDL‐C: LDL cholesterol; Lp(a): lipoprotein(a); TC: total cholesterol; TG: triglyceride.
3.2. Association between Lp(a) and CAD
Patients with CAD had significantly higher level of Lp(a) and Lp(a) year score as well as prevalence of Lp(a) ≥ 300 mg/L (all P < .05) as shown in Table 1. In turn, as shown in Figure 1, patients with Lp(a) ≥ 300 mg/L had higher prevalence of CAD (69.7% vs 50%, P = .016) and GS (median 27 vs 3, P = .006) significantly. Furthermore, using Spearman's correlation analysis (Table 3), we observed that there were positive correlations between GS with Lp(a) and Lp(a) year score (r = .223, P = .01; r = .276, P = .001, respectively). After adjusting confounding risk factors including age, male sex, BMI, family history of CAD, statin, HT, DM, smoke, and HDL‐C, multivariable regression analysis (Table 4) revealed that Lp(a), Lp(a) year score, and Lp(a) ≥ 300 g/L were all significantly independent predictor for the presence of CAD (OR = 4.99, P = .007; OR = 4.73, P = .009; OR = 4.46, P = .006, respectively). Besides, family history of CAD, HT, and current smoker were also predictors for the presence of CAD.
Figure 1.
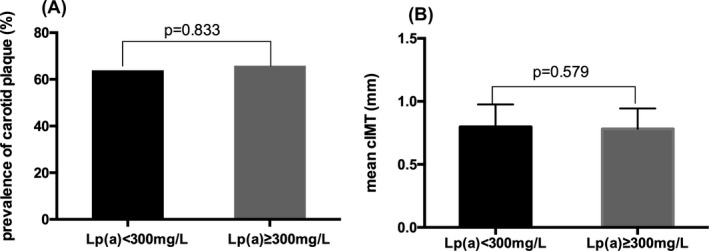
Association between Lp(a) level with carotid plaque (A) and cIMT (B). Lp(a): lipoprotein(a); cIMT: carotid intima‐media thickness
Table 3.
Correlation between risk factors with GS and mean cIMT
| Variables | GS | Mean cIMT | ||
|---|---|---|---|---|
| r | P‐value | r | P‐value | |
| Age, year | .198 | .022 | .231 | .007 |
| LDL‐C, mmol/L | −.107 | .216 | .054 | .531 |
| HDL‐C, mmol/L | −.203 | .018 | −.134 | .121 |
| ApoA, g/L | −.14 | .112 | .005 | .959 |
| ApoB, g/L | −.06 | .499 | .055 | .534 |
| Non‐HDL‐C, mmol/L | −.114 | .188 | .11 | .203 |
| BMI kg/(m2) | .227 | .017 | .044 | .641 |
| HbA1C (%) | .352 | <.001 | .161 | .082 |
| Lp(a)a, mg/L | .223 | .01 | −.073 | .398 |
| Lp(a) year scorea, (g y/L) | .276 | .001 | −.01 | .908 |
Bold values indicate statistical significance.
BMI, body mass index; cIMT, carotid intima‐media thickness; GS, Gensini score; HbA1C, glycated hemoglobin; HDL‐C, HDL cholesterol; LDL‐C, LDL cholesterol; Lp(a), lipoprotein(a).
Log‐transformed data.
Table 4.
Predictive value of Lp(a) for the presence of CAD and carotid plaque
| Lp(a)c | Lp(a) year scorec | Lp(a) ≥ 300 g/L | ||
|---|---|---|---|---|
| CAD | ||||
| Univariate | OR (95% CI) | 3.02 (1.38‐6.61) | 3.60 (1.7‐7.62) | 2.30 (1.16‐4.56) |
| P‐value | .006 | .001 | .017 | |
| Multivariatea | OR (95% CI) | 4.99 (1.55‐16.06) | 4.73 (1.48‐15.14) | 4.46 (1.55‐12.83) |
| P‐value | .007 | .009 | .006 | |
| Carotid plaque | ||||
| Univariate | OR (95% CI) | 1.33 (0.617‐2.88) | 1.79 (0.87‐3.62) | 1.08 (0.54‐2.14) |
| P‐value | .466 | .113 | .833 | |
| Multivariateb | OR (95% CI) | 1.47 (0.54‐4.04) | 1.44 (0.53‐3.92) | 1.07 (0.56‐2.53) |
| P‐value | .451 | .480 | .887 | |
Bold values indicate statistical significance.
CAD: coronary artery disease; Lp(a): lipoprotein(a).
The analysis was adjusted for age, sex, BMI, family history of CAD, statin, HT, DM, smoke, and HDL‐C.
The analysis was adjusted for age, statin, HT, HDL‐C, and ApoB.
Log‐transformed data.
3.3. Association between Lp(a) and carotid lesions
Patients with carotid plaques did not have significant differences regarding Lp(a) and Lp(a) year score nor the prevalence of Lp(a) ≥ 300 mg/L (P = .675, P = .122 and P = .833, respectively) as shown in Table 2. The presence of carotid plaque between patients with high or low level of Lp(a) also did not differ significantly (Figure 2). What is more, there was no difference in mean cIMT between high Lp(a) group and low Lp(a) group (0.782 ± 0.16 mm vs 0.798 ± 0.18 mm, P = .579, respectively). In the Pearson's correlation analysis (Table 3), there were no statistically significant associations between Lp(a), Lp(a) year score, and cIMT (r = −.073, P = .398 and r = −.01, P = .908). Only age showed an association with mean cIMT. Further, multivariable regression analysis (Table 4) demonstrated that Lp(a), Lp(a) year score, and Lp(a) ≥ 300 g/L were all not independent risk factors for the presence of carotid plaque (OR = 1.47, P = .451; OR = 1.44, P = .48; OR = 1.07, P = .887, respectively).
Table 2.
Clinical characteristic of patients with FH according to the presence of carotid plaque
| Variables | Carotid plaque absent N = 51 | Carotid plaque present N = 97 | P‐value |
|---|---|---|---|
| Age, year | 43 ± 13 | 52 ± 12 | <.001 |
| Male, n (%) | 25 (49%) | 52 (53.6%) | .595 |
| Female, n (%) | 26 (51%) | 45 (46.4%) | .595 |
| BMI, kg/(m2) | 23.92 ± 3.33 | 25.07 ± 3.16 | .063 |
| Family history of CAD, n (%) | 19 (38%) | 38 (41.3%) | .701 |
| Xanthoma, n (%) | 2 (4%) | 9 (9.3%) | .249 |
| Statin, n (%) | 26 (51%) | 68 (70.1%) | .022 |
| HT, n (%) | 11 (21.6%) | 42 (45.2%) | .005 |
| DM, n (%) | 6 (11.8%) | 16 (17.2%) | .386 |
| Current smoking, n (%) | 7 (13.7%) | 21 (22.6%) | .199 |
| Alcohol drinker, n (%) | 5 (9.8%) | 15 (16.1%) | .294 |
| Glucose, mmol/L | 5.32 ± 1.19 | 5.36 ± 1.2 | .857 |
| HbA1C (%) | 5.9 ± 1 | 6 ± 0.8 | .387 |
| TC, mmol/L | 6.75 ± 1.49 | 7.1 ± 1.87 | .244 |
| HDL‐C, mmol/L | 1.23 ± 0.45 | 1.09 ± 0.34 | .033 |
| LDL‐C, mmol/L | 4.77 ± 1.71 | 5.35 ± 1.67 | .049 |
| ApoA, g/L | 1.34 ± 0.35 | 1.27 ± 0.33 | .189 |
| ApoB, g/L | 1.35 ± 0.44 | 1.58 ± 0.43 | .003 |
| Non‐HDL‐C, mmol/L | 5.52 ± 1.54 | 6.01 ± 1.85 | .103 |
| TG, mmol/L | 1.5 (0.94‐2.2) | 1.63 (1.15‐2.09) | .349 |
| Lp(a), mg/L | 275.92 (96.3‐562.6) | 250.45 (133.7‐524.3) | .675 |
| Lp(a) year score, (g y/L) | 11.55 (4.16‐30.93) | 13.89 (6.24‐30.21) | .122 |
| Lp(a) ≥ 300 mg/L | 22 (44%) | 44 (45.8%) | .833 |
Data are expressed as mean ± SD, median (25th‐75th percentile) or n (%). Bold values indicate statistical significance.
BMI, body mass index; CAD, coronary artery disease; DM, diabetes mellitus; FH, familial hypercholesterolemia; HbA1C, glycated hemoglobin; HDL‐C, HDL cholesterol; HT, hypertension; LDL‐C, LDL cholesterol; Lp(a), lipoprotein(a); TC, total cholesterol; TG, triglyceride.
Figure 2.
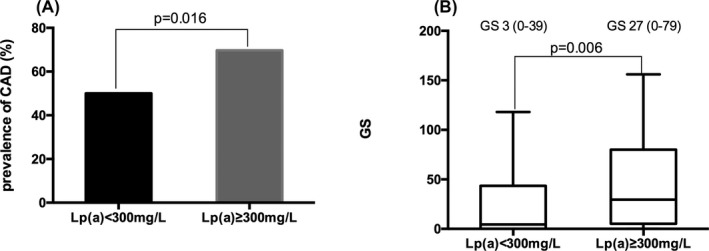
Association between Lp(a) level with CAD (A) and its severity (B). Lp(a): lipoprotein(a); CAD: coronary artery disease; GS: Gensini score
4. DISCUSSION
The present study, for the first time, indicated that Lp(a) level was associated with the presence and severity of CAD but not with carotid atherosclerosis in patients with HeFH, suggesting that Lp(a) may play diverse roles in the pathological process of atherosclerosis in different arteries of the same individual with HeFH.
During past decades, Lp(a) has been considered as a risk factor for CVD including myocardial infarction, aortic stenosis, peripheral arterial disease, and stroke in general population.4, 14, 15, 16 With the expanding knowledge of residual risk for CVD, Lp(a) has become a re‐emerging risk factor attracting much more attention.17 Similarly, the deleterious effect of elevated Lp(a) level on the development of CVD in patients with FH has also been demonstrated in recent studies.18, 19 A retrospective analysis conducted by Holmes DT et al evaluated the effect of Lp(a) on CVD outcomes in 388 patients with possible, probable, and definite HeFH and found that Lp(a) concentration above 560 mg/L was an independent risk factor (HR 2.59, 95% CI 1.53‐4.39, P < .001).20 Unfortunately, this earlier study did not adopt molecular diagnosis. A large cross‐sectional analysis including 1960 patients with genetically confirmed FH and 957 non‐FH relatives as control revealed that patients with Lp(a) level ≥500 mg/L carrying LDLR null mutations had the highest risk for CVD.21 Furthermore, Tada H el al found that Lp(a) level raised in patients with FH irrespective of LDLR or PCSK9 gene mutation and was independently associated with the presence of CAD (OR 1.146, 95% CI 1.026‐1.268, P = .015) in Japanese population.22 Although the data were generally consistent with them, several key points should be emphasized with regard to our study. Firstly, the HeFH patients recruited were diagnosed using DLCN criteria and screened for gene mutations. Furthermore, we used the Lp(a)‐related indexes including Lp(a), Lp(a) year score, and Lp(a) ≥ 300 mg/L and demonstrated that they were all significantly independent risk factors for the presence of CAD. More importantly, we expanded the knowledge of the positive correlation between Lp(a) with GS, which has been well recognized as a powerful tool to reflect the severity of coronary lesions and assess the cardiovascular outcome.23 Besides, considering the lower level of Lp(a) in Chinese population compared to Caucasian populations due to the ethnic heterogeneity, the current study chose the cut value of 300 mg/L instead of 500 mg/L in western studies as one of the risk indicators according to published data.3, 24
Compared with its hazardous role in the development of CAD, the associations of Lp(a) with carotid IMT and plaques are still controversial. Some previous studies found no association between Lp(a) concentrations and mean IMT as well as plaques, while others showed that elevated serum Lp(a) was a significant determinant of the presence and extent of carotid atherosclerosis.25, 26, 27 Notably, the carotid artery manifestation in patients with FH has been much less examined up to date. It has been reported that children with FH had significantly increased carotid IMT than their unaffected siblings as early as 8 years old, with sex, age, and LDL‐C as risk factors.28 Whereas, Bos S et al also suggested that the presence of carotid plaques and IMT in adult patients with FH with long‐term statin therapy did not differ from those of healthy controls, indicating that timely lipid‐lowering therapy may also benefit the regression of carotid atherosclerosis.29 Moreover, the association between Lp(a) and carotid atherosclerosis was scarcely reported in patients with FH. In our present study, we found that Lp(a) level was not only unrelated to carotid IMT but also could not predict the presence of carotid plaques, which was similar to the previous cross‐sectional study including 191 statin‐treated patients with FH conducted by Bos S et al.30 However, their study did not provide information regarding CAD. In addition, similar to prior study, we have found that apoB was associated with the presence of carotid plaques.31 Unquestionably, more studies about the irrelevant results between Lp(a) and carotid lesions in our observation may be needed considering our small sample size.
In fact, we firstly queried that whether Lp(a) played equally important roles in the presence of coronary and carotid lesions according to reviewing previous publications. Unfortunately, there has been no relevant data available in patients with HeFH before as far as we know. Currently, we found that Lp(a) level was associated with coronary but not carotid atherosclerosis. The exact mechanism with regarding this disparity in carotid and coronary arteries is unknown, while a large number of studies have suggested that Lp(a) exerts atherogenic property due to its cholesterol‐rich particle, the prothrombotic, and proinflammatory effects.32 In other words, in spite of the similarities between coronary and carotid atherosclerosis including sharing partial conventional risk factors, the exact correspondence between the two arteries remains unclear and thus the specific effects of Lp(a) on coronary and carotid arteries may be different.33 Therefore, how Lp(a) was involved in the pathogenic mechanism of coronary and carotid atherosclerosis needs to be further established.
It is imperative to note several limitations in the present study when interpreting the results. First, our study was a cross‐sectional study and the association between Lp(a) and carotid atherosclerosis needs further robust evidence from prospective studies. Second, this was a single‐center study with relatively small sample size.
In conclusion, the present study suggested that Lp(a) level was associated with the presence and severity of CAD but not with carotid atherosclerosis in patients with HeFH. Notwithstanding the mechanism remains uncertain, the current study may provide further insights into the role of Lp(a) in the development of different atherosclerosis in patients with FH and help improve future specific preventions.
ETHICS APPROVAL
The current study was in accordance with the Helsinki Declaration and approved by the hospital's ethical review board (Fu Wai Hospital & National Center for Cardiovascular Diseases, Beijing, China). Informed consent was obtained from all patients for being included in the study.
AUTHOR CONTRIBUTIONS
Dr Di Sun completed the project, analyzed the data, and wrote the article. Dr Jian‐Jun Li designed the study,interpreted the data, and contributed to critically revising the article. Dr Bing‐Yang Zhou, Sha Li, and Xi Zhao contributed to data collection and genetic analysis. Drs Zhu, Guo, and Wu contributed to recruitment of patients. Drs Gao, Qing, Liu, and Dong contributed to the collections of clinical data and procedure of laboratory examination. All authors have approved the final article.
ACKNOWLEDGMENTS
This work was supported by the Capital Health Development Fund (201614035) and CAMS Major Collaborative Innovation Project (2016‐I2M‐1‐011) awarded to Dr. Jian‐Jun Li, MD, PhD.
Sun D, Zhou B‐Y, Zhao X, et al. Lipoprotein(a) level associates with coronary artery disease rather than carotid lesions in patients with familial hypercholesterolemia. J Clin Lab Anal. 2018;32:e22442 10.1002/jcla.22442
REFERENCES
- 1. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–90a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Santos RD, Gidding SS, Hegele RA, Cuchel MA, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850‐861. [DOI] [PubMed] [Google Scholar]
- 3. Nordestgaard BG, Chapman MJ, Ray K, Boren J, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844‐2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kamstrup PR, Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176‐184. [DOI] [PubMed] [Google Scholar]
- 5. Nave AH, Lange KS, Leonards CO, Siegerink B, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta‐analysis. Atherosclerosis. 2015;242:496‐503. [DOI] [PubMed] [Google Scholar]
- 6. Clarke R, Peden JF, Hopewell JC, Kyriakou T, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518‐2528. [DOI] [PubMed] [Google Scholar]
- 7. Chan DC, Pang J, Hooper AJ, Burnett JR, et al. Elevated lipoprotein(a), hypertension and renal insufficiency as predictors of coronary artery disease in patients with genetically confirmed heterozygous familial hypercholesterolemia. Int J Cardiol. 2015;201:633‐638. [DOI] [PubMed] [Google Scholar]
- 8. Langsted A, Kamstrup PR, Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577‐587. [DOI] [PubMed] [Google Scholar]
- 9. Vongpromek R, Bos S, Ten Kate GJ, Yahya R, et al. Lipoprotein(a) levels are associated with aortic valve calcification in asymptomatic patients with familial hypercholesterolaemia. J Intern Med. 2015;278:166‐173. [DOI] [PubMed] [Google Scholar]
- 10. Haralambos K, Whatley SD, Edwards R, Gingell R, et al. Clinical experience of scoring criteria for Familial Hypercholesterolaemia (FH) genetic testing in Wales. Atherosclerosis. 2015;240:190‐196. [DOI] [PubMed] [Google Scholar]
- 11. Li JJ, Li S, Zhu CG, Wu NQ, et al. Familial hypercholesterolemia phenotype in chinese patients undergoing coronary angiography. Arterioscler Thromb Vasc Biol. 2017;37:570‐579. [DOI] [PubMed] [Google Scholar]
- 12. Li S, Wu NQ, Zhu CG, Zhang Y, et al. Significance of lipoprotein(a) levels in familial hypercholesterolemia and coronary artery disease. Atherosclerosis. 2017;260:67‐74. [DOI] [PubMed] [Google Scholar]
- 13. Gensini GG. A more meaningful scoring system for determining the severity of coronary heart disease. Am J Cardiol. 1983;51:606. [DOI] [PubMed] [Google Scholar]
- 14. Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Donoghue ML, Morrow DA, Tsimikas S, Sloan S, et al. Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol. 2014;63:520‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nsaibia MJ, Mahmut A, Boulanger MC, Arsenault BJ, et al. Autotaxin interacts with lipoprotein(a) and oxidized phospholipids in predicting the risk of calcific aortic valve stenosis in patients with coronary artery disease. J Intern Med. 2016;280:509‐517. [DOI] [PubMed] [Google Scholar]
- 17. Khera AV, Everett BM, Caulfield MP, Hantash FM, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129:635‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nenseter MS, Lindvig HW, Ueland T, Langslet G, et al. Lipoprotein(a) levels in coronary heart disease‐susceptible and ‐resistant patients with familial hypercholesterolemia. Atherosclerosis. 2011;216:426‐432. [DOI] [PubMed] [Google Scholar]
- 19. Alonso R, Mata P, Muniz O, Fuentes‐Jimenez F, et al. PCSK9 and lipoprotein (a) levels are two predictors of coronary artery calcification in asymptomatic patients with familial hypercholesterolemia. Atherosclerosis. 2016;254:249‐253. [DOI] [PubMed] [Google Scholar]
- 20. Holmes DT, Schick BA, Humphries KH, Frohlich J. Lipoprotein(a) is an independent risk factor for cardiovascular disease in heterozygous familial hypercholesterolemia. Clin Chem. 2005;51:2067‐2073. [DOI] [PubMed] [Google Scholar]
- 21. Alonso R, Andres E, Mata N, Fuentes‐Jimenez F, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982‐1989. [DOI] [PubMed] [Google Scholar]
- 22. Tada H, Kawashiri MA, Yoshida T, Teramoto R, et al. Lipoprotein(a) in Familial Hypercholesterolemia With Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Gain‐of‐Function Mutations. Circ J. 2016;80:512‐518. [DOI] [PubMed] [Google Scholar]
- 23. Sinning C, Lillpopp L, Appelbaum S, Ojeda F, et al. Angiographic score assessment improves cardiovascular risk prediction: the clinical value of SYNTAX and Gensini application. Clin Res Cardiol. 2013;102:495‐503. [DOI] [PubMed] [Google Scholar]
- 24. Cai DP, He YM, Yang XJ, Zhao X, Xu HF. Lipoprotein (a) is a risk factor for coronary artery disease in Chinese Han ethnic population modified by some traditional risk factors: a cross‐sectional study of 3462 cases and 6125 controls. Clin Chim Acta. 2015;451(Pt B):278‐286. [DOI] [PubMed] [Google Scholar]
- 25. Calmarza P, Trejo JM, Lapresta C, Lopez P. Relationship between lipoprotein(a) concentrations and intima‐media thickness: a healthy population study. Eur J Prev Cardiol. 2012;19:1290‐1295. [DOI] [PubMed] [Google Scholar]
- 26. Boras J, Ljubic S, Car N, Metelko Z, et al. Lipoprotein(a) predicts progression of carotid artery intima‐media thickening in patients with type 2 diabetes: a four‐year follow‐up. Wien Klin Wochenschr. 2010;122:159‐164. [DOI] [PubMed] [Google Scholar]
- 27. Watts GF, Mazurkiewicz JC, Tonge K, Nelson V, Warburton FG, Slavin BM. Lipoprotein(a) as a determinant of the severity of angiographically defined carotid atherosclerosis. QJM. 1995;88:321‐326. [PubMed] [Google Scholar]
- 28. Kusters DM, Wiegman A, Kastelein JJ, Hutten BA. Carotid intima‐media thickness in children with familial hypercholesterolemia. Circ Res. 2014;114:307‐310. [DOI] [PubMed] [Google Scholar]
- 29. Bos S, Duvekot MH, Ten Kate GR, Verhoeven AJ, et al. Carotid artery plaques and intima medial thickness in familial hypercholesteraemic patients on long‐term statin therapy: a case control study. Atherosclerosis. 2017;256:62‐66. [DOI] [PubMed] [Google Scholar]
- 30. Bos S, Duvekot MH, Touw‐Blommesteijn AC, Verhoeven AJ, et al. Lipoprotein (a) levels are not associated with carotid plaques and carotid intima media thickness in statin‐treated patients with familial hypercholesterolemia. Atherosclerosis. 2015;242:226‐229. [DOI] [PubMed] [Google Scholar]
- 31. Walus‐Miarka M, Czarnecka D, Wojciechowska W, Kloch‐Badelek M, et al. Carotid Plaques Correlates in Patients With Familial Hypercholesterolemia. Angiology. 2016;67:471‐477. [DOI] [PubMed] [Google Scholar]
- 32. Imhof A, Rothenbacher D, Khuseyinova N, Hoffmeister A, et al. Plasma lipoprotein Lp(a), markers of haemostasis and inflammation, and risk and severity of coronary heart disease. Eur J Cardiovasc Prev Rehabil. 2003;10:362‐370. [DOI] [PubMed] [Google Scholar]
- 33. Jashari F, Ibrahimi P, Nicoll R, Bajraktari G, Wester P, Henein MY. Coronary and carotid atherosclerosis: similarities and differences. Atherosclerosis. 2013;227:193‐200. [DOI] [PubMed] [Google Scholar]