

Abstract
To navigate within the geomagnetic field, magnetotactic bacteria synthesize magnetosomes, which are unique organelles consisting of membrane-enveloped magnetite nanocrystals. In magnetotactic spirilla, magnetosomes become actively organized into chains by the filament-forming actin-like MamK and the adaptor protein MamJ, thereby assembling a magnetic dipole much like a compass needle. However, in Magnetospirillum gryphiswaldense, discontinuous chains are still formed in the absence of MamK. Moreover, these fragmented chains persist in a straight conformation indicating undiscovered structural determinants able to accommodate a bar magnet-like magnetoreceptor in a helical bacterium.
Here, we identify MamY, a membrane-bound protein that generates a sophisticated mechanical scaffold for magnetosomes. MamY localizes linearly along the positive inner cell curvature (the geodetic cell axis) likely by self-interaction and curvature sensing. In a mamY deletion mutant, magnetosome chains detach from the geodetic axis and fail to accommodate a straight conformation coinciding with reduced cellular magnetic orientation. Co-deletion of mamKY completely abolishes chain formation, whereas upon synthetic tethering of magnetosomes to MamY, the chain configuration is regained, emphasizing the structural properties of the protein. Our results suggest MamY as membrane-anchored mechanical scaffold essential to align the motility axis of magnetotactic spirilla with their magnetic moment vector and to perfectly reconcile magnetoreception with swimming direction.
The ability to sense and navigate along the Earth’s magnetic field is widely distributed from microbes to animals1,2. The best studied organisms are magnetotactic bacteria, ubiquitous aquatic microorganisms that synthesize unique organelles (magnetosomes) to perform magnetotaxis in search for growth-favoring microoxic zones in chemically stratified water columns or sediments1,3. Magnetosomes consist of magnetite nanocrystals biomineralized within vesicles of a dedicated membrane. To build an efficient magnetoreceptor, magnetosomes must be assembled into a linear chain against their inherent tendency to agglomerate4, so that the magnetic moments of individual particles sum up to form a dipole (much like a compass needle) sufficiently strong to align the whole bacterium in the weak geomagnetic field5.
In the alphaproteobacterium Magnetospirillum gryphiswaldense (Mgryph) and related magnetospirilla, the magnetosome chain (MC) is assembled by two dedicated proteins: the actin-like MamK polymerizes into dynamic cytomotive filaments to which magnetosomes are connected via the acidic MamJ protein. Both proteins organize and concatenate magnetosomes into a chain of ~45 particles, which becomes evenly split upon cytokinesis and actively repositioned from the new pole to midcell by treadmilling action of MamK6–8.
Absence of MamJ disrupts the chain configuration resulting in irregular magnetosome clusters due to their unconstrained mutual magnetostatic interactions4,9. However and by contrast, lack of MamK does not entirely abolish MC formation, but merely results in shorter and fragmented MCs10. These distinct phenotypes suggest the existence of additional determinants that mechanically scaffold a MC preventing its collapse even in the absence of actin-like MamK filaments. Yet, the identity of such structures has remained elusive.
Another, but largely unrecognized proficiency of magnetospirilla is to reconcile linear locomotion of helical cells with the magnetic moment of their magnetoreceptor to achieve maximum efficiency of magnetotaxis. For an actively swimming spirillum propelled by its polar flagella, this means that the MC must always be accommodated straight (comparable to a bar magnet) within the highly curved rotating cell11, and the magnet’s dipole must be constantly and precisely maintained parallel to the cell’s motility axis. This is intriguing since unlike rod-shaped bacteria, spirilla lack any straight cell surface to support a rod-like magnetoreceptor. However, this question has remained widely unappreciated, and again, structures and mechanisms for three-dimensional MC positioning have been previously unknown.
Here, we demonstrate that MamY, a membrane-bound structural protein encoded within a genomic magnetosome island12 (i) localizes as a cell-spanning linear structure that identifies the cellular geodetic axis likely by curvature sensing, and (ii) acts as an organelle-scaffolding backbone able to support a MC in the absence of MamK. Thus, we demonstrate that the function of MamY is to perfectly superimpose the magnetic moment of the receptor on the swimming direction of the cell. Overall, we suggest MamY as a curvature-sensing structural element dedicated to accommodate a straight magnetoreceptor perfectly in a helical cell ensuring most efficient magnetotaxis.
Results
MamY positions the magnetosome chain along the cellular geodetic axis
Within the genomic magnetosome island, MamY is encoded in the mamXY operon where the adjacent genes (mamX, mamZ and ftsZm) exert functions in the biomineralization of magnetite13–16. To investigate a putative role of MamY in MC formation, we deleted the gene, but at a glance, the mutant presented chains reminiscent of those in the WT. On closer inspection however, we noticed a chain mislocalization. While WT MCs always appear straight, MCs of ΔmamY cells seemed bent and adapted to the negative inner curvature (Fig. 1a). This subtle and unexpected phenotype prompted us to re-assess MC positioning in WT cells in more detail. Thorough examination of TEM images suggested that WT MCs trace a well-defined path parallel to the long cell axis (Fig. 1b). Despite the highly curved cell shape, the chain itself appeared straight and regularly touching sections of positive cell curvature, which was evident even in the fragmented chains of ΔmamK cells10. However, as TEM microscopy only generates a 2D projection of the highly helical cells, we visualized their 3D morphology by cryo-electron tomography (cryo-ET)17. 3D-rendering of the tomograms revealed that WT MCs do not approach the cytoplasmic membrane punctually or randomly as suggested by TEM, but always associate tightly with regions of highest positive (convex) curvature (Fig. 1c-e. Supplementary Fig.1a and Video 1), which in helical cells coincides with their geodetic axis18, i.e. the shortest path connecting the cell poles on the curved inner cell surface (Fig. 1f).
Figure 1. MamY determines the localization of the magnetosome chain at the geodetic axis of Magnetospirillum gryphiswaldense.

(a) TEM micrograph of a ΔmamY cell with magnetosome chain (black electron-dense magnetite crystals) mispositioned at the negative curvature (100% of cells. N = 126). The model (lower-left corner) indicates the theoretical WT chain position (red-dashed line).
(b) TEM of a WT cell. The magnetosome chain seemingly touches sites of positive curvature (100% of cells. N = 167).
Insets in (a) and (b): magnification of magnetite crystals. Scale bar: 500 nm (inset: 100 nm).
(c-e) Cryo-ET of a WT cell. (c) A 15.7 nm tomographic slice through the center of the cell showing magnetosome vesicles (black-double arrowheads) and magnetite crystals (black arrowhead). Inset: tomographic slice at different z-depth displays magnetosome chain continuity and close proximity to the positively-curved cytoplasmic membrane. Scale bars: 200 nm. (d) 3D-rendering of the tomogram from the cell in (c). Magnetite crystals: red; magnetosome vesicles: yellow; MamK filament: green; Inner and outer membranes: blue. (e) 3D-rendering views from the angle indicated by the red and light-blue arrowheads in (d). Curved-white-dashed lines: membrane projection (approximation, missing wedge). White-curved arrows: magnetosome chain track inside the cell along the positive cytoplasmic membrane curvature.
(f) SEM image of two Mgryph cells (of different length) highlighting their helical morphology and geodetic axis (red-dashed line). Scale bars: 500 nm.
(g-j) Cryo-ET of a ΔmamY cell. (g) A 15.7 nm tomographic slice with the magnetosome chain shifted to the negative membrane curvature. Inset: tomographic slice of another ΔmamY cell. Scale bars: 200 nm. (h) 3D-rendering from the tomogram of the cell from (g). (i) 3D-rendering views -indicated by the red and light-blue arrowheads in (h)- shows misplacement of the magnetosome chain apart from the geodetic axis. (j) 3D-rendering and tomogram slice of the same cell in perspective. White-dashed-straight line in (h) and (j): projection of the geodetic axis in 2D. Grey-curved arrow in (i -right panel): theoretical geodetic axis. Similar tomograms (for WT and ΔmamY strains) have been obtained from at least three experiments using independent preparations of cells.
By contrast, cryo-ET of ΔmamY cells confirmed that MCs were detached from the geodetic axis (Fig. 1g, Supplementary Fig. 1b and Video 2) following the negative curvature of the cytoplasmic membrane (Fig. 1h-j and Supplementary Fig. 1b), and hence the longest possible distance contrary to a geodetic path. Although spiral cell morphology remained unaffected, loss of MamY correlated with a reduced cellular magnetic orientation (Cmag19: 64% of WT, Supplementary Fig. 2a), suggesting that MC positioning along the geodetic axis is necessary to achieve maximum magnetic cell alignment. MC positioning could be restored in ΔmamY cells by trans-complementation with mamY (Supplementary Fig. 2b), indicating that MamY is an essential factor to properly localize the MC within Mgryph cells.
Double deletion of mamY-mamK abolishes magnetosome chain formation
We next investigated the role of MamY in the ΔmamK mutant (Fig. 2a). Deletion of mamY in the ΔmamK strain, ΔmamKY, resulted in complete loss of the fragmented MCs and agglomeration of magnetosomes (Fig. 2b), strikingly reminiscent of the ΔmamJ strain phenotype (Fig. 2c)9. The Cmag of ΔmamKY cells was reduced to 20% of the WT (Supplementary Fig. 2a) similar to the ΔmamJ strain (10%). Trans-complementation of the ΔmamKY strain with either mamY or mamK restored the ΔmamK and ΔmamY phenotype, respectively (Supplementary Fig. 2c-d). Trans-complementation of ΔmamKY cells with mamK-mamY recovered the WT MC arrangement along the geodetic axis and the Cmag (92% of WT, Supplementary Fig. 2a, e). These results support the hypothesis that MamY is able to scaffold the fragmented MCs in the mamK mutant.
Figure 2. MamY is essential to support the fragmented magnetosome chains in the mamK deletion mutant.

(a) TEM images of ΔmamK cells displaying fragmented magnetosome chains that follow a path along the positive cell curvature (indicated by the red-dashed line - lower inset).
(b) TEM micrographs of the mamY-mamK double deletion mutant, which is unable to organize magnetosomes into chains and displays aggregated magnetosomes.
(c) TEM image of a ΔmamJ cell that presents aggregated magnetosomes. Insets: magnification of the magnetite crystals. TEM images showing corresponding magnetosome configurations were obtained from at least five independent experiments and cell preparations. Scale bars: 500 nm. Insets: 200 nm (except lower inset in A: 500 nm).
MamY localizes in a cell-spanning linear pattern
Fluorescence microscopy of functional (Supplementary Fig. 3a-b) N- and C-terminal mCherry fusions revealed a linear localization signal of MamY contacting the positive membrane curvatures across the cell (Fig. 3a and Supplementary Fig. 3c). In agreement with the predicted MamY TM domain, we also observed weaker fluorescence dispersed at the cellular membrane. By immunodetection, MamY was found in the membrane and magnetosome fractions, supporting the notion that MamY is associated with the cytoplasmic as well as magnetosome membranes (Fig. 3b).
Figure 3. MamY localizes as a linear structure along the positive curvature in M. gryphiswaldense cells.
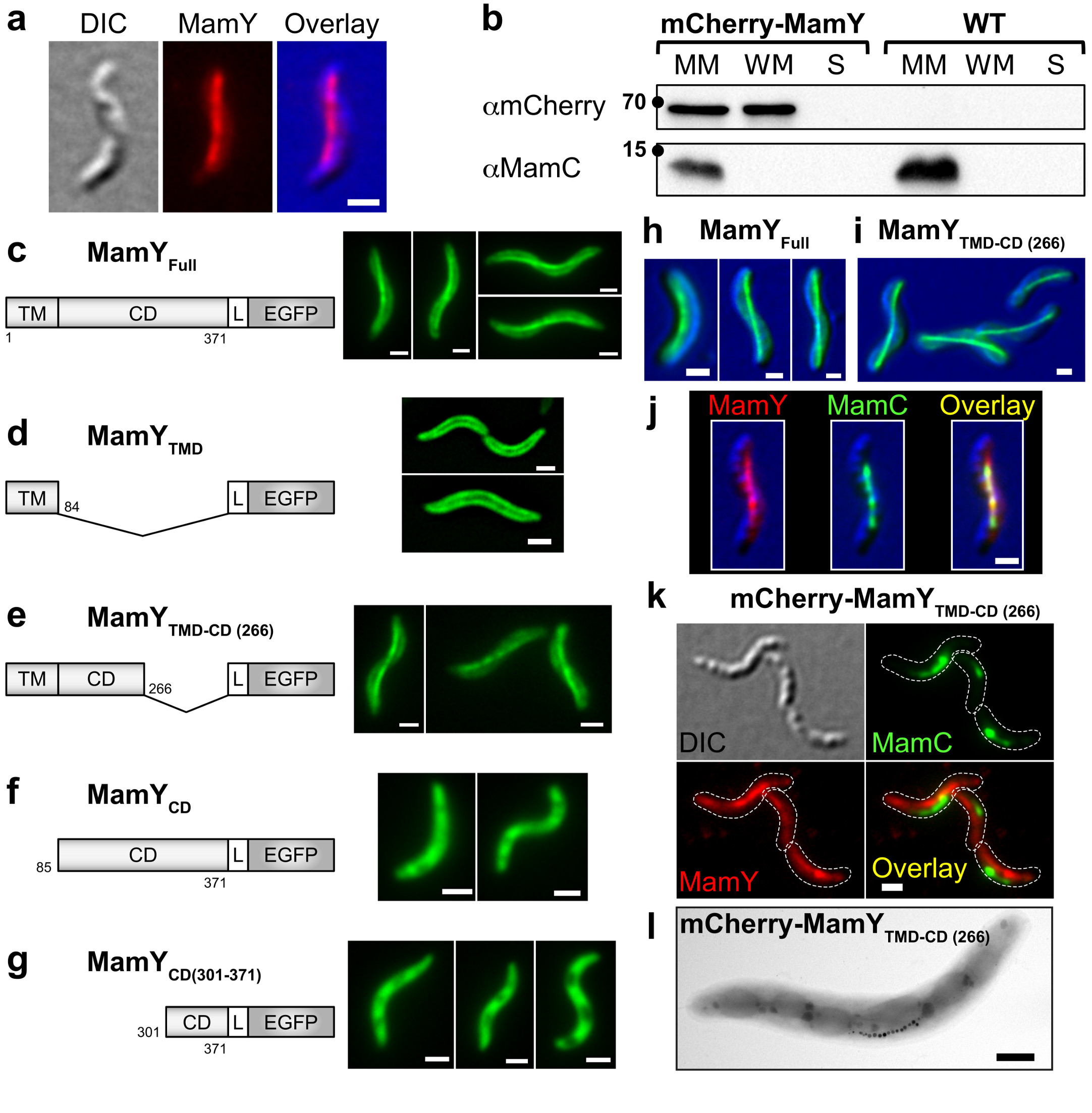
(a) Subcellular localization of a markerless chromosomal native-site N-terminal mCherry fusion to MamY seen by widefield fluorescence microscopy. mCherry (red); DIC (blue).
(b) Cell fractionation and immunodetection of mCherry-MamY and MamC (control for cross-contamination of cell fractions) in the magnetosome membrane (MM), whole membrane (WM) and soluble (S) fractions. Molecular weight standards are indicated by a dot (kDa). MamC ∼12 kDa, mCherry-MamY ∼68 kDa.
(c-g) Localization patterns of EGFP-labeled full length and truncated versions of MamY in the WT strain. (c) MamYFull: consists of a transmembrane (TM) plus the cytoplasmic domain (CD) equivalent to 371 amino acids. (d) MamYTMD: harbors only the TM domain (amino acids 1 to 84). Micrographs are central slices through the cell of a deconvolved z-stack. (e) MamYTMD-CD(266): N-terminal 266 amino acids (including the TM domain and part of the cytoplasmic domain, from the amino acid position 85 to 266). (f) MamYCD: cytoplasmic domain only. (g) MamYCD(301-371): C-terminal 71 amino acids. Schemes of the protein fusions are not to scale (L: linker), numbers indicate amino acids of MamY.
(h-i) Localization patterns of (h) MamY full length and (i) C-terminal truncated MamYTMD-CD(266) are conserved in the heterologous host R. rubrum. EGFP (green); DIC (blue).
(j) Magnetosome chain and MamY co-localize in Mgryph. mCherry (red); MamC (magnetosome marker, green); DIC (blue).
(k-l) MamY C-terminal domain is essential to recruit the magnetosome chain to the positive cell curvature. (k) The MamC-EGFP fluorescence signal is shifted to the negative curvature in the mCherry-mamYTMD-CD(266) mutant. (l) TEM image showing displacement of the magnetosome chain to the negative cell curvature in the mCherry-mamYTMD-CD(266) mutant. Scale bars: 1 μm (l: 500 nm). All subcellular localization patterns were observed in at least three independent experiments.
To assign functionalities to MamY protein domains, we designed distinct N- and C-terminal EGFP-tagged MamY truncations and analyzed their localization by fluorescence microscopy. Unlike the full-length protein (MamYFull-EGFP, Fig. 3c), the TM domain (MamYTMD: amino acids 1-84-EGFP, Fig. 3d) localized exclusively to the cellular membrane. However, a longer variant encompassing the TM domain and part of the cytoplasmic domain (MamYTMD-CD(266): amino acids 1-266-EGFP, Fig. 3e) regained the linear positioning. A truncation consisting only of the cytoplasmic domain (amino acids 85-371: MamYCD-EGFP), or harboring only the C-terminal amino acids 301-371 (MamYCD(301-371)-EGFP) both showed disperse cytoplasmic signals (Fig. 3f-g). This suggests that the N-terminal first 266 amino acids (including the TM domain) are necessary for the linear localization pattern whereas the C-terminal 71 amino acids are dispensable. In the ΔmamY and Mgryph-1B (lacking most magnetosome genes) mutants, the MamY variants localized similarly suggesting that neither native MamY nor other magnetosome-related genes are needed for the detected patterns (Supplementary Fig. 4a-e).
To analyze whether the distinct MamY localization can be evoked in other spirillum-shaped bacteria, we used the heterologous host Rhodospirillum rubrum, a helical α-proteobacterium that lacks homologs of magnetosome-related genes but exhibits positively bent membranes with the potential to integrate a discrete geodetic axis. In R. rubrum, MamYFull-EGFP and MamYTMD-CD(266)-EGFP showed the typical geodetic localization observed in Mgryph (Fig. 3h-i). In contrast, in the rod-shaped Escherichia coli (lacking positively-curved membranes) MamYFull-EGFP was found exclusively membrane-associated, and no linear structures were observed (Supplementary Fig. 4f). These results suggest that the geodetic localization is an intrinsic property of MamY, independent of other magnetosome-related proteins and the host.
MamY interacts with itself and the magnetosome chain
Based on these localization patterns, we hypothesized that the MamY N-terminal region may act in membrane tethering, putative oligomerization and positive curvature sensing, whereas the C-terminal region could recruit magnetosomes. Therefore, we tested for localization of mCherry-MamY and MamC-EGFP (a magnetosome-specific protein used to visualize MCs7,9) and found that both fluorescence signals co-localized (Fig. 3j). When we replaced mamY by the truncated mamYTMD-CD(266) allele, mCherry-MamYTMD-CD(266) still showed a WT-like localization pattern along the inner positive cell curvature. However, MamC-EGFP was now found shifted to the negatively-curved membrane (Fig. 3k), resembling the ΔmamY phenotype, which was confirmed by TEM (Fig. 3l). This suggests that the C-terminal amino acids 267-371 are not involved in MamY localization but rather in magnetosome recruitment.
To corroborate the notion that the TM domain is essential for curvature sensing and geodetic localization, we exchanged it for the TM domain of an arbitrary Mgryph membrane protein (a non-magnetosome-related kinase), generating the chimeric TMHK-MamYCD protein. In the magnetosome-free Mgryph-1B strain, TMHK-MamYCD-EGFP showed only membrane localization, but no linear patterns (Supplementary Fig. 4g). In agreement, the same fusion in R. rubrum localized at the cytoplasmic membrane, but lost the original enrichment at the geodetic axis (Supplementary Fig. 4h). However, in Mgryph WT cells, TMHK-MamYCD-EGFP co-localized specifically with the MC (MamC-mCherry), but the signal did not extend to the cell poles as for WT MamY, rather suggesting a recruitment of the chimeric protein to magnetosomes (Supplementary Fig. 4i). The TMHK itself (TMHK-EGFP), used as negative control, localized exclusively to the membrane in WT cells (Supplementary Fig. 4j). We also examined whether the presence of MamY on the magnetosome surface alone is sufficient to recruit magnetosomes to the geodetic axis. To this, we substituted the MamY TM-domain for MamC to confine MamY exclusively to the magnetosome membrane and analyzed the position of magnetosomes in WT and the mamY mutant. The results suggest that MamY must be present at the cytoplasmic membrane to properly localize magnetosomes (see Supplementary Information and Supplementary Fig. 5a-b) again suggesting that the TM domain is implicated in curvature sensing and/or oligomerization, whereas the C-terminal region is involved in magnetosome recruitment.
Because MamJ was shown to be essential for MC formation9, we interrogated a potential MamY-MamJ interplay by quantification of mCherry-MamY in cell fractions using MamC as a reference20,21. The results suggest that magnetosome-associated MamY is reduced by ~50% in the absence of MamJ (MamY:MamC ratio: 0.8 and 1.5 for ΔmamJ and WT strains, respectively (Supplementary Fig. 5c). This together with the ΔmamJ mutant phenotype (agglomerated magnetosomes in the presence of MamY) supports the notion that MamJ is needed for MamY-magnetosome interaction. Nevertheless, an in vivo MamY-MamJ interplay could not be detected unambiguously by bacterial two-hybrid (BTH) assays22 (Supplementary Fig. 5d), suggesting an only weak or indirect interaction.
MamY assembles into a geodetically localizing linear structure
To determine whether MamY self-assembles a distinct membrane-associated structure, we first applied 3D-SIM, where mCherry-MamY displayed a cell-spanning linear localization from positive-to-positive curvature in 2D (Fig. 4a and Supplementary Fig. 6). Remarkably, 3D inspection of the signal revealed a sinusoidal wave-like structure, which continuously followed the inner positive curvature (Fig. 4b-c and Supplementary Video 3). Thus, 3D-SIM confirmed the linear geodetic positioning, supporting that a putative MamY structure may act as a landmark to position the MC inside helical cells.
Figure 4. 3D-SIM and PALM reveal that MamY forms a linear structure along the positive curvature.

(a-c) 3D-SIM of a markerless mCherry-MamY translational fusion expressed from the mamY locus. (a) Maximum intensity projection of the mCherry-MamY signal combined with the brightfield channel (blue). (b-c) Z-axis inspection of the cell in Ai shows the continuity of the structure and its localization along the positive cell curvature, i.e., along the geodetic axis (see Supplementary Video S3). Micrographs: maximum intensity projections from the indicated angles. Scale bars: 1 μm.
(d-f) Single-molecule detection of Dendra2-MamY in WT (native MamY present). (d) PSF rendering: micrograph generated by density rendering of the PSF FWHM of all single events. Inset: brightfield channel. (e) Local density: each dot represents the geometric center position of a fluorophore. Density of events is depicted in color-coded values. Upper inset: nearest neighbor distances. Data were edge-corrected using the border method (red line), Kaplan-Meier (black line) or Hanisch (green line) estimators yielding similar results. A theoretical random distribution (blue line) considers identical area and amounts of events per cell as the experimental data. (f) Clustering: algorithm that considers the localization precision area of individual events and the intermolecular distances. Two molecules are clustered if the distance of their localization precision areas are ≤0 nm. Light blue: single molecules and clusters <5 molecules; Blue: clusters of 5 to 50 molecules; Dark blue: clusters of >50 molecules. Insets: magnification of the indicated area showing clusters of molecules rendered with their corresponding localization precision area. Scale bars: 500 nm.
(g-l) PALM of Dendra2-MamY in ΔmamY strain. (g-k) Local density rendering of single events from cells with different amounts of MamY. The color-coded scale in (k) applies to (g-k) (l) Nearest neighbor distances of the events for each depicted cell. Blue to red color gradient of the plot lines represent the lowest and highest MamY molecule counts per cell, respectively. Experimental distribution data was edge-corrected using the Hanisch estimator (continuous lines). Theoretical distribution: dashed lines. PALM data are representative of at least three independent experiments.
We next performed PALM to localize single Dendra2-MamY molecules at a higher resolution. PSF rendering of Dendra2-MamY single events suggests that the molecules localize as a linear structure extending throughout the long cell axis from positive-to-positive curvature in 2D, likely following a geodetic path (Fig. 4d, average localization precision: 26.8 nm. See Methods). While few molecules outside the geodetic axis and likely residing at the cytoplasmic membrane were observed, a local density map of the events showed that the putative structural element along the geodetic axis comprised an evidently higher density of MamY molecules (Fig. 4e).
Nearest neighbor distances of the experimental data displayed a cumulative distribution with a hyperbolic behavior distinct from that of a theoretical distribution (Fig. 4e inset, and Supplementary Information), indicating a strong tendency of MamY molecules to be closer than expected for a random distribution. This suggests that MamY molecules become enriched at specific areas of the cell membrane where they localize tightly and potentially interact. To support this hypothesis, we designed an algorithm considering the localization precision to cluster events that are, theoretically, able to interact. Only single molecules with precision areas in direct contact or overlap became clustered. The clustering showed that the cell-spanning MamY element is the most prominent structure containing the largest clusters (Fig. 4f), indicating that highest molecule densities that promote direct interactions are found along the geodetic path.
PALM of Dendra2-MamY in ΔmamK cells (precision average ~27 nm) demonstrated that the MamY localization pattern remained unchanged compared to WT (Supplementary Fig. 7a-e), indicating that formation and positioning of the MamY structure is independent of MamK filaments. We validated our clustering algorithm pipeline using the well-characterized actin-like MamK protein, which was reliably identified as a continuous filamentous structure (Supplementary Fig. 8a).
To assess how these MamY assemblies behave upon loss of positive membrane curvature in Mgryph, we generated spheroplasts, which seemed to retain their pre-existing membrane-associated linear structures (Supplementary Fig. 4k-l). Identical results were obtained in the ΔMAI strain, again suggesting that MamY localization is independent of other magnetosome-related factors (Supplementary Fig. 4m). The preserved linear pattern in spheroplasts also suggests that the MamY structure, once formed, is fairly stable even in the absence of positive membrane curvature.
To further corroborate the assumed self-interaction properties of MamY, we analyzed its intermolecular interaction capability in vitro using dynamic light scattering (DLS). We therefore purified the cytoplasmic domain of MamY (MamYCD) and tested for polymerization under different pH and ionic conditions. DLS results suggest that MamY oligomerization can be mediated by its cytoplasmic domain; controlled by pH changes within a physiological range and is fully reversible (Supplementary Information and Supplementary Fig. 9a-b). Self-interaction capabilities of MamYFull and the truncated versions (MamYTMD-CD(266) and MamYCD) were confirmed in vivo by a BTH assay (Supplementary Fig. 5e), indicating that MamY, as other structural proteins, has the potential to form higher-order homo-oligomeric complexes.
Altogether, these results substantiate that MamY forms a scaffolding structural element localizing preferably along the geodetic cell axis in vivo.
Localized MamY assemblies depend on the transmembrane domain
As the TM domain seemed dispensable for MamY homo-oligomerization yet fundamental to generate a cell-spanning structure, we performed PALM of MamYCD and TMHK-MamYCD to search for short linear structures unresolved by widefield microscopy. Analysis of Dendra2-MamYCD molecules in the ΔmamKY strain confirmed the tendency of MamYCD to cluster in vivo (Supplementary Fig. 10a-f) consistent with DLS and BTH data. However, neither linearly extending densities nor geodetic cluster enrichments were detected, suggesting that MamYCD molecules are incapable to generate linear structures in the cytoplasm. This also indicates that membrane anchoring of MamY may be necessary to restrict its interactions to a single plane as prerequisite to become curvature sensitive and to generate a linear structure.
Interestingly, replacement of the native TM domain for TMHK (TMHK-MamYCD) also abolished formation of linearly extended structures in the ΔmamKY strain, leading to only membrane-associated molecule clusters but no geodetic enrichment (Supplementary Fig. 10g-l). Thus, the native MamY TM domain (along with the N-terminal 266 amino acids) is essential to form a long-range, membrane-associated and geodetically positioned structural element.
MamY oligomers nucleate stochastically and become enriched along the geodetic axis
To study how such geodetically cell-spanning structure is formed, we followed the de novo emergence of the MamY structure upon induced gene expression in ΔmamY cells by PALM. Analysis of cells with different Dendra2-MamY molecule counts showed that, initially, the protein enriched stochastically in few discrete areas, likely by oligomerization (Fig. 4g). These putative oligomeric structures became more evident with increasing amounts of protein (Fig. 4g-k), producing regions of higher density specifically along the geodetic path. Such MamY-enriched regions remained evidently detectable even at a high protein overdose (~17,500 events, Fig. 4k).
Nearest neighbor distances of single-molecule events quantitatively displayed a clear tendency of MamY to cluster (distinct from a random distribution), denoting a directed self-interaction at low and high cellular concentrations (Fig. 4l). Thus, MamY oligomerization generates discrete clusters of molecules at the membrane, which become preferentially positioned at the geodetic axis and further enriched upon increasing amounts of protein (Fig. 4g-k), presumably localizing by a membrane topology sensing mechanism.
MamY can sustain a synthetic magnetosome chain
To unambiguously confirm that MamY is able to support the alignment of a linear MC, we aimed to rebuild an artificial MC on MamY in vivo circumventing the function of MamJ and MamK. We employed an intracellular nanobody-based Nanotrap23–25 (engineered antibodies expressed in Mgryph to generate intracellular tethers by antibody-antigen interactions) consisting of an mCherry-binding nanobody (MBN) located on the magnetosome surface by fusion to MamC (MamC-MBN25), and mCherry-MamY to recruit MBN-decorated magnetosomes to MamY, termed Nanotrap-to-Y. To exclude any influence of MamK, we generated a mamJ-mamK double mutant, ΔmamJK, which exhibited aggregated magnetosomes (Fig. 5a, c) due to the dominant mamJ mutant phenotype. Strikingly, ΔmamJK cells expressing the Nanotrap-to-Y regained the ability to align magnetosomes into fragmented chains located at the geodetic cellular axis (Fig. 5b, d), resembling the mamK mutant phenotype. Similar results were obtained in the ΔmamJ strain that showed reconstituted MCs (Supplementary Fig. 11a-b) overriding the magnetosome-cluster phenotype of ΔmamJ cells. This indicates that the MamY structure is indeed capable to provide a mechanical scaffold for MCs.
Figure 5. Reconstruction of a synthetic MamY-supported magnetosome chain.

(a) TEM micrograph of the mamJK double deletion mutant displaying clustered magnetosomes.
(b) Several ΔmamJK cells transformed with the Nanotrap-to-Y (MamY-mCherry and MamC-MBN) displaying artificially restored magnetosome chains onto the MamY structure. Red line: projection of the geodetic axis in 2D.
(c) Model of a ΔmamJK cell with clustered magnetosomes localized at the positive curvature. Blue: MamY structure. Note that this is a 2D projection resembling the cells observed by TEM as in (a), therefore, the MamY structure seemingly detaches from the membrane. However, it is continuously associated to the inner positive curvature of the cell.
(d) Model of a ΔmamJK cell plus the Nanotrap-to-Y (mCherry-MamY and MamC-MBN), where the antigen-antibody interaction (mCherry-MBN) allows the artificial recruitment of the magnetosomes to the MamY structural element. Scale bars: 500 nm. Similar magnetosome configurations were observed in cells from three experiments using independent mutant strains and cell preparations.
We next tried to construct an artificial MC on the MamK filament as a control, using MamC-MBN and mCherry-MamK (Nanotrap-to-K). The Nanotrap-to-K caused the mamJ mutant to regain a single synthetic MC from their agglomerated magnetosomes. However, the MC mislocalized to the negative curvature (Supplementary Fig. 11c-d), resembling the mamY mutant phenotype although WT-mamY was present. The latter again supports a MamJ-dependent interaction between MamY and magnetosomes.
To rule out that MamY influences the assembly of an artificial MC on the MamK filament, we used a mamJ-mamY double mutant (ΔmamJY), which displays clustered magnetosomes because of the absence of MamJ (Supplementary Fig. 11e). Nanotrap-to-K reconstituted MCs in ΔmamJY cells, albeit mislocalized to the negative curvature (Supplementary Fig. 11f), confirming that the reconstruction of MCs onto either MamK or MamY structures is independent from each other. These results further support that MamY interacts with magnetosomes, probably via MamJ, and demonstrate that the MamY structural element is able to support a MC mechanically by itself.
Discussion
A decade after identification of the actin-like MamK filament9,10,26 and the adaptor MamJ9,27, the discovery of MamY as a core constituent of the MC scaffold uncovers an additional structural protein with unique properties. We propose a model where MamY generates a topological landmark and essential mechanical support for MCs underneath the cytoplasmic membrane (Fig. 6a). Magnetosomes are proposed to be connected via MamJ to the scaffolds formed by MamY and MamK (Fig. 6b). This highlights that magnetotactic bacteria employ a dedicated and interacting set of structural and scaffolding elements for magnetotaxis. We therefore introduce the term “magnetoskeleton” to define such magnetosome-specific structural proteins, now encompassing the curvature-sensing MamY, the actin-like MamK, and the adaptor MamJ.
Figure 6. Model for MamY molecular interaction, intracellular polymer formation and localization, and its function as part of the magnetoskeleton.

(a) 3D-rendered illustration of a helical Mgryph cell body (purple) highlighting the localization of the MamY structure (blue) which continuously follows the inner positively-curved membrane. Cross sections depict the position of the MamY structure at different positions along the cell. The MamY structure recruits, scaffolds and organizes the magnetosome chain (membrane-bounded grey crystals) along the geodetic axis.
(b) The magnified model depicts MamY as a transmembrane protein assembling into a continuous structure (blue molecules) attached to the cytoplasmic membrane. An indirect MamJ-MamY interplay is also suggested. This model represents a 2D-projection of a longitudinal section of a helical Mgryph cell, causing the membrane to appear bent. MamY cytoplasmic domain: blue; MamY TM domain: light-blue; MamJ: orange; MamK filament: green; magnetite crystal: grey; magnetosome and cytoplasmic membranes: dark green.
(c) Mechanistic model of MamY monomer interaction by 3D domain swapping. Interactions result in formation of either soluble open-ended oligomers (in the absence of the TM domain) or a stable patch-like layer when anchored to the cytoplasmic membrane (lower scheme) further described in the Supplementary Discussion.
(d) Model for de novo assembly of long-range structures and curvature sensing by MamY. First, MamY oligomers are formed, generating membrane patches large enough to become curvature sensitive. Consequently, MamY oligomers become enriched along the inner positive cell curvature, which promotes further directed interactions thereby generating a geodetically positioned MamY structure. Models are not to scale.
Known bacterial cytoskeletal elements include several tubulin-, actin- and intermediate filament-like proteins with diverse functions, such as cell shape and polarity determination, segregation, cytokinesis and differentiation28. Also, few cell-cycle-related membrane curvature sensing and/or landmark proteins have been described (e.g., DivIVA, PopZ, bactofilins)29–32. Unlike those, MamY is not involved in controlling cell morphology, cycle or division. Instead, our data strongly suggest that MamY forms a structural element underneath the cytoplasmic membrane along the geodetic cell axis, providing a scaffold to assemble, position, and hold a chain of magnetosomes (Fig. 6a). This notion is further supported by the observation that linear MamY assemblies remained upon a dramatic cell shape deformation in spheroplasts. This suggests that the linear MamY structure is sufficiently rigid (locally stable33) to physically maintain magnetosomes as chains, preventing them from agglomeration. However, the nature of the structure remains to be resolved. We currently consider that a filament-like assembly is rather unlikely, but suggest that densely packed MamY oligomers form stable cluster interactions. We explain the capability of MamY to oligomerize by a mechanism called 3D-domain swapping, known for polymerizing proteins and mediated by interchange of corresponding protein domains between a variable number of monomers34–38 (Fig. 6c). However, whereas MamYCD alone failed to form linearly extending structures, the full length protein did. We hypothesize that restricting diffusion to two dimensions by membrane attachment not only promotes self-interaction but also localization, because a sufficiently large polymeric structure may well become curvature sensitive and localize to the geodetic axis where it eventually extends in only one dimension (Fig. 6d and Supplementary Discussion).
Furthermore, synthetic reconstruction of a MC on either MamY or MamK demonstrates that the magnetosome scaffolding function of both elements is not interdependent. We argue that MamY represents a membrane-anchored and curvature-sensing structural and scaffolding element, which can serve as landmark for positioning of prokaryotic organelles and as mechanical support for their spatial arrangement.
But why are MamK filaments alone insufficient to build a straight MC? Floating and continuously growing filaments can bend and twist, particularly when they grow longer than the cell. MamK filaments can also turn and curve, which becomes evident upon overexpression10, and highlights that such filaments are no rigid structures. In contrast, functionality and localization of MamY are rather robust, even upon high protein overdose (Fig. 4k). Therefore, the distinct oligomerization and localization properties of MamY seem superior to filamentously growing cytoskeletal proteins in terms of steadiness and scaffolding properties. A skeleton dyad of a cytomotive filament -to provide MC concatenation and repositioning after cytokinesis- and a static, curvature sensing membrane anchor -to capture the dynamic organelle assembly- seems an optimal combination to support both dynamics and stability of the magnetoreceptor.
With that, magnetotactic spirilla solve a morphology dilemma: (i) they accommodate a bar-like magnet in the helical cell body and, most notably (ii), maintain the cellular magnetic moment parallel to the motility axis to avoid gyration around the linear swimming path by MC misalignment during the rotational cell movement. This effect becomes visible when strains impaired in chain assembly/localization (ΔmamK, J and Y) are compelled to be non-motile27. In contrast to the magnetic orientation of WT cells, where the Cmag of motile and non-motile cells is equal because of perfect alignment between cell axis and MC, the Cmag of the mutants decreases strongly upon stopping motility because cells remain misaligned to the magnetic field. We conclude that the biological role of MamY is to precisely force and immobilize the magnetoreceptor to the (inner) positive surface of curved cells. This, in turn, perfectly aligns the vector of the cellular magnetic-dipole moment with the motility axis of a helical cell body and maximizes magnetotaxis efficiency.
Methods
Bacterial strains, plasmids and culture conditions
Bacterial strains, plasmids (construction procedure) and oligonucleotides generated and used in this work are listed in Table 1, Table 2 and Table 3, respectively. Mgryph strains were grown under microoxic conditions in 2% oxygen aerated modified flask standard medium39 (FSM) containing 50 μM ferric citrate at 30ºC and moderate agitation (120 rpm). Rhodospirillum rubrum was grown in YPS medium40 photoheterotrophically in closed screw cap tubes with illumination from a light bulb at 28°C.
E. coli strains DH5α, BW29427 and WM3064 were grown in LB medium at 37ºC41. E. coli strain BW29427 and WM3064 used for conjugations of plasmids into Mgryph were supplemented with 1 mM DL-α,ε-diaminopimelic acid (DAP). For strains carrying recombinant plasmids, media were supplemented with kanamycin at 25 g ml-1 for E. coli, and 5 g ml-1 for Mgryph and R. rubrum.
Molecular and genetic techniques
The draft genome sequence of M. gryphiswaldense (GenBank accession number CU459003) was used for oligonucleotide design. Oligonucleotides were purchased from Sigma-Aldrich. All constructs were sequenced on an ABI 3700 capillary sequencer (Applied Biosystems), utilizing BigDye Terminator v3.1 or by Macrogen Europe (Amsterdam, Netherland). Sequence data were analyzed with CLC Main Workbench Software (Qiagen) or Geneious (Biomatters).
Plasmid construction
Plasmids were constructed by amplifying the DNA fragments of interest with the Phusion High Fidelity DNA Polymerase (Thermo Scientific). Plasmid description and oligonucleotides are listed in Table 2 and Table 3, respectively (see below). For details of the in-frame insertion or deletion mechanism with the pORFM-galK plasmid refer to Raschdorf et al.42. All plasmids were introduced into M. gryphiswaldense and R. rubrum by conjugation.
Plasmid pMT005 was constructed to delete the mamY gene. The mamY up- and downstream region were amplified (upstream: oMTN025-026, downstream: oMTN027-028, producing 700 and 900 bp fragments, respectively). Both regions were fused by overlap PCR (oMTN025-028) and the fused fragment was cloned into pORFM-galK between the SalI-NotI restriction sites. This vector was used to create the in-frame deletion of mamY by homologous recombination in the WT, ΔmamJ and ΔmamK genetic backgrounds generating the strains MT001, MT004 and MT015, respectively.
To construct pMT099, the mamY gene was amplified (oMTN310-312) and further cloned into the vector pJH2 under the control of a tetracycline-inducible promoter (Ptet) using the restriction sites NdeI-XbaI.
For creation of pMT102, the mamY gene was amplified (oMTN311-312) and NheI-XbaI digested and cloned in-frame downstream of the dendra2 gene into the identically digested pMT065 vector.
To generate pMT104, the genes mamC (oMTN328-345), RBP (anti-RFP nanobody – oMTN346-329), mamY-mCherry fusion (oMTN330-331) were amplified. Next, mamC and RBP were fused by overlap PCR (oMTN328-329). Subsequently, the mamC-RBP and mamY-mCherry PCR products were fused by overlap PCR (oMTN328-331) generating the mamC-RBP_mamY-mCherry, which was NdeI-XbaI digested and ligated into the identically digested pMT099 vector.
The plasmid pMT105 was generated by amplifying the mamK gene (oMTN040-350) and further NdeI-XbaI digested and cloned into identically digested pMT099 vector.
For construction of pMT106, the mamK (oFM564-565) and mamY (oFM566-567) genes were amplified and the PCR products were fused by overlap PCR (oFM564-567). The mamK_mamY fused fragment as well as the pMT099 vector were NdeI-XbaI digested and ligated.
The plasmid pFM251 (chromosomal mamC-mamYCD fusion) was derived from pFM23742. mCherry was cut out from pFM237 with KpnI and EcoRI and replaced by the 3` mamY fragment amplified with oFM435-436.
pFM252 (C-terminal markerless mamY-mCherry fusion) was constructed as follows: mamY was amplified with primers oFM439-440 and ligated into pORFM galK blu after digestion with SalI and SpeI. mCherry was amplified with oFM441-442 and the mamY downstream fragment was amplified with oFM443-444. Both fragments were fused by overlap PCR and ligated into the same vector after digestion with XmaI and SpeI.
To generate pFM253 (N-terminal markerless fusion of mCherry to mamY) the upstream fragment of mamY and mCherry were amplified with oFM446-447 producing a 1 kB and a 705 bp fragment respectively. Both fragments were fused by overlap PCR and cloned into pORFM galK blu after digestion with SalI and BamHI. The 5` region of mamY was amplified with oFM450-451, producing a 1,01 kB fragment which was cloned behind the fused upstream-mCherry fragment using NotI and SpeI restriction sites.
The pFM269a-g vectors are pAP160 based20 and were constructed to analyze localization of MamY full length and truncated proteins by EGFP tagging. For pFM269 (mamY full length) the gene was amplified with primers oFM484-487, for pFM269a with oFM484-482, for pFM269g with oFM484-483a, for pFM269c with oFM485-487 and for pFM269d with oFM486-487. pFM269h was used to exchange the TM domain of MamY by that of a histidine kinase (acc. no. CAM76561, Locus tag Mgr_1233). Therefore, the TM encoding section of mgr_1233 was amplified with primers oFM556-557 and the 3’ part of mamY was amplified with oFM558-487. Both fragments were fused by overlap extension PCR. All amplified fragments were cloned into pAP160 using NdeI and HindIII restriction sites. pFM269j served to analyze localization of M. magnet MamY-EGFP in M. gryph. To this end, mamY from M. magnet was amplified (oMTN324-325), NdeI-BamHI digested and cloned under the control of the Ptet promoter into the identically digested pFM269 backbone.
To generate pFM272, the upstream and 5’ region of mamY was amplified with oFM439-514 generating a truncated mamY version. The mamY-mCherry region from pFM252 was cut out with AvrII and exchanged by the PCR product generating a truncated allele for allelic replacement.
To construct pFM283, mgr_1233 was amplified with oFM560-561, digested with NdeI and cloned into pAP160.
Finally, to construct the vector pOR035, the PmamXY was amplified (oOR087-088) and cloned into pBBR1-MCS2 between the sites BamHI and HindIII. Subsequently, the mamY gene was amplified (oOR089-090) and inserted downstream the PmamXY promoter between NdeI and XhoI restriction sites. Then, the mCherry gene was amplified (oOR100-101) and cloned in-frame downstream the mamY gene between the sites NheI-NsiI.
Bacterial two-hybrid (BTH) plasmids
To clone mamY and its truncated versions in-frame with the adenylate cyclase domain of p25 and p18 expression vectors, the respective gene fragments were PCR amplified and inserted into PstI and EcoRI restriction sites of the p25 and p18 vectors. Primer pairs for the full-length versions were BTH_Yf1-BTH_Yr5, BTH_Yf1-BTH_Yr3, BTH_Yf5-BTH_Yr5 and BTH_Yf1-BTH_Yr3. For the MamYTM-CD(266)-version, primer pairs were BTH_Yf1-BTH_Yr6, BTH_Yf1-BTH_Yr4, BTH_Yf5-BTH_Yr6 and BTH_Yf1-BTH_Yr4 and for the MamYCD version, primer pairs were BTH_Yf9-BTH_Yr5, BTH_Y_Yf9-BTH_Yr3, BTH_Yf13-BTH_Yr5 and BTH_Yf9-BTH_Yr3.
Cell fractionation, SDS-gel electrophoresis and immunological protein detection
Cell lysis and cell fractionation with magnetosome purification of M. gryph were performed essentially as previously described 21,43. Briefly, after cell disruption, the lysate was subjected to centrifugation at 4000 g for 60 min to remove residual cell debris. The supernatant was applied on a magnetized separation column to retain magnetosomes and to isolate the magnetic fraction. To separate soluble and insoluble non-magnetic fractions, the flow-trough was subjected to 60 min of centrifugation at 160 000 g. The sedimented membrane fraction was resuspended (100 mM Tris-HCl, pH 8,0) and both fractions were centrifuged a second time at 160 000 g for 120 min. The resulting supernatant contained the non-magnetic soluble and the resuspended pellet the non-magnetic insoluble fraction. For SDS-PAGE, samples were supplemented with electrophoresis buffer, heated to 95°C for 10 min and applied to 11% (for mCherry-MamY) or 15% (for MamC) polyacrylamide gels. For Western blot, proteins were semi-dry electro-blotted on PVDF membranes. Immunological protein detection was performed using Anti-RFP (6G6αRed, ChromoTek GmbH, Martinsried, Germany) and Anti-MamC21 primary antibodies. HRP conjugated secondary antibodies and chemiluminescence substrate (Quant HRP substrate, Takara Biotech) were used to generate signals which were detected with a ChemiDoc™ XRS+ system (Biorad).
Western blotting and immunodetection of proteins
Fractionation of Mgryph cells, SDS-PAGE and western blotting were performed as described previously20. All samples were loaded on the same polyacrylamide gel and blotted on a PVDF membrane. Immunodetection was performed by chemiluminescence, using an anti-MamC44 and an anti-MCherry primary antibody simultaneously and a horseradish peroxidase conjugated anti-rabbit IgG secondary antibody. The luminescence signal was generated with Western Blot Quant substrate (Takara) and detected using a ChemiDoc™ XRS+ system with ImageLab software (Biorad). Densiometric analyses were performed with ImageLab software only on images without saturated pixels.
Bacterial Two-Hybrid (BTH) assays
Two hybrid analyses were carried out essentially as described22. E. coli BTH101 was transformed with complementary two-hybrid plasmids and grown at 37°C in LB broth containing 100 μg/ml ampicillin and 50 μg/ml kanamycin. Interaction analyses were performed according to manufacturer instructions (Euromedex). Briefly, cells were spotted onto M63 minimal medium agar plates (supplemented with: 0.2 % Maltose, 50 μg/ml ampicillin and 25 μg/ml kanamycin, 80 μg/ml X-Gal and 0.5 mM IPTG) and incubated at 30°C for 48 h. Since cya+ cells are mal+, they are able to grow on a minimal medium supplemented with maltose as a unique carbon sources. Furthermore, the lacZ gene encoding β-galactosidase is positively controlled by cAMP/CAP. Therefore, bacteria expressing interacting hybrid proteins will be detected by the presence of growth (mal+ colonies) and also by their blue coloring (lac+ colonies). The mamY gene variants (full length, C-terminally truncated and without TM domain (N-terminally truncated) were PCR-amplified and cloned into the expression vectors pKT25, pKNT25, pUT18 and pUT18C, respectively, and sequenced. T25 and T18 vectors with and without (as control) mamY variants in all combinations were co-transformed into E. coli strain BTH101 (Euromedex). Transformants were transferred on LB medium supplemented with ampicillin, kanamycin and X-Gal and incubated at 30°C for 24 hours prior to screen for blue colonies.
Protein purification
For the overexpression of the MamY cytoplasmic domain the vector pET28-MamYCD, and the E. coli BL21 (DE3) pLysS strain were used. Plasmid DNA was transformed followed by overnight incubation; cells were inoculated in a pre-culture and grown to an optical density (OD600nm) of approximately 1. Next, a 0.5L culture was inoculated to an OD600nm of 0.05 and incubated at 37ºC with shaking at 200 rpm. Once an OD600nm of 0.5 was reached, the culture was induced by addition of 500 μM IPTG and grown for 4h at 37°C with shaking at 200 rpm. After incubation, cells were harvested by centrifugation at 4,000 rpm for 20 min at 4ºC. The supernatant was removed and the cell pellet was flash frozen in liquid nitrogen and stored at –80ºC.
Frozen pellets containing the protein of interest were thawed on ice and resuspended in (5 times the volume of the cell pellet) binding buffer (50 mM sodium phosphate, 150 mM NaCl, 10 mM imidazole, pH 7.4) supplemented with DNase I and protease inhibitor. The resuspended cultures were lysed in a cell disruptor (5 passages) and centrifuged at 4,000 rpm for 20 min to remove cell debris. The supernatant was then spun in an ultracentrifuge at 45,000 rpm for 45 min to remove membranes. The cleared cell lysate was applied to an Äkta FPLC Ni-NTA agarose column (1 mL Protino-Ni-NTA column, Macherey and Nagel, Germany). Elution was done in elution buffer (50 mM sodium phosphate, 150 mM NaCl, 500 mM imidazole, pH 7.4) and fractionated in volumes of 1 ml. Following elution, samples from each fraction were examined by SDS-PAGE.
Dynamic Light Scattering
DLS was used to study polymerization of the purified MamY cytoplasmic domain. All buffers and protein samples were passed through a 0.02 μm pore size filter to exclude any aggregates or other debris that might interfere with the measurements. 20 μL of protein (0.3 to 0.5 μg/ml equivalent to ~10 to 15 μM, respectively) was added to a 100 μL-volume cuvette. The sample was measured using the DynaPro NanoStar (Wyatt Technology) with a 634 nm laser. The system was set to 25ºC and the sample was measured for a period of 30 min before or after changing the pH conditions. Each measurement corresponds to 10 acquisitions (every 10s).
For increasing the pH from 7.4 to 8.0 a 1.5 μL of NaOH 50 mM was added. Subsequently, 3 μL of HCl 1% were used to decrease the pH to 6.0. In a second experiment, to increase the pH from 7.4 to 7.6, 8 μL of Buffer A (50 mM sodium phosphate, 150 mM NaCl, pH 8.0) were added. Next, 3 μL of Buffer B (Buffer A at pH 6.0) were used to lower the pH from 7.6 to 7.4. Again, to increase the pH from 7.4 to 7.6, 12.5 μL of Buffer A were added.
To study protein polymerization in the absence of phosphate, the protein was dialyzed overnight in 500-fold the sample volume using HEPES buffer (10 mM HEPES, 150 mM NaCl, pH 7.4). To increase the pH of the HEPES buffer from 7.4 to 8.0, 1.75 μL of 50 mM NaOH were used. Subsequently, 1 μL of 0.5% HCl was added to decrease the pH from 8.0 to 6.0.
The estimation of the molecular weight was carried out in the DYNAMICS software (version 7.1.9) and based on a model for globular proteins (Mw-R Model) and spherical molecules (Rg Model). Experiments were made with three independent freshly purified protein samples and performed under presence or absence (protein dialyzed in 500-fold the sample volume using Buffer A) of imidazole, yielding similar and reproducible results.
Fluorescence microscopy
We used either (i) an Olympus BX81 microscope equipped with a 100x/1.40 Oil UPLSAPO100XO objective and an Orca-ER camera (Hamamatsu) or (ii) a Delta Vision Elite (GE Healthcare, Applied Precision) Olympus IX71 microscope with a CoolSnap HQ2 CCD camera (Photometrics), a 100x/1.40 Oil PSF objective (U-PLAN S-APO 100x Oil, 0.12 WD) and equipped with a four color standard set Insight SSITM illumination module. Mgryph cells were spotted onto a 1% “Mgryph agarose pad” as per Toro-Nahuelpan et al (2016), whereas R. rubrum was spotted onto a 1% PBS (NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM, KH2PO4 1.8 mM, pH 7.3) agarose pad. Imaging was performed at room temperature (25ºC, in the Olympus BX81 microscope) or at 30ºC (Delta Vision Elite microscope).
When stated, deconvolution was carried out from optical sections using the softWoRx software (version 6.1.1) and the Ratio (conservative) method (GE Healthcare, Applied Precision) or Deconvolution of Z-stacks taken with the Olympus BX81 microscope was done with the deconvolution plug in of the CellM software package (Olympus) using “no neighbor” filtering and appropriate channel settings such as emission wavelength and refractive index used.
3D Structured Illumination Microscopy
3D-SIM was performed on a DeltaVision OMX V4 Blaze fluorescence microscope (GE Healthcare) using a 60x/1.42 Oil PlanApo N UIS2 objective lens (Olympus), giving 80 nm pixel spacing in raw images with striped illumination at 3 angles and 5 phases. High precision coverslips of 170 μm thickness, and immersion oil with a refractive index of either 1.514 or 1.516 at 593 nm were used to eliminate sample induced spherical aberration, matching the measured PSF of the calibrated system, for high quality image reconstructions at around 120 nm lateral (x,y) spatial resolution with 40 nm reconstructed image pixel spacing. Fast piezo stage Z-series images were taken at 1.5-3.0 μm total thickness with 0.125 μm Z-step spacing with raw frame exposure times of 100 ms, avoiding detector saturation of the 15 bit 1.6 electron read noise pco.Edge sCMOS camera (PCO), using the mCherry/Alexa Fluor 658 fluorescence emission filter (581-636 nm) and fluorescence excitation with a 568 nm laser. Cells expressing mCherry-mamY from the mamY locus were spotted onto a 1% Mgryph agarose pad as per Toro-Nahuelpan et al (2016). 3D-SIM image reconstruction was performed in SoftWoRx (version 6.1.1, GE Healthcare) according to the method of Gustafsson and colleagues45.
Photo-Activated Localization Microscopy
Sample Preparation
Matrix preparation: A 1.5% low melting point agarose solution (Biozym), prepared in 0.22 μm-pore filtered Milli-Q water, was incubated at 80ºC for 1.5 h. Gas-tight Gene FrameΔ (1.5 x 1.6 cm, 65 μL volume, Thermo Fisher) was filled with the agarose solution and covered with 18 x 18 mm high precision coverslips of 170 ± 0.005 μm thickness (Zeiss). Glass slides and coverslips used for these experiments were plasma-cleaned for 5 min and kept sealed in a plasma-cleaned box until use. Agarose pads were freshly prepared, incubated 20 min at 4ºC and 30 min at room temperature before use.
Cell preparation: Mgryph cells were grown and induced in darkness. The expression of the gene fusion dendra2-mamY under control of the tetracycline promoter (Ptet) was induced by addition of anhydrotetracycline to 50 ng mL–1 for 3 to 5 hours, which proved sufficient to produce fusion protein localizing as a line in Mgryph cells. Then, 10 mL cell culture was fixed in 1% of formaldehyde (0.22 μm-pore filtered) for 30 min at room temperature. Next, cells were spun down at 3,000 x g for 10 min and the cell pellet was gently resuspended in 10 mL of HEPES buffer (10 mM, pH 7) and 100 mM glycine (0.22 μm-pore filtered), incubated for 10 min and spun at 3,000 x g for 10 min. Then, the cell pellet was washed with 10 mL of HEPES (10 mM, pH 7) and again spun at 3,000 x g for 10 min. Finally, cells were resuspended in 300 μL of HEPES (10 mM, pH 7).
Mounting: 2 μL of concentrated cells were mixed with 6 μL of a 1:1,000 dilution of TetraSpeck beads (Thermo Fisher) used as fiducial markers for further alignment purposes. From the mixture, 2 μL were spotted onto the agarose pad, incubated for 2 min to evaporate the excess of water and subsequently sealed with a coverslip. Using this ratio, an optimal concentration was reached, yielding three to five beads and sufficient cells per field of view. Moreover, the matrix preparation generated cells and beads found close to the coverslip at similar z-distance, ideal for imaging.
Imaging conditions
A Zeiss ElyraP1 microscope was used for PALM imaging with an Alpha Plan-Apochromat 100x/1.46 oil objective DIC M27 and equipped with an Andor iXon 897 EM-CCD camera. The Dendra2 fluorophore was imaged in epifluorescence illumination mode in a sample of approximately 500 nm thickness. A 405 nm (50 mW) laser was used for the photo-conversion of the fluorophore. The 405 nm laser power started at zero and was gradually increased until most fluorophores were detected (the power was manually changed during the experiments in order to keep the photo-conversion frequency as stable as possible). A 561 nm laser line (15% power of a 200 mW laser line) combined with a Long Pass 570 nm (LP570) emission filter and an exposure time of 50 ms were used to detect and bleach the blinking fluorophores. The EMCCD camera gain was set to 200. Prior to each imaging, the sample was exposed to the 561 nm laser to bleach pre-activated molecules and the fluorescent beads, the latter in order to avoid pixel saturation of the EMCCD camera, which impairs the Gaussian fitting to localize the marker. The pre-bleaching time was conditioned to the fluorescent bead brightness and was approximately in the order of one minute. Throughout the imaging, the blinking events per cell were commonly one per frame and kept below three.
We acquired 15,000 frames for imaging of Dendra2-MamY in the WT and ΔmamK strains and 100,000 frames for imaging of Dendra2 fused to: MamY (in ΔmamY cells), MamK (in WT cells), MamYCD and TMHK-MamYCD (in the ΔmamKY strain). Concerning PALM of Dendra2-MamY in the ΔmamY strain, since a maximum of ~17,500 single events per cell were detected, it can be assumed that virtually all events were imaged. Under that premise, the total single event counts can be directly correlated to the amount of protein per cell.
Data post-processing and quality check:
PALM images were calculated and lateral frame drift was corrected by alignment of the fluorescent beads as reference points using the Zeiss ZenBlack software. Data coordinates were exported and further analyzed in R-studio. Subsequently, a stringent data filtering was applied in order to eliminate multiple events and to isolate only resolved single-molecule events. Sorting by PSF width at half-maximum intensity (80 to 160 nm) and photon number (100 to 500)46–48, multiple events that cannot be resolved and molecules that do not correspond to the Dendra2 photon emission range, respectively, were filtered out (Supplementary Fig. 12a-b). Moreover, to avoid unresolvable multiple events, imaging of one molecule per cell per frame was aimed, maintaining below thee events per cell per frame (Supplementary Fig. 12c). After application of the filtering parameters, a localization precision area average of ~25-30 nm (ranging from 5 to 50 nm) per imaged cell was obtained. Importantly, the linear structure remained unchanged and consistent (Supplementary Fig. 12d-g). In addition, the background from the agarose matrix (0.3 events/μm2) and Mgryph WT cells (82 events/cell, distributed throughout the cell (n= 5), Supplementary Fig. 8b) were negligible and did not contribute to the linear structure.
Local density is defined as events present in a pixel of 50 nm side, centered on each event. The density of events is rendered in color-coded values and illustrates the number of events per defined area (pixel) in order to represent single-molecule event enriched areas in an unbiased manner. The nearest neighbor distances distribution function (Gest function, R documentation) was edge-corrected using the border method estimator, the spatial Kaplan-Meier estimator49 as well as the Hanisch estimator50 in R programming language. Due to the high amount of experimental data, the edge correction using these three methods resulted in no differences with the experimental data. Density and centroid maps were generated using R51.
Clustering algorithm and image rendering
R programming language52 and RStudio53 were used to create a molecule clustering script. First, data was loaded into R-studio and filtered by photon number and PSF as above-mentioned. Next, each single event was tested for putative interaction with its neighboring events by a local search and taking into consideration their localization precision area. To this aim, interactions between the event and its neighbors were tested only within a square area (lateral side: 200 nm) centered on the tested event. Two molecules were clustered together only if the distance between the two localizations was smaller than the sum of the precision of the two molecules divided by two, i.e., only molecules whose localization precision areas were in direct contact or overlapped (≤ 0 nm) were clustered.
Clusters were further classified by size, thus, single-molecules and clusters smaller than 5 molecules were colored as light blue (R color: lightskyblue1), clusters between 5 to 50 molecules were blue (R color: skyblue2), and cluster with more than 50 molecules as dark blue (R color: royalblue4).
The centroid is the geometric center or arithmetic mean position of each fluorophore, which is taken from the contour map of intensities obtained from its PSF. Finally, for plots displaying the clustering results were generated using plotrix package54, the localization precision area of each molecule was rendered and colored based on the size of the cluster they belonged to.
A computed fluorescent image (density rendering) was generated by rendering of the PSF FWHM in the Zeiss ZenBlack software. The PSF FWHM multiplicator was 0.71 and the dynamic range a 100%.
Scanning electron microscopy
Cells were harvested and resuspended in 10 mM HEPES buffer (pH 7,2). Fixation was carried out by addition of fresh glutaraldehyde to 4% and incubation at 4°C for 4 h. Fixed cells were mounted onto poly-L-lysine coated coverslips and dehydrated through an ascending series of ethanol baths (35%, 50%, 70%, 80%, 95%, 100% and 100% for 10 min each) followed by critical point drying. Samples were mounted onto stubs, sputter coated with platinum and examined by using a Zeiss Ultra plus FE-SEM (acceleration voltage: 3 kV; aperture size: 30 μm, working distance: 6.5 mm).
Transmission electron microscopy
For conventional TEM analysis, cells were grown at 28°C under microaerobic conditions, fixed in formaldehyde (1%) and ten-times concentrated. Next, cells were adsorbed on carbon coated copper mesh grids (Plano, Wetzlar). Bright field TEM was performed on a FEI CM200 (FEI; Eindhoven, The Netherlands) transmission electron microscope using an accelerating voltage of 160 kV. Images were captured with an Eagle 4k CCD camera using EMMenu 4.0 (Tietz). Alternatively, a FEI Tecnai F20 operated at 200 kV and an Eagle 4k CCD camera was used, and micrograph acquisition was performed using SerialEM software55. For data analysis, the Fiji software was used.
Plunge-freezing vitrification for cryo-electron tomography
5 μL of Mgryph culture were mixed with 2 μL of (two-fold concentrated) BSA-coated 15 nm colloidal gold clusters (Sigma) used for subsequent alignment purposes. The mixture was added on glow-discharged Quantifoil R 2/1 holey carbon molybdenum grids (Quantifoil Micro Tools GmbH, Jena), manually blotted for 4 s and embedded in vitreous ice by plunge freezing into liquid ethane (< –170°C). The grids were stored in sealed boxes in liquid nitrogen until used.
Cryo-electron tomography
Tomography was performed under low-dose conditions using a FEI Tecnai F30 G2 Polara equipped with a 300 kV field emission gun, and a Gatan GIF 2002 post-column energy filter. A 3838 x 3710 Gatan K2 Summit Direct Detection Camera operated in counting and dose-fractionation mode was used for imaging. Data collection was performed at 300 kV, with the energy filter operated in the zero-loss mode (slit width of 20 eV). Tilt series were acquired using Serial EM software55. The specimen was tilted about one axis with 1.5° increments over a typical total angular range of ± 60°. The cumulative electron dose during the tilt series was kept below 150 e- Å-2. To account for the increased specimen thickness at high tilt angles, the exposure time was multiplied by a factor of 1/cos α. Pixel size at the specimen level was 5.22 Å at an EFTEM magnification of 22,500x. Images were recorded at nominal –5 μm defocus.
Tomogram reconstruction and segmentation
Tomograms were reconstructed in the IMOD package (Kremer et al, 1996). Tomographic reconstructions from tilt series were performed with the weighted back-projection with IMOD software using particles as a fiducial marker. Aligned images were binned to the final pixel size of 31.32 Å. For tomographic reconstruction, the radial filter options were cut off: 0.5 and fall off: 0.05. The dataset for this study consisted of 7 tomograms for the WT strain and 9 tomograms for ΔmamY strain. Tomograms were treated with an anisotropic nonlinear diffusion denoising algorithm to improve signal-to-noise ratio.
Segmentation of the tomogram was done with Amira software on binned volumes with a voxel size of 31.32 Å. Membrane segmentation was done using the software TomoSegMemTV and a complementary package, SynapSegTools, both for Matlab56. Tomogram slices were obtained using 3dmod software from the IMOD package.
Supplementary Material
Acknowledgments
We are grateful to Günter Pfeifer (MPI of Biochemistry) for constant support with TEM and Cryo-ET, also to Marin Turk for help with TEM. We thank Brigitte Melzer, Isabelle Mai and Marius Klein for technical assistance, and we acknowledge Theresa Zwiener for providing the Mgryph ΔMAI strain. We are thankful to Jan Peychl and Bert Nitzsche at the MPI-CBG, light microscopy facility, Dresden, Germany, for access to a GE Deltavision OMX v4 Blaze microscope and to Daniel J. White of GE Healthcare - Cell Analysis, for applications support, technical specifications for publication and assistance with microscopy on the OMX. We also thank Raz Zarivach, Ben-Gurion University of the Negev Beer Sheva, Israel for helpful discussions. We are also grateful to Martina Heider, Bayreuth Institute for Macromolecule Research for technical assistance with scanning electron microscopy and we thank ChromoTek GmbH, Planegg-Martinsried, Germany for providing the mCherry-binding (RBP-) nanobody. Finally, we are deeply thankful to Nadine Albrecht for protein purification and help with DLS analysis. This work was supported by the Deutsche Forschungsgemeinschaft Grants Schu1080/9-2 (DS), INST 86/1452 (MB), INST 160/646-1, TRR174 project 5 (MB) and has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No 692637 (DS)).
Abbreviations
To ease reading, the following abbreviations are used throughout the manuscript:
- 3D-SIM
three-dimensional structured illumination microscopy
- Mmagnet
Magnetospirillum magneticum AMB-1
- Cmag
cellular magnetic response
- Cryo-ET
cryo-electron tomography
- DLS
dynamic light scattering
- FWHM
full-with half-maximum
- MC(s)
magnetosome chain(s)
- MBN
mCherry-binding nanobody
- Mgryph
Magnetospirillum gryphiswaldense MSR 1
- PALM
photo-activated localization microscopy
- PSF
point spread function
- TEM
transmission electron microscopy
Footnotes
Data and code availability
The computer code used to analyze the PALM data is publicly available on https://github.com/GiacomoGiacomelli/mamy-cluster-analysis. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
MT-N, DS and FDM conceived and designed research. MT-N, GG, OR, SB, and FDM performed experiments. MT-N and FDM performed SIM imaging. MT-N and FDM performed TEM and fluorescence microscopy. GG and MT-N established and carried out PALM. GG created the single-molecule clustering algorithm and generated the PALM images. GG, MT-N and MB analyzed PALM data. MT-N performed cryo-ET; MT-N and JMP analyzed the data. FDM performed scanning electron microscopy with help of Martina Heider. MT-N and MB analyzed DLS data. MT-N and FDM analyzed the whole dataset and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Uebe R, Schüler D. Magnetosome biogenesis in magnetotactic bacteria. Nat Rev Microbiol. 2016;14:621–637. doi: 10.1038/nrmicro.2016.99. [DOI] [PubMed] [Google Scholar]
- 2.Nordmann GC, Hochstoeger T, Keays DA. Magnetoreception-A sense without a receptor. PLoS Biol. 2017;15:e2003234. doi: 10.1371/journal.pbio.2003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popp F, Armitage JP, Schüler D. Polarity of bacterial magnetotaxis is controlled by aerotaxis through a common sensory pathway. Nat Commun. 2014;5 doi: 10.1038/ncomms6398. 5398. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi A, et al. Experimental observation of magnetosome chain collapse in magnetotactic bacteria: Sedimentological, paleomagnetic, and evolutionary implications. Earth Planet Sci Lett. 2006;245:538–550. [Google Scholar]
- 5.Frankel R, Blakemore R. Navigational compass in magnetic bacteria. J Magnetism Magnet Mat. 1980;15–18:1562–1564. [Google Scholar]
- 6.Zahn C, et al. Measurement of the magnetic moment of single Magnetospirillum gryphiswaldense cells by magnetic tweezers. Sci Rep. 2017;7 doi: 10.1038/s41598-017-03756-z. 3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toro-Nahuelpan M, et al. Segregation of prokaryotic magnetosomes organelles is driven by treadmilling of a dynamic actin-like MamK filament. BMC Biol. 2016;14:88. doi: 10.1186/s12915-016-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katzmann E, et al. Magnetosome chains are recruited to cellular division sites and split by asymmetric septation. Mol Microbiol. 2011;82:1316–1329. doi: 10.1111/j.1365-2958.2011.07874.x. [DOI] [PubMed] [Google Scholar]
- 9.Scheffel A, et al. An acidic protein aligns magnetosomes along a filamentous structure in magnetotactic bacteria. Nature. 2006;440:110–114. doi: 10.1038/nature04382. [DOI] [PubMed] [Google Scholar]
- 10.Katzmann E, Scheffel A, Gruska M, Plitzko JM, Schüler D. Loss of the actin-like protein MamK has pleiotropic effects on magnetosome formation and chain assembly in Magnetospirillum gryphiswaldense. Mol Microbiol. 2010;77:208–224. doi: 10.1111/j.1365-2958.2010.07202.x. [DOI] [PubMed] [Google Scholar]
- 11.Orue I, et al. Configuration of the magnetosome chain: a natural magnetic nanoarchitecture. Nanoscale. 2018;10:7407–7419. doi: 10.1039/C7NR08493E. [DOI] [PubMed] [Google Scholar]
- 12.Ullrich S, Kube M, Schübbe S, Reinhardt R, Schüler D. A hypervariable 130-kilobase genomic region of Magnetospirillum gryphiswaldense comprises a magnetosome island which undergoes frequent rearrangements during stationary growth. Journal of Bacteriology. 2005;187:7176–7184. doi: 10.1128/JB.187.21.7176-7184.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raschdorf O, Müller FD, Posfai M, Plitzko JM, Schüler D. The magnetosome proteins MamX, MamZ and MamH are involved in redox control of magnetite biomineralization in Magnetospirillum gryphiswaldense. Mol Microbiol. 2013;89:872–886. doi: 10.1111/mmi.12317. [DOI] [PubMed] [Google Scholar]
- 14.Ding Y, et al. Deletion of the ftsZ-like gene results in the production of superparamagnetic magnetite magnetosomes in Magnetospirillum gryphiswaldense. Journal of Bacteriology. 2010;192:1097–1105. doi: 10.1128/JB.01292-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siponen MI, et al. Structural insight into magnetochrome-mediated magnetite biomineralization. Nature. 2013;502:681–684. doi: 10.1038/nature12573. [DOI] [PubMed] [Google Scholar]
- 16.Müller FD, et al. The FtsZ-like protein FtsZm of Magnetospirillum gryphiswaldense likely interacts with its generic homolog and is required for biomineralization under nitrate deprivation. J Bacteriol. 2014;196:650–659. doi: 10.1128/JB.00804-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tocheva EI, Li Z, Jensen GJ. Cold Spring Harb Perspect Biol. In: Shapiro L, Losick R, editors. Cell Biology of Bacteria. Vol. 2. 2010. pp. 1–20. [Google Scholar]
- 18.Kürner J, Frangakis AS, Baumeister W. Cryo-electron tomography reveals the cytoskeletal structure of Spiroplasma melliferum. Science. 2005;307:436–438. doi: 10.1126/science.1104031. [DOI] [PubMed] [Google Scholar]
- 19.Schüler D, Uhl R, Baeuerlein E. A simple light scattering method to assay magnetism in Magnetospirillum gryphiswaldense. FEMS Microbiol Lett. 1995;132:139–145. [Google Scholar]
- 20.Borg S, Hofmann J, Pollithy A, Lang C, Schüler D. New vectors for chromosomal integration enable high-level constitutive or inducible magnetosome expression of fusion proteins in Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2014;80:2609–2616. doi: 10.1128/AEM.00192-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grünberg K, et al. Biochemical and proteomic analysis of the magnetosome membrane in Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2004;70:1040–1050. doi: 10.1128/AEM.70.2.1040-1050.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothbauer U, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3:887–889. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- 24.Borg S, et al. An intracellular nanotrap redirects proteins and organelles in live bacteria. MBio. 2015;6 doi: 10.1128/mBio.02117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollithy A, et al. Magnetosome expression of functional camelid antibody fragments (Nanobodies) in Magnetospirillum gryphiswaldense. Applied and Environmental Microbiology. 2011;77:6165–6171. doi: 10.1128/AEM.05282-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Komeili A, Li Z, Newman D, Jensen G. Magnetosomes are cell membrane invaginations organized by the actin-like protein MamK. Science. 2006;311:242–245. doi: 10.1126/science.1123231. [DOI] [PubMed] [Google Scholar]
- 27.Scheffel A, Schüler D. The acidic repetitive domain of the Magnetospirillum gryphiswaldense MamJ protein displays hypervariability but is not required for magnetosome chain assembly. J Bacteriol. 2007;189:6437–6446. doi: 10.1128/JB.00421-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagstaff J, Löwe J. Prokaryotic cytoskeletons: protein filaments organizing small cells. Nature Reviews Microbiology. 2018;16:187–201. doi: 10.1038/nrmicro.2017.153. [DOI] [PubMed] [Google Scholar]
- 29.Oliva MA, et al. Features critical for membrane binding revealed by DivIVA crystal structure. EMBO J. 2010;29:1988–2001. doi: 10.1038/emboj.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowman GR, et al. A polymeric protein anchors the chromosomal origin/ParB complex at a bacterial cell pole. Cell. 2008;134:945–955. doi: 10.1016/j.cell.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebersbach G, Briegel A, Jensen GJ, Jacobs-Wagner C. A self-associating protein critical for chromosome attachment, division, and polar organization in caulobacter. Cell. 2008;134:956–968. doi: 10.1016/j.cell.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kühn J, et al. Bactofilins, a ubiquitous class of cytoskeletal proteins mediating polar localization of a cell wall synthase in Caulobacter crescentus. EMBO J. 2010;29:327–339. doi: 10.1038/emboj.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gittes F, Mickey B, Nettleton J, Howard J. Flexural rigidity of microtubules and actin filaments measured from thermal fluctuations in shape. J Cell Biol. 1993;120:923–934. doi: 10.1083/jcb.120.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci U S A. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janowski R, Abrahamson M, Grubb A, Jaskolski M. Domain swapping in N-truncated human cystatin C. J Mol Biol. 2004;341:151–160. doi: 10.1016/j.jmb.2004.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Yang F, et al. Crystal structure of cyanovirin-N, a potent HIV-inactivating protein, shows unexpected domain swapping. J Mol Biol. 1999;288:403–412. doi: 10.1006/jmbi.1999.2693. [DOI] [PubMed] [Google Scholar]
- 37.Chinthalapudi K, Rangarajan ES, Brown DT, Izard T. Differential lipid binding of vinculin isoforms promotes quasi-equivalent dimerization. Proc Natl Acad Sci U S A. 2016;113:9539–9544. doi: 10.1073/pnas.1600702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prutzman KC, et al. The focal adhesion targeting domain of focal adhesion kinase contains a hinge region that modulates tyrosine 926 phosphorylation. Structure. 2004;12:881–891. doi: 10.1016/j.str.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 39.Heyen U, Schüler D. Growth and magnetosome formation by microaerophilic Magnetospirillum strains in an oxygen-controlled fermentor. Appl Microbiol Biotechnol. 2003;61:536–544. doi: 10.1007/s00253-002-1219-x. [DOI] [PubMed] [Google Scholar]
- 40.Falk G, Johansson B. Complementation of a nitrogenase Fe-protein mutant of Rhodospirillum rubrum with the nif-plasmid pRD1 containing nif genes of Klebsiella pneumoniae. FEMS Microbiology Letters. 1983;19:145–149. doi: 10.1111/j.1574-6968.1983.tb00530.x. [DOI] [Google Scholar]
- 41.Bertani G. Studies on lysogenesis I: the mode of phage liberation by lysogenic Escherichia coli. J Bacteriol. 1951;62:293–300. doi: 10.1128/jb.62.3.293-300.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raschdorf O, Plitzko JM, Schüler D, Müller FD. A tailored galK counterselection system for efficient markerless gene deletion and chromosomal tagging in Magnetospirillum gryphiswaldense. Appl Environ Microbiol. 2014;80:4323–4330. doi: 10.1128/AEM.00588-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uebe R. Mechanism and regulation of magnetosomal iron uptake and biomineralisation in Magnetospirillum gryphiswaldense. Dr. rer. nat thesis, Ludwig-Maximilians-Universität; 2011. [Google Scholar]
- 44.Lang C, Schüler D. Expression of green fluorescent protein fused to magnetosome proteins in microaerophilic magnetotactic bacteria. Applied and Environmental Microbiology. 2008;74:4944–4953. doi: 10.1128/AEM.00231-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustafsson MG, et al. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Moffitt JR, Dempsey GT, Xie XS, Zhuang X. Characterization and development of photoactivatable fluorescent proteins for single-molecule-based superresolution imaging. Proc Natl Acad Sci U S A. 2014;111:8452–8457. doi: 10.1073/pnas.1406593111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bach JN, Giacomelli G, Bramkamp M. Sample Preparation and Choice of Fluorophores for Single and Dual Color Photo-Activated Localization Microscopy (PALM) with Bacterial Cells. Methods Mol Biol. 2017;1563:129–141. doi: 10.1007/978-1-4939-6810-7_9. [DOI] [PubMed] [Google Scholar]
- 49.Baddeley AJ, Gill RD. Kaplan-Meier estimators of interpoint distance distributions for spatial point processes. Annals of Statistics. 1997;25:263–292. [Google Scholar]
- 50.Hanisch K-H. Some remarks on estimators of the distribution function of nearest-neighbour distance in stationary spatial point patterns. Mathematische Operationsforschung und Statistik, series Statistics. 1984;15:409–412. [Google Scholar]
- 51.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2009. [Google Scholar]
- 52.R Development Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna, Austria: 2014. http://www.R-project.org/ [Google Scholar]
- 53.RStudio Team. RStudio: Integrated Development for R. RStudio, Inc; Boston, MA: 2015. http://www.rstudio.com/ [Google Scholar]
- 54.Lemon J. Plotrix: a package in the red light district of R. R-News. 2006;6:8–12. [Google Scholar]
- 55.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 56.Martinez-Sanchez A, Garcia I, Asano S, Lucic V, Fernandez JJ. Robust membrane detection based on tensor voting for electron tomography. J Struct Biol. 2014;186:49–61. doi: 10.1016/j.jsb.2014.02.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.