

Abstract
Background
Different methods available for extraction of human genomic DNA suffer from one or more drawbacks including low yield, compromised quality, cost, time consumption, use of toxic organic solvents, and many more. Herein, we aimed to develop a method to extract DNA from 500 μL of fresh or frozen human blood.
Methods
Five hundred microliters of fresh and frozen human blood samples were used for standardization of the extraction procedure. Absorbance at 260 and 280 nm, respectively, (A260/A280) were estimated to check the quality and quantity of the extracted DNA sample. Qualitative assessment of the extracted DNA was checked by Polymerase Chain reaction and double digestion of the DNA sample.
Results
Our protocol resulted in average yield of 22±2.97 μg and 20.5±3.97 μg from 500 μL of fresh and frozen blood, respectively, which were comparable to many reference protocols and kits.
Conclusion
Besides yielding bulk amount of DNA, our protocol is rapid, economical, and avoids toxic organic solvents such as Phenol. Due to unaffected quality, the DNA is suitable for downstream applications. The protocol may also be useful for pursuing basic molecular researches in laboratories having limited funds.
Keywords: cost effective, DNA extraction, high yield, human blood
1. Introduction
The increasing demand of genome based analyses in modern evolutionary and disease researches have also increased the need for bulk amount of pure genomic DNA1, 2 which should also be free from protein and RNA contaminants. It is indeed the primary requirement of various molecular biological techniques such as Polymerase Chain Reaction (PCR), restriction enzyme analysis, mutation detection, genotyping, and linkage analysis as well as determination of genetic abnormalities, epigenetic studies, and various diagnostic and preventive tests.1, 2, 3, 4 Moreover, it would become much more research friendly if the DNA extraction method becomes rapid and cost effective.
DNA can be extracted from many biological samples such as hair, blood, semen, saliva, skin cells, and many more. Among these, the one that has gained astounding importance in biological researches is blood. Blood has become an integral part of biochemistry, hematology and clinical studies and forensic investigations. It serves as an important source of genomic DNA because of the presence of nucleated white blood cells.
Many protocols have been published regarding DNA isolation from blood.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 Some of these published protocols applied enzymes and organic solvents for yielding high quality DNA, devoid of PCR inhibitors, while others including salting out procedure targeted toward higher DNA yields.15, 16 Thus some protocols are expensive and time consuming17 while others compromised with the DNA quality.18, 19, 20
Therefore, in order to fulfill the demand of a rapid and cost effective procedure for obtaining high quality genomic DNA, hereby we have aimed to develop a protocol free from costly enzymes and toxic organic solvents for extracting pure DNA from fresh and frozen human blood samples.
2. Materials and Methods
2.1. Blood samples
Blood samples were collected in EDTA‐containing vacutainer tubes from 20 healthy individuals randomly chosen from the localities in and around the campus of University of North Bengal, Siliguri, West Bengal, India. The volunteers provided their prior informed consent in becoming a part of this study. All the donors were interviewed and a questionnaire concerning their health conditions was completed with the assistance of medical staff to ensure that none of the volunteers has any prevailing disease conditions. The investigation was approved by the Human Ethics Committee of the Department of Zoology, University of North Bengal, India and was performed in accordance with the with the Helsinki Declaration of 1975.21 Fresh blood samples were used for DNA extraction after 1 hour from the time of collection and for frozen blood sample, the extraction was done generally after 15‐21 days from the date of collection. In case of the frozen blood, the samples were refrigerated at −20°C for future use. Blood may sometimes act as potential biohazard and therefore suitable care was taken during handling of the blood samples.
2.2. Reagents and solutions
RBC lysis Buffer (RLB): 0.155 mol/L NH4Cl, 10 mmol/L KHCO3 and 0.1 mol/L EDTA (Na2) in 1000 mL of distilled H2O. The pH was adjusted to 7.6.
Extraction Buffer: 1.5 mol/L Tris pH 7.6, 0.4 mol/L disodium salt of ethylenediaminetetra acetic acid (Na2EDTA; Merck, Darmstadt, Germany), 2.5 mol/L NaCl, 2% Cetyl trimethyl ammonium bromide (CTAB; Merck, Germany) 850 mL H2O. Adjust the pH to 8.0 and make the final volume to 1 L.
10% SDS (Sodium dodecyl sulfate).
β‐Mercaptoethanol.
Chloroform: Isoamyl alcohol (24:1).
Isopropanol.
70% and 90% ethanol (Merck, Germany);
2.3. DNA extraction procedure
The extraction procedure was standardized both for fresh and frozen blood samples.
Step 1. 500 μL of blood sample was transferred from the vacutainer to an eppendorf tube. In case of frozen blood, the sample was thawed at room temperature for 20‐30 minutes before transferring the blood to the eppendorf tube.
Step 2. Plasma was aspirated out carefully by centrifuging the sample at 2664 RCF for 7 minutes at 4°C.
Step 3. 1 mL of RLB was added to the precipitate, mixed gently and was allowed to stand at room temperature for 1‐2 minutes.
Step 4. The mixture was then centrifuged at 2664 RCF for 6 minutes at room temperature.
Step 5. The supernatant was discarded. This step may be repeated 1‐2 times until a white colored pellet is obtained.
Step 6. 500 μL of prewarmed DNA extraction buffer was added to the pellet followed by 30 μL of 10% SDS and 2 μL of B‐Mercaptoethanol respectively and mixed gently. The mixture was then incubated at 56‐60°C for 1 hour.
Step 7. 500 μL of Chloroform: isoamylalcohol (24:1) was added to the mixture after incubation and shaken well. The mixture was then centrifuged at 10 656 RCF for 12 minutes at 4°C.
Step 8. The supernatant was pipetted out in another fresh sterilized centrifuge tube containing chilled ethanol. The tube was shaken for a while until fine white threads appeared in the solution. The sample tube may be kept at −20°C for 20 minutes instead of shaking.
Step 9. The sample was then centrifuged at 10 656 RCF for 12 minutes at 4°C.
Step 10. The supernatant was discarded without disturbing the pellet and 500 μL of 90% alcohol was added to it.
Step 11. The sample was then centrifuged at 10 656 RCF for 12 minutes at 4°C.
Step 12. Step 10 and 11 were repeated with 500 μL of 70% alcohol.
Step 13. The supernatant was discarded and the pellet was allowed to dry at 37°C.
Step 14. The pellet was then dissolved overnight in 100 μL of TE buffer.
Step 15. The DNA solution was then stored at −20°C for future use.
2.4. DNA assessment
Ratio of the absorbance at 260 and 280 nm respectively (A260/A280) were estimated to check the quality and quantity of the extracted DNA sample (Table 1). The absorbance ratio was measured using UV spectrophotometer (Rayleigh UV‐2100, Beifen‐Ruili Analytical Instrument (Group) Co., Ltd., Beijing, China). DNA concentration was measured based on A260 values. Absorbance value of 1 at 260 nm equals to 50 μg/mL of pure dsDNA. This concentration was multiplied with the total eluted volume to provide the total yield of DNA. Furthermore, electrophoresis of 5 μL of each extracted DNA sample was done on 1% agarose gel in order to trace any degradation of the DNA sample during the extraction procedure (Figure 1).
Table 1.
Optical density (OD) ratios and DNA yield (μg) per 500 μL of blood extracted by the three protocols respectively. ST1 and ST2 denote Standard Protocol 1 and 2 respectively. Both OD ratios and DNA yield were expressed as mean±SD
| Experimental protocol | ST1 | ST2 | ||||
|---|---|---|---|---|---|---|
| Fresh | Frozen | Fresh | Frozen | Fresh | Frozen | |
| OD260/OD280 ratio | 1.88±0.07 | 1.86±0.06 | 1.96±0.03 | 1.90±0.05 | 1.72±0.11 | 1.7±0.09 |
| Yield (μg) per 500 mL | 22±2.97 | 20.5±3.97 | 19.5±3.95 | 17±2.12 | 25.2±3.32 | 24±4.28 |
Figure 1.
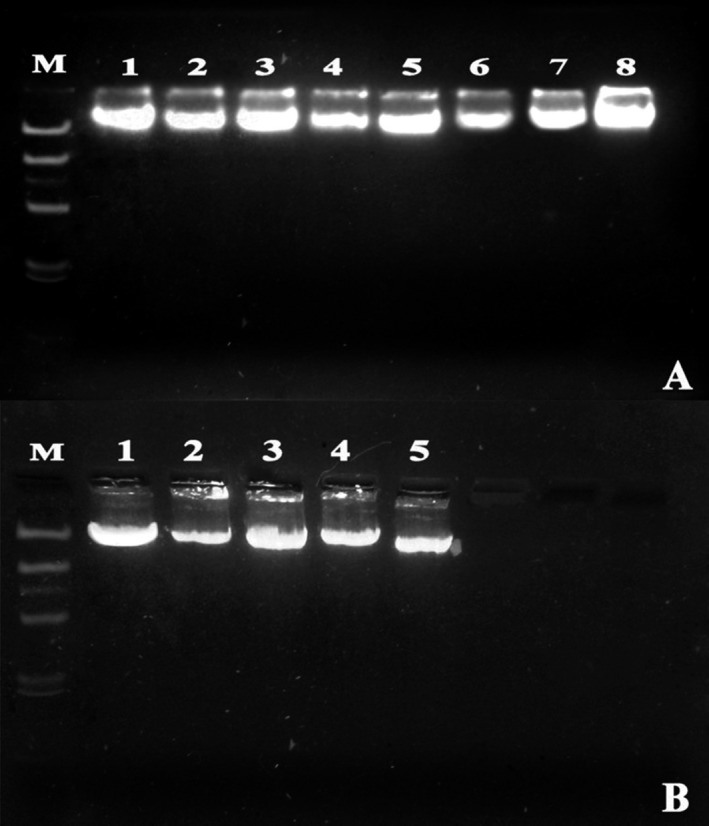
One percent agarose gel electrophoresis to demonstrate the extracted DNA. (A) Fresh Bloods and (B) Frozen Bloods. M stands for Marker (λ‐DNA/Hind III Marker)
Polymerase Chain Reactions were carried out using the extracted DNA samples in order to check the proficiency of the extracted DNA in gene amplification studies and also to check whether any inhibitory component were present in the samples which may hinder the participation of the DNA in PCR reactions. A fragment of Growth Hormone (GH) gene was amplified using the extracted DNA sample for which the forward and reverse primers were 5′‐CTT CCC AAC CAT TCC CTT A‐ 3′ and 5′‐CGG ATT TCT GTT GTG TTT C‐3′ respectively. Each 25 μL PCR reaction contained 2.5 μL 10× PCR buffer (Bangalore Genei, Bangalore, India), 0.2 mmol/L of deoxynucleoside triphosphate (dATP, dCTP, dGTP, dTTP; (Bangalore Genei), 1.0 μmol/L each of forward and reverse primers (Imperial LifeSciences, Gurgaon, India), 50 ng of template DNA and 1 U of Taq polymerase (Bangalore Genei). The PCR reactions were carried out in MJ Mini Gradient Thermal Cycler (Bio‐Rad PTC 1148, BioRad, CA, USA). The amplification program consisted of an initial denaturation step at 94°C for 3 minutes, followed by 30 cycles of 30 seconds at 95°C, 50 seconds at 58°C for primer annealing, and 60 seconds at 72°C for extension and finally terminating with a final extension of 10 minutes at 72°C. Electrophoresis of the PCR products were done on ethidium bromide (0.5 μg/mL) prestained 1% agarose gel after which a photograph of the agarose gel was taken over a UV‐transilluminator (Spectroline TVD‐1000R, Spectronics Corporation, Westbury, NY, USA; Figure 2A).
Figure 2.
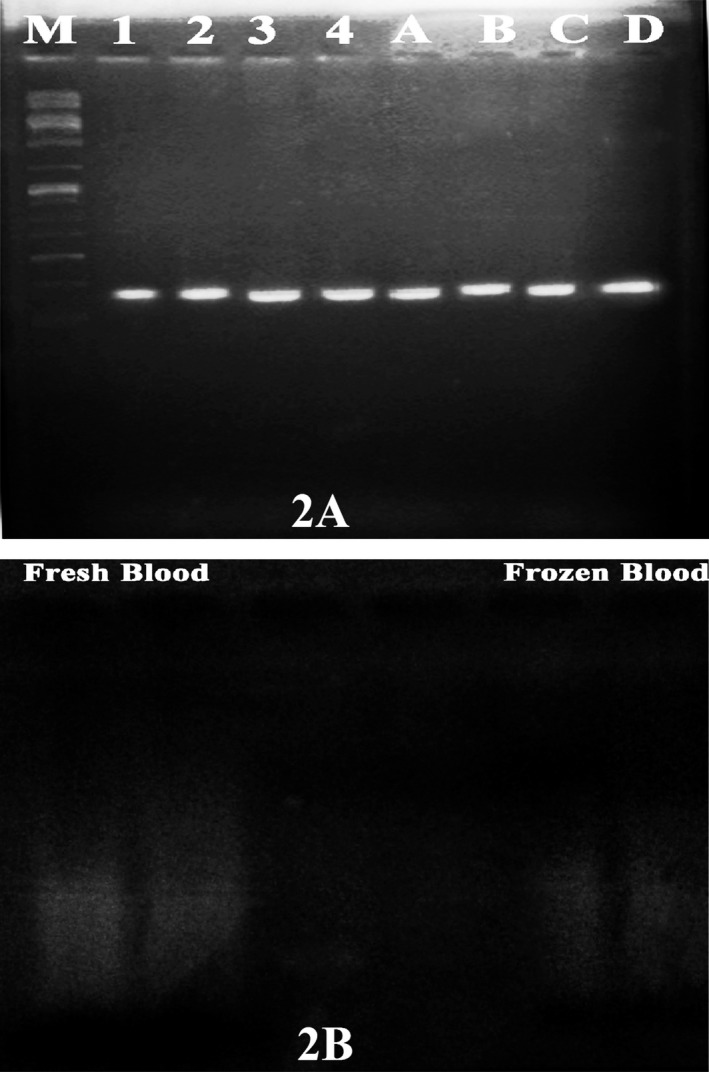
(A) One percent agarose gel electrophoresis showing the PCR amplification products of the GH gene from fresh and frozen blood samples. Lanes 1‐4 corresponds to fresh blood from samples 1, 3, 7,10, respectively, while lanes A‐D corresponds to frozen blood from the same samples. (B) Double digested fresh and frozen blood samples run on 1% agarose gel. Eco RI and Hind III were used for the digestion
Qualitative assessment of the extracted DNA was also checked by double digesting the DNA with restriction enzymes: Eco RI and Hind III (Genei; Figure 2B). Briefly, each reaction mixture contained: 1.5 μg DNA, 2 μL of 10x assay buffer D (Genei) enzyme specific, 1 μL of restriction enzymes, EcoR1(10 U/μL) and Hind III (10 U/μL), 0.2 μL of 100x acetylated BSA and ddH2O to make the final volume to 20 μL. After brief centrifugation, the mixture was incubated at 37°C for overnight. Gel electrophoresis of the digested product was carried out on 1% agarose gel, prestained with ethidium bromide and visualized on a UV transilluminator.
Our data were also compared with some of the previously published protocols10, 12, 22, 23, 24 and commercially available kits which includes Wizard Genomic DNA purification kit (Promega, Madison, WI, USA), QIAamp DNA Investigator Kit (Qiagen, Hilden, Germany), Archivepure™ DNA purification kit (5 Prime, Hilden, Germany) and GenElute™ Blood Genomic DNA kit (Sigma‐Aldrich, St. Louis, MO, USA). In case of the reference protocols, average DNA yields, wherever mentioned, were considered for the analyses, else the maximum yield was considered if a range of value was provided (Figure 3). Bar graphs and scatter plots were constructed using MS Excel software (Redmond, WA, USA). The box plot was constructed using SPSS ver. 15.0 (Chicago, IL, USA).
Figure 3.
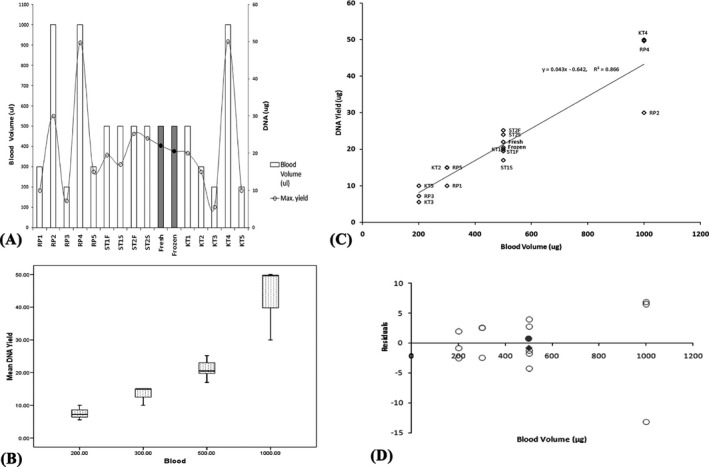
(A) Bar graph to compare the yield of DNA and the amount of blood volume required in our study protocols with other published protocols and kits. R1‐5(Reference Protocols), KT1‐5 (Reference kits), ST1‐2(Standard Protocol) F or S (fresh or frozen). The results of the experimental protocol are mentioned “Fresh” or “Frozen” in bold. (B) Box plot to compare the average yield of DNA from different blood volumes as reported in our study and other published reports. (C) Scatterplot to compare DNA yield from our protocol with that of published reports. R1‐5 (Reference Protocols), KT1‐5 (Reference kits), ST1‐2(Standard Protocol) F or S (fresh or frozen). The results of the experimental protocol are mentioned “Fresh” or “Frozen” in bold. (D) Residual Plot to show whether the linear regression model is appropriate for the dataset Residual values for DNA from fresh and frozen blood samples following our protocol are symbolized as (●) and (♦) respectively
2.5. Standard protocols
Furthermore, two standard protocols were also used to extract DNA from the collected blood samples both in fresh and frozen conditions, namely the phenol‐chloroform DNA extraction procedure by Sambrook et al.25 and the high salt DNA extraction procedure by Miller et al.14 respectively. Both the protocols were standardized for 500 μL of blood.
As per the phenol‐chloroform method, after removal of the RBCs (by EDTA) nucleated cell pellets from both fresh and frozen blood samples were suspended in extraction buffer (10 mmol/L Tris HCl [pH 8.0], 0.1 mmol/L EDTA, 0.5% (w/v) SDS and 20 μg/mL pancreatic Ribonuclease A) along with Proteinase K (20 μg/mL). This step was followed by phenol‐chloroform‐isoamylalcohol (25:24:1) extraction and ethanol precipitation, finally dissolving the DNA in TE buffer.
On the other hand, in high salt precipitation method, after removal of the RBCs, the WBCs were treated with nuclei lysis buffer (10 mmol/L Tris‐HCl, 400 mmol/L NaCl and 2 mmol/L Na2EDTA, pH 8.2) following overnight digestion at 37°C with 10% SDS and 20 mg/mL Proteinase K. This was followed by addition of 6 mol/L saturated NaCl. Protein pellets and DNA in the supernatant were procured after shaking the mixture vigorously followed by centrifugation. This was followed by ethanol precipitation and dissolving the DNA in TE buffer. The total DNA yield (μg) and the absorbance ratio (A260/A280) estimated from these two standard protocols were then compared with our experimental protocol.
3. Results
A260/A280 absorbance ratio in case of both fresh and frozen human whole blood samples ranged consistently between 1.8 and 2.0 averaging at 1.88±0.07 and 1.86±0.06 for fresh and frozen blood respectively, which efficiently signified purity and successful deproteinization of the samples (Table 1). It has also suggested that the extracted DNA samples were also free from RNA contaminations. Following our protocol, the yield of genomic DNA per 500 μL of fresh blood ranged between 15 and 29 μg while that for 500 μL of frozen blood ranged between 14 and 28 μg respectively. Thus, the average yields for fresh and frozen blood samples were calculated to be 22±2.97 μg and 20.5±3.97 μg, respectively, which were comparable to the outcomes of both the standard protocols (Table 1).
DNA samples, run in 1% agarose gel, demonstrated bands of varying intensities, which were more or less comparable in case of fresh and frozen, blood (Figure 1). However, all the DNA bands were prominent and unified with very negligible smearing in the lanes. This suggests that no degradation has occurred in the extracted DNA inspite of the exposure to several chemical washes. Furthermore, agarose gel containing PCR products showed very prominent bands and therefore further demonstrating the quality and purity of the extracted DNA (Figure 2A). Double digestion by restriction enzymes (Figure 2B) showed that the extracted DNA samples were free from any inhibitory and interfering compounds.
It was observed that our standardized protocol yielded sufficient amount of high quality human genomic DNA, as was represented by the Bar graph and scatterplot (Figure 3A and C) when compared with other published protocols and commercially available kits. The mean DNA yields (μg) of different sample volumes were shown by box plot constructed in SPSS version 15.0 software (Figure 3B). The R‐Squared value was estimated to be 0.866 which signified that our data fitted the line of the linear regression model with other reference data following the equation y=0.043x‐0.642 (Figure 3C).
4. Discussion
Pure and intact genomic DNA is the first and foremost requirement for many modern applications in molecular biology. Therefore, the efficiency of a DNA extraction protocol will be affected by its robustness and ability to yield bulk amount of clean and unblemished genomic DNA. A number of DNA extraction protocols have already been published by several workers around the globe, which were verified to be reproducible and efficient in yielding sufficient amount of high quality DNA.4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 However, use of expensive enzymes and toxic solvents in these extraction procedures raised questions on their competence. Therefore, search for an inexpensive but efficient genomic DNA extraction methodology is still in progress. Herein, we have aimed to establish and standardize a simple, inexpensive yet useful procedure devoid of costly enzymes and toxic reagents for extraction of genomic DNA from whole blood samples.
Clear understanding of the chemistry and function of different reagents and buffer helped researchers to construct alternative methodologies for genomic DNA extraction from different sample sources. While designing the protocol for DNA extraction, the compositions of the reagents were determined based on the chemical effect of each reagent on various cellular organelles. Heme protein present in RBC is a strong inhibitor of Taq DNA polymerase such that even 1 μL blood can completely inhibit the PCR in a 100 μL PCR reaction. Therefore, sufficient attention should be paid toward the effective removal of hemoglobin and other contaminating proteins present in the blood sample. The Red Cell Lysis Buffer contained NH4Cl, KHCO3, and Tris buffer. NH4Cl results in increased osmotic pressure inside the RBC until the cells burst from water influx. However, it has least effect on other cellular contents of the blood. It did not affect other cell types especially leucocytes due to the absence of Cl‐/HCO3‐ trans‐membrane anion exchanger in leucocytes which are present in case of RBC. KHCO3 increases the rate of swelling of RBC and can serve as a buffer component. On the other hand, low concentration of Tris Buffer would fasten the erythrocyte lysis process without having considerable deleterious effect on WBCs. Moreover, it also helps to maintain the pH of the buffer at a steady state. In case of the DNA extraction buffer, Tris buffer was used in higher concentration. This was followed by the addition of EDTA in the extraction buffer, which binds divalent cations such as calcium and magnesium. These ions help to maintain the membrane integrity. Their binding with EDTA destabilizes the membrane. CTAB is a cationic surfactant, which helps to lyse the cell membrane. It may also help to precipitate and remove all the unnecessary junk materials such as membrane debris, denatured proteins, polysaccharides etc. However, due to its positive charge, CTAB may form complex with DNA and precipitate it. This is undesirable and therefore NaCl is added which provides Na+ ions into the reaction. These Na+ ions neutralizes the negative charges on phosphates of DNA by forming ionic bond which otherwise would cause the DNA molecules to repel each other. Moreover, when present at higher ionic strength, NaCl disturbs the formation of CTAB‐DNA complex and helps to keep the DNA in solution. SDS used along with the extraction buffer acted as strong anionic detergent that can solubilize the proteins and lipids of the membranes. This will help the cell membranes and nuclear envelopes to break down and expose the chromosomes that contain the DNA. In addition to removing the membrane barriers, SDS may also be useful in releasing the DNA from histones and other DNA binding proteins by denaturing them. Β‐Mercaptoethanol was used along with the DNA extraction buffer in the digestion step because it is a very strong reducing agent. It breaks down disulfide bonds between the cysteine residues of protein molecules, resulting in denaturation of the proteins by linearizing them. These linearized proteins were entangled, messed up and finally removed during centrifugation along with CTAB. After this step, Chloroform: Isoamyl alcohol was added which help in binding and precipitation of protein and lipids of cell membrane This step resulted in the formation of an aqueous phase containing DNA and a non‐aqueous phase containing lipids and proteins. At this stage, DNA molecules are surrounded by water molecules forming the shell of hydration. Therefore, isopropanol is added at this stage as it may act as a dehydrating agent and disrupts the hydration shell resulting in precipitation of the DNA, which can then be separated, from the remaining soluble components through centrifugation.
The average yield of DNA per 500 μL of fresh and frozen blood was found to be 22±2.97 and 20.5±3.97 μg. As per the recent estimates, the sizes of diploid human female and male genomes are 6.406 × 109 bp and 6.294 × 109 bp respectively. Following Doležel et al., 2003 the mean molecular weight of 1 base pair was estimated to be 1.023 × 109 pg. Based on these values, diploid human female and male nuclei in G1 phase of the cell cycle should contain approximately 6.55 and 6.436 pg of DNA respectively.26 As total count of WBC in a normal adult human being ranged from 4.5‐10 × 103/μL of human blood, the total amount of DNA per μL of blood was calculated to be within the range of 29.48‐65.5 ng approximately. Thus, it was seen that using our protocol, the average DNA yield was within the normal range. Furthermore, the protocol roughly consumes two and a half hours for successful completion.
An earlier protocol of DNA extraction using CTAB was published by Thomas et al.,27 but it was found that the buffer compositions and the sequences of the steps differed considerably from our experimental protocol. When compared with other published protocols and commercially available kits, it was observed that our protocol yielded comparable amount of high quality human genomic DNA, as was represented by the scatterplot. We also tested the appropriateness of the model by examining residual plots (Figure 3D). It was observed that the points in the residual plot were more or less randomly dispersed around the horizontal axis and thereby suggesting that the linear regression model is appropriate for the data.
At last, it can be said that the present method is unique as it cuts down both time and expenditure of DNA extraction per human blood sample more robustly when compared to many other available protocols. It is simple and can be carried out successfully even by a nonprofessional. Furthermore, this protocol does not contain any toxic reagents and therefore provides safety while performing the extraction procedure. Lastly, although the protocol is simple and inexpensive, but the protocol did not compromises with the quality and integrity of the extracted DNA.
5. Conclusion
The protocol mentioned in this study may prove to be efficient in yielding considerable amount of genomic DNA from both fresh and frozen human blood samples. Furthermore, the elimination of time consuming steps such as enzymatic incubation (for Proteinase K and RNAase) and avoiding the use of toxic organic solvents such as Phenol made the protocol time‐saving and economical without affecting the quality of the DNA samples which could be reliable enough for applications in advanced molecular biological techniques. Moreover, it may prove to be useful for laboratories with limited funds to pursue basic molecular biological researches.
Acknowledgments
The authors gratefully acknowledge the volunteers who provided their consent in becoming a part of the study. We also gratefully acknowledge Dr. Biswajit Halder, Department of Pathology, North Bengal Medical College and Hospital for providing us a trained medical staff who assisted us during the process of sample collection.
Guha P, Das A, Dutta S, Chaudhuri TK. A rapid and efficient DNA extraction protocol from fresh and frozen human blood samples. J Clin Lab Anal. 2018;32:e22181 10.1002/jcla.22181
References
- 1. Phillips HA, Howard GC, Miller WR. p53 mutations as a marker of malignancy in bladder washing samples from patients with bladder cancer. Br J Cancer. 2000;82:136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang SS, Thornton K, Kuhn AM, Nadeau JG, Hellyer TJ. Homogeneous real‐time detection of single‐nucleotide polymorphisms by strand displacement amplification on the BD ProbeTec ET system. Clin Chem. 2003;49:1599–1607. [DOI] [PubMed] [Google Scholar]
- 3. Lewis CM, Cler LR, Bu DW, et al. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin Cancer Res. 2005;11:166–172. [PubMed] [Google Scholar]
- 4. Angelini A, Di Febbo C, Rullo A, Di Ilio C, Cuccurullo F, Porreca E. New method for the extraction of DNA from white blood cells for the detection of common genetic variants associated with thrombophilia. Pathophysiol Haemost Thromb. 2002;32:180–183. [DOI] [PubMed] [Google Scholar]
- 5. Albarino CG, Romanowski V. Phenol extraction revisited: a rapid method for the isolation and preservation of human genomic DNA from whole blood. Mol Cell Probes. 1994;8:423–427. [DOI] [PubMed] [Google Scholar]
- 6. Parzer S, Mannhalter C. A rapid method for the isolation of genomic DNA from citrated whole blood. Biochem J. 1991;273:229–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Robbins V, Aguinaga MP, Valenzuela MS. Efficient isolation of whole genomic DNA from cell cultures and blood samples. Biotechniques. 1995;18:414–416, 418. [PubMed] [Google Scholar]
- 8. Rudbeck L, Dissing J. Rapid, simple alkaline extraction of human genomic DNA from whole blood, buccal epithelial cells, semen and forensic stains for PCR. Biotechniques. 1998;25:588–590, 592. [DOI] [PubMed] [Google Scholar]
- 9. Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor; 2001. [Google Scholar]
- 10. Wang L, Hirayasu K, Ishizawa M, Kobayashi Y. Purification of genomic DNA from human whole blood by isopropanol‐fractionation with concentrated Nal and SDS. Nucleic Acids Res. 1994;22:1774–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elgort MG, Herrmann MG, Erali M, Durtschi JD, Voelkerding KV, Smith RE. Extraction and amplification of genomic DNA from human blood on nanoporous aluminum oxide membranes. Clin Chem. 2004;50:1817–1819. [DOI] [PubMed] [Google Scholar]
- 12. Lahiri DK, Nurnberger JI Jr. A rapid non‐enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nucleic Acids Res. 1991;19:5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lahiri DK, Bye S, Nurnberger JI Jr, Hodes ME, Crisp M. A non‐organic and non‐enzymatic extraction method gives higher yields of genomic DNA from whole‐blood samples than do nine other methods tested. J Biochem Bioph Methods. 1992;25:193–205. [DOI] [PubMed] [Google Scholar]
- 14. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castella V, Dimo‐Simonin N, Brandt‐Casadevall C, Mangin P. Forensic evaluation of the QIAshredder/QIAamp DNA extraction procedure. Forensic Sci Int. 2006;156:70–73. [DOI] [PubMed] [Google Scholar]
- 16. Cattaneo C, Craig OE, James NT, Sokol RJ. Comparison of three DNA extraction methods on bone and blood stains up to 43 years old and amplification of three different gene sequences. J Forensic Sci. 1997;42:1126–1135. [PubMed] [Google Scholar]
- 17. Nasiri H, Forouzandeh M, Rasaee MJ, Rahbarizadeh F. Modified salting‐out method: high‐yield, high‐quality genomic DNA extraction from whole blood using laundry detergent. J Clin Lab Anal. 2005;19:229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El Bali L, Diman A, Bernard A, Roosens NH, De Keersmaecker SC. Comparative study of seven commercial kits for human DNA extraction from urine samples suitable for DNA biomarker‐based public health studies. J Biomol Tech. 2014;25:96–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chacon‐Cortes D, Haupt LM, Lea RA, Griffiths LR. Comparison of genomic DNA extraction techniques from whole blood samples: a time, cost and quality evaluation study. Mol Biol Rep. 2012;39:5961–5966. [DOI] [PubMed] [Google Scholar]
- 20. Santos EM, Paula JF, Motta PM, et al. Comparison of three methods of DNA extraction from peripheral blood mononuclear cells and lung fragments of equines. Genet Mol Res. 2010;9:1591–1598. [DOI] [PubMed] [Google Scholar]
- 21. Shephard DA. The 1975 Declaration of Helsinki and consent. Can Med Assoc J. 1976;115:1191–1192. [PMC free article] [PubMed] [Google Scholar]
- 22. Suguna S, Nandal D, Kamble S, Bharatha A, Kunkulol R. Genomic DNA isolation from human whole blood samples by non enzymatic salting out method. Int J Pharm Pharm Sci. 2014;6:198–199. [Google Scholar]
- 23. Subbarayan PR, Sarkar M, Ardalan B. Isolation of genomic DNA from human whole blood. Biotechniques. 2002;33:1231–1234. [DOI] [PubMed] [Google Scholar]
- 24. Gong R, Li S. Extraction of human genomic DNA from whole blood using a magnetic microsphere method. Int J Nanomedicine. 2014;9:3781–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sambrook J, Fritsh EF, Maniatis T. Molecular Cloning: A Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 26. Dolezel J, Bartos J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry A. 2003;51:127–128; author reply 129. [DOI] [PubMed] [Google Scholar]
- 27. Thomas JC, Khoury R, Neeley CK, Akroush AM, Davies EC. A fast CTAB method of human DNA isolation for polymerase chain reaction applications. Biochem Educ. 1997;25:233–235. [Google Scholar]