

Abstract
Objective.
Systemic lupus erythematosus (SLE) is characterized by antibody production against self-antigens. However, the events underlying autoantibody formation in SLE remains unclear. This study investigated the role of plasma autoantibody levels, microbial translocation, and the microbiome in SLE.
Methods.
Plasma samples from two cohorts, one with 18 unrelated healthy controls (UHCs) and 18 first-degree relatives (FDRs) and the other with 19 healthy controls and 21 SLE patients were assessed for autoantibody levels by autoantigen microarrays, lipopolysaccharide (LPS) levels by limulus amebocyte assay and microbiome composition by microbial 16S rDNA sequencing.
Results.
FDRs and SLE patients exhibited increased plasma autoantibodies compared to their control groups. Parents and children of lupus patients exhibited elevated plasma LPS levels in comparison to controls (p = 0.02). Plasma LPS levels positively correlated with plasma anti-dsDNA IgG levels in FDRs (r=0.51, p=0.03) but not in SLE patients. Circulating microbiome analysis revealed that FDRs (Observed species, p=0.004; Chao1 index, p=0.005) but not patients had significantly reduced microbiome diversity compared to their controls. The majority of differentially abundant bacteria identified between UHCs and FDRs were in the Firmicutes phylum, while bacteria from several different phyla were identified between HCs and SLE patients. Bacteria in the Paenibacillus genus was the only overlapping differentially abundant bacteria in both cohorts, and it was reduced in FDRs (p.adj = 2.13 x 10-12) and SLE patients (p.adj = 0.008) but elevated in controls.
Conclusions.
These results indicate a possible role for plasma microbial translocation and microbiome composition in influencing autoantibody development in SLE.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease characterized by loss of tolerance to self-antigens and autoantibody production [1]. Genetic factors are linked to SLE development with certain genes correlating to an increased risk of SLE [2, 3], and a tendency for clustering of SLE within families [4, 5]. First-degree relatives (FDRs) of SLE patients are thirteen times more likely to develop SLE compared to the general population with 5–10% of SLE patients having a second family member with SLE [6].
Several years prior to the manifestation of SLE clinical symptoms and diagnosis, antinuclear antibodies (ANAs) and other autoantibodies can be detected [7]. FDRs of SLE patients have a higher prevalence of autoantibodies compared to the general population [5, 8–10]. The observed increase of autoantibodies prior to SLE onset suggests that insights into SLE pathogenesis can be gained from studies of FDRs, especially in the absence of immunosuppressive therapy, along with studies on SLE patients.
The exact etiology of autoantibody production in SLE remains unknown, but a combination of factors is suggested as playing a role in disease pathogenesis including genetic, environmental, immunologic, and hormonal factors. Recently, there is increasing evidence to support the concept of increased intestinal permeability contributing to the pathogenesis of autoimmune diseases such as SLE [11]. Under normal conditions, the intestinal epithelial lining and factors secreted from it create a barrier separating the host from environmental antigens [11]. However, in various disease states, this barrier may be compromised leading to the translocation of microbial products into the systemic circulation, which may induce chronic inflammation and systemic tissue damage [11, 12]. In SLE, recent work shows evidence of translocating gut microbes including Enterococcus gallinarum and Lactobacillus reuteri in influencing disease pathogenesis in genetic backgrounds predisposed to autoimmunity [13, 14]. However, the role of microbial translocation in SLE and its correlation with autoantibody levels is unclear.
In this study, we show a direct correlation between levels of microbial translocation and plasma autoantibodies in FDRs but not in patients. We also identify differences in the circulating microbiome composition of SLE patients and their first-degree relatives compared to healthy controls. These results indicate that plasma microbial translocation may play a role in autoantibody development and immune suppressive therapy may affect this association.
PATIENTS AND METHODS
Study participants
We examined two cohorts in this study (Supplementary Tables S1 and S2). The first cohort consisted of 18 unrelated healthy controls (UHCs) and 18 first-degree relatives (FDRs) of SLE patients. First-degree relatives were either a sibling, parent, or child of a SLE patient, and the UHCs were often a friend brought by an SLE patient or FDR. All 36 participants in the first cohort were African American males or females. The second cohort consisted of 19 healthy controls (HCs) and 21 SLE patients. All 40 participants in the second cohort were female with 20 Caucasian (12 HCs, 8 SLE patients) and 20 African-American (7 HCs, 13 SLE patients) participants. The inclusion criteria for enrolled subjects was age eighteen or older and able to provide informed consent. The exclusion criteria included pregnancy or breastfeeding, recent severe illness, contraindications for blood draws and use of antibiotics within the past ninety days prior to participation in the study. The Institutional Review Board of the Medical University of South Carolina approved this study.
Autoantigen array
Plasma autoantibody reactivities were analyzed on a slide-based protein array containing a panel of 122 autoantigens and 6 controls in the Genomics and Microarray Core facility at the University of Texas Southwestern Medical Center in Dallas, TX [15]. Plasma was diluted 1:50 and added to each array in duplicate. The levels of IgG autoantibodies were analyzed by Cy3-labelled anti-human IgG. Mean fluorescence intensities (MFI) represent the signal intensity of each autoantibody [15]. Heat maps were generated using Cluster and Treeview software [15].
Plasma LPS level
Plasma samples were diluted to 10% with endotoxin-free water and heated to 80°C for 10 minutes to inactivate inhibitory plasma proteins. Lipopolysaccharide (LPS) level in plasma was then quantified using the limulus amebocyte lysate QCL-1000 kit (Lonza, Walkersville, USA) as described in our previous studies [16].
DNA extraction
Bacterial DNA was extracted from 400 μl of plasma or endotoxin-free water controls using the QIAamp UCP Pathogen Mini Kit according to the manufacturer’s instructions (Qiagen, Valencia, CA). The V4 variable region of bacterial 16S rDNA gene was amplified using PCR primers 515/806 in HotStarTaq Plus Master Mix (Qiagen) under the following conditions: 94°C for 3 min, followed by 28 cycles of 94°C for 30 sec, 53°C for 40 sec, and 72°C for 1 min, and a final elongation step at 72°C for 5 min. Sequencing was performed at MR DNA on a 454 Roche FLX Titanium pyrosequencing system following the manufacturer’s guidelines (MR DNA, Shallowater, TX, USA).
Sequence processing and taxonomic assignment
Using the Quantitative Insights Into Microbial Ecology (QIIME) 1 pipeline [17], sequences were demultiplexed and poor-quality sequences removed using the default settings of the QIIME 1 script split_libraries.py (minimum average quality score=25, minimum/maximum sequence length=200/1000 base pairs, no ambiguous base calls and no mismatches allowed in the primer sequence). Following demultiplexing and quality filtering, the QIIME 1 script pick_de_novo_otus.py was used to cluster sequences into de novo operational taxonomic units (OTUs) based on 97% sequence similarity, and representative sequences for each OTU were assigned taxonomy based on fully-sequenced microbial genomes (IMG/GG GreenGenes) [17]. Chimeric sequences identified by ChimeraSlayer [18] were removed, as were sequences that failed alignment and singleton OTUs.
To account for potential bacterial 16S rDNA contamination from molecular biological reagents, OTUs observed in the blank water controls were filtered from the experimental samples using several scripts in QIIME 1. Briefly, an OTU table with just the blank water controls was created using the filter_samples_from_otu_table.py script. OTU ids with zero counts in the water controls were then filtered out using the filter_otus_from_otu_table.py script. OTU ids determined to be present in the water controls were then filtered from the experimental samples using the filter_otus_from_otu_table.py script followed by the filter_samples_from_otu_table.py script.
Finally, to focus on the prominent taxa in the samples, only OTUs with at least a mean 0.01% total abundance across all samples were retained in the final OTU tables (Supplementary Tables S3 and S4). The final total data set comparing UHCs to FDRs contained 212,828 sequences (mean ± s.d.: 5911.889 ± 3974.339 sequences per sample) and 910 OTUs (Supplementary Table S3), while the final total data set comparing HCs to SLE patients contained 591,393 sequences (mean ± s.d.: 14784.825 ± 17982.308 sequences per sample) and 473 OTUs (Supplementary Table S4). The data set containing UHCs and FDRs was analyzed independently from the data set containing HCs and SLE patients.
Statistical analysis
The OTU table of raw counts was normalized to an OTU table of relative abundances, and taxa of the same type were aggregated at the phylum, class, order, family, genus, and species levels. The non-parametric Mann-Whitney U test was used in QIIME 1 to compare abundances and p-values were adjusted for multiple comparisons by the false discovery rate. The microbiome species diversity within each sample (α-diversity) was computed using the phyloseq package in R for the observed OTUs and Chao1 diversity indices. The Wilcoxon rank sum test in R was used for testing significance of the alpha diversity estimates. Beta diversity analyses were performed on unweighted UniFrac distances using the phyloseq package in R [19] and visualized on principal coordinate analysis plots using the ade4 package in R [20]. Permutational MANOVA (‘adonis’ function, vegan package, R) was used to test the statistical significance of variances in microbiome composition between groups. Differential abundance testing was done using the DESeq2 R package to test for differentially abundant taxa among UHCs/FDRs or HCs/SLE at the phylum to genus level and at OTU level [21, 22]. P-values were adjusted for multiple comparisons by the Benjamin-Hochberg false discovery rate.
RESULTS
Plasma levels of SLE-related autoantibodies are increased in SLE patients and FDRs of SLE patients compared to controls
To determine the levels of autoantibodies in FDRs compared to UHCs and in SLE patients compared to HCs, we examined plasma reactivities to a panel of 122 autoantigens and 6 controls in the two cohorts. The autoantigen array showed greater plasma levels of a large spectrum of autoantibodies in FDRs compared to UHCs and in SLE patients compared to HCs (Figures 1A-B). These autoantibodies not only included the anti-nuclear antibodies (ANAs), but also those directly related to cellular and extracellular antigens in the skin, kidney, thyroid and joints. Four representative SLE-related IgG autoantibodies including anti-double stranded DNA (anti-dsDNA), anti-nucleosome, anti-single stranded DNA (anti-ssDNA), and anti-chromatin [23, 24] were increased in FDRs compared to UHCs (Figures 1C-F), and in SLE patients compared to HCs (Figures 1G-J). These results are consistent with previous publications from our group, demonstrating that certain SLE-related autoantibodies are increased in FDRs of patients compared to UHCs [10, 25]. In addition, the results are consistent with many prior studies showing elevated autoantibodies in SLE patients compared to HCs.
Figure 1. Plasma levels of autoantibodies in UHCs and FDRs and HCs and SLE patients.

(A, B) Plasma samples from UHCs and FDRs and HCs and SLE patients were tested for reactivities to a variety of autoantigens in an autoantigen array. A heat map with clustering of IgG autoantibodies was generated from the autoantigen array. Intensities higher than the mean were colored red (A) or yellow (B), those below the mean were colored green (A) or blue (B), and cells with signals close to the mean were colored black (A, B). Gray was used to denote missing data. (C-J) The median mean fluorescence intensities (MFIs) of antibodies against dsDNA (C, G), nucleosome antigen (D, H), ssDNA (E, I), and chromatin antigen (F, J) were shown in the two groups. The non-parametric Mann-Whitney U-test was used for comparison and horizontal lines represent medians.
Presence of the microbial TLR4 ligand LPS in plasma is associated with autoreactive IgGs in FDRs
Most studies of autoantibodies in humans have focused on the role of toll-like receptor (TLR)7 and TLR9, while studies of TLR2 and TLR4 in autoimmunity were performed in animal models [26–28]. We believe that as a result of microbial translocation, microbial products, such as universal TLR ligands, promote heightened inflammation and autoantibody production in genetically predisposed high-risk individuals (e.g., FDRs of SLE patients) and SLE patients. One such TLR ligand is LPS, a known TLR4 agonist considered to be a representative marker of microbial translocation [29]. We found that compared to UHCs, parent or child FDRs of SLE patients displayed increased microbial translocation, as reflected by plasma LPS levels, compared to sibling FDRs and UHCs (Figure 2A). Similarly, SLE patients had elevated plasma LPS levels compared to healthy controls (Figure 2B). Plasma LPS levels were positively correlated with plasma levels of anti-dsDNA IgG when UHCs and FDRs were combined (Figure 2C), and when HCs and SLE patients were combined (Figure 2D). Individually, there was a significant positive correlation between plasma LPS levels and anti-dsDNA levels in FDRs (Figure 2C), but not in SLE patients (Figure 2D).
Figure 2. Plasma LPS level is associated with anti-dsDNA autoantibody production.

(A, B) Plasma LPS was tested by the limulus amebocyte assay in a study including 18 UHCs, 11 sibling FDRs, and 7 parent or child FDRs (A), and another study including 19 HCs and 21 SLE patients (B). (C, D) Plasma LPS level correlation with plasma level of anti-dsDNA autoantibody in UHCs and FDRs (C) and in HCs and SLE patients (D). Non-parametric Mann-Whitney U tests and Spearman’s correlation tests were used for comparison and correlation respectively and horizontal lines represent medians.
Distinct circulating microbiome in UHCs and FDRs but not in HCs and SLE patients
To investigate further the role of microbial products in autoantibody production and SLE disease, we examined the circulating microbiome of UHCs compared to FDRs and HCs compared to SLE patients. The increased translocation of bacterial products into the systemic circulation from the permeable mucosa suggests that insights into autoimmune pathology can be gained from studying the circulating microbiome as opposed to other sites. Profiling of the circulating microbiome in UHCs and FDRs and HCs and SLE patients revealed some similarities and differences in the microbiome composition of the two cohorts. At the phylum level, all individuals in the UHC and FDR cohort have similar bacteria found in the top five based on relative abundance when compared to all individuals in the HC and SLE cohort (Figures 3A and4A). Shared bacteria in the top five at the phylum level include Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes. Similarly, at the class level, Alphaproteobacteria and Bacilli appear in the top five bacteria in the two cohorts (Figures 3B and4B). Beyond the class level, there were no overlapping bacteria found in the top five at the various taxa levels between the two cohorts (Figures 3C-E and Figures 4C-E).
Figure 3. Circulating microbiome relative abundance in UHCs and FDRs.

(A-E) The top five bacteria at the taxonomic levels of phylum (A), class (B), order (C), family (D) and genus (E) based on relative abundance in both UHCs and FDRs. Non-parametric Mann-Whitney U test was used in QIIME 1 to compare abundances and p-values were adjusted for multiple comparisons by the false discovery rate (p.fdr).
Figure 4. Circulating microbiome relative abundance in HCs and SLE patients.

(A-E) The top five bacteria at the taxonomic levels of phylum (A), class (B), order (C), family (D) and genus (E) based on relative abundance in both HCs and SLE patients. Non-parametric Mann-Whitney U test was used in QIIME 1 to compare abundances and p-values were adjusted for multiple comparisons by the false discovery rate (p.fdr).
Another significant aspect of the circulating microbiome that we examined in the two cohorts was diversity. Compared to UHCs, FDRs have decreased species diversity within each sample (alpha-diversity) as evaluated by the observed OTUs and Chao1 species-richness (Figure 5A). In addition, diversities of the circulating microbiome of SLE patients tended to decrease compared to controls but the difference did not reach significance (Figure 5B). To determine whether overall microbiome composition differed according to health status (UHC vs. FDR or HC vs. SLE) as well as between sample diversity (beta-diversity), we conducted principal coordinate analysis based on unweighted UniFrac phylogenetic distances. Health status was significantly associated with overall circulating microbiome composition when comparing UHCs and FDRs (Figure 5C), however, this association was not seen when comparing HCs and SLE patients (Figure 5D).
Figure 5. Alpha and beta diversity analyses of the circulating microbiome in FDRs and SLE patients compared to their controls.
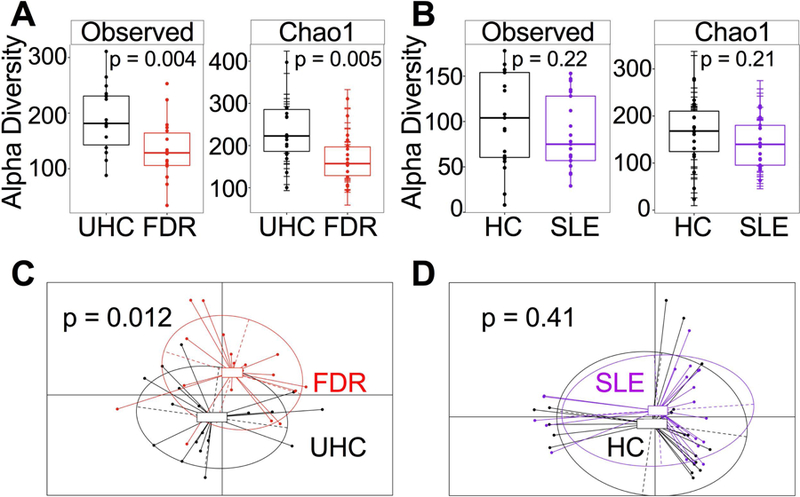
(A, B) Observed OTUs and the Chao1 species-richness metric were evaluated using the phyloseq package in R to assess alpha diversity. Statistical significance was determined using the Wilcoxon rank sum test in R. (C, D) Principal coordinate analysis (PCoA) was conducted based on the unweighted UniFrac distance to determine beta diversity using the phyloseq and ade4 packages in R. Statistical significance testing of beta diversity was done through permutational MANOVA (‘adonis’ function, vegan package, R).
To identify bacteria that were differentially abundant between UHCs and FDRs or between HCs and SLE patients, we used DESEq2 [21, 22]. In comparing UHCs to FDRs, Firmicutes was the predominant phylum containing several differentially abundant bacteria between the two groups (Table 1). Bacteria belonging to the Firmicutes phylum were identified as differentially abundant between UHCs and FDRs at the family, genus, and species levels. Of the bacteria found in the Firmicutes phylum, Thermoanaerobacterium saccharolyticum and Lactobacillus iners were two species with elevated mean relative abundances in FDRs compared to UHCs (Table 1). The remaining bacteria identified as differentially abundant between UHCs and FDRs belonged to the WPS-2, Actinobacteria, and Proteobacteria phyla and their mean relative abundances were primarily elevated in UHCs compared to FDRs (Table 1). In contrast, the analysis between HCs and SLE patients yielded differentially abundant bacteria belonging to several phyla including Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Planctomycetes, and Gemmatimonadetes (Table 1). Out of the twenty-two bacteria identified in the differentially abundant analysis between controls and patients, fifteen bacteria had elevated mean relative abundances in SLE patients compared to controls (Table 1). An overlapping bacterium identified as differentially abundant in both the UHCs versus FDRs analyses and the HCs versus patients’ analyses is the Paenibacillus genus belonging to the Firmicutes phylum, Bacilli class, Bacillales order, and Paenibacillaceae family. In both comparisons, the mean relative abundance of the Paenibacillus genus is increased in controls compared to FDRs or SLE patients. Therefore, alterations in the circulating microbiome may be influencing autoantibody production in SLE disease.
Table 1.
Differentially abundant taxa between UHCs and FDRs and HCs and SLE patients. The DESEq2 R package was used to identify differentially abundant taxa at the phylum to species level. P-values were adjusted for multiple comparisons by the false discovery rate.
| Groups compared | Taxa Level | UHC Mean | FDR Mean | p | padj | Phylum | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UHC vs. FDR | Class | 49.44 | 0 | 2.18E-13 | 7.62E-12 | WPS-2 | [Unassigned] | ||||
| Order | 49.44 | 0 | 4.84E-13 | 2.61E-11 | WPS-2 | [Unassigned] | [Unassigned] | ||||
| Family | 1.89 | 357.83 | 3.61E-15 | 4.00E-13 | Actinobacteria | Actinobacteria | Actinomycetales | Geodermatophilaceae | |||
| 46.06 | 0 | 7.90E-15 | 4.39E-13 | Actinobacteria | Actinobacteria | Actinomycetales | Actinomycetaceae | ||||
| 48.00 | 0 | 3.58E-14 | 1.33E-12 | Firmicutes | Bacilli | Bacillales | Paenibacillaceae | ||||
| 88.50 | 0 | 1.63E-04 | 3.63E-03 | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | ||||
| 49.44 | 0 | 5.65E-14 | 1.57E-12 | WPS-2 | [Unassigned] | [Unassigned] | [Unassigned] | ||||
| Genus | 46.06 | 0 | 1.54E-14 | 1.04E-12 | Actinobacteria | Actinobacteria | Actinomycetales | Actinomycetaceae | Actinomyces | ||
| 3.11 | 76.78 | 5.88E-15 | 9.58E-13 | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Thermoanaerobacterium | |||
| 21.94 | 2.5 | 1.92E-14 | 1.04E-12 | Firmicutes | Clostridia | Clostridiales | [Tissierellaceae] | Anaerococcus | |||
| 48.00 | 0 | 6.42E-14 | 2.62E-12 | Firmicutes | Bacilli | Bacillales | Paenibacillaceae | Paenibacillus | |||
| 77.28 | 0.06 | 1.75E-03 | 4.76E-02 | Firmicutes | Bacilli | Lactobacillales | Streptococcaceae | Streptococcus | |||
| 49.44 | 0 | 9.99E-14 | 3.26E-12 | WPS-2 | [Unassigned] | [Unassigned] | [Unassigned] | [Unassigned] | |||
| Species | 3.11 | 76.78 | 3.16E-16 | 5.53E-14 | Firmicutes | Clostridia | Clostridiales | Clostridiaceae | Thermoanaerobacterium | saccharolyticum | |
| 0.00 | 169.11 | 3.32E-15 | 2.91E-13 | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae | Lactobacillus | iners | ||
| 21.94 | 2.5 | 1.01E-14 | 5.87E-13 | Firmicutes | Clostridia | Clostridiales | [Tissierellaceae] | Anaerococcus | [Unassigned] | ||
| 48.00 | 0 | 4.87E-14 | 2.13E-12 | Firmicutes | Bacilli | Bacillales | Paenibacillaceae | Paenibacillus | [Unassigned] | ||
| 15.17 | 0.11 | 1.23E-03 | 3.58E-02 | Proteobacteria | Gammaproteobacteria | Pseudomonadales | Moraxellaceae | Acinetobacter | johnsonii | ||
| 49.44 | 0 | 7.46E-14 | 2.61E-12 | WPS-2 | [Unassigned] | [Unassigned] | [Unassigned] | [Unassigned] | [Unassigned] | ||
| Groups compared | Taxa Level | HC Mean | SLE Mean | p | padj | Phylum | Class | Order | Family | Genus | Species |
| HC vs. SLE | Class | 198.84 | 0.19 | 0.00006 | 0.003 | Actinobacteria | Thermoleophilia | ||||
| 63.05 | 93.38 | 0.00296 | 0.032 | Bacteroidetes | Flavobacteriia | ||||||
| 493.11 | 0.38 | 0.00023 | 0.004 | Gemmatimonadetes | Gemmatimonadetes | ||||||
| 0.63 | 113.71 | 0.00028 | 0.004 | Planctomycetes | Planctomycetia | ||||||
| 0.42 | 40.19 | 0.00031 | 0.004 | Proteobacteria | Epsilonproteobacteria | ||||||
| Order | 82.58 | 0.24 | 0.00013 | 0.008 | Actinobacteria | Thermoleophilia | Solirubrobacterales | ||||
| 63.05 | 93.38 | 0.00019 | 0.008 | Bacteroidetes | Flavobacteriia | Flavobacteriales | |||||
| 10.42 | 409.67 | 0.00026 | 0.008 | Firmicutes | Bacilli | Lactobacillales | |||||
| 0.42 | 40.19 | 0.00037 | 0.008 | Proteobacteria | Epsilonproteobacteria | Campylobacterales | |||||
| Family | 94.74 | 0.76 | 0.00057 | 0.025 | Actinobacteria | Actinobacteria | Actinomycetales | Streptomycetaceae | |||
| 27.84 | 13.62 | 0.00035 | 0.025 | Actinobacteria | Actinobacteria | Actinomycetales | Nocardioidaceae | ||||
| 19.84 | 44.00 | 0.00082 | 0.028 | Bacteroidetes | Sphingobacteriia | Sphingobacteriales | Sphingobacteriaceae | ||||
| 0.11 | 123.57 | 0.00059 | 0.025 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | ||||
| 0.42 | 40.19 | 0.00012 | 0.021 | Proteobacteria | Epsilonproteobacteria | Campylobacterales | Campylobacteraceae | ||||
| Genus | 27.84 | 13.62 | 0.00060 | 0.026 | Actinobacteria | Actinobacteria | Actinomycetales | Nocardioidaceae | [Unassigned] | ||
| 25.11 | 109.76 | 0.00014 | 0.008 | Bacteroidetes | Bacteroidia | Bacteroidales | Marinilabiaceae | [Unassigned] | |||
| 37.68 | 127.71 | 0.00007 | 0.008 | Firmicutes | Clostridia | Clostridiales | Peptococcaceae | [Unassigned] | |||
| 0.32 | 78.38 | 0.00010 | 0.008 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | [Unassigned] | |||
| 232.11 | 10.43 | 0.00008 | 0.008 | Firmicutes | Bacilli | Bacillales | Paenibacillaceae | Paenibacillus | |||
| 59.11 | 166.62 | 0.00113 | 0.041 | Firmicutes | Clostridia | Clostridiales | [Acidaminobacteraceae] | WH1–8 | |||
| Species | 37.68 | 127.71 | 0.00015 | 0.017 | Firmicutes | Clostridia | Clostridiales | Peptococcaceae | [Unassigned] | [Unassigned] | |
| 0.32 | 78.38 | 0.00014 | 0.017 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae | [Unassigned] | [Unassigned] | ||
DISCUSSION
A hallmark feature of SLE is the production of autoantibodies against a large number of self-antigens; however, the events underlying this process remain unclear [30]. One hypothesis is that translocation of microbial components from the gastrointestinal (GI) tract or other mucosal sites into the systemic circulation can lead to activation of the immune system [12, 31]. In this study, we examined relationships between microbial translocation and autoantibody levels in SLE patients, FDRs of SLE patients, and healthy controls. The nature of familial aggregation in SLE and the accumulation of autoantibodies prior to disease onset make FDRs an ideal model for understanding autoantibody development in the absence of immunosuppressive therapy in SLE.
We first determined the plasma levels of a variety of autoantibodies in FDRs compared to UHCs and in HCs compared to SLE patients using autoantigen arrays. In agreement with the results from previous studies [5, 10, 25], FDRs had significantly elevated levels of autoantibodies including anti-dsDNA, anti-ssDNA and anti-nucleosome compared to UHCs. Similarly, SLE patients exhibited increased plasma autoantibody levels in contrast to healthy controls. Accompanying the increases in autoantibodies in FDRs was an increase in plasma LPS levels. We noted that FDRs with a parent or child relationship with a SLE patient had elevated plasma LPS levels compared to UHCs and siblings of patients, which has not been shown previously. This increase in plasma LPS level in parent or child FDRs compared to sibling FDRs may be explained by past studies that documented significant similarities in clinical manifestations of SLE in parent/offspring relationships as opposed to sibling relationships [32, 33]. Plasma LPS levels were also higher in SLE patients than in healthy controls which is consistent with previous observations of higher serum endotoxin levels in lupus patients [34, 35]. Overall, the increases in plasma LPS level exhibited a positive correlation with anti-dsDNA levels when all individuals are combined in both cohorts. The direct correlation between plasma LPS and anti-dsDNA levels is significant for FDRs, suggesting a role for microbial TLR ligands in immune activation resulting in autoantibody production in genetically pre-disposed high-risk populations. However, this trend was not significant for SLE patients which could be due to a variety factors including immunosuppressive medications and SLE disease activity. Despite differences in the significance of the correlation between FDRs and SLE patients, the results appear consistent with prior research linking infection and autoimmunity via mechanisms of molecular mimicry or bystander activation [36–38], and with a recent study demonstrating the existence of antibodies to antigens from the gut commensal Ruminococcus gnavus that are cross-reactive with anti-dsDNA autoantibodies in SLE patients [39]. Further investigation is needed to establish causality between bacterial TLR ligands such as LPS and the autoantibody generation and systemic inflammation seen in SLE.
To better understand how translocation of microbial products such as LPS may impact autoantibody levels, we utilized plasma 16S rDNA analysis to determine the circulating microbiome composition of UHCs in comparison to FDRs and HCs in comparison to SLE patients. The detection of 16S rRNA in the plasma does not indicate the presence of whole organisms, but rather the presence of fragments of DNA. The predominant phyla of bacteria found in both UHCs and FDRs and HCs and SLE were Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes with the dominant phylum being Proteobacteria. This is in contrast to prior studies on the microbiome composition in SLE in which Firmicutes and Bacteroidetes were the dominant phyla with smaller contributions from Proteobacteria and Actinobacteria [40, 41]. Our study profiled the circulating microbiome while past studies profiled the gut microbiome; the difference in composition is likely due to differences in sampling sites and other factors such as diet, donor, therapy regimen and disease stage. Differences in composition could also be attributed to the high variation of the circulating microbiome between individuals, similar to what is seen in the gut microbiome [42]. Two different studies, one in a Spanish population and another in a Chinese population noted a lower Firmicutes/Bacteroidetes ratio in the gut microbiota of SLE patients compared to controls [40, 41]. In contrast, we did not identify any significant differences in the Firmicutes/Bacteroidetes ratio in UHCs and FDRs or HCs and SLE patients (data not shown) which is in agreement with a more recent study on the gut microbiota in SLE [43].
Previous studies have shown reduced gut microbiota diversity in SLE patients compared to healthy controls, suggesting a dysbiosis or imbalance in the microbiome of SLE patients [40, 41, 44, 45]. However, in this study, FDRs of SLE patients had reduced diversity in their circulating microbiome compared to UHCs, while no significant difference was found in the diversity of the circulating microbiome of SLE patients compared to HCs. The divergence in the results between FDRs and SLE patients could be attributed to several factors, but a major one is immunosuppressive therapies or the effect of the disease itself. The FDRs examined were not taking any immunosuppressive therapies in contrast to the SLE patients who were all receiving immunosuppressive treatments. In addition, SLE patients are more likely to develop infections and thus get antibiotics, which can greatly influence microbiome composition [46]. Future studies examining the microbiome of SLE patients pre and post initiation of therapy will better delineate the role of the microbiome in autoantibody production and subsequent development of SLE as well as the effect of specific immunosuppressive therapies on the microbiome.
Despite differences in the circulating microbiome diversity between UHCs and FDRs and between HCs and SLE patients, we were able to identify differentially abundant bacteria in the two cohorts. Most of the bacteria found to be differentially abundant between UHCs and FDRs belonged to the Firmicutes phylum with specific bacteria at the family, genus and species levels being primarily elevated in UHCs in relation to FDRs. The two bacteria belonging to the Firmicutes phylum that were elevated in FDRs and not UHCs were Thermoanaerobacterium saccharolyticum and Lactobacillus iners. T. saccharolyticum is an anaerobic, thermophilic, gram-positive bacteria known primarily for its ability to ferment a wide number of carbohydrates [47], but not much is known about T. saccharolyticum in the clinical setting nor in the context of SLE disease. L. iners is more widely studied and is a gram-variable, anaerobic bacteria found in the lower reproductive tract of women [48]. However, the role of L. iners in vaginal health is currently unclear as it is detected in normal conditions and during states of dysbiosis such as bacterial vaginosis [48]. The presence of these bacteria in the circulating microbiome suggests their translocation into the systemic circulation, but more studies are needed to determine if and how they play a contributing role to SLE development. One recent study identifies Enterococcus gallinarium as a pathobiont that translocates from the gut to the liver and lymphoid organs in the (NZW x BXSB)F1 mice model and subsequently triggers autoimmune responses in backgrounds with a genetic predisposition for autoimmunity [13]. In another study, Lactobacillus reuteri is shown to worsen systemic autoimmunity by translocating from the gut to the liver, spleen, and mesenteric lymph nodes in lupus-prone mice [14]. These studies support a role for translocating microbes in the development of autoimmune diseases such as SLE, but further investigation is needed. Furthermore, why certain bacteria translocate and others do not is unknown, but changes to gut permeability and a microbe’s genomically encoded capacity to produce flagella could play role [49], however further studies are needed to determine translocation mechanisms. Future studies comparing paired circulating microbiota composition to the microbiota composition from various sites such as the gut, vaginal tract, oral cavity, or skin will help to elucidate the sites from which bacteria translocate, and which translocating bacteria contribute to SLE disease pathogenesis.
In the HCs and SLE microbiome comparison, the identified differentially abundant bacteria belong to several different phyla including Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Planctomycetes, and Gemmatimonadetes, but no phylum was particularly dominant. Most of the differentially abundant bacteria detected were increased in SLE patients compared to HCs. However, the family level was the lowest taxonomic level that most of the bacteria could be identified, making it difficult to draw any strong conclusions. One unique finding in the analysis of differentially abundant bacteria is the identification of bacteria belonging to the Paenibacillus genus in both cohorts. The Paenibacillus bacteria was reduced in FDRs and SLE patients but elevated in controls. Bacteria in the Paenibacillus genus have been isolated from many different sources and comprise many species pertaining to humans, animals, plant, and the environment [50]. Paenibacillus species are known to produce antimicrobial compounds that are beneficial in medicine, on the other hand, some Paenibacillus species were identified as causing opportunistic infections in humans [50]. The significance of the elevated presence of Paenibacillus in controls, but not in FDRs or SLE patients, is yet to be elucidated and future work is needed to clarify if it has a protective role in autoantibody production or SLE pathogenesis.
Overall, this study establishes previously unknown direct relationships between plasma microbial translocation and autoantibody levels in FDRs of SLE patients. In addition, this study establishes a circulating microbiome profile for FDRs of SLE patients that is reduced in diversity in comparison to UHCs, while SLE patients on medication have a circulating microbiome profile with a diversity that is similar to healthy controls. Several bacteria were also identified as distinctive to SLE patients and their FDRs, and further studies are needed to explore their direct or indirect roles in autoantibody formation and SLE disease pathogenesis. The findings of this study are not without limitations including, the small sample size in both cohorts, the specific patient demographics, and the use of clustering analysis generating OTUs instead of amplicon sequencing variants. Despite these limitations, this study provides a framework for future exploration of how changes in the microbiome can shape autoantibody formation and inflammation. An understanding of the mechanism of autoantibody induction in SLE can lead to the development of therapeutic targets that prevent autoantibody production thereby slowing disease onset, mitigating downstream inflammation and reducing tissue damages.
Supplementary Material
Acknowledgments
Funding: The funding support was from the National Institute of Arthritis and Musculoskeletal and Skin Diseases grant P60 AR062755 (Gilkeson, Kamen and Oates), UL1 RR029882, the Medical Research Service at the Ralph H. Johnson VA Medical Center Merit grant VA CSRD MERIT (CX001211, Gilkeson), the Biorepository & Tissue Analysis Shared Resource, and Hollings Cancer Center, Medical University of South Carolina (P30 CA138313), R01LM012517 (Alekseyenko), R21TR002513 (Alekseyenko), P30AR072582 (Gilkeson, Kamen and Oates), AR067459 (Kamen), AR068406 (Kamen), RR001070 (Kamen), UL1 TR001450, R01CA164964, U54CA210962, and P50AR070591. Prof. Macedo receives productivity grant from the Brazilian Institution CNPq.
Footnotes
Conflicts of interest: The authors have no conflicts of interest to disclose.
REFERENCES
- 1.Tsokos GC, Lo MS, Costa Reis P, and Sullivan KE, New insights into the immunopathogenesis of systemic lupus erythematosus. Nature reviews. Rheumatology, 2016. 12(12): p. 716–730. [DOI] [PubMed] [Google Scholar]
- 2.Moser KL, Kelly JA, Lessard CJ, and Harley JB, Recent insights into the genetic basis of systemic lupus erythematosus. Genes and Immunity, 2009. 10(5): p. 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghodke-Puranik Y and Niewold TB, Immunogenetics of systemic lupus erythematosus: A comprehensive review. Journal of autoimmunity, 2015. 64: p. 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, et al. , Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis & Rheumatism, 2005. 52(4): p. 1138–1147. [DOI] [PubMed] [Google Scholar]
- 5.Navarra SV, Ishimori ML, Uy EA, Hamijoyo L, Sama J, James JA, et al. , Studies of Filipino patients with systemic lupus erythematosus: Autoantibody profile of first-degree relatives. Lupus, 2011. 20(5): p. 537–543. [DOI] [PubMed] [Google Scholar]
- 6.Lawrence JS, Martins CL, and Drake GL, A family survey of lupus erythematosus. 1. Heritability. The Journal of rheumatology, 1987. 14(5): p. 913–921. [PubMed] [Google Scholar]
- 7.Arbuckle MR, McClain MT, Rubertone MV, Scofield HR, Dennis GJ, James JA, et al. , Development of Autoantibodies before the Clinical Onset of Systemic Lupus Erythematosus. The New England Journal of Medicine, 2003. 349(16): p. 1526–1533. [DOI] [PubMed] [Google Scholar]
- 8.Pollak VE, Antinuclear Antibodies in Families of Patients with Systemic Lupus Erythematosus. The New England Journal of Medicine, 1964. 271(4): p. 165–171. [DOI] [PubMed] [Google Scholar]
- 9.Corporaal S, Bijl M, and Kallenberg CGM, Familial Occurrence of Autoimmune Diseases and Autoantibodies in a Caucasian Population of Patients with Systemic Lupus Erythematosus. Clinical Rheumatology, 2002. 21(2): p. 108–113. [DOI] [PubMed] [Google Scholar]
- 10.Bruner BF, Guthridge JM, Lu R, Vidal G, Kelly JA, Robertson JM, et al. , Comparison of autoantibody specificities between traditional and bead‐based assays in a large, diverse collection of patients with systemic lupus erythematosus and family members. Arthritis & Rheumatism, 2012. 64(11): p. 3677–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mu Q, Kirby J, Reilly CM, and Luo XM, Leaky Gut As a Danger Signal for Autoimmune Diseases. Frontiers in Immunology, 20178: p. 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenchley JM and Douek DC, Microbial translocation across the GI tract. Annual review of immunology, 2012. 30: p. 149–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vieira MS, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. , Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science, 2018. 359(6380): p. 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zegarra-Ruiz DF, Beidaq A, Iñiguez AJ, Ricco M, Vieira S, Ruff WE, et al. , A Diet-Sensitive Commensal Lactobacillus Strain Mediates TLR7-Dependent Systemic Autoimmunity. Cell host & microbe, 2018. 25(1): p. 113–127000000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li QZ, Karp DR, Quan J, Branch VK, Zhou J, Lian Y, et al. , Risk factors for ANA positivity in healthy persons. Arthritis Res Ther, 2011. 13(2): p. R38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang W, Lederman MM, Hunt P, Sieg SF, Haley K, Rodriguez B, et al. , Plasma Levels of Bacterial DNA Correlate with Immune Activation and the Magnitude of Immune Restoration in Persons with Antiretroviral-Treated HIV Infection. The Journal of Infectious Diseases, 2009. 199(8): p. 1177–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. , QIIME allows analysis of high-throughput community sequencing data. Nature methods, 2010. 7(5): p. 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. , Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Research, 2011. 21(3): p. 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McMurdie PJ and Holmes S, phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one, 2013. 8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dray S and of statistical software D-AB, The ade4 package: implementing the duality diagram for ecologists. Journal of statistical software, 2007. [Google Scholar]
- 21.McMurdie PJ and Holmes S, Waste not, want not: why rarefying microbiome data is inadmissible. PLoS computational biology, 2014. 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology, 2014. 15(12): p. 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schett G, Smole J, Zimmermann C, Hiesberger H, Hoefler E, Fournel S, et al. , The autoimmune response to chromatin antigens in systemic lupus erythematosus: autoantibodies against histone H1 are a highly specific marker for SLE associated with increased disease activity. Lupus, 2002. 11(11): p. 704–15. [DOI] [PubMed] [Google Scholar]
- 24.Reveille JD, Predictive value of autoantibodies for activity of systemic lupus erythematosus. Lupus, 2004. 13(5): p. 290–7. [DOI] [PubMed] [Google Scholar]
- 25.Kamen DL, Barron M, Parker TM, Shaftman SR, Bruner GR, Aberle T, et al. , Autoantibody prevalence and lupus characteristics in a unique African American population. Arthritis Rheum, 2008. 58(5): p. 1237–47. [DOI] [PubMed] [Google Scholar]
- 26.Chauhan SK, Singh VV, Rai R, Rai M, and Rai G, Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol, 2013. 33(5): p. 954–64. [DOI] [PubMed] [Google Scholar]
- 27.Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, et al. , Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol, 2009. 183(10): p. 6207–16. [DOI] [PubMed] [Google Scholar]
- 28.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, and Bolland S, Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science, 2006. 312(5780): p. 1669–72. [DOI] [PubMed] [Google Scholar]
- 29.Marchetti G, Tincati C, and Silvestri G, Microbial Translocation in the Pathogenesis of HIV Infection and AIDS. Clinical Microbiology Reviews, 2013. 26(1): p. 2–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham KL and Utz PJ, Sources of autoantigens in systemic lupus erythematosus. Current opinion in rheumatology, 2005. 17(5): p. 513–517. [DOI] [PubMed] [Google Scholar]
- 31.Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. , Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nature Medicine, 2006. 12(12): p. 1365–1371. [DOI] [PubMed] [Google Scholar]
- 32.Aett FC and Shulman LE, Studies in familial systemic lupus erythematosus. Medicine, 1976. 55(4): p. 313. [DOI] [PubMed] [Google Scholar]
- 33.Tsao BP, Grossman JM, Riemekasten G, Strong N, Kalsi J, Wallace DJ, et al. , Familiality and co‐occurrence of clinical features of systemic lupus erythematosus. Arthritis & Rheumatism, 2002. 46(10): p. 2678–2685. [DOI] [PubMed] [Google Scholar]
- 34.Issara-Amphorn J, Surawut S, Worasilchai N, Thim-uam A, Finkelman M, Chindamporn A, et al. , The Synergy of Endotoxin and (1→3)-β-D-Glucan, from Gut Translocation, Worsens Sepsis Severity in a Lupus Model of Fc Gamma Receptor IIb-Deficient Mice. Journal of Innate Immunity, 2018. (0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi L, Zhang Z, Yu AM, Wang W, Wei Z, Akhter E, et al. , The SLE Transcriptome Exhibits Evidence of Chronic Endotoxin Exposure and Has Widespread Dysregulation of Non-Coding and Coding RNAs. PLoS ONE, 2014. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujinami RS, von Herrath MG, Christen U, and Whitton JL, Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev, 2006. 19(1): p. 80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yadav R, Zammit DJ, Lefrancois L, and Vella AT, Effects of LPS-mediated bystander activation in the innate immune system. J Leukoc Biol, 2006. 80(6): p. 1251–61. [DOI] [PubMed] [Google Scholar]
- 38.Sanderson NS, Zimmermann M, Eilinger L, Gubser C, Schaeren-Wiemers N, Lindberg RL, et al. , Cocapture of cognate and bystander antigens can activate autoreactive B cells. Proc Natl Acad Sci U S A, 2017. 114(4): p. 734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Azzouz D, Omarbekova A, Heguy A, Schwudke D, Gisch N, Rovin BH, et al. , Lupus nephritis is linked to disease-activity associated expansions and immunity to a gut commensal. Annals of the rheumatic diseases, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hevia A, Milani C, López P, Cuervo A, Arboleya S, Duranti S, et al. , Intestinal Dysbiosis Associated with Systemic Lupus Erythematosus. mBio, 2014. 5(5): p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He Z, Shao T, Li H, Xie Z, and Wen C, Alterations of the gut microbiome in Chinese patients with systemic lupus erythematosus. Gut pathogens, 2016. 8: p. 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium T, Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, et al. , Structure, function and diversity of the healthy human microbiome. Nature, 2012. 486(7402). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antal‐Szalmás P, Szöllősi I, Lakos G, Kiss E, Csípő I, Sümegi A, et al. , A novel flow cytometric assay to quantify soluble CD14 concentration in human serum. Cytometry, 2001. 45(2): p. 115–123. [DOI] [PubMed] [Google Scholar]
- 44.Luo XM, Edwards MR, Mu Q, Yu Y, Vieson MD, Reilly CM, et al. , Gut Microbiota in Human Systemic Lupus Erythematosus and a Mouse Model of Lupus. Applied and Environmental Microbiology, 2017. 84(4): p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodríguez-Carrio J, López P, Sánchez B, González S, Gueimonde M, Margolles A, et al. , Intestinal Dysbiosis Is Associated with Altered Short-Chain Fatty Acids and Serum-Free Fatty Acids in Systemic Lupus Erythematosus. Frontiers in Immunology, 2017. 8: p. 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Langdon A, Crook N, and Dantas G, The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Medicine, 2016. 8(1): p. 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Currie DH, Raman B, Gowen CM, Tschaplinski TJ, Land ML, Brown SD, et al. , Genome-scale resources for Thermoanaerobacterium saccharolyticum. BMC systems biology, 2015. 9: p. 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Petrova MI, Reid G, Vaneechoutte M, and Lebeer S, Lactobacillus iners: Friend or Foe? Trends in microbiology, 2017. 25(3): p. 182–191. [DOI] [PubMed] [Google Scholar]
- 49.Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, et al. , Innate and Adaptive Immunity Interact to Quench Microbiome Flagellar Motility in the Gut. Cell host & microbe, 2013. 14(5): p. 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grady EN, MacDonald J, Liu L, Richman A, and Yuan Z-CC, Current knowledge and perspectives of Paenibacillus: a review. Microbial cell factories, 2016. 15(1): p. 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.