

Abstract
Volume-regulated anion channel (VRAC) is a glutamate-permeable channel that is activated by physiological and pathological cell swelling and promotes ischemic brain damage. However, because VRAC opening requires cytosolic ATP, it is not clear if and how its activity is sustained in the metabolically compromised CNS. In the present study, we used cultured astrocytes – the cell type which shows prominent swelling in stroke – to model how metabolic stress and changes in gene expression may impact VRAC function in the ischemic and post-ischemic brain. The metabolic state of primary rat astrocytes was modified with chemical inhibitors and examined using luciferin-luciferase ATP assays and a Seahorse analyzer. Swelling-activated glutamate release was quantified with the radiotracer D-[3H]aspartate. The specific contribution of VRAC to swelling-activated glutamate efflux was validated by RNAi knockdown of the essential subunit, leucine-rich repeat-containing 8A (LRRC8A); expression levels of VRAC components were measured with qRT-PCR. Using this methodology, we found that complete metabolic inhibition with the glycolysis blocker 2-deoxy-D-glucose and the mitochondrial poison sodium cyanide reduced astrocytic ATP levels by >90% and abolished glutamate release from swollen cells (via VRAC). When only mitochondrial respiration was inhibited by cyanide or rotenone, the intracellular ATP levels and VRAC activity were largely preserved. Bypassing glycolysis by providing the mitochondrial substrates pyruvate and/or glutamine led to partial recovery of ATP levels and VRAC activity. Unexpectedly, the metabolic block of VRAC was overridden when ATP-depleted cells were exposed to extreme cell swelling (≥50% reduction in medium osmolarity). 24-h anoxic adaptation caused a moderate reduction in the expression levels of the VRAC component LRRC8A, but no significant changes in VRAC activity. Overall, our findings suggest that (1) astrocytic VRAC activity and metabolism can be sustained by low levels of glucose and (2) the inhibitory influence of diminishing ATP levels and the stimulatory effect of cellular swelling are the two major factors that govern VRAC activity in the ischemic brain.
Keywords: volume-regulated anion channel, LRRC8, metabolic inhibition, cerebral ischemia, stroke
Graphical Abstract
In this issue:
In stroke, pathological cell swelling promotes ischemic brain damage via activation of the glutamate-permeable, volume-regulated anion channel (VRAC). Because VRAC activity requires cytosolic ATP, we explored in rat astrocyte cultures whether metabolic inhibition limits or prevents the VRAC-mediated glutamate release, which was traced with a radiolabeled glutamate analogue. Our findings suggest that in the ischemic penumbra, astrocytic VRAC activity can be fully sustained by glycolysis even when glucose and oxygen levels are dramatically reduced.
Introduction
Ischemic stroke is a highly prevalent neurological disorder that arises as a result of reduced blood flow to the whole, or a portion of the brain. Focal cerebral ischemia develops due to embolic or thrombotic occlusion of major brain arteries, while the less common global ischemia results from cardiac arrest or a precipitous drop in systemic blood pressure (Dirnagl et al. 1999; White et al. 2000; Moskowitz et al. 2010). Because brain metabolism is nearly exclusively fueled by the oxidation of blood-derived glucose, the location and extent of tissue damage correlate with the degree of the reduction of blood flow (Siesjo 1978; Hossmann 1994; Hossmann 2006; Hossmann 2008). In focal ischemia, pathological tissue is divided into two functionally distinct areas: the ischemic core and penumbra (Astrup et al. 1981; Hossmann 1994). In the ischemic core, cerebral blood flow is reduced to less than 15–20% of its normal levels, and ATP levels drop close to zero in the minutes following occlusion [reviewed in (Siesjo 1978; Hossmann 1994; Lipton 1999)]. The collapse of cellular metabolism leads to the dissipation of transmembrane ionic gradients, massive anoxic depolarization, and irreversible injury and death of neural cells. In the surrounding ischemic penumbra, where collateral circulation partially sustains blood supply at 20–50% of its normal levels, the brain tissue is electrically silent, but cells are capable of maintaining their membrane integrity and ionic gradients due to residual metabolism. The average measured levels of ATP in the penumbra are 50–70% of those in the healthy tissue but may be even higher in the regions distal to the infarction [reviewed in (Lipton 1999; Hossmann 2006)]. Over time, waves of peri-infarct depolarizations extend the volume of brain injury into the territory of the ischemic penumbra, with irreversible brain damage occurring within hours or up to several days (Hossmann 1994; Dirnagl et al. 1999; Moskowitz et al. 2010). Due to the timing of tissue loss, translational and clinical studies largely focus on finding a way to preserve the viability of the penumbral tissue.
Stroke damage is strongly associated with excessive release of numerous neurotransmitters, including the excitatory amino acids glutamate and aspartate [e.g., (Benveniste et al. 1984; Wahl et al. 1994; Phillis et al. 1997; Seki et al. 1999)]. The ensuing sustained stimulation of ionotropic and metabotropic excitatory amino acid receptors drives pathological increases in intracellular Ca2+ levels and initiates a plethora of irreversible degradative processes, which are collectively termed ‘excitotoxicity’ [reviewed in (Rothman and Olney 1986; Choi and Rothman 1990; Choi 1992; Lipton 1999)]. The release of excitotoxins involves a number of mechanisms, which differ between the core and penumbra. In the anoxic core, glutamate and aspartate accumulate in the extracellular space in a Ca2+-independent manner, largely via the reversal of Na+/K+-dependent glutamate transporters, especially glial GLT-1 (Wahl et al. 1994; Seki et al. 1999; Rossi et al. 2000). The minor contribution of Ca2+-dependent vesicular glutamate release has also been noted, but only at the onset of ischemia (Katayama et al. 1991; Wahl et al. 1994). In the ischemic penumbra, partially preserved metabolism sustains normal function of glutamate transporters but extracellular glutamate levels are nonetheless elevated due to the activation of alternative, incompletely defined release mechanisms (Dawson et al. 2000; Feustel et al. 2004; Zhang et al. 2008).
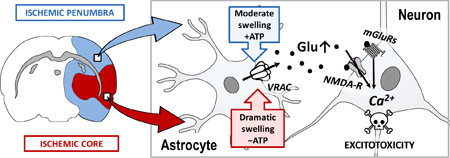
One pathway, which is quantitatively important for glutamate release in the penumbra, is the ubiquitously expressed volume-regulated anion channel (VRAC) (Feustel et al. 2004). The primary physiological role of VRAC is protection against excessive cell swelling (Strange et al. 1996; Nilius et al. 1997; Okada 1997; Pedersen et al. 2016). In the ischemic brain, swelling is detected predominantly in astrocytes, particularly in their perivascular endfeet, but is also characteristic for dendritic processes [see reviews (Kimelberg 1995; Mongin and Kimelberg 2005a; Wilson and Mongin 2018)]. This swelling is counteracted by the compensatory release of inorganic and small organic anions (Cl−, bicarbonate, glutamate, and aspartate) and a variety of small zwitterionic and uncharged organic molecules (e.g., taurine, glutamine, GABA, myo-inositol) [reviewed in (Strange et al. 1996; Okada 1997; Nilius et al. 1997)]. However, because several of the released compounds are excitotoxins, this intrinsically protective mechanism poses a danger for the brain (Mongin and Kimelberg 2005a; Kimelberg 2005; Mongin 2016). The broad-spectrum anion channel blocker tamoxifen and the more selective VRAC inhibitor DCPIB reduce ischemic brain damage in rodent and canine models of stroke by as much as 70–80% (Kimelberg et al. 2000; Kimelberg et al. 2003; Zhang et al. 2008; Boulos et al. 2011).
The promising pharmacological findings in animal stroke models strongly support the idea of VRAC as a viable therapeutic target in cerebral ischemia (Kimelberg 2005; Mongin 2007; Mongin 2016). Yet, there are two “chinks in the armor” of the existing experimental evidence for this hypothesis. One caveat is related to the limited selectivity of existing VRAC blockers. Tamoxifen and other broad spectrum Cl−/anion channels inhibitors poorly discriminate between VRAC, other Cl− channels, and connexin and pannexin hemichannels (Ye et al. 2009; Benfenati et al. 2009; Friard et al. 2017) and have multiple off-target effects (Allen et al. 1998; Hardy et al. 1998; Osuka et al. 2001; He et al. 2003). Even the most selective compound, DCPIB, blocks connexins, glutamate transporters, and proton pumps, and activates two-domain K+ channels (Bowens et al. 2013; Minieri et al. 2013; Fujii et al. 2015). The uncertainty associated with the specificity of VRAC blockers could not be addressed due to the second major obstacle: the unknown molecular nature of VRAC (Strange et al. 1996; Okada 1997; Pedersen et al. 2015). This second problem has recently been resolved in two genome-wide siRNA screens in model cell lines. The laboratories of A. Patapoutian and T.J. Jentsch independently discovered that VRAC is formed by the heteromeric assembly of proteins belonging to the leucine-rich repeat-containing family 8 (LRRC8A through LRRC8E), with one of these proteins, LRRC8A, playing an indispensable role in channel function (Qiu et al. 2014; Voss et al. 2014). Our group was the first to uncover the critical role of LRRC8A in forming the endogenous, glutamate-permeable VRAC in brain astrocytes, which are prone to pathological swelling (Hyzinski-Garcia et al. 2014). Furthermore, we found that astrocytes possess multiple functionally distinct LRRC8 heteromers, one of which is preferentially permeable to glutamate vs. other organic osmolytes (Schober et al. 2017).
The impetus for the present work stems from another major question about the role of VRAC in stroke: because VRAC activation requires cytosolic ATP, how can it be active in the ischemic tissue? Metabolic inhibition precludes VRAC opening and, when introduced acutely, causes the rapid rundown of VRAC currents [reviewed in (Strange et al. 1996; Okada 1997; Nilius et al. 1997)]. Because this phenomenon is prevented by the introduction of non-hydrolysable ATP or GTP analogues into the cytosol, the prevailing theory in the field suggests that VRAC opening requires non-hydrolytic ATP binding (Diaz et al. 1993; Jackson et al. 1994; Oike et al. 1994; Oiki et al. 1994). On the surface, these data suggest that VRAC activity cannot be sustained in the ischemic core and is likely severely limited in the ischemic penumbra. In the present work, we performed detailed analyses of the effects of acute metabolic inhibition and 24-h adaptation to anoxia on VRAC-mediated glutamate release in rat astrocyte cultures. Our objective was to resolve the discrepancy between the ATP dependency of VRAC and its purported role in the release of glutamate and excitotoxic damage in the ischemic tissue.
Materials and methods
Ethics statement
All animal procedures in this study were approved by the Institutional Animal Care and Use Committee of Albany Medical College (ACUP #18–05004), and were carried out in strict adherence to the Guide for the Care and Use of Laboratory Animals as adopted by the U.S. National Institutes of Health (https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-Use-of-Laboratory-Animals.pdf).
Materials
All cell culture media, sera, antibiotics, and solution of the protease TrypLE Express were of the Gibco brand and obtained from Thermo Fisher Scientific (Waltham, MA, USA). Radiotracer D-[3H]aspartate (cat. # NET581001MC, specific activity ~55 mCi/mmol) was acquired from Perkin Elmer (Waltham, MA, USA). Seahorse XF Cell Mito Stress assay reagents (cat. # 103015–100) were from Agilent (Santa Clare, CA, USA). 4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (DCPIB, cat. # 1540) and 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059, cat. # 1213) were from Biotechne-Tocris (Minneapolis, MN, USA). Trametinib (GSK1120212, cat. # S2673) was from Selleckchem (Houston, TX, USA). Rotenone (cat. # R8875), sodium cyanide (NaCN, cat. # 205222), 2-deoxy-d-glucose (DDG, cat. # D8375), DNAse I (cat. # D5025), l-glutamine (cat. # 49419), sodium pyruvate (cat. # P2256), and all other reagents and solvents were purchased from Sigma-Millipore (St. Louis, MO, USA), unless otherwise specified, and were of the highest purity available. All reagents were acquired within the last 1–3 years and stored at the temperatures specified by manufacturers, with the exception of rotenone and NaCN, which were ~10-year old and stored at room temperature.
Primary rat astrocyte cultures
Primary rat astrocytes were isolated from newborn (0–2-day old) Sprague Dawley rat pups (RRID: RGD 734476), as described previously (Mongin et al. 2011; Schober et al. 2017). Briefly, brains from pups of both sexes were collected via rapid decapitation, and the cortices were dissected from the meninges and subcortical structures in a sterile tissue culture hood. The tissue was moved into ice-cold optimized minimal essential medium (Opti-MEM, cat. # 31985–070), minced with sharp scissors, and subsequently transferred into a solution of the protease TrypLE (cat. # 12605–028) diluted with Opti-MEM (1:1 v/v). Three enzymatic extractions were performed in the TrypLE solution additionally containing DNAse I (1 mg/mL) at 37oC, with slow stirring. Supernatants from the last two extractions were combined and the dissociated cells were sedimented by brief centrifugation (1.5 min at ~1,000 g) and resuspended in Eagle’s low glucose MEM supplemented with 2 mM glutamine (cat # 11095–080), 10% heat inactivated horse serum (HIHS, cat. # 26050–070), 50 U/mL penicillin and 50 µg/mL streptomycin (P/S, cat. 15140–122). Isolated brain cells were plated at low density (250,000 cells/flask), which promotes astrocytic selection, in MEM+HIHS+P/S on T-75 culture flasks pre-treated with poly-D-lysine (cat. # P6407). Cells were grown for 2–3 weeks at 37°C in a humidified atmosphere containing 5% CO2/balance air. Culture medium was changed twice per week. Confluent cultures contained 95–98% astrocytes, as routinely verified by staining with monoclonal antibodies against the astrocyte cell marker glial fibrillary acidic protein (anti-GFAP, Sigma-Aldrich, cat. no. G3893, RRID: AB 477010).
siRNA knockdown of LRRC8A
To verify the involvement of VRAC in glutamate transport, we downregulated the expression of the indispensable VRAC subunit LRRC8A using an siRNA approach, as described in detail in our previous work (Hyzinski-Garcia et al. 2014; Schober et al. 2017). We used the previously validated by us siRNA construct siLRRC8A_4 (Qiagen, Valencia, CA, USA, cat. # SI01725360) with the target sequence 5′-CTCTACCTGAACCGCAACAAA-3′. As a control, we used the proprietary scrambled AllStars negative control siRNA (Qiagen, cat. # SI03650318). The potency and specificity of the RNAi knockdown was validated by qRT-PCR and additionally tested by comparing the effects of several structurally independent siRNA constructs (Hyzinski-Garcia et al. 2014). Briefly, primary astrocytes were grown to 80–90% confluency and transfected with siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific, cat. # 13778–075). siRNA/Lipofectamine complexes were prepared in Opti-MEM as per the manufacturer’s protocol, further diluted with Opti-MEM to the final siRNA concentration of 50 nM, and added to cultured cells. After 24 h, the transfection medium was replaced with fresh MEM+HIHS+P/S. Changes in the mRNA expression levels were measured after 48 h, and functional assays were performed 96 h post transfection.
qRT-PCR measurements of mRNA expression levels
Relative expression of LRRC8 genes following RNAi knockdown (LRRC8A only) or 24-h anoxia (LRRC8A-E), and the induction of the hypoxia marker VEGFA were measured using quantitative reverse-transcriptase PCR (qRT-PCR) as previously described (Schober et al. 2017). Primary astrocytes, grown on 60-mm Petri dishes, were transfected with siRNA as described above. 48 h after transfection, mRNA species were isolated using an RNAqueous-4PCR kit (Thermo Fisher Scientific, cat # AM1914). The mRNA was immediately transcribed to cDNA using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA, USA, cat. #170–8891). Gene expression was quantified by qPCR using SYBR Green Master Mix (Bio-Rad, cat. # 170–8880) and gene-specific qPCR primers in a CFX96 Real Time PCR set-up (Bio-Rad). The qRT-PCR parameters were: 50°C for 10 min → 95°C for 5 min → 40 cycles (95°C for 10 sec, 60°C for 30 sec) → 95°C for 10 sec → melt curve from 65°C to 95°C with the rate of 0.5°C per 5 sec. Three housekeeping genes, RPL13a, RPS20, and GAPDH, were used for the within-sample normalization of expression levels. All primers used are listed in Table 1.
Table 1.
qPCR primers used in this study
| Target Gene | Qiagen cat no. for qPCR primer |
|---|---|
| LRRC8A | QT01575483 |
| LRRC8B | QT00434805 |
| LRRC8C | QT01583897 |
| LRRC8D | QT00370111 |
| LRRC8E | QT01591352 |
| GAPDH | QT00199633 |
| RPL13a | QT00178675 |
| RPS20 | QT00431333 |
Radiotracer assays of VRAC activity
VRAC activity in astrocytes was measured by tracing the release of the non-metabolizable glutamate analogue, d-[3H]aspartate, as specified previously (Mongin and Kimelberg 2005b; Bowens et al. 2013; Schober et al. 2017). We used two variations of this approach, as briefly described below: (1) the kinetics of swelling-activated glutamate release were resolved in a Lucite perfusion chamber, and (2) integral glutamate release measurements (the bulk release over 10-min period) were performed in a 12-well plate format to increase experimental throughput.
In the Lucite chamber experiments, astrocytes were plated and grown on poly-d-lysine treated 18×18 mm glass coverslips. Cells were loaded overnight in MEM+HIHS+P/S medium supplemented with 2 μCi/mL d-[3H]aspartate, washed from the extracellular radiotracer and culture medium components in Basal isosmotic medium (for composition, see below), and then transferred into a Lucite superfusion chamber. This chamber has a depression on the bottom, which was cut to precisely accommodate the glass coverslip, and a Teflon screw lid that leaves approximately 200 μm of space above the cells. The chamber was perfused with isosmotic or hypoosmotic media at a rate of ~1.2 mL/min, which allows for the complete exchange of medium at least 5 times/min. One-minute superfusate fractions were collected into scintillation vials using an automated fraction collector Spectra/Chrom CF-1 (Religen, Waltham, MA, USA). At the end of the experiment, cells were lysed with a solution of 2% SDS plus 8 mM EDTA. The [3H] content in each sample and cell lysates was measured off-line in a TriCarb 2900 scintillation counter (Perkin Elmer), after adding Ecoscint A scintillation liquid (Atlanta Biologicals, Atlanta, GA, USA, cat. # LS-273). The fractional release rates were analyzed relative to the “retrospectively” calculated total cellular [3H] isotope content at each time point, using an Excel-based computer program.
The Basal isosmotic medium contained (in mM): 135 NaCl, 3.8 KCl, 1.2 MgSO4, 1.3 CaCl2, 1.2 KH2PO4, 10 HEPES, and 10 d-glucose (pH 7.4, osmolarity 290 ±2 mOsm). To induce cell swelling and VRAC opening, astrocytes were exposed to hypoosmotic media, in which NaCl concentration was reduced by 50 mM (“−30% hypoosmotic”, 200 ±2 mOsm), 80 mM (“−50% hypoosmotic”, 145 ±2 mOsm), or 113 mM (“−70% hypoosmotic”, 87 ±2 mOsm). All osmolarity values were verified with a freezing-point µOsmette osmometer (Precision Systems, Natick, MA, USA).
In the plate efflux experiments, cells grown in poly-d-lysine treated 12-well plates were loaded overnight with 1 μCi/mL d-[3H]aspartate in MEM+HIHS+P/S. Prior to the initiation of efflux experiments, cells were washed from extracellular radiotracer and culture media with isosmotic Basal medium and preincubated in fresh Basal for 10 min at 37oC. After preincubation, Basal medium was replaced with pre-warmed LiCl-containing isosmotic media (for composition, see below). NaCl was substituted with LiCl to prevent the re-uptake of d-[3H]aspartate through the Na+-dependent glutamate transporter GLAST (Bowens et al. 2013). To measure VRAC activity, we replaced LiCl media in each well at 10-min intervals with either isosmotic or hypoosmotic media as specified in the figure legends. All steps were performed at 37oC. The fractional d-[3H]aspartate release rates were calculated by comparing [3H]radiotracer amounts in each 10-min fraction to the total isotope remaining in cells at the end of the incubation period, as described above. The remaining isotope at the end of experiment was extracted by lysing the cells in 2% SDS + 8 mM EDTA.
The isosmotic LiCl medium contained 120 mM LiCl, and the remaining components were the same as in Basal (pH 7.4 adjusted with Trizma, osmolarity 290 ±2 mOsm). In the hypoosmotic LiCl media, the LiCl concentrations were reduced to achieve the desired osmolarities of 200, 145 and 87 mOsm, which were verified with a µOsmette osmometer.
Luciferin-luciferase ATP assays
As an index of metabolic state, intracellular ATP levels ([ATP]i) were measured after exposure of astrocytes to isosmotic media containing various metabolic inhibitors. Cells plated on poly-d-lysine-treated 24-well plates were grown to confluency in MEM+HIHS+P/S. They were washed from culture media components with Basal medium, and then exposed for 20 min to Basal or glucose-free Basal medium with or without inhibitors, as indicated. [ATP]i were determined using an ATP-lite Luminescence ATP Detection Assay Kit (Perkin Elmer, cat.# 6016943) according to the manufacturer’s protocol and a Victor plate reader (Perkin Elmer) for quantification of bioluminescence signal. Intracellular ATP content was calibrated against the ATP calibration curve prepared on the same day and subsequently normalized to protein content determined using a Bicinchoninic (BCA) Assay Kit (Thermo Fisher Scientific/Pierce, cat. # 23227). Because calibrated [ATP]i values in control cells somewhat varied between experimental days, the effects of metabolic inhibitors were normalized to the within-experiment control values obtained on the same experimental plate.
Seahorse Mito Stress Test Assay
To validate the effects of mitochondrial inhibitors on metabolic state, we measured rates of astrocytic respiration using an Agilent Seahorse XFp setup and a Seahorse XF Cell Mito Stress Test Kit. Astrocytes were plated and grown to confluency in Seahorse XFp Cell Culture Miniplates, and the Cell Mito Stress assay was performed according to the manufacturer’s protocol. Before the assay, cells were preincubated in Agilent Seahorse XF Base medium containing 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine (pH 7.4). They were subsequently exposed to the commercial reagents of the Cell Mito Stress kit (oligomycin, FCCP, and antimycin A plus rotenone). In the test samples, the commercial mix of antimycin A plus rotenone was replaced with NaCN or rotenone that were used in all functional assays, as specified in figures and figure legends. The acquired oxygen consumption rate values were normalized to the protein content in each well, which was determined using a BCA Assay kit as described in the prior section.
Western Blotting
The efficacy of MAP kinase inhibitors, Trametinib or PD98059, was validated by visualizing the levels of phosphorylated Erk1/2 using Western blotting. Confluent astrocyte cultures grown in 60-mm Petri dishes, were washed from cell culture media with Basal medium, pretreated with inhibitors or vehicle, and exposed to Basal or hypoosmotic media as specified in the Results section. Cells were lysed in the solution of 2% SDS + 8 mM EDTA supplemented with phosphatase and protease inhibitor cocktails (ThermoFisher Scientific). Lysates were further diluted with the reducing 4× Laemmeli buffer and proteins were separated using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE, 10% acrylamide-polyacrylamide). Separated proteins were transferred to a nitrocellulose membrane. The membranes were blocked in either 5% BSA, 0.1% Tween 20 in PBS for probing pErk1/2, or 3% milk in 0.1% Tween-20 in PBS for the total Erk1/2 assays. Blocked membranes were incubated with primary antibody overnight at 4°C. Rabbit polyclonal pErk1/2 Thr202/Tyr204 antibody (Cell Signaling Technology, Danvers, MA, U.S.A, cat. # 9101, RRID: AB 331646) was used at 1: 500 dilution. The total Erk1/2 immunoreactivity was probed with rabbit polyclonal p44/42 MAPK (Erk1/2) ERK antibody (Cell Signaling, cat. # 9102, RRID: AB 330744) at 1:1000 dilution. After washes, the membranes were probed with the secondary horseradish peroxidase-conjugated goat anti-rabbit IGG antibody (Jackson ImmunoResearch laboratories, West Grove, PA, U.S.A., cat. # 111–035-003, RRID: AB 2313567) at 1:10,000 dilution in matching blocking solution for 1 h. Immunoreactivity was visualized using the BioRad Clarity ECL reagent (cat. # 170–5061) in a LAS-3000 imager (Fujifilm, Tokyo, Japan). Band intensities were quantified by a densitometric analysis software Multi Gauge 3.0 (FujiFilm, RRID: SCR 014299).
Anoxia treatment
To determine the long-term effects of hypoxia or anoxia on VRAC activity and expression levels of VRAC subunits, we exposed cultured astrocytes to experimental anoxia. Confluent astrocyte cultures grown in 12-well plates or 60-mm Petri dishes were placed into an anoxia incubator chamber (STEMCELL Technologies, Cambridge, MA, USA). Oxygen and CO2-containing atmosphere was purged with 100% nitrogen gas, and the chamber was sealed and incubated for 1 h at 37°C in a thermostated incubator. After 1 h, the chamber was purged with N2 gas again to remove any residual oxygen from the cell culture media, resealed, and returned to the incubator for an additional 23 h. After completion of the 24-h incubation period, cells were removed from the chamber and either immediately (within 3–5 min) lysed or subjected to functional VRAC activity plate assays at normal partial O2 pressure as described above. The glutamate efflux assays were completed within 20 min of removing the cells from the hypoxia chamber. As a positive control for anoxic exposure, we measured mRNA expression levels of the Hif-1alpha (HIF1A)-dependent gene, vascular endothelial growth factor A (VEGFA).
Statistical analyses
All data are presented as means ±SEM with the number of independent experiments indicated in figure legends. An independent experiment in this study represents an independent assay in independently plated and treated cells. The group numbers were selected empirically, based on variability in our prior work. They were 4–6 independent experiments per group for superfusion efflux assays and 3–4 experiments for all assays performed in a plate format. All plate assays were run in triplicate or quadruplicate, and their results were averaged within experiment (n=1) and further statistically compared as described above. Generally, this approach allows for detection of 25% difference at 80% power and the 0.05 significance level, however no specific sample size calculations have been performed. To account for the variability between cell cultures, all experiments (with the exception of those presented in Fig. 5C and 8) were performed in at least two different astrocyte cultures, which were prepared on a monthly basis. No data were excluded from statistical analyses. In most cases, statistical differences between groups were analyzed using Student’s t-test or a one-way ANOVA, followed by a Bonferroni post hoc test for multiple comparisons. Normalized expression and [ATP]i values were compared against unity (control) using Student’s one-sample t-test and a Bonferroni post hoc test for multiple comparisons according to the formula p*=1-(1-p)n, where p* represents the p value corrected for multiple comparisons and n stand for the number of comparisons. For glutamate efflux assays, the analysis was broken down into several components. We compared (1) maximum release values under hypoosmotic conditions, which represent maximal VRAC activity in swollen cells, and (2) the total integral hypoosmotic release (area under the curve), which reflects compounded glutamate release via VRAC. All statistical analyses were performed using Prism 5.0 (GraphPad Software, San Diego, CA, USA; RRID: SCR 002798), and graphing was done in Origin 8.1 (OriginLab, Northampton, MA, USA; RRID: SCR002815).
Results
Astrocytic VRAC activity is determined by [ATP]i, which can be fully sustained through glycolysis
VRAC opening and sustained activity require non-hydrolytic binding of cytosolic ATP in multiple cell types [reviewed in (Strange et al. 1996; Okada 1997; Nilius et al. 1997)]. Therefore, we started by testing the effects of metabolic inhibition on VRAC-mediated glutamate release in primary astrocytes.
To determine the parameters of metabolic inhibition, we first measured the intracellular ATP levels in astrocytes which were exposed to inhibitors of glycolysis and mitochondrial respiration. To block glycolysis, we transferred astrocytes into glucose-free medium supplemented with the non-metabolizable glucose analogue, 2-deoxy-d-glucose (DDG, 10 mM). After a 20-min incubation, the [ATP]i was reduced by ~65% (Fig. 1a). To accomplish complete metabolic inhibition, we combined DDG with the complex IV respiratory chain blocker NaCN (2.5 mM). This combinatory treatment reduced [ATP]i by 90% (Fig 1a). Surprisingly, treatments with NaCN or another respiratory chain inhibitor, the complex I blocker rotenone (5 μM), did not affect [ATP]i (Fig. 1a).
Fig. 1. Effect of metabolic inhibition on intracellular ATP levels and swelling-activated glutamate release in rat astrocyte cultures.

(a) Astrocytic ATP content was quantified using a luciferin/luciferase assay. Cells were exposed to (i) control Basal medium, (ii) glucose-free Basal medium containing 10 mM 2-deoxy-D-glucose (DDG), (iii) 2.5 mM sodium cyanide (NaCN) in the presence of glucose, (iv) 5 µM rotenone (Roten.) in the presence of glucose, or (v) glucose-free Basal medium with DDG and NaCN (DDG+NaCN). Data are the normalized mean values ±SEM of 3–4 independent experiments in two astrocyte cultures. **p<0.01, ***p<0.001, vs. Control. (b) Kinetics of swelling-activated d-[3H]aspartate release in astrocytes exposed to the same metabolic conditions as in (a). Data are the mean values ±SEM of 5–6 experiments per group performed in two astrocyte cultures. ***p<0.001, maximal release values vs. Control; ##p<0.01, ###p<0.001, integral 20-min release values vs. Control. (left inset) Graphical representation of the metabolic processes that were targeted in the presented experiments.
Because both mitochondrial inhibitors showed no impact on [ATP]i, we tested their effects on mitochondrial respiration, which was measured as oxygen consumption using a Seahorse Analyzer. We compared the actions of the commercially available Mito Stress Test inhibitors, antimycin A plus rotenone, to the effects of our NaCN and rotenone, which were used in the [ATP]i assays presented above. The Cell Mito Stress Test allowed for measurement of the baseline cellular respiration, the ATP-dependent respiration (sensitive to the ATP synthetase inhibitor oligomycin), the maximal respiration rate (after uncoupling electron transport from ATP synthesis with the protonophore FCCP), and the residual non-mitochondrial respiration after mitochondrial electron transport chain and oxygen consumption are completely suppressed (following treatment with the combination of the complex I inhibitor rotenone and the complex III inhibitor antimycin A). These results, presented in Fig. 2, show that the rotenone and NaCN used in ATP experiments suppressed mitochondrial function as effectively as the standard commercial mix of rotenone and antimycin A.
Fig. 2. Validation of the efficacy of mitochondrial metabolic inhibitors.

The efficacy of the mitochondrial blockers, cyanide and rotenone, was confirmed using a Seahorse Cell Mito Stress Test in an Agilent Seahorse XFp setup. Oxygen consumption rate (OCR) in astrocytes was measured upon addition of indicated inhibitors and mitochondrial respiration was blocked using the same agents (2.5 mM NaCN or 5 µM rotenone) as in ATP and VRAC activity assays. As a positive control we used combination of antimycin A and rotenone from the Cell Mito Stress kit. Data are the mean values ±SEM of six independent wells in three independent cell preparations. (top inset) Graphical representation of mitochondrial processes targeted by the Cell Mito Stress Test components. Abbreviations: Ant. A, antimycin; FCCP, mitochondrial uncoupling agent; NaCN, sodium cyanide; Oligo., oligomycin; Rot, rotenone.
Next, we explored how metabolic inhibitors modulate VRAC activity in hypoosmotically swollen astrocytes exposed to a 30% reduction in medium osmolarity, which is in the typical stimulus range for activation of astrocytic VRAC [e.g., (Olson and Li 1997; Abdullaev et al. 2006; Liu et al. 2006; Ramos-Mandujano et al. 2007)]. Consistent with prior studies, cell swelling caused prompt (peaking within 2–3 min) and dramatic (~20-fold) elevation in glutamate release rates, which was followed by a gradual decay over the next 20 min, likely reflecting cell volume regulation (Fig. 1b). The return to isosmotic conditions reversed glutamate release to pre-hypoosmotic values. When astrocytes were completely metabolically inhibited, swelling-activated glutamate release was fully suppressed (Fig. 1b). DDG alone, which is expected to completely halt glycolysis and starve mitochondria of their major metabolite pyruvate, inhibited volume-sensitive glutamate efflux nearly completely (>95%) despite the fact that intracellular ATP levels were around 35% of their control values (compare Fig 1a and 1b). In contrast, when mitochondrial function was inhibited by adding NaCN or rotenone, glutamate release was either not affected or strongly potentiated, respectively (Fig. 1b). Potential reasons for the stimulatory actions of rotenone are provided in the Discussion.
To determine to what extent our astrocytes could sustain their [ATP]i using glycolytic flux alone, we exposed cells to media containing NaCN, to inhibit mitochondrial function, and decreasing concentrations of extracellular glucose. Surprisingly, even in the presence of sodium cyanide, a 100-fold drop in the extracellular glucose levels (from 10 to 0.1 mM) did not reduce cytosolic [ATP] (Fig 3). However, the complete removal of glucose and supplementation with DDG, caused the precipitous inhibition of cellular metabolism (Fig. 3). Because we were not able to titer the [ATP]i while varying extracellular glucose, we did not pursue functional analyses of VRAC activity in low-glucose media.
Fig. 3. Effect of decreasing concentrations of glucose on intracellular ATP levels in primary rat astrocytes treated with the mitochondrial inhibitor sodium cyanide.
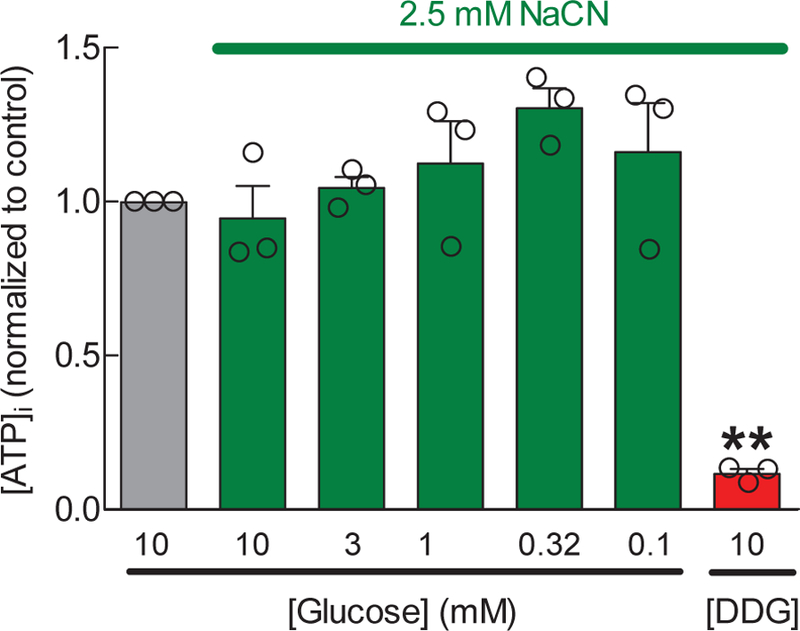
Astrocytic ATP content was quantified using a luciferin/luciferase assay in cells that were exposed for 20 min to 2.5 mM sodium cyanide and various concentrations of glucose, as shown. Data are the normalized mean values ±SEM of 3 experiments. DDG+NaCN data are reproduced from the experiments in Fig. 1 for reference purposes. **p<0.01, vs. Control.
Glucose-deprived astrocytes cannot sustain [ATP]i and VRAC function using mitochondrial substrates pyruvate and glutamine
Based on the above-mentioned results, it became apparent that cultured rat astrocytes can effectively cover their energetic demands via glycolysis and require nearly normal ATP levels for sustaining VRAC function. Next, we wanted to test if VRAC activity can be decoupled from glycolytic ATP flux by circumventing glycolysis with the addition of the mitochondrial substrates, pyruvate and/or glutamine (see mechanistic inset in Fig. 4). Astrocytes were preincubated for 20 min in glucose-free media plus DDG and additionally supplemented with 5 mM pyruvate or 5 mM L-glutamine or their combination. As compared to the glucose-free media with DDG, alternative mitochondrial substrates, when given individually, increased [ATP]i, to ≥50% of control levels (Fig. 4a). The supplementation with pyruvate recovered ATP levels to almost 75% of control (Fig. 4a) but caused only a partial rescue of the swelling-activated glutamate release (to 30% of the hypoosmotic controls, Fig. 4b). The combination of pyruvate and glutamine did not show additivity in rescuing [ATP]i or swelling-activated glutamate release (Fig. 4a, b). In sum, these experiments suggest that VRAC activity requires near normal ATP levels (>80% of control values), and that ATP production in astrocytes can be sustained by glycolysis.
Fig. 4. Are astrocytic mitochondria capable of sustaining VRAC activity without glycolytic flux?

(a) Intracellular ATP levels in cells exposed to control Basal medium, or glucose-free media containing 10 mM DDG, with or without 5 mM sodium pyruvate, 5 mM L-glutamine, or combination of pyruvate and glutamine. Data are the means ± SEM of 3–5 experiments per group in two astrocytic cultures, normalized to control ATP levels. *p<0.05, vs. control. (b) Kinetics of swelling-activated d-[3H]aspartate release in astrocytes exposed to the same conditions as in (a). Data are the means ± SEM of 6–7 experiments per group performed in two different astrocytic cultures. ***p< 0.001, maximal release values vs. control; ###p < 0.001, integral 20-min release values vs. control. +p<0.05, maximal release values vs. DDG. (left inset) Graphical representation of the relevant metabolic processes. Abbreviations: DDG, 2-deoxy-d-glucose; Gln, glutamine. αKG, α-ketoglutarate; Pyr, pyruvate; TCA, the tricarboxylic acid cycle.
Swelling-activated glutamate release does not require activity of MAP kinases
Present findings clearly demonstrate that VRAC activity requires cytosolic ATP. One publication suggested that hypoosmotic activation of VRAC currents requires MAP kinase cascade activity, since it was completely blocked by the MEK inhibitor PD98059 (Crepel et al. 1998). To test if this is the case for swelling-activated glutamate release, we probed the effects of the selective MEK kinase inhibitors, PD98059 and Trametinib (Alessi et al. 1995; Gilmartin et al. 2011). We first measured the effects of the MEK inhibitors on intracellular ATP levels. Trametinib (0.1 μM) did not alter [ATP]i (Fig. 5a). The effects of PD98059 (20 and 50 µM) could not be determined accurately because they interfered with the luciferase assay giving artificially low ATP readings (Supplemental Fig. 1). To confirm the efficacy of Trametinib and PD98059, we performed Western blotting analysis of phosphorylation of the downstream Erk1/2 kinases in control and hypoosmotic media. Serum-treated cells were used as a positive control. Under all conditions, Trametinib and PD98059 strongly reduced Erk1/2 phosphorylation, with Trametinib being more efficacious (Fig. 5b). Full Western blot images and additional loading controls (β-actin) are presented in Supplemental Fig. 2. In VRAC activity assays, 0.1 µM Trametinib or 20 µM PD98059 were completely ineffective in inhibiting swelling-activated glutamate release (Fig. 5c and Supplemental Fig. 3, for Trametinib and PD98059, respectively). These results indicate that in primary rat astrocytes, VRAC-mediated glutamate release does not require MAP kinase activity. The potential reasons for the discrepancy between our findings and the results of the prior electrophysiology experiments are addressed in the Discussion.
Fig. 5. MAP kinase phosphorylation is not required for VRAC activity.

(a) Intracellular ATP levels in cells exposed to control Basal medium, Basal medium +vehicle (0.05% DMSO), or 0.1 µM Trametinib (Tram.). Data are the means ± SEM of 3 different experiments per group in two astrocytic cultures, normalized to control ATP levels. (b) Western blot analysis of Erk1/2 activation in Basal, 30% hypoosmotic, or Basal medium supplemented with 10% FBS. Cells were pretreated with vehicle, 0.1 µM Trametinib (TR), or 20 µM PD98059 (PD) for 20 min and then exposed to Basal, hypoosmotic, or serum-containing media supplemented with the same inhibitors, as indicated. Erk1/2 phosphorylation was quantified as the ratio of pErk1/2 to total Erk immunoreactivity in the same lysates. Data are the means ± SEM of three Western blot analyses in two cell culture preparations. *p<0.05, **p<0.01, vs. no inhibitor under the same conditions. Insets show representative immunoreactivity images for pErk1/2 and total Erk1/2. Molecular weight standard in the first lane =37 kD. (c) Kinetics of swelling-activated d-[3H]aspartate release in astrocytes pretreated with vehicle or 0.1 µM Trametinib and perfused with isosmotic and hypoosmotic (HYPO) media. Data are the means ± SEM of 4 experiments per group performed in one astrocytic culture.
Metabolic inhibition of VRAC activity can be overridden by strong cell swelling
A few electrophysiology studies found an exception for metabolic inhibition of VRAC: strong cell swelling could override the metabolic block of VRAC activity, at least in select cell types – the human lung H69AR cell line, rat kidney IMCD cells, and mouse N1E115 neuroblastoma cells (Jirsch et al. 1994; Volk et al. 1996; Bond et al. 1999). Therefore, we tested if a similar phenomenon exists in rat astrocytes and is applicable to swelling-activated glutamate release. In medium-throughput plate assay experiments, we compared side-by-side swelling-activated d-[3H]aspartate efflux from astrocytes exposed to 30%, 50%, or 70% reductions in medium osmolarity. Consistent with the results presented in Fig. 1, complete metabolic inhibition (DDG + NaCN) eliminated glutamate release from moderately swollen cells (−30% hypoosmotic medium, central panel in Fig. 6). In cells exposed to a larger degree of hypoosmolarity (−50% hypoosmotic medium), the trend for metabolic inhibition was largely preserved. However, when medium osmolarity was reduced by 70%, swelling-activated glutamate release from metabolically-inhibited cells was almost rescued (central panel in Fig. 6).
Fig 6. Dramatic cell swelling overrides the metabolic block of VRAC activity.

(central panel) Effects of complete metabolic inhibition on d-[3H]aspartate release in astrocytes exposed to isosmotic medium or various degrees of hypoosmolarity (−30%,−50%, or −70%). Cells were incubated in control glucose-containing media (black bars), or glucose-free media supplemented with 10 mM DDG and 2.5 mM NaCN (red bars). The results are the mean 10-min integral release values ±SEM of 6 experiments per group performed in two different astrocytic cultures. **p <0.01, ***p <0.001, vs isosmotic conditions. ###p < 0.001, metabolically competent vs. metabolically inhibited cells in media with the same osmolarity. (left inset) Molecular biology evidence for VRAC involvement in glutamate release from cells exposed to moderate swelling (−30%). Astrocytes were transfected with negative control siRNA (siNC) or the siRNA targeting the essential VRAC subunit LRRC8A (siLRRC8A). Data are the mean values ±SEM of 3 independent transfections in two different astrocytic cultures. ***p < 0.001, maximal release values vs. siNC. ##p<0.01, integral release values, vs. siNC. (right inset) Molecular biology evidence for VRAC involvement in glutamate release from metabolically inhibited cells exposed to dramatic cell swelling (70% reduction in medium osmolarity). Astrocytes were transfected with siNC or siLRRC8A. Composition of the hypoosmotic media and concentrations of metabolic inhibitors were the same as in central panel. Data are the mean values ±SEM of 4–6 experiments per group performed in two different astrocytic cultures. ***p<0.001, maximal release values vs. siNC. ###p<0.001 integral release values vs. siNC.
Because extreme cell swelling liberates nearly 90% of the preloaded d-[3H]aspartate (central panel in Fig. 6), we were concerned with (a) the possibility of osmotic cell lysis and non-specific changes in membrane permeability, (b) the prospect of activation of alternative, non-VRAC release mechanisms, and (c) the potential ceiling effect of the excessive cell swelling on VRAC activity. To address these concerns, we repeated these experiments in metabolically-inhibited cells using a Lucite chamber perfusion assay, which captures the release values with high temporal resolution and accounts for changes in the intracellular isotope content. We also implemented knockdown of the indispensable VRAC subunit, LRRC8A, to probe for VRAC involvement. As a positive control, we tested the effect of LRRC8A knockdown on the swelling-activated glutamate release with 30% reduction in media osmolarity and 10 mM glucose. The control VRAC activity was reduced in siRNA-treated cells by ≥70% (see left panel in Fig. 6). The incomplete inhibition of release is explained by the <100% downregulation of the LRRC8A mRNA and relatively slow turnover of the LRRC8A protein (Hyzinski-Garcia et al. 2014). We next superfused metabolically-inhibited cells with −70% hypoosmotic medium. In the cells, which were treated with negative control siRNA and subjected to metabolic stress, we found dramatic swelling-activated d-[3H]aspartate efflux. In the cells treated with LRRC8A-siRNA, this response was inhibited by >90% (right panel in Fig. 6). These results unequivocally demonstrate that extreme cell swelling activates glutamate release via VRAC and that the contribution of cell lysis and/or non-VRAC pathways is negligible.
Effect of 24-h anoxia on VRAC activity and expression of VRAC subunits
Our next experimental question was whether protracted periods of anoxia trigger adaptive changes in the expression levels of VRAC subunits and VRAC activity. Such changes would be relevant to modulation of extracellular glutamate levels during reperfusion and/or recovery after an ischemic episode. To explore any potential effects, we exposed astrocytes, grown in glucose-containing culture media, to 24-h anoxia at 37oC and then measured changes in LRRC8 subunit expression levels and VRAC activity. In the experiments presented in Fig. 7a, we reproduced our previously published data exploring the relative expression of all five LRRC8 subunits in cell cultures utilized in the present work. Consistent with our prior report (Schober et al. 2017), we found very similar, practically stochiometric expression of LRRC8A, LRRC8B, LRRC8C, and LRRC8D mRNA species, while the abundance of LRRC8E mRNA was approximately 20-times lower. After exposure to 24-h anoxia, the expression levels for LRRC8A were significantly reduced by 45% (p=0.019). There was a weak trend for reduction for LRRC8B (−33%, p=0.157) and LRRC8C (−29%, p=0.071), and no meaningful difference for LRRC8D (−25%, p=0.358) and LRRC8E (−7%, p=0.713) (Fig. 7b). To confirm that cells were responding to the anoxic conditions, we measured in the same samples the expression of the HIF1A-dependent gene VEGFA, which was upregulated by >20-fold (right panel in Fig. 7b). To further test if anoxia causes changes in VRAC activity, either via translation-dependent or independent mechanisms, we measured the swelling-activated release of D-[3H]aspartate within 20 min after the removal of cells from the anoxia chamber. As shown in Fig. 8, 24-h anoxia had minimal impact on VRAC activity: we found <10% inhibition of radiotracer release in radiotracer efflux assays, which was statistically significant but unlikely to be physiologically important.
Fig. 7. Effect of 24-h anoxia on expression of LRRC8 subunits.

(a) Relative abundance of mRNA species for five members of the LRRC8 family (LRRC8A-E) in cultured rat astrocytes, quantified using qRT-PCR. Data are the mean values ± SEM normalized to the housekeeping gene ribosomal RPL13a and then LRRC8A within the same samples in four astrocyte cultures. (b) Changes in the LRRC8A-E subunit expression following 24-h anoxia normalized to expression of the same gene in control (normoxic) cells (∆∆Ct values). and validation of anoxia-induced gene expression changes in the same cells. Expression of the HIF-1 dependent gene VEGFA hypoxia/anoxia marker was quantified immediately after 24 h anoxia. Data are the mean values ± SEM from 4 experiments. *p<0.05 vs. normoxic control.
Fig. 8. Effect of 24-h anoxia on VRAC-mediated glutamate release.
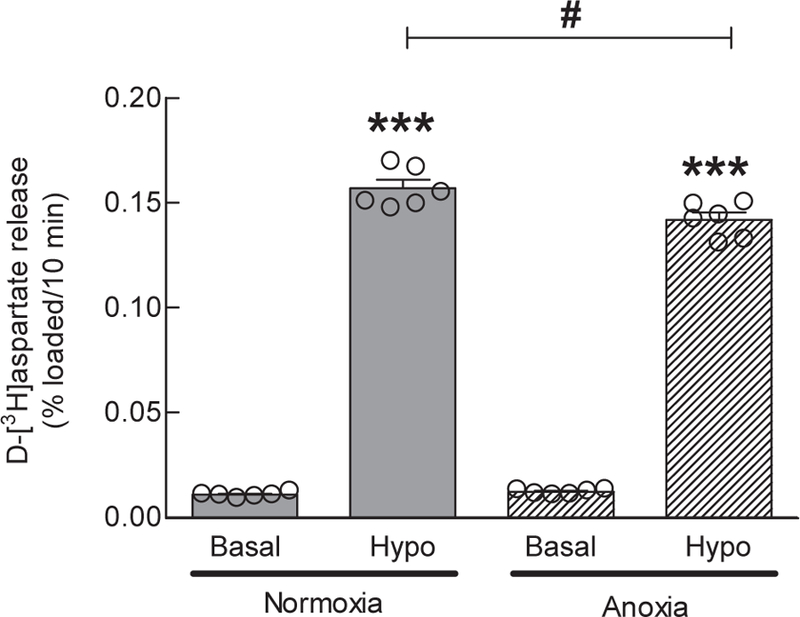
Swelling-activated release of D-[3H]aspartate was measured after 24-h anoxia, by exposing cells to 30% hypoosmotic media immediately after removal of cells from the anoxia chamber. Data are the mean 10-min integral release values ±SEM of 6 assays in one astrocytic culture. ***p<0.001, Hypo vs. Basal. #p<0.05 Hypo anoxia vs. Hypo normoxia.
Discussion
The objective of the present work was to reconcile the purported role of VRAC in ischemic brain damage with the known dependence of this channel on intracellular ATP. If VRAC cannot be open without cytosolic ATP, then how can its activity be sustained in stroke tissue? The main findings of this study are: (1) Complete metabolic inhibition (chemical ischemia) abolishes swelling-activated glutamate release through VRAC; (2) Metabolically compromised astrocytes can sustain their [ATP]i and VRAC function via glycolysis even when mitochondrial respiration is blocked; (3) Remarkably, the dependence of VRAC-mediated glutamate release on [ATP]i, is not absolute: it can be overridden when cells are exposed to extreme degrees of cell swelling. As discussed below, we believe that our present results are highly instructive for understanding how VRAC activity and VRAC-mediated glutamate release are modulated in the ischemic tissue and sustained in the ischemic penumbra.
VRAC-mediated glutamate release requires high cytosolic ATP levels
The requirement of intracellular ATP for VRAC function is a well-established concept in the field (Strange et al. 1996; Okada 1997; Nilius et al. 1997). This signature channel property was initially identified in diverse cell types – HeLa, glioma C6, intestinal epithelial, and endothelial cells (Diaz et al. 1993; Jackson et al. 1994; Oike et al. 1994; Oiki et al. 1994), and subsequently reproduced in many other primary and immortalized cells. The majority of the prior studies found that VRAC cannot be activated in cells which are metabolically depleted, and that acute metabolic inhibition leads to the rapid rundown of VRAC currents. This phenomenon is also characteristic for different classes of neural cells, including malignant glial C6 cells (Jackson et al. 1994), cerebellar granule neurons and neuroblastoma N1E115 cells (Patel et al. 1998; Bond et al. 1999), and microglia (Schlichter et al. 2011). As shown in our prior work in primary rat astrocytes, in the present study, and publications from the K. Strange laboratory, swelling-activated release of organic osmolytes, including taurine, myo-inositol, and excitatory amino acids, is also abolished by metabolic inhibition [e.g., (Jackson et al. 1994; Rutledge et al. 1999) and Fig. 1]. The congruency between metabolic dependency of swelling-activated Cl− fluxes and organic osmolyte release is consistent with the idea that both processes are mediated by the same pathway. Recent molecular biology studies identified proteins belonging to the LRRC8 family as components of the VRAC channel, which is permeable to both Cl− and various organic osmolytes (Qiu et al. 2014; Voss et al. 2014; Hyzinski-Garcia et al. 2014). Emerging work points to the existence of functionally distinct LRRC8 heteromers, which are either (a) preferentially responsible for the conductance of Cl− and the negatively charged aspartate and glutamate, or (b) favor the movement of uncharged or the net-neutral osmolytes, such as myo-inositol, taurine, and GABA (Planells-Cases et al. 2015; Lutter et al. 2017; Schober et al. 2017).
Despite extensive work on metabolic dependence, very few studies have evaluated the quantitative [ATP]i requirements of VRAC activity. In early electrophysiological experiments performed in intestinal 407 cells, the minimal [ATP] in the pipette solution needed to sustain VRAC activity was 10 µM, but this [ATP]i threshold strongly varied with changes in intracellular [Mg2+] (Oiki et al. 1994). In mouse neuroblastoma N1E115 cells, the half-maximal [ATP]i for VRAC activation was 0.32 mM (Bond et al. 1999). In transformed rat C6 glioma cells treated with increasing concentrations of metabolic blockers, the half-maximal [ATP]i for swelling-activated release of [3H]taurine and myo-[3H]inositol was ~1.7–1.8 mM, or ~45% of the “normal” [ATP]i ≈ 4 mM (Jackson et al. 1994). In our prior work in primary rat astrocytes, the estimated EC50 for ATP sustaining swelling-activated d-[3H]aspartate efflux was ~3.5 mM, or ~70% of the average control [ATP]i values calculated to be ~5 mM (Rutledge et al. 1999). In the present study, the apparent EC50(ATP) for VRAC activity was even higher. Replacement of carbohydrates with pyruvate and/or glutamine restored [ATP]i to 50–70% of control values but was not sufficient for the half-maximal activation of glutamate release via VRAC (Fig. 4).
Can VRAC-mediated glutamate release be sustained in the ischemic penumbra?
Keeping in mind that high levels of intracellular ATP are needed for VRAC activation, one may doubt whether astrocytes can sustain swelling-activated glutamate release in the ischemic penumbra. In the penumbra, the levels of blood flow and the derived glucose and oxygen supply are reduced to a mere 20–30% of pre-ischemia levels (Astrup et al. 1981; Hossmann 1994). Although diverse VRAC blockers suppressed glutamate release in the penumbral tissue (Phillis et al. 1997; Feustel et al. 2004; Zhang et al. 2008), the selectivity of previously tested compounds has been called into question (Ye et al. 2009; Bowens et al. 2013). The present study provides a potential explanation for how astrocytic VRAC can still be active in the penumbral tissue. In our in vitro model, astrocytes effectively covered their energetic needs and sustained full VRAC activity with completely blocked mitochondrial respiration and media glucose levels as low as 0.1 mM (i.e., 100-fold reduction from control values, see Fig. 3). In line with our findings, genetic ablation of astrocytic mitochondrial respiration in vivo, in the inducible COX10 knockout mice, does not modify morphology and function of astrocytes and related cerebellar Bergman glia and does not produce signs of neurodegeneration or inflammation for up to one year after COX10 deletion (Supplie et al. 2017). Interestingly, our attempt to replace glycolytic flux of ATP by providing the mitochondrial substrates pyruvate and/or glutamine did not succeed in fully restoring [ATP]i and VRAC activity (Fig. 4). Taken together, the present and the previously published data support the idea of the high glycolytic potential of astroglia and explain why astrocytic VRAC can be active and contribute to glutamate release in the ischemic penumbra.
Granted, our present in vitro approach is rather simplistic and does not take into account other biochemical processes that occur in the penumbra. For example, reactive oxygen and nitrogen species, which are produced in the penumbral tissue and during reperfusion, synergize with cell swelling in triggering astrocytic glutamate release in vitro and in vivo (Haskew et al. 2002; Haskew-Layton et al. 2005; Haskew-Layton et al. 2008). In the present study, this can be indirectly seen with the inhibitor of mitochondrial complex I, rotenone, which stimulated rather than inhibited astrocytic VRAC (Fig. 1b). The likely explanation for this phenomenon is the known increase in the mitochondrial production of ROS by rotenone (Li et al. 2003; Lopez-Fabuel et al. 2016). Furthermore, thrombin, bradykinin, adenosine, and ATP, which are all known to be liberated in ischemia, can also potentiate VRAC activity in swollen cells (Mongin and Kimelberg 2002; Mongin and Kimelberg 2005b; Ramos-Mandujano et al. 2007; Rudkouskaya et al. 2008; Liu et al. 2009).
Phosphorylation vs. non-hydrolytic ATP binding in the [ATP]i-dependent activation of VRAC
While the importance of ATP in VRAC has been well established, the exact mechanism by which ATP acts on the channel is not agreed upon. The pioneer studies from the groups of F.V. Sepulveda, K. Strange, Y. Okada, and B. Nilius found that swelling-activated Cl− currents require non-hydrolytic ATP binding because VRAC activity could be sustained upon replacement of ATP with non-hydrolysable ATP analogues (Diaz et al. 1993; Jackson et al. 1994; Oiki et al. 1994; Oike et al. 1994). On the contrary, several publications have proposed a role for protein phosphorylation in VRAC activation [reviewed and discussed in (Okada 1997; Nilius et al. 1997; Hoffmann et al. 2009)]. In astrocytes, Crepel et al. found a strong dependence of swelling-activated Cl− currents on the activity of receptor tyrosine kinases and the MAP kinase cascade (Crepel et al. 1998). The present study argues against the necessity of MAP kinase signaling for swelling-activated glutamate release via VRAC. In our hands, the specific MEK kinase inhibitor Trametinib potently blocked the activity of the MAPK cascade (Erk1/2 phosphorylation) without any impact on volume-sensitive glutamate release (Fig. 5). The less potent and specific PD98059 was also ineffective (Supplemental Fig. 3). There are a few potential reasons for the discrepancy between the present data and findings of Crepel et al. PD98059, U0126, and other first generation MEK inhibitors have been reported to interfere with cell metabolism and produce other non-specific effects, complicating the interpretation of their actions (Yung et al. 2004; Dokladda et al. 2005). Another factor to consider is that VRAC activity can be modulated by extracellular signaling molecules such as ATP, adenosine, and bradykinin, which act via GPCR receptors and protein phosphorylation [for astrocyte studies, see (Mongin and Kimelberg 2005b; Akita and Okada 2011). Although GPCR activation cannot open VRAC directly, it can act synergistically with cell swelling, potentially confounding conclusions of electrophysiology studies [reviewed in (Fisher et al. 2008; Franco et al. 2008)]. This question will be definitively resolved by future studies performing mutagenesis of prospective phosphorylation and ATP binding sites.
Paradoxical, ATP-independent VRAC activation
On the surface, complete metabolic inhibition, as would be expected in the ischemic core, should prevent swelling-activated glutamate release in the compromised tissue. The bulk of the literature and our present data argue against VRAC activation in the ischemic core. Yet, there are two lines of in vitro and in vivo evidence which point to the intriguing possibility that VRAC may be activated in an ATP-independent fashion. (1) At least three electrophysiology studies in lung cancer H69AR cell line, kidney IMCD, and neuroblastoma N1E115 cells (Jirsch et al. 1994; Volk et al. 1996; Bond et al. 1999) have found that under select conditions, when medium osmolarity is reduced by ≥50%, VRAC shows incomplete or very weak dependence on [ATP]i. (2) In vivo, microdialysis experiments have revealed that a large fraction of glutamate release in the ischemic core is inhibited by the non-selective VRAC blocker DNDS (Seki et al. 1999). The latter study can be interpreted as indirect evidence for the [ATP]i-independent VRAC activation in vivo. Our present work may shed a light on these unexpected findings.
We are the first to provide the molecular biological evidence for activation of the LRRC8-containing VRAC in metabolically inhibited astroglial cells, or any other cell type. This is critical because metabolic inhibition can activate alternative glutamate permeability pathways, for example, the swelling-activated maxi-Cl− channel [see (Liu et al. 2006) and review (Okada et al. 2018)]. The siRNA knockdown experiments presented in Fig. 6, suggest that VRAC dominates the release from dramatically swollen cells, even when they are completely metabolically depleted. Of course, the extreme degree of cell swelling required in our in vitro experiments to switch from the [ATP]i-dependent to the [ATP]i-independent mode of VRAC activity is hard to envision in vivo. Because brain tissue is confined within the rigid skull, the extracellular space cannot accommodate more than a 20–30% increase in the volume of brain cells [see (Wilson and Mongin 2018) for extended discusussion]. Yet, it is fathomable that such extreme swelling might occur in a cell region-specific manner. A postmortem stereology study found that in the ischemic core, astrocytic endfeet can swell up to 10 times their original size (Ito et al. 2011).
Conclusion
This is the first study to use a molecular approach for exploring the activation of heteromeric LRRC8 channels upon metabolic inhibition in a native cellular environment. Overall, our work in cellular models strongly suggests that two opposing factors – the inhibitory influence of diminishing ATP levels and the stimulatory impact of cellular swelling – are likely determinants of VRAC activity in the ischemic brain. We propose that the ability to sustain energetic metabolism (ATPi levels) is the main constraint for VRAC opening in ischemia. In the penumbra, astrocytes are the most likely contributors for the VRAC-mediated glutamate release because of their propensity to swell and high glycolytic capacity. VRAC activation in the ischemic core is less likely. Yet, the discovery that, at least in vitro, the metabolic block of VRAC can be overridden by significant cell swelling opens an intriguing possibility that a similar mechanism may exist in stroke.
Supplementary Material
Highlights.
In stroke, pathological cell swelling is thought to promote ischemic brain damage via activation of the glutamate-permeable volume-regulated anion channel (VRAC)
Since VRAC activity requires cytosolic ATP, we explored whether metabolic inhibition limits or prevents the VRAC-mediated release of a radiolabeled glutamate analogue
Complete metabolic inhibition with cyanide (NaCN) and 2-deoxy-D-glucose (DDG) abolished the VRAC-mediated glutamate release from rat astrocytes
When only mitochondrial respiration was blocked, astrocytes maintained normal intracellular ATP levels and VRAC activity in a broad range of glucose concentrations
Our study strongly suggests that in the ischemic penumbra, astrocytic VRAC activity can be fully sustained by glycolysis
Acknowledgements
This work was supported in part by NIH grants R01 NS061953, R01 NS111943 (to A.A.M.). and R01 GM124133 (to A.P.A.). The Authors are grateful to Dr. David Jourd’heuil and Frances L. Jourd’heuil for helpful advice on anoxia experiments and Dr. Paul J. Feustel for help with statistical analyses.
Abbreviations used:
- DCPIB
4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid
- DDG
2-deoxy-d-glucose
- FBS
fetal bovine serum
- NaCN
sodium cyanide
- PBS
phosphate-buffered saline
- RRID
Research Resource Identifier (scicrunch.org)
- VEGFA
vascular endothelial growth factor A
- VRAC
volume-regulated anion channel
Footnotes
conflict of interest disclosure
The Authors have no conflict of interest to declare.
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK and Mongin AA (2006) Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in rat cultured astrocytes. J. Physiol 572, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akita T and Okada Y (2011) Regulation of bradykinin-induced activation of volume-sensitive outwardly rectifying anion channels by Ca2+ nanodomains in mouse astrocytes. J. Physiol 589, 3909–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT and Saltiel AR (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem 270, 27489–27494. [DOI] [PubMed] [Google Scholar]
- Allen MC, Newland C, Valverde MA and Hardy SP (1998) Inhibition of ligand-gated cation-selective channels by tamoxifen. Eur. J. Pharmacol 354, 261–269. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjo BK and Symon L (1981) Thresholds in cerebral ischemia - the ischemic penumbra. Stroke 12, 723–725. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Nicchia GP, Rossi A, Dovizio M, Cervetto C, Nobile M and Ferroni S (2009) Carbenoxolone inhibits volume-regulated anion conductance in cultured rat cortical astroglia. Channels (Austin. ) 3, 323–336. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Drejer J, Schousboe A and Diemer NH (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem 43, 1369–1374. [DOI] [PubMed] [Google Scholar]
- Bond T, Basavappa S, Christensen M and Strange K (1999) ATP dependence of the ICl,swell channel varies with rate of cell swelling. Evidence for two modes of channel activation. J. Gen. Physiol 113, 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulos AS, Deshaies EM, Dalfino JC, Feustel PJ, Popp AJ and Drazin D (2011) Tamoxifen as an effective neuroprotectant in an endovascular canine model of stroke. J. Neurosurg 114, 1117–1126. [DOI] [PubMed] [Google Scholar]
- Bowens NH, Dohare P, Kuo YH and Mongin AA (2013) DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol. Pharmacol 83, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1992) Excitotoxic cell death. J. Neurobiol 23, 1261–1276. [DOI] [PubMed] [Google Scholar]
- Choi DW and Rothman SM (1990) The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13, 171–182. [DOI] [PubMed] [Google Scholar]
- Crepel V, Panenka W, Kelly ME and MacVicar BA (1998) Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J. Neurosci 18, 1196–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson LA, Djali S, Gonzales C, Vinegra MA and Zaleska MM (2000) Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res. Bull 53, 767–776. [DOI] [PubMed] [Google Scholar]
- Diaz M, Valverde MA, Higgins CF, Rucareanu C and Sepulveda FV (1993) Volume-activated chloride channels in HeLa cells are blocked by verapamil and dideoxyforskolin. Pflugers Arch 422, 347–353. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C and Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22, 391–397. [DOI] [PubMed] [Google Scholar]
- Dokladda K, Green KA, Pan DA and Hardie DG (2005) PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP:ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett 579, 236–240. [DOI] [PubMed] [Google Scholar]
- Feustel PJ, Jin Y and Kimelberg HK (2004) Volume-regulated anion channels are the predominant contributors to release of excitatory amino acids in the ischemic cortical penumbra. Stroke 35, 1164–1168. [DOI] [PubMed] [Google Scholar]
- Fisher SK, Cheema TA, Foster DJ and Heacock AM (2008) Volume-dependent osmolyte efflux from neural tissues: regulation by G-protein-coupled receptors. J. Neurochem 106, 1998–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Panayiotidis MI and Ochoa de la Paz LD (2008) Autocrine signaling involved in cell volume regulation: the role of released transmitters and plasma membrane receptors. J. Cell. Physiol 216, 14–28. [DOI] [PubMed] [Google Scholar]
- Friard J, Tauc M, Cougnon M, Compan V, Duranton C and Rubera I (2017) Comparative effects of chloride channel inhibitors on LRRC8/VRAC-mediated chloride conductance. Front Pharmacol 8, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Takahashi Y, Takeshima H, Saitoh C, Shimizu T, Takeguchi N and Sakai H (2015) Inhibition of gastric H+,K+-ATPase by 4-(2-butyl-6,7-dichloro-2-cyclopentylindan-1-on-5-yl)oxybutyric acid (DCPIB), an inhibitor of volume-regulated anion channel. Eur. J. Pharmacol 765, 34–41. [DOI] [PubMed] [Google Scholar]
- Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D and Laquerre SG (2011) GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin. Cancer Res 17, 989–1000. [DOI] [PubMed] [Google Scholar]
- Hardy SP, deFelipe C and Valverde MA (1998) Inhibition of voltage-gated cationic channels in rat embryonic hypothalamic neurones and C1300 neuroblastoma cells by triphenylethylene antioestrogens. FEBS Lett 434, 236–240. [DOI] [PubMed] [Google Scholar]
- Haskew RE, Mongin AA and Kimelberg HK (2002) Peroxynitrite enhances astrocytic volume-sensitive excitatory amino acid release via a src tyrosine kinase-dependent mechanism. J. Neurochem 82, 903–912. [DOI] [PubMed] [Google Scholar]
- Haskew-Layton RE, Mongin AA and Kimelberg HK (2005) Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem 280, 3548–3554. [DOI] [PubMed] [Google Scholar]
- Haskew-Layton RE, Rudkouskaya A, Jin Y, Feustel PJ, Kimelberg HK and Mongin AA (2008) Two distinct modes of hypoosmotic medium-induced release of excitatory amino acids and taurine in the rat brain in vivo. PLoS ONE 3, e3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Kargacin ME, Kargacin GJ and Ward CA (2003) Tamoxifen inhibits Na+ and K+ currents in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol 285, H661–H668. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Lambert IH and Pedersen SF (2009) Physiology of cell volume regulation in vertebrates. Physiol. Rev 89, 193–277. [DOI] [PubMed] [Google Scholar]
- Hossmann KA (1994) Viability thresholds and the penumbra of focal ischemia. Ann. Neurol 36, 557–565. [DOI] [PubMed] [Google Scholar]
- Hossmann KA (2006) Pathophysiology and therapy of experimental stroke. Cell Mol. Neurobiol 26, 1055–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA (2008) Cerebral ischemia: models, methods and outcomes. Neuropharmacology 55, 257–270. [DOI] [PubMed] [Google Scholar]
- Hyzinski-Garcia MC, Rudkouskaya A and Mongin AA (2014) LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J. Physiol 592, 4855–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito U, Hakamata Y, Kawakami E and Oyanagi K (2011) Temporary [corrected] cerebral ischemia results in swollen astrocytic end-feet that compress microvessels and lead to delayed [corrected] focal cortical infarction. J. Cereb. Blood Flow Metab 31, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson PS, Morrison R and Strange K (1994) The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am. J. Physiol 267, C1203–C1209. [DOI] [PubMed] [Google Scholar]
- Jirsch JD, Loe DW, Cole SP, Deeley RG and Fedida D (1994) ATP is not required for anion current activated by cell swelling in multidrug-resistant lung cancer cells. Am. J. Physiol 267, C688–C699. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Kawamata T, Tamura T, Hovda DA, Becker DP and Tsubokawa T (1991) Calcium-dependent glutamate release concomitant with massive potassium flux during cerebral ischemia in vivo. Brain Res 558, 136–140. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK (1995) Current concepts of brain edema. Review of laboratory investigations. J. Neurosurg 83, 1051–1059. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK (2005) Astrocytic swelling in cerebral ischemia as a possible cause of injury and target for therapy. Glia 50, 389–397. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Feustel PJ, Jin Y, Paquette J, Boulos A, Keller RW Jr. and Tranmer BI (2000) Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 11, 2675–2679. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Jin Y, Charniga C and Feustel PJ (2003) Neuroprotective activity of tamoxifen in permanent focal ischemia. J. Neurosurg 99, 138–142. [DOI] [PubMed] [Google Scholar]
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA and Robinson JP (2003) Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem 278, 8516–8525. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79, 1431–1568. [DOI] [PubMed] [Google Scholar]
- Liu HT, Akita T, Shimizu T, Sabirov RZ and Okada Y (2009) Bradykinin-induced astrocyte-neuron signalling: glutamate release is mediated by ROS-activated volume-sensitive outwardly rectifying anion channels. J. Physiol 587, 2197–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y and Sabirov RZ (2006) Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 54, 343–357. [DOI] [PubMed] [Google Scholar]
- Lopez-Fabuel I, Le DJ, Logan A, James AM, Bonvento G, Murphy MP, Almeida A and Bolanos JP (2016) Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. U. S. A 113, 13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter D, Ullrich F, Lueck JC, Kempa S and Jentsch TJ (2017) Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci 130, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Minieri L, Pivonkova H, Caprini M, Harantova L, Anderova M and Ferroni S (2013) The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br. J. Pharmacol 168, 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA (2007) Disruption of ionic and cell volume homeostasis in cerebral ischemia: The perfect storm. Pathophysiology 14, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA (2016) Volume-regulated anion channel--a frenemy within the brain. Pflugers Arch 468, 421–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA, Hyzinski-Garcia MC, Vincent MY and Keller RW Jr. (2011) A simple method for measuring intracellular activities of glutamine synthetase and glutaminase in glial cells. Am. J. Physiol. Cell Physiol 301, C814–C822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongin AA and Kimelberg HK (2002) ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am. J. Physiol. Cell Physiol 283, C569–C578. [DOI] [PubMed] [Google Scholar]
- Mongin AA and Kimelberg HK (2005a) Astrocytic swelling in neuropathology, in Neuroglia, (Kettenmann H and Ransom BR eds), pp. 550–562. Oxford University Press, Oxford/New York. [Google Scholar]
- Mongin AA and Kimelberg HK (2005b) ATP regulates anion channel-mediated organic osmolyte release from cultured rat astrocytes via multiple Ca2+-sensitive mechanisms. Am. J. Physiol. Cell Physiol 288, C204–C213. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH and Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V and Droogmans G (1997) Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol 68, 69–119. [DOI] [PubMed] [Google Scholar]
- Oike M, Droogmans G and Nilius B (1994) The volume-activated chloride current in human endothelial cells depends on intracellular ATP. Pflugers Arch 427, 184–186. [DOI] [PubMed] [Google Scholar]
- Oiki S, Kubo M and Okada Y (1994) Mg2+ and ATP-dependence of volume-sensitive Cl- channels in human epithelial cells. Jpn. J. Physiol 44 Suppl 2, S77–S79. [PubMed] [Google Scholar]
- Okada Y (1997) Volume expansion-sensing outward-rectifier Cl- channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. Cell Physiol 273, C755–C789. [DOI] [PubMed] [Google Scholar]
- Okada Y, Okada T, Islam MR and Sabirov RZ (2018) Molecular identities and ATP release activities of two types of volume-regulatory anion channels, VSOR and Maxi-Cl. Curr. Top. Membr 81, 125–176. [DOI] [PubMed] [Google Scholar]
- Olson JE and Li GZ (1997) Increased potassium, chloride, and taurine conductances in astrocytes during hypoosmotic swelling. Glia 20, 254–261. [DOI] [PubMed] [Google Scholar]
- Osuka K, Feustel PJ, Mongin AA, Tranmer BI and Kimelberg HK (2001) Tamoxifen inhibits nitrotyrosine formation after reversible middle cerebral artery occlusion in the rat. J. Neurochem 76, 1842–1850. [DOI] [PubMed] [Google Scholar]
- Patel AJ, Lauritzen I, Lazdunski M and Honore E (1998) Disruption of mitochondrial respiration inhibits volume-regulated anion channels and provokes neuronal cell swelling. J. Neurosci 18, 3117–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Klausen TK and Nilius B (2015) The identification of a volume-regulated anion channel: an amazing Odyssey. Acta Physiol (Oxf) 213, 868–881. [DOI] [PubMed] [Google Scholar]
- Pedersen SF, Okada Y and Nilius B (2016) Biophysics and physiology of the volume-regulated anion channel (VRAC)/volume-sensitive outwardly rectifying anion channel (VSOR). Pflugers Arch 468, 371–383. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Song D and O’Regan MH (1997) Inhibition by anion channel blockers of ischemia-evoked release of excitotoxic and other amino acids from rat cerebral cortex. Brain Res 758, 9–16. [DOI] [PubMed] [Google Scholar]
- Planells-Cases R, Lutter D, Guyader C, Gerhards NM, Ullrich F, Elger DA, Kucukosmanoglu A, Xu G, Voss FK, Reincke SM, Stauber T, Blomen VA, Vis DJ, Wessels LF, Brummelkamp TR, Borst P, Rottenberg S and Jentsch TJ (2015) Subunit composition of VRAC channels determines substrate specificity and cellular resistance to Pt-based anti-cancer drugs. EMBO J 34, 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, Reinhardt J, Orth AP and Patapoutian A (2014) SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157, 447–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Mandujano G, Vazquez-Juarez E, Hernandez-Benitez R and Pasantes-Morales H (2007) Thrombin potently enhances swelling-sensitive glutamate efflux from cultured astrocytes. Glia 55, 917–925. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T and Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403, 316–321. [DOI] [PubMed] [Google Scholar]
- Rothman SM and Olney JW (1986) Glutamate and the pathophysiology of hypoxic--ischemic brain damage. Ann. Neurol 19, 105–111. [DOI] [PubMed] [Google Scholar]
- Rudkouskaya A, Chernoguz A, Haskew-Layton RE and Mongin AA (2008) Two conventional protein kinase C isoforms, alpha and betaI, are involved in the ATP-induced activation of volume-regulated anion channel and glutamate release in cultured astrocytes. J. Neurochem 105, 2260–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge EM, Mongin AA and Kimelberg HK (1999) Intracellular ATP depletion inhibits swelling-induced D-[3H]aspartate release from primary astrocyte cultures. Brain Res 842, 39–45. [DOI] [PubMed] [Google Scholar]
- Schlichter LC, Mertens T and Liu B (2011) Swelling activated Cl- channels in microglia: Biophysics, pharmacology and role in glutamate release. Channels (Austin. ) 5, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober AL, Wilson CS and Mongin AA (2017) Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J. Physiol 595, 6939–6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y, Feustel PJ, Keller RW Jr., Tranmer BI and Kimelberg HK (1999) Inhibition of ischemia-induced glutamate release in rat striatum by dihydrokinate and an anion channel blocker. Stroke 30, 433–440. [DOI] [PubMed] [Google Scholar]
- Siesjo BK (1978) Brain Energy Metabolism John Wiley and Sons, New York, NY. [Google Scholar]
- Strange K, Emma F and Jackson PS (1996) Cellular and molecular physiology of volume-sensitive anion channels. Am. J. Physiol 270, C711–C730. [DOI] [PubMed] [Google Scholar]
- Supplie LM, Duking T, Campbell G, Diaz F, Moraes CT, Gotz M, Hamprecht B, Boretius S, Mahad D and Nave KA (2017) Respiration-deficient astrocytes survive as glycolytic cells in vivo. J. Neurosci 37, 4231–4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk KA, Zhang C, Husted RF and Stokes JB (1996) Cl- current in IMCD cells activated by hypotonicity: time course, ATP dependence, and inhibitors. Am. J. Physiol 271, F552–F559. [DOI] [PubMed] [Google Scholar]
- Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, Andrade-Navarro MA, von Kries JP, Stauber T and Jentsch TJ (2014) Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344, 634–638. [DOI] [PubMed] [Google Scholar]
- Wahl F, Obrenovitch TP, Hardy AM, Plotkine M, Boulu R and Symon L (1994) Extracellular glutamate during focal cerebral ischaemia in rats: time course and calcium dependency. J. Neurochem 63, 1003–1011. [DOI] [PubMed] [Google Scholar]
- White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI, Rafols JA and Krause GS (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J. Neurol. Sci 179, 1–33. [DOI] [PubMed] [Google Scholar]
- Wilson CS and Mongin AA (2018) Cell volume control in healthy brain and neuropathologies. Curr. Top. Membr 81, 385–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZC, Oberheim N, Kettenmann H and Ransom BR (2009) Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia 57, 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Wyttenbach A and Tolkovsky AM (2004) Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem. Pharmacol 68, 351–360. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang H, Feustel PJ and Kimelberg HK (2008) DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp. Neurol 210, 514–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.