

Summary
Wnt signaling plays a key role in regulating bone remodeling. In vitro studies suggest that sclerostin's inhibitory action on Lrp5 is facilitated by the membrane-associated receptor Lrp4. We generated an Lrp4 R1170W knockin mouse model (Lrp4KI), based on a published mutation in patients with high bone mass (HBM). Lrp4KI mice have an HBM phenotype (assessed radiographically), including increased bone strength and formation. Overexpression of a Sost transgene had osteopenic effects in Lrp4-WT but not Lrp4KI mice. Conversely, sclerostin inhibition had blunted osteoanabolic effects in Lrp4KI mice. In a disuse-induced bone wasting model, Lrp4KI mice exhibit significantly less bone loss than wild-type (WT) mice. In summary, mice harboring the Lrp4-R1170W missense mutation recapitulate the human HBM phenotype, are less sensitive to altered sclerostin levels, and are protected from disuse-induced bone loss. Lrp4 is an attractive target for pharmacological targeting aimed at increasing bone mass and preventing bone loss due to disuse.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
Highlights
-
•
Missense mutation in the third beta-propeller of Lrp4 improve bone properties
-
•
The R1170W mutation in Lrp4 interferes with sclerostin inhibition in vivo
-
•
The R1170W Lrp4 mutation alters the bone wasting effects of mechanical disuse
Biological Sciences; Molecular Biology; Cell Biology
Introduction
Wnt signaling plays a key role in regulating bone modeling and remodeling. Perturbations in the Wnt pathway can have profound effects on bone properties, in both positive (osteosclerosis) and negative (osteopenia) directions. Those effects are most easily appreciated by the handful of skeletal conditions that are directly linked to mutations in the Wnt pathway. For example, patients with loss-of-function mutations in the Wnt co-receptor low-density lipoprotein receptor-related protein 5 (Lrp5) present with very low bone mass and density, a condition known as Osteoporosis Pseudoglioma. Conversely, patients with gain-of-function mutations in LRP5 can present with very high bone mass and density, a condition known as Endosteal Hyperostosis (Little et al., 2002, Boyden et al., 2002, Costantini et al., 2017, Van Wesenbeeck et al., 2003). The Lrp5 inhibitor sclerostin (Sost) is another key regulator of bone properties. Patients with homozygous loss-of-function mutations in Sost have sclerosteosis, characterized by dramatically increased bone mass and density (Balemans et al., 2001, Balemans et al., 2002, Staehling-Hampton et al., 2002).
The skeletal phenotypes associated with mutations in the Wnt pathway, particularly the high-bone-mass causing mutations, have highlighted the potential utility of targeting Wnt pathway components for therapeutic benefit in patients with low bone mass. Specifically, the observation of increased bone mass in patients with sclerosteosis and the relatively restricted expression of Sost to bone tissue suggest that sclerostin targeting is an attractive strategy for improving bone properties. In both pre-clinical animal models and human clinical trials, treatment with antibodies raised against sclerostin has consistently improved bone properties beyond any currently approved therapies. At the time of this writing, the sclerostin monoclonal antibody Evenity was very recently approved in the United States and Japan for treatment of osteoporotic patients at high risk of fracture. A better understanding of sclerostin's mechanism of action might reveal other targets or accessory agents that could make this approach safer, more efficacious, or more cost effective.
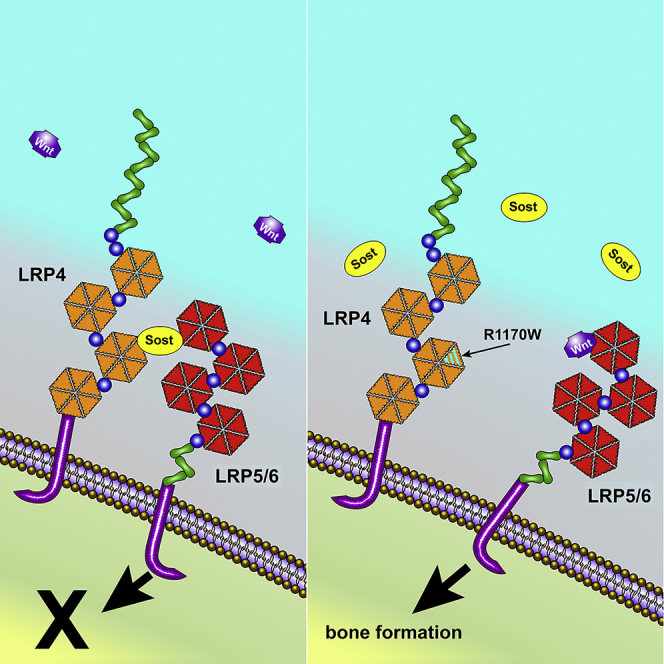
One of the more recent discoveries regarding sclerostin biology is the observation that sclerostin's inhibitory action on Lrp5 is facilitated by the membrane-associated receptor Lrp4. Lrp4 is most widely studied for its role in the neuromuscular junction (NMJ), where it serves as a receptor for motor neuron-derived agrin/Wnt and facilitates acetylcholine receptor clustering on the myocyte. Loss of Lrp4 prevents the formation of NMJs and, consequently, leads to perinatal lethality (Barik et al., 2014). However, Lrp4 also plays a role in bone metabolism, and patients with certain NMJ-sparing missense mutations in LRP4 exhibit a sclerosteosis-like phenotype (SOST2; see Leupin et al., 2011). Deeper investigation revealed a sclerostin-interacting pocket in the central domain of the receptor's third β-propeller (3βCD), mutation of which (e.g., R1170W, R1170Q, and W1186S) does not appear to alter the Agrin/LRP4/MuSK axis. However, these 3βCD mutations are proposed to impair interaction with sclerostin and, ultimately, sclerostin's efficacy in inhibiting Lrp5/6. Accordingly, this domain of Lrp4 represents a potential opportunity to augment the sclerostin-Lrp5/6 interaction for therapeutic value (Chang et al., 2014).
In addition to regulating baseline bone mass and bone metabolism, the Wnt signaling pathway is critical for orchestrating the response of bone tissue to changes in the mechanical environment. Reduced mechanical input to bone (e.g., disuse) leads to decreases in bone mass and increased bone resorption. Conversely, increased mechanical stimulus to bone (e.g., loading) leads to increases in bone mass and osteogenesis. Sost/sclerostin plays a significant regulatory role in both of these phenomena (Tu et al., 2012, Lin et al., 2009), but it is unclear whether and how Lrp4 modifies sclerostin action in these processes.
In this communication, we further elucidate the interaction between Lrp4 and sclerostin using a new Lrp4 3βCD mutant mouse model (R1170W knockin). We examine the role of 3βCD in sclerostin efficacy by overexpressing or neutralizing sclerostin in Lrp4 R1170W mice using genetic and pharmacologic tools. We also probe the role of Lrp4 in disuse mechanotransduction, to evaluate its translational potential as bone-sparing therapy for disuse. Our results suggest that Lrp4 plays a significant role in bone metabolism and likely does so through interaction with sclerostin. Furthermore, Lrp4 might serve as a high-yield target for the development of compounds that can protect skeletal integrity during mechanodeprivation.
Results
Mice with the Lrp4 R1170W Mutation Exhibit Increased Bone Mass
To better understand the cellular mechanisms driving the high bone mass (HBM) phenotype in patients with SOST2, we generated a mouse model recapitulating the mutation found in one of the families (LRP4 R1170W) using a CRISPR/Cas9 approach (Figure 1A). A C→T point mutation was knocked into the first position of the codon for aa.1170 (confirmed by Sanger sequencing, Figure 1B), which resulted in the predicted Arg→Trp amino acid substitution. Western blotting for Lrp4 indicated that the missense mutation had no effect on Lrp4 protein expression in cortical bone (Figure 1C).
Figure 1.

Generation of Lrp4 R1170W Knockin Mouse Model
(A) Mouse Lrp4 genomic DNA schematic, showing the exon 25/intron 25 targeting area. A CRISPR/Cas9 approach was used to replace a 135-bp sequence with a donor oligo containing a C→T mutation at position 1 of aa1170, as well as two silent mutations in the flanking codons to facilitate genotyping.
(B) PCR products from exon 25 were sequenced to confirm mutations. A representative Sanger electropherogram from a heterozygous knockin mouse shows equal expression of the three individual base substitutions in the three consecutive codons.
(C) Western blot of cortical bone tissue protein extract from 6- to 8-week-old Lrp4 WT and Lrp4KI mice, immunoreacted for Lrp4 (∼240kDa), indicating comparable Lrp4 protein expression levels in Lrp4KI versus Lrp4 WT mice. Lane loading equivalency was assessed by stripping and reprobing for the focal adhesion protein vinculin (lower panel) and via Ponceau-S staining of the membrane for total protein before blotting (right panel). Two control lanes were removed between molecular markers and wild-type lanes for clarity.
We next sought to determine whether the Lrp4-R1170W knockin mice (hereafter referred to as Lrp4KI) recapitulate the skeletal phenotype reported for patients with SOST2 with centrally located third β-propeller missense mutations (R1170W, R1170Q, W1186S). The Lrp4KI colony was expanded, along with wild-type (WT) and heterozygous littermates, and subjected to a battery of skeletal phenotyping endpoints. The mutation did not affect body mass or femur length (Figures 2B-2C), but Lrp4KI mice displayed significant increases in dual-energy X-ray absorptiometry (DEXA)-derived whole-body bone mineral density (BMD) and content (BMC) as early as 6 weeks of age (Figure 2A, Tables S1 and S2). At 18 weeks of age, μCT-derived femur and vertebral cortical and cancellous bone mass were significantly increased in Lrp4KI as compared with both WT and heterozygous littermates (Figures 2D–2G and Tables S1 and S2). pQCT-derived cortical BMC and trabecular BMD in the proximal tibia of 8-week-old Lrp4KI mice were also significantly elevated (Figures 2H–2J). Three-point bending tests conducted on femora from 18-week-old mice revealed improved biomechanical properties in Lrp4KI mice, including significantly increased ultimate force and energy absorption (Figures 2K–2M).
Figure 2.

Lrp4KI Mice Have a High Bone Mass Phenotype
(A and B) (A) Whole-body bone mineral density (BMD) was significantly elevated in female Lrp4KI mice versus WT (+/+) and heterozygous (+/KI) littermates, despite (B) normal body mass.
(C–G) (D–G) Trabecular bone volume fraction (Tb.BV/TV) in the distal femur and fifth lumbar vertebra (L5) and cortical thickness (Ct.Th) in the mid-diaphyseal femur were significantly increased in 18-week-old female Lrp4KI mice compared with WT (+/+) and heterozygous (+/KI) littermates, despite (C) normal femur length. (E) The center panel depicts μCT reconstructions from the midshaft femur (upper), distal femur (middle) and vertebral body (lower) from representative +/+, +/KI, and KI/KI mice. Scale bar, 1 mm.
(H–J) Peripheral quantitative computed tomography (pQCT) measurements through the proximal tibia of 8-week-old female +/+, +/KI, and KI/KI mice reveal an increase in trabecular BMD and cortical BMC among KI/KI mice.
(K–M) Mechanical testing of femora from 18-week-old female +/+, +/KI, and KI/KI mice reveal increased ultimate force and energy to ultimate force (FU) among knockin mice.
(N–P) Parietal thickness and planar area of foramen ovale in 18-week-old female +/+ and KI/KI female crania were not affected by the mutation. Male data are presented in Table S2. For (A) and (B), *p < 0.05 for comparison to +/+ by repeated measures ANOVA; for (C)–(P), *p < 0.05 for comparison to +/+ mice, using one-way ANOVA followed by Fisher's protected least significant differences (PLSD) post hoc tests. n = 10/group. Data are presented as means ±SEM.
Patients with SOST2 display calvarial sclerosis, which is recapitulated in Lrp4KI mice, although much more robustly in male mice (Table S2) compared with female mice (Figures 2N–2P). We previously reported stenosis of cranial nerve foramina in the basicranium of Lrp5-HBM and Sost knockout mouse models (Niziolek et al., 2015), which, at least in patients with sclerosteosis, can lead to cranial nerve deficits. However, the foramen ovale area was not significantly affected by the Lrp4KI mutation (Figure 2P). Although dental anomalies have not been reported in patients with SOST2, we also found dental anomalies in Lrp4KI mice, including supernumerary incisors and molars, with altered cusp morphology (Figure S1). In summary, Lrp4KI mice recapitulate the human HBM phenotype associated with SOST2.
Lrp4KI Mice Have Increased Bone Formation, Sclerostin Expression, and Mildly Impaired Muscle Function
Understanding of the mechanism of action (increased osteoanabolism or decreased osteocatabolism) for the observed HBM phenotype in Lrp4KI mice can guide translational applications for Lrp4 targeting. To investigate cellular activity, we performed quantitative cortical bone histomorphometry on Lrp4KI and WT control mice. Bone formation parameters were increased in 16- to 17-week-old but not 8- to 11-week-old mutant mice (Figures 3A–3G). No changes in the resorption marker CTx were detected in 8-week-old wild-type or Lrp4KI mice (Figure 4H). Taken together those observations suggest that the increased bone mass observed in Lrp4KI mice is due to increased osteoblast-mediated bone formation, positioning Lrp4 as a target for anabolic action in bone.
Figure 3.

Lrp4KI Mice Have Increased Bone Formation, Increased Circulating and Local Sclerostin, and Reduced Skeletal Muscle Function
(A–G) (A) Quantitative endocortical bone histomorphometry of the midshaft femur of 18-week-old +/+, +/KI, and KI/KI female mice, measured using labels injected at 8 (orange) and 11 (red) weeks of age (B–D), and again using labels injected at 16 (green) and 17 (red) weeks of age (E–G), indicate a significant increase in the bone formation parameters mineralizing surface (MS/BS), mineral apposition rate (MAR), and bone formation rate (BFR/BS) at the later time point. Scale bar 500 μm.
(H) Western blot of cortical bone tissue protein extract from 6- to 8-week-old Lrp4 WT and Lrp4KI mice, immunoreacted for sclerostin (∼30 kDa), indicating increased sclerostin protein expression levels in Lrp4KI versus Lrp4 WT mice. Lane loading equivalency was assessed by Ponceau-S staining of the membrane for total protein before blotting (right panel). Molecular weight markers were moved to right side of blot for clarity with Ponceau-S staining labeling.
(I) Serum sclerostin was also significantly elevated in the circulation of Lrp4 KI mice, compared with +/+ and +/KI mice.
(J–M) (J) In vivo muscle function of the anterior compartment musculature (left panel) in 10-week-old female mice revealed a deficiency in maximum torque (K), but not the time to maximum torque (L) or half muscle relaxation time (M), among Lrp4KI mice, compared with WT mice. For (B)–(I), p < 0.05 for comparison to +/+ mice, using one-way ANOVA followed by Fisher's PLSD post hoc tests; for (K)–(M), *p < 0.05 for comparison to +/+ mice by repeated measures ANOVA. n = 6/group (B–G); 3–8/group (H–I); and 9–10/group (K–M). Data are presented as means ±SEM.
Figure 4.

Lrp4KI Mice Are Protected from the Osteopenic Effects of Sost Overexpression
(A) Whole-body bone mineral density (BMD) in female Lrp4+/+ mice was significantly reduced by the Dmp1-hSost transgene, which induces Sost overexpression in late-stage osteoblasts and osteocytes, whereas Lrp4KI mice maintained high BMD regardless of Dmp1-hSost presence.
(B–E) (B–D) Trabecular bone volume fraction (Tb.BV/TV) in the distal femur and fifth lumbar vertebra (L5) were significantly reduced in 17-week-old female Lrp4 WT that carried the hSost transgene but not in Lrp4KI mice that carried the hSost transgene. Scale bar, 1 mm. (E) Cortical thickness (Ct.Th) was not affected by the hSost transgene in either Lrp4 genotype.
(F and G) Quantitative endocortical bone histomorphometry of the tibial cortex (I), calculated from 16 to 17 weeks, revealed no effect of the transgene on the bone formation parameters mineralizing surface (MS/BS) bone formation rate (BFR/BS) in either Lrp4 genotype. Scale bar 250 μm.
(H) The serum resorption marker C-terminal telopeptide (CTx) was reduced by the hSost transgene in Lrp4 mice, but not wild-type mice. Data from male mice are in Table S4. For (A), *p < 0.05 by repeated measures ANOVA for within Lrp4 genotype comparison of transgene presence, n = 8–10/group. For (C)–(H), data were tested using two-way ANOVA with Lrp4 genotype and the Sost transgene as main effects. Inset at the top of each graph indicates significance of the main effects and interaction (# = Lrp4 genotype p < 0.05; @ = Dmp1-hSost p < 0.05; † = interaction p < 0.05). When at least one term was significant, Fisher's PLSD post hoc tests were conducted and are indicated as *p < 0.05. For (C)–(E), n = 8–10/group. For (F) and (G), n = 4–6/group. For (H), n = 6–7/group. Data are presented as means ±SEM.
Lrp4 has been proposed to serve as a sequestering protein for sclerostin, where it might serve as a molecular net to keep sclerostin levels high in the perimembrane region of bone cells (Fijalkowski et al., 2016). Serum levels of sclerostin were significantly greater in Lrp4KI mice than in WT and heterozygous mice (Figure 3I), which is consistent with patient data (Leupin et al., 2011) and supports the premise that mutant Lrp4 has an impaired ability to retain sclerostin at the cell surface. However, cortical bone tissue lysates from Lrp4KI mice yielded greater levels of sclerostin than lysates from WT mice (Figure 3H). That observation runs counter to the proposal that osteocytes with mutant Lrp4 are less efficient at retaining sclerostin locally, but it could also be explained by increased Sost expression in response to impaired Wnt inhibition (Taylor et al., 2018, Holdsworth et al., 2018).
Lrp4 is critical for NMJ formation and muscle function, where it serves as a receptor for motoneuron-derived Agrin, triggering interaction with MuSK (Shen et al., 2015). Mutations in the third β-propeller of Lrp4 can affect Agrin and MuSK binding (Ohkawara et al., 2014), so we performed in vivo muscle force testing in 10-week-old female WT and Lrp4KI mice (Figure 3J) to determine whether muscle function was affected by the R1170W missense mutation. Muscle mass, time to maximum torque, and half relaxation time were not different between WT and Lrp4KI mice, but maximum torque was significantly lower in Lrp4KI mice (Figures 3K–3M). However, a change in muscle function does not explain the HBM phenotype in Lrp4KI mice since the mutation was associated with reduced rather than enhanced muscle forces on the skeleton. In summary, Lrp4KI mice have HBM owing to increased anabolic activity, despite increased local and systemic sclerostin and compromised muscle function.
Overexpression of Sost in Bone Tissue Causes Osteopenia in WT but Not Lrp4KI Mice
Some reports suggest that Lrp4 acts as a facilitator of sclerostin inhibition of Lrp5/6, a proposition that is supported by direct interaction between the Lrp4 third β-propeller and sclerostin and by suppression of sclerostin-mediated Wnt inhibition by Lrp4 knockdown (Leupin et al., 2011). To determine whether compromise of the sclerostin-Lrp4 interaction can account for the HBM phenotype of Lrp4KI, we bred Lrp4KI mice to transgenic mice that overexpress human Sost in osteocytes (8kbDmp1-hSost). As expected, whole-body BMD and BMC were decreased in Lrp4 WT mice overexpressing Sost, but Lrp4KI mice were unaffected by Sost overexpression (Figures 4A and Tables S3 and S4). Lrp4KI mice were protected from the osteopenic effects of Sost overexpression in the cancellous compartment of both the femur and spine (Figures 4B–4D), but the protective effects in the cortical compartment could not be evaluated because Lrp4 WT mice did not manifest a Sost transgene effect when evaluated by μCT (Figure 4E) or bone formation parameters (Figures 4F and 4G). The Sost transgene had no effect on the resorption marker CTx in WT mice. CTx was slightly but significantly suppressed in Lrp4KI mice carrying the Sost transgene (Figure 4H). In summary, the otherwise bone-suppressive effects of excessive sclerostin in bone were largely absent in Lrp4KI mice, suggesting that the R1170W mutation confers resistance to the inhibitory effects of sclerostin in vivo.
Inactivation of Sclerostin Elicits a Blunted Osteogenic Effect in Lrp4KI Mice
In the previous section we evaluated the ability of Lrp4KI mice to maintain high bone mass in the presence of excessive sclerostin. Next, we evaluated the contraposition regarding the sclerostin-Lrp4 interaction, i.e., whether Lrp4KI mice can elicit a full anabolic response when sclerostin is inactivated pharmacologically. If the R1170W mutation mimics a sclerostin-depleted environment, sclerostin neutralization should have minimal effects. Four weeks of treatment with sclerostin monoclonal antibody (Scl-mAb) increased whole-body BMD significantly compared with saline treatment in both WT and Lrp4KI female mice, but the antibody-induced BMD gain exhibited by Lrp4KI mice was only about half of that exhibited by WT mice (Figure 5E). Large gains in μCT-derived parameters (trabecular and cortical bone mass in the femur and spine) were measured among WT mice in response to Scl-mAb, but a blunted response (roughly a third to a half of WT gains, yet still significant) was observed in Lrp4KI mice (Figures 5A–5D). With the exception of mineral apposition rate (MAR), Scl-mAb treatment increased bone formation parameters significantly in WT mice but not in Lrp4KI mice (Figures 5F–5H, Table S5). In summary, the strong anabolic effects of sclerostin neutralization were compromised or absent in Lrp4KI mice, likely due to the mutation having similar functional (i.e., redundant) effects as sclerostin inactivation.
Figure 5.

Response to Pharmacologic Inhibition of Sost Is Blunted in Lrp4KI Mice
Cortical thickness (Ct.Th.) (A) and trabecular bone volume fraction (Tb.BV/TV) (B) in the mid-diaphyseal and distal femur and fifth lumbar vertebra (L5) (C) were significantly increased in 16-week-old female Lrp4KI and WT (+/+) mice treated with sclerostin monoclonal antibody (Scl-mAb), but the gains were more pronounced in WT mice.
(D) μCT reconstructions from the midshaft femur (upper) distal femur (middle) and vertebral body (lower) from representative +/+ and KI/KI mice treated with vehicle (−) or Scl-mAb (+). Scale bar, 1 mm.
(E) Percent change in whole-body bone mineral density (BMD) during the 4-week antibody treatment period was greater in WT mice compared with Lrp4KI mice.
(F–H) The periosteal bone formation parameters mineralizing surface (MS/BS), mineral apposition rate (MAR), and bone formation rate (BFR/BS) were significantly increased in WT but not Lrp4KI mice in response to sclerostin antibody. Scale bar 100 μm. For all data panels, data were tested using two-way ANOVA with Lrp4 genotype and the Scl-mAb injection as main effects. Inset at the top of each graph indicates significance of the main effects and interaction (# = Lrp4 genotype p < 0.05; @ = Scl-mAb p < 0.05; † = interaction p < 0.05). When at least one term was significant, Fisher's PLSD post hoc tests were conducted and are indicated as *p < 0.05. For all panels, n = 6–10/group.
Data are presented as means ±SEM. See also Table S5.
Lrp4KI Mice Are Partially Protected from Disuse-Induced Bone Wasting
Bone is a highly mechanosensitive tissue, and previous studies highlighted the importance of canonical Wnt signaling, particularly sclerostin, in this process. Mechanical disuse induces an increase in Sost expression, and Sost genetic deletion or sclerostin pharmacologic neutralization prevented disuse-induced bone loss (Robling et al., 2016). Given our accumulating evidence that the Lrp4 R1170W mutation confers properties to bone tissue that are similar to sclerostin neutralization, we explored the role of Lrp4KI in response to disuse mechanotransduction. Lrp4KI mice (12 weeks of age) were subjected to 4 weeks of unilateral mechanical disuse of the hindlimb using the Botulinum toxin-induced muscle paralysis model (Warner et al., 2006), and the degree of bone wasting was evaluated radiographically. The paralyzed limb of WT mice lost ∼10% BMD, but Lrp4KI mice did not lose BMD in response to Botox (Figure 6A). However, although bone loss was spared in the paralyzed limb of Lrp4KI mice, we detected a significant failure to gain bone as was observed in the saline-treated limb of Lrp4KI mice (Figure 6A). Proximal tibial trabecular bone mass was significantly reduced by Botox in both WT and Lrp4KI mice, but the Lrp4KI mice lost roughly half as much bone as WT mice in response to paralysis (Figures 6B and 6D). Cortical bone loss was significant in Botox-treated WT mice as expected, but paralyzed (Botox-treated) limbs from Lrp4KI mice exhibited no difference from saline-treated limbs (Figure 6C). Periosteal bone formation was nearly abolished in the paralyzed limbs of WT mice, but Lrp4KI mice maintained unparalyzed levels of MAR but not MS/BS or BFR (Table S6). In summary, Lrp4KI mice exhibit less severe bone wasting in response to mechanical disuse than WT mice, suggesting that pharmacologically targeting the third β-propeller of Lrp4 during a disuse event might have therapeutic value in reducing bone loss.
Figure 6.

Lrp4KI Mice Are Partially Protected From Disuse-Induced Bone Loss
(A) Percent change in hindlimb bone mineral density (BMC) following 4 weeks of Botox-induced disuse.
(B and C) Percent difference (from contralateral limb) in trabecular bone volume fraction (Tb.BV/TV) in the proximal tibia and cortical thickness (Ct.Th) in the mid-diaphyseal tibia were significantly greater in Botox-treated WT mice compared with Botox-treated Lrp4KI mice.
(D) μCT reconstructions at the proximal tibia from representative +/+ and KI/KI mice treated with Botox (+) or vehicle (−). Scale bars, 1 mm. Data were tested using two-way ANOVA using Lrp4 genotype and Botox injection as main effects. Inset at the top of each graph indicates significance of the main effects and interaction (# = Lrp4 genotype p < 0.05; @ = Botox effect p < 0.05; † = interaction p < 0.05). When at least one term was significant, Fisher's PLSD post hoc tests were conducted and are indicated as *p < 0.05. For all panels, n = 9–10/group.
Data are presented as means ±SEM. See also Table S6.
Discussion
High bone mass (HBM)-causing mutations in genes coding for secreted or plasma membrane-bound Wnt signaling components represent rare glimpses into molecular targeting opportunities to improve bone health and fracture resistance in osteoporotic patients. Targets that can be modulated to produce anabolic action in the skeleton are particularly attractive, given the paucity of approved and pipeline osteoanabolic agents. The identification of patients with a sclerosteosis-like condition harboring missense mutations in LRP4 (Leupin et al., 2011) has furthered our understanding of accessory proteins involved in extracellular Wnt inhibition and also provided additional avenues for therapeutic development. We engineered an orthologous mouse model to one of the HBM human families identified with an HBM-causing LRP4 missense mutation, which yielded an HBM phenotype and provided a tool to study more mechanistic questions beyond those suited to clinical studies (e.g., bone strength, tissue protein levels). We found that the Lrp4 R1170W knockin mice recapitulate the skeletal phenotype observed in patients with SOST2 and that the phenotype is driven largely by anabolic action.
Moreover, we were able to identify several additional traits associated with Lrp4 3βCD mutation that have not been reported in patients with SOST2, including changes to the dentition (also observed in other Lrp4 models [Ahn et al., 2017]), compromised muscle function, and increased levels of sclerostin in osteocyte-enriched protein lysates from bone tissue. Conversely, we failed to find some traits in the murine model that have been identified in patients (e.g., skull thickness, phenotype in the heterozygous state). The reduction in muscle function in Lrp4KI mice is surprising. Certain missense mutations in the third β-propeller of LRP4 (e.g., E1233K and R1277H) can cause congenital myasthenic syndrome (CMS), a non-autoimmune disorder associated with progressive muscle weakness. CMS mutations located on the outer edge of the third β-propeller impair Lrp4 interaction with both Agrin and MuSK—key interactions for NMJ assembly and maintenance (Ohkawara et al., 2014). However, cell culture models show that overexpression of R1170W impairs Wnt signaling but has no effect on Agrin-induced MuSK signaling, suggesting that centrally located missense mutations (e.g., R1170W) affect Wnt but not Agrin/MuSK, whereas those located on the propeller edge have the inverse effect. Our data do not identify a direct role for the R1170W mutation in regulating muscle function; other indirect explanations (e.g., R1170W-induced increase in circulating sclerostin might impair muscle function) cannot be ruled out.
Another interesting finding was the increased levels of sclerostin protein in bone extracts. Human patients with LRP4 HBM (Fijalkowski et al., 2016), other mouse models of Lrp4 mutation (Xiong et al., 2015, Boudin et al., 2017), and WT mice treated with Lrp4 neutralizing antibody (Chang et al., 2014) all have increased levels of circulating sclerostin, which we confirmed in our R1170W model. Additionally, we also found increased sclerostin protein in bone, which was surprising given (1) Lrp4's purported role in retaining sclerostin at high levels in the local bone environment (which is presumably inhibited by the Lrp4 HBM mutations) and (2) previous reports of decreased sclerostin in the osteocytes of Lrp4 knockin mice (R1170Q), as assessed by immunohistochemistry (Boudin et al., 2017). One explanation for the increase in tissue sclerostin despite a lack of its primary retention mechanism (functional Lrp4) that is consistent with our previous data is the self-regulation of sclerostin expression observed when sclerostin signaling is inhibited genetically or pharmacologically (Witcher et al., 2018).
Investigation of the interaction between Lrp4 and sclerostin has thus far been restricted to cell culture experiments involving overexpression of various signaling components, which have the potential to be problematic or misleading specifically in Wnt-related in vitro studies (Goel et al., 2012). We designed in vivo experiments to better understand this interaction in a more physiologic context and did so using excessive (hSost transgene) and depleted (Scl-Ab) sclerostin levels in Lrp4KI mice. Lrp4KI mice were largely immune to changes in levels of sclerostin, where Lrp4KI mice overexpressing Sost failed to manifest the osteopenic effects of the transgene and antibody-treated Lrp4KI failed to gain as much bone as treated WT mice (although they did exhibit significant gains for several parameters). Although those experiments do not prove a direct role in binding alterations, they do suggest that Lrp4 3βCD mutations disturb the normal function of sclerostin in regulating bone mass in an in vivo context, both positively and negatively.
To provide a translational context to Lrp4 clinical utility, we explored the role of Lrp4 in mediating disuse osteoporosis. Wnt is a major regulator of mechanical signaling in bone, and we have previously reported therapeutic effects of modulating Lrp5, Sost, and β-catenin in mechanical models (Niziolek et al., 2015, Robling et al., 2016, Bullock et al., 2019). Here, we evaluated the utility of Lrp4 modulation in a muscle paralysis model of bone loss. Lrp4KI mice exhibited significant protection from the bone wasting effects of lower limb paralysis, suggesting that pharmacologic targeting of Lrp4 during or soon after a disuse injury might have clinical benefit.
Recent regulatory approval for clinical use of sclerostin-neutralizing antibody in several countries (United States, Japan, Canada, South Korea, Australia) has opened the door for widespread use of the first Wnt-based therapy for bone health. Whether other components (e.g., Lrp4) alone or in conjunction with sclerostin targeting can provide additional benefits to patients remains to be determined, but Lrp4 targeting for low bone mass states appears attractive from a preclinical perspective.
Limitations of the Study
The experiments performed have several limitations. First, the R1170W mutation was expressed globally. Although that approach provided an orthologous model of the human patients with HBM-causing LRP4 mutations, it did not allow for cell-selective Lrp4 impairment as could be achieved with a conditional knockin approach (Cui et al., 2011). Therefore, we do not know the cell type in which Lrp4 is most critical for regulating sclerostin signaling. Second, we did not perform assays to assess sclerostin-Lrp4 interaction in vivo, with and without the R1170W mutation. Those experiments have been performed in overexpression studies in vitro, but it is possible that, in the context of the in vivo environment, the interaction is modified. Lastly, the Lrp4 mutant mice had altered Lrp4 function since conception. For translational purposes, it would be more appropriate to understand the acute effects of disrupting the Lrp4-sclerostin axis, as could be accomplished with neutralizing antibody or a small molecule inhibitor (Chang et al., 2014).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Thanks to Emily Pemberton for assistance with colony maintenance. Thank you to Amgen/UCB Pharma for providing sclerostin antibody. This work was supported by NIH grants AR070624 (to W.A.B.), AR065971 (to W.A.B.), AR069029 (to F.M.P.), DK075730 (to G.G.L.) and AR053237 (to A.G.R.); and by VA grants BX001478 and BX003783 (to A.G.R.). G.G.L. performed work under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344. This publication was made possible, in part, with support from the Indiana Clinical and Translational Sciences Institute funded, in part by Award Number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author Contributions
W.A.B. designed and performed experiments. A.M.H., D.J.H., A.J.E., and A.Y.S. – data collection. T.B., G.G.L., and F.M.P. designed experiments. A.G.R. designed experiments and is the Lead Contact.
Declaration of Interests
The authors declare no competing financial interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.023.
Supplemental Information
References
- Ahn Y., Sims C., Murray M.J., Kuhlmann P.K., Fuentes-Antras J., Weatherbee S.D., Krumlauf R. Multiple modes of Lrp4 function in modulation of Wnt/beta-catenin signaling during tooth development. Development. 2017;144:2824–2836. doi: 10.1242/dev.150680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balemans W., Ebeling M., Patel N., Van Hul E., Olson P., Dioszegi M., Lacza C., Wuyts W., Van Den Ende J., Willems P. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum. Mol. Genet. 2001;10:537–543. doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- Balemans W., Patel N., Ebeling M., Van Hul E., Wuyts W., Lacza C., Dioszegi M., Dikkers F.G., Hildering P., Willems P.J. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002;39:91–97. doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik A., Zhang B., Sohal G.S., Xiong W.C., Mei L. Crosstalk between Agrin and Wnt signaling pathways in development of vertebrate neuromuscular junction. Dev. Neurobiol. 2014;74:828–838. doi: 10.1002/dneu.22190. [DOI] [PubMed] [Google Scholar]
- Boudin E., Yorgan T., Fijalkowski I., Sonntag S., Steenackers E., Hendrickx G., Peeters S., Mare A., Vervaet B., Verhulst A. The Lrp4 R1170Q homozygous knock-in mouse recapitulates the bone phenotype of sclerosteosis in humans. J. Bone Miner. Res. 2017;32:1739–1749. doi: 10.1002/jbmr.3160. [DOI] [PubMed] [Google Scholar]
- Boyden L.M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M.A., Wu D., Insogna K., Lifton R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- Bullock W.A., Hoggatt A., Horan D.J., Yokota H., Sebastian A., Loots G.G., Pavalko F.M., Robling A.G. Expression of a degradation-resistant beta-catenin mutant in osteocytes protects the skeleton from mechanodeprivation-induced bone wasting. J. Bone Miner. Res. 2019 doi: 10.1002/jbmr.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M.K., Kramer I., Huber T., Kinzel B., Guth-Gundel S., Leupin O., Kneissel M. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc. Natl. Acad. Sci. U S A. 2014;111:E5187–E5195. doi: 10.1073/pnas.1413828111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini A., Kekalainen P., Makitie R.E., Makitie O. High bone mass due to novel LRP5 and AMER1 mutations. Eur. J. Med. Genet. 2017;60:675–679. doi: 10.1016/j.ejmg.2017.09.001. [DOI] [PubMed] [Google Scholar]
- Cui Y., Niziolek P.J., Macdonald B.T., Zylstra C.R., Alenina N., Robinson D.R., Zhong Z., Matthes S., Jacobsen C.M., Conlon R.A. Lrp5 functions in bone to regulate bone mass. Nat. Med. 2011;17:684–691. doi: 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fijalkowski I., Geets E., Steenackers E., Van Hoof V., Ramos F.J., Mortier G., Fortuna A.M., Van Hul W., Boudin E. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J. Bone Miner. Res. 2016;31:874–881. doi: 10.1002/jbmr.2782. [DOI] [PubMed] [Google Scholar]
- Goel S., Chin E.N., Fakhraldeen S.A., Berry S.M., Beebe D.J., Alexander C.M. Both LRP5 and LRP6 receptors are required to respond to physiological Wnt ligands in mammary epithelial cells and fibroblasts. J. Biol. Chem. 2012;287:16454–16466. doi: 10.1074/jbc.M112.362137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth G., Greenslade K., Jose J., Stencel Z., Kirby H., Moore A., Ke H.Z., Robinson M.K. Dampening of the bone formation response following repeat dosing with sclerostin antibody in mice is associated with up-regulation of Wnt antagonists. Bone. 2018;107:93–103. doi: 10.1016/j.bone.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Leupin O., Piters E., Halleux C., Hu S., Kramer I., Morvan F., Bouwmeester T., Schirle M., Bueno-Lozano M., Fuentes F.J. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011;286:19489–19500. doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., Jiang X., Dai Z., Guo X., Weng T., Wang J., Li Y., Feng G., Gao X., He L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 2009;24:1651–1661. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- Little R.D., Carulli J.P., Del Mastro R.G., Dupuis J., Osborne M., Folz C., Manning S.P., Swain P.M., Zhao S.C. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niziolek P.J., Bullock W., Warman M.L., Robling A.G. Missense mutations in LRP5 associated with high bone mass protect the mouse skeleton from disuse- and ovariectomy-induced Osteopenia. PLoS One. 2015;10:e0140775. doi: 10.1371/journal.pone.0140775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkawara B., Cabrera-Serrano M., Nakata T., Milone M., Asai N., Ito K., Ito M., Masuda A., Ito Y., Engel A.G., Ohno K. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2014;23:1856–1868. doi: 10.1093/hmg/ddt578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robling A.G., Kang K.S., Bullock W.A., Foster W.H., Murugesh D., Loots G.G., Genetos D.C. Sost, independent of the non-coding enhancer ECR5, is required for bone mechanoadaptation. Bone. 2016;92:180–188. doi: 10.1016/j.bone.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C., Xiong W.C., Mei L. LRP4 in neuromuscular junction and bone development and diseases. Bone. 2015;80:101–108. doi: 10.1016/j.bone.2015.05.012. [DOI] [PubMed] [Google Scholar]
- Staehling-Hampton K., Proll S., Paeper B.W., Zhao L., Charmley P., Brown A., Gardner J.C., Galas D., Schatzman R.C., Beighton P. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am. J. Med. Genet. 2002;110:144–152. doi: 10.1002/ajmg.10401. [DOI] [PubMed] [Google Scholar]
- Taylor S., Hu R., Pacheco E., Locher K., Pyrah I., Ominsky M.S., Boyce R.W. Differential time-dependent transcriptional changes in the osteoblast lineage in cortical bone associated with sclerostin antibody treatment in ovariectomized rats. Bone Rep. 2018;8:95–103. doi: 10.1016/j.bonr.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X., Rhee Y., Condon K.W., Bivi N., Allen M.R., Dwyer D., Stolina M., Turner C.H., Robling A.G., Plotkin L.I., Bellido T. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–217. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wesenbeeck L., Cleiren E., Gram J., Beals R.K., Benichou O., Scopelliti D., Key L., Renton T., Bartels C., Gong Y. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 2003;72:763–771. doi: 10.1086/368277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner S.E., Sanford D.A., Becker B.A., Bain S.D., Srinivasan S., Gross T.S. Botox induced muscle paralysis rapidly degrades bone. Bone. 2006;38:257–264. doi: 10.1016/j.bone.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher P.C., Miner S.E., Horan D.J., Bullock W.A., Lim K.E., Kang K.S., Adaniya A.L., Ross R.D., Loots G.G., Robling A.G. Sclerostin neutralization unleashes the osteoanabolic effects of Dkk1 inhibition. JCI Insight. 2018;3 doi: 10.1172/jci.insight.98673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong L., Jung J.U., Wu H., Xia W.F., Pan J.X., Shen C., Mei L., Xiong W.C. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc. Natl. Acad. Sci. U S A. 2015;112:3487–3492. doi: 10.1073/pnas.1419714112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.