

Summary
MicroRNAs (miRNAs) play crucial roles in biological processes involved in diseases. The associations between diseases and protein-coding genes (PCGs) have been well investigated, and miRNAs interact with PCGs to trigger them to be functional. We present a computational method, DimiG, to infer miRNA-associated diseases using a semi-supervised Graph Convolutional Network model (GCN). DimiG uses a multi-label framework to integrate PCG-PCG interactions, PCG-miRNA interactions, PCG-disease associations, and tissue expression profiles. DimiG is trained on disease-PCG associations and an interaction network using a GCN, which is further used to score associations between diseases and miRNAs. We evaluate DimiG on a benchmark set from verified disease-miRNA associations. Our results demonstrate that DimiG outperforms the best unsupervised method and is comparable to two supervised methods. Three case studies of prostate cancer, lung cancer, and inflammatory bowel disease further demonstrate the efficacy of DimiG, where top miRNAs predicted by DimiG are supported by literature.
Subject Areas: Disease, Gene Network, Biocomputational Method, Computer Modeling
Graphical Abstract
Highlights
-
•
Formulate disease-miRNA association prediction as node classification in a network
-
•
Semi-supervised graph convolutional network is used to infer miRNA-associated diseases
-
•
Introduce domain knowledge based on tissue expression profiles into model training
-
•
Evaluate DimiG using a strictly independent way; DimiG is superior to other methods
Disease; Gene Network; Biocomputational Method; Computer Modeling
Introduction
MicroRNAs (miRNAs) are a type of small non-coding RNAs with a size of about 22 nucleotides, and they interact with other RNAs to play important roles in transcriptional and post-transcriptional gene regulation (Bartel, 2004). It is estimated that over 60% of all human protein-coding genes (PCGs) are regulated by miRNAs (Friedman et al., 2009), and these miRNAs have been implicated in diseases. To date, the associations between diseases and PCGs are well investigated; many disease-PCG associations have been discovered and collected in public databases, e.g., DISEASES (Pletscher-Frankild et al., 2015), OUGene (Pan and Shen, 2016), and DisGeNET (Pinero et al., 2017). Compared with PCG's well-known important roles in diseases, the studies of effects of miRNAs are increasing. With increasing high-throughput sequencing data generated, more and more miRNAs are being discovered, and experimentally identifying their functions is costly and time consuming. Thus, it is imperative to develop computational methods to identify functional miRNA biomarkers associated with diseases, especially using rich information buried in disease-associated PCGs.
Some miRNAs are mainly expressed in certain tissues and show tissue specificity (Ludwig et al., 2016), which have certain tissue-specific expression patterns associated with diseases (Baker et al., 2017). They are expected to behave similarly to other disease-associated genes like PCGs or long non-coding RNAs (lncRNAs). Thus, several existing computational methods have used tissue expression data to infer gene-disease associations. For instance, GeneTIER makes use of disease-tissue associations to prioritize disease candidate genes (Antanaviciute et al., 2015). NetWAS identifies disease-associated genes by combining tissue-specific interaction networks and genome-wide association studies (Greene et al., 2015). Especially, some methods use tissue expression profiles with machine learning models to infer disease-associated lncRNAs. For example, DislncRF trains machine learning models on tissue expression profiles of disease-associated PCGs and further applies the trained models to infer disease-associated lncRNAs (Pan et al., 2019). All the above-mentioned studies demonstrated that tissue expression profiles indeed can facilitate the detection of disease-gene associations.
On the other hand, interaction networks contain rich clues for linking miRNAs to diseases. Many computational methods have been developed under the context of gene-gene networks (Chen et al., 2019). For example, Jiang el al. integrate miRNA and disease similarity network and miRNA-disease association to prioritize disease candidate miRNAs using a network-based approach (Jiang et al., 2010); midp applies random walk on the interaction network to infer disease-associated miRNAs (Xuan et al., 2015). Similarly, RWRMDA implements random walk on the miRNA functional similarity network to link miRNAs to diseases (Chen et al., 2012); the MDHGI integrates the predicted association score based on sparse learning method to infer disease-associated miRNAs (Chen et al., 2018c). More closely related studies are as follows: DRMDA applies stacked autoencoder to learn deep representation for predicting miRNA-disease association (Chen et al., 2018a), LRSSLMDA and PBMDA use Laplacian regularized sparse subspace learning and path-based computational model for miRNA-disease association prediction (Chen and Huang, 2017, You et al., 2017), BNPMDA uses Bipartite Network Projection based on the known miRNA-disease associations (Chen et al., 2018b), and KBMF-MDI employs kernelized Bayesian matrix factorization to score miRNA-disease associations by integrating disease and miRNA similarity (Lan et al., 2018). Similarly, DLRMC infers disease-associated miRNAs using dual Laplacian regularized matrix completion (Tang et al., 2019). FamCluRank applies non-negative matrix factorization on the heterogeneous network with node attributes to predict disease-associated miRNAs (Xuan et al., 2018b). Especially deep learning has been utilized to extract deep representation for disease-miRNA association prediction (Xuan et al., 2018a).
A common hypothesis for the above methods is they assume that similar miRNAs can be associated with the same disease and similar diseases would be associated with the same miRNA. Thus, they commonly train and evaluate the models with representations of miRNAs and diseases as inputs on verified disease-miRNA associations through cross-validation approach.
However, as pointed out in Lehtinen et al. (2015), in the context of gene function prediction, cross-validation may be problematic because some gene-function associations are not independent in the benchmark set. There exists the same issue for disease-miRNA associations due to the following: (1) miRNAs from the same family may be associated with the same disease, (2) disease-associated miRNAs from miRNA-target assay may be derived from the targets that these miRNAs interact with, and (3) the associated miRNAs of child diseases are related to the miRNAs of parent diseases in disease ontology. When training and evaluating the models using cross-validation, randomly dividing the disease-miRNA associations may cause dependent associations to be separated into the training and test sets, potentially leading to an overestimated predictive performance. Cross-validating miRNA-disease associations may not actually reflect the method's ability to predict new miRNA-disease associations, but rather which information is dissipated in the benchmark set. In addition, as reported in Park and Marcotte (2012), there may exist flaws in cross-validation for computational pair-input prediction. One disease or one miRNA may be associated with multiple miRNAs or diseases, so randomly dividing disease-miRNA pairs into training and test sets will make some pairs in the test set share either the miRNA or the disease with the pairs in the training set, which causes the trained models to not generalize well to unseen disease-miRNA associations.
Thus, during cross-validation, complicated steps are required to make sure that dependent samples are divided into the same training set or the same test set and that pairs in the training and test sets do not share the miRNA or disease. It is almost impossible to construct a completely independent test set. An alternative strategy is that we do not use disease-miRNA associations for model training. For instance, instead of using miRNA-disease associations, the miRPD approach combines PCG-disease associations and miRNA-PCG network to score miRNAs and diseases (Mork et al., 2014). This has triggered us to further investigate disease-miRNA associations based on an interaction network. To date, there exist many high-confidence disease-PCG associations, and one miRNA may share the same disease with its PCG targets; we will be capable of transferring PCG-associated diseases to miRNAs on an interaction network under a new semi-supervised framework.
Recently, deep learning has achieved remarkable results in computational biology (Angermueller et al., 2016, Ching et al., 2018), especially convolutional neural networks (CNNs) (Lecun et al., 1998). CNNs can capture local correlation buried in data and mainly consist of convolutional layers, pooling layers, and fully connected layers. Many studies have demonstrated that the CNN networks are powerful in learning the hidden patterns from complicated biological data. For example, DeepBind (Alipanahi et al., 2015) and DeepSEA (Zhou and Troyanskaya, 2015) apply CNNs to predict preference of DNA/RNA-binding proteins and the impact of non-coding variants, respectively. iDeep (Pan and Shen, 2017) and iDeepE (Pan and Shen, 2018) further improve the performance of predicting RNA-binding protein (RBP)-binding sites and motifs using hybrid CNNs. The iDeepS (Pan et al., 2018) identifies binding sequence and structure preferences of RBPs simultaneously using CNNs and long short-term memory network.
Although the CNN has shown its power, it cannot handle structured datasets, like gene-gene networks. To analyze these types of network data, graph convolutional networks (GCNs) have been developed (Defferrard et al., 2016, Hamilton et al., 2017, Kipf and Welling, 2017). Under the framework of spectral graph convolutions, it encodes both local graph structure and features of nodes. The GCNs have been used on the graph data to predict polypharmacy side effects, where the graph is a multimodal graph constructed from protein-protein interactions, drug-protein interactions, and the polypharmacy side effects (Zitnik et al., 2018). The GCN is a graph-based semi-supervised learning method that does not require labels for all nodes. This setting is especially powerful for inferring miRNA-associated diseases, because many miRNAs are not well investigated about their associations with diseases and many disease-PCG associations are available. Compared with traditional semi-supervised methods (Jia et al., 2016, Wan and Wang, 2019, Zhang et al., 2018, Zoidi et al., 2018), GCNs can capture the structural information within the node's local network, similar to CNNs in images. In addition, one PCG or miRNA can be associated with multiple diseases. Thus, we can formulate the prediction of disease-miRNA associations as a multi-label classification problem.
In this study, we present a new semi-supervised multi-label learning method, DimiG, based on GCNs to integrate multiple networks of PCG-PCG interactions, PCG-miRNA interactions, PCG-disease associations, and tissue expression profiles to infer miRNA-associated diseases. The DimiG does not require the disease-miRNA associations, and it is trained on the graph consisting of PCG-PCG and miRNA-PCG interactions, where only PCGs have labeled diseases. Then DimiG is further used to score associations between diseases and miRNAs.
This study has made the following four major contributions for understanding disease-miRNA associations. (1) We further demonstrate that cross-validation performance of methods trained on known disease-miRNA associations could be overestimated and may not be able to reflect the method's actual ability to predict new disease-miRNA associations. We have proposed a network-based knowledge transfer approach for this problem. Considering that an miRNA may share the same disease with its PCG targets and there exist many high-confidence disease-PCG associations, we will be able to transfer the PCG-associated diseases to miRNAs in an interaction network framework. (2) We have formulated disease-miRNA association prediction as a semi-supervised multi-label node classification in a graph, which can help learn the complex networks composed of unlabeled miRNAs and labeled PCGs and the multi-label associations. This is a new prediction protocol for this problem. (3) We use semi-supervised GCN to learn patterns from PCG-associated diseases on an interaction network, which are further used to score diseases and miRNAs. This GCN-based approach combines the advantages of deep learning for representation learning and network-based methods. (4) We have further incorporated the domain knowledge into our model construction. Considering that miRNAs are often expressed in a tissue-specific way, we integrate the expression profiles across tissues into our GCN framework. Our results demonstrate that informative signals in more tissues can be captured for aiding the inference of disease-associated miRNAs.
Results
In this study, we first evaluate the prediction performance of DimiG on four tissue expression data with different number of tissues. Then we compare DimiG with other baseline methods that do not use disease-miRNA associations for model training on the independent test set, and further compare with BNPMDA and DRMDA trained using disease-miRNA associations on the unseen disease-miRNA set. Last, we present three case studies for prostate cancer, lung cancer, and inflammatory bowel disease (IBD).
DimiG Pipeline
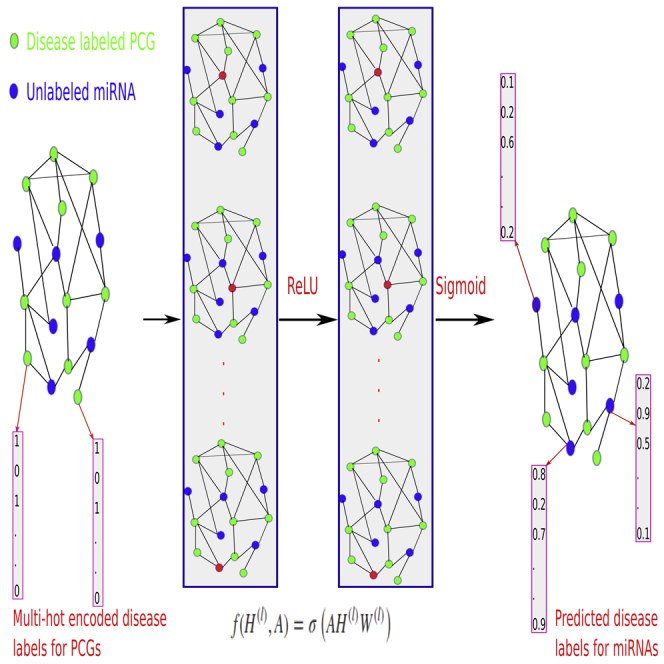
DimiG integrates gene network, expression profiles, and PCG-disease associations (Figure S1) using semi-supervised multi-label GCN. DimiG only requires PCGs with associated diseases (Figure S2) as labels during the model training, and then it propagates the node embedding to those miRNAs and further infers their associated labels. DimiG consists of two layers of GCNs, which require a node feature matrix, an adjacency matrix, and a label matrix. Each node (gene) is represented as a vector of expression profiles across tissues from GTEx (Lonsdale et al., 2013). The adjacency matrix is derived from the PCG-PCG and PCG-miRNA interactions. Only PCGs have assigned labels, which are a multi-hot vector corresponding to the presence of 248 associated diseases. We train GCN models on labeled PCGs and the interaction network, and the trained GCN model is used to score associations between diseases and unlabeled miRNAs. In the end, DimiG outputs a 1,034 × 248 score matrix, where 1,034 is the number of miRNAs and 248 is the number of diseases. More details are shown in Figure 1.
Figure 1.

The Flowchart of DimiG with Two-Layer GCN
Each node (gene) is represented as a vector of expression values across tissues with its sum across tissues from GTEx, and the network is constructed from PCG-PCG and PCG-miRNA interactions. When doing forward propagation, the embedding of the red node in each network is the weighted sum of the embedding of its neighbors, where all nodes in the network are updated simultaneously. The label is a multi-hot vector indicating the presence of diseases. In the end, DimiG can infer the probability between diseases and unlabeled miRNAs.
The Performance Comparison of DimiG on Four Tissue Expression Datasets
We first checked the impact of the number of epochs on training DimiG. As shown in Figure 2A, the training and validation loss converge to the same as the number of epochs approaches 50. Thus, in this study, we use 50 epochs for our below-mentioned experiments. It should be noted that the training loss is larger than validation loss during the first 30 epochs, which is also observed for the citation network data in the GCN article (Kipf and Welling, 2017). One possible reason is regularization (e.g., dropout), which is used during training, but not during validation. Another potential reason is that the features of the genes have certain discriminative power and they behave similarly to other genes in the training set. After several epochs, the learned node features are propagated well from interacting neighbor nodes for both training and validation genes.
Figure 2.

The Training and Performance of DimiG
(A) The training and validation loss change with the number of epochs on GTEx dataset.
(B) The ROC curve of DimiG using expression profiles from different datasets, where GTEx, E-MTAB-513, GSE30352, and GSE43520 cover 53, 16, 6, and 4 human tissues, respectively. We train DimiG for each expression dataset separately.
Grid search approach is used to select the best parameters for DimiG, where we search the learning rate with values of [0.001, 0.005, 0.0001, 0.0005], number of neurons in hidden layer with values of [248, 496, 744, 992, 1984], weight_decay with values of [0.001, 0.005, 0.0001] and Dropout with values of [0.5, 0.7, 0.8]. We yield the best area under receiver operating characteristic (ROC) curve (AUC) when learning rate = 0.0001, number of neurons = 744, weight_decay = 0.005, and Dropout = 0.8, which are finally used in our DimiG model.
As shown Figure 2B, DimiG yields the AUCs of 0.748, 0.729, 0.707, and 0.707 on GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. It yields the best performance on GTEx, which covers 53 tissues and thus contains more complete tissues. Compared with GTEx, DimiG achieves a lower performance on GSE43520. It is presumably because GSE43520 only covers four tissues and many tissues associated with certain diseases are missing.
When expression profiles from fewer tissues represent node features, DimiG easily suffers from noises that may be caused by sequencing errors. Of more interest, the area of the ROC curve with low false-positive rate for GTEx is much bigger than those for other three datasets; this region is especially important for evaluating predictive models. The results indicate that expression profiles across more tissues as node features can improve the prediction performance for disease-miRNA associations.
Comparing DimiG with Other Baseline Methods that Do Not Use Disease-miRNA Associations for Model Training
In DimiG, tissue expression profiles from GTEx are used as node features. Its variant DimiG-I just uses the one-hot encoding as the node features. As shown in Figure 3A, the proposed final DimiG yields an AUC of 0.748, which is an increase of 6% over AUC 0.706 of DimiG-I. These results demonstrate that expression profiles across tissues are very informative for inferring disease-miRNA associations. Another variant DimiG-C combines the expression profiles and one-hot encoding of nodes as node features and yields an AUC of 0.728, which is better than DimiG-I, but still worse than DimiG. These results demonstrate that simply concatenating the expression profile and one-hot encoding of nodes not only introduces computational burden but also may decrease the prediction performance. One possible reason is that the one-hot encoding of nodes has no correlations with the expression profiles, making the GCN unable to encode the node features well.
Figure 3.

The Performance of DimiG Using Expression Profiles as Node Features from GTEx and Baseline Methods
(A) ROC curve.
(B) Precision-recall curve. AUC is the area under ROC curve, and AUPRC is the area under precision-recall curve.
We also compare DimiG with another published method miRPD, which provides association scores between diseases and miRNAs derived from three different sources of miRNA-PCG interactions. As shown in Figure 3A, DimiG outperforms the AUC 0.673 of miRPD-T by 11.1%. Of the three miRPD-based methods, miRPD -T yields the best AUC 0.673, and it is based on the miRNA-PCGs predicted by TargetScan. The miRPD-C and miRPD-M yield similar AUC, which is much worse than miRPD-T. One potential reason is that miRPD-C infers association scores only based on a small number of verified miRNA-PCG. miRPD-M uses a similar number of miRNA-PCG interactions, but it may suffer higher false-positives than TargetScan. To make a fair comparison, we rerun the miRPD method using the same miRNA-PCG interactions and disease-PCG associations as DimiG. As shown in Figure 3, miRPD-I-sum and miRPD-I-max yield an AUC of 0.682 and 0.716, respectively, and both are superior to miRPD-T, miRPD-C, and miRPD-M, but they still perform worse than DimiG.
In addition, our results show that the coexpression-based coding-non-coding co-expression (CNC) method yields the performance AUC = 0.518, suggesting the co-expressed PCGs with miRNAs are not enough for identifying high-confidence disease-miRNA associations. It is because CNC is only based on expression profiles and interaction information of miRNAs is completely ignored.
We also calculate the area under precision-recall curve (AUPRC) of different methods. As shown in Figure 3B, DimiG yields the best AUPRC of 0.765, which is also better than its two variants DimiG-I and DimiG-C. In addition, compared with state-of-the-art method miPRD, the miRBD-T yields an AUPRC of 0.708, which is ∼9% worse than the AUPRC 0.765 of DimiG. However, miRPD-I-sum and miRPD-I-max yield much lower AUPRCs than other methods.
All the above results show that DimiG is are able to achieve better performance for inferring disease-miRNA associations, which is not a surprise because GCNs can better integrate interaction network data and tissue expression profiles, can operate on graphs similarly to CNNs on images, and can take the features and connectivity of nearby nodes into account.
Performance Comparison of DimiG on the Unseen Disease-miRNA Set
We also compare DimiG with other two state-of-the-art supervised methods BNPMDA and DRMDA on the unseen disease-miRNA set; both BNPMDA and DRMDA use disease-miRNA associations for model training. As shown in Figure 4, on this unseen disease-miRNA set, DimiG yields an AUC of 0.710 and an AUPRC of 0.724, which are better than the performance of BNPMDA with an AUC of 0.686 and an AUPRC of 0.698 and the performance of DRMDA with an AUC of 0.708 and an AUC of 0.715. The results indicate that DimiG outperforms the state-of-the-art supervised methods on inferring new disease-miRNA associations. The AUC 0.687 of BNPMDA on this unseen disease-miRNA set is lower than the reported 5-fold cross-validation AUC of 0.898. Similarly, the AUC 0.708 of DRMDA is also lower than the reported 5-fold cross-validation AUC of 0.916. It should be noted that there still exist some possible dependent disease-miRNA pairs with the training set derived from HMDD v2.0. The results demonstrate that the methods trained on disease-miRNA associations may yield biased cross-validation performance, which could not reflect the methods' actual ability to predict unseen miRNA-disease associations and generalize well to new miRNA-disease associations, as observed in Park and Marcotte (2012). As DimiG does not use any disease-miRNA associations, the performance of DimiG (an AUC of 0.710) on this unseen disease-miRNA set is consistent with the performance (an AUC of 0.748) on the full independent test set constructed from HMDD v3.0. The results indicate that the reported performance reflects DimiG's ability to infer new disease-miRNA associations.
Figure 4.

The Performance of DimiG, BNPMDA, and DRMDA on the Unseen Disease-miRNA Dataset
Here, DimiG uses expression profiles as node features from GTEx; BNPMDA and DRMDA are trained on known disease-miRNA associations.
(A) ROC curve.
(B) Precision-recall curve.
Case Studies
We present three case studies of miRNAs associated with prostate cancer, lung cancer, and IBD to demonstrate the applicability of DimiG for inferring disease-associated miRNAs. The top predicted candidates for these three diseases are checked with verified associations from literature and public databases, HMDD v3.0 and dbDEMC v2.0. In addition, we evaluate the disease-specific prediction performance of DimiG.
Prostate Cancer
We first investigate the prediction of prostate cancer-associated miRNAs from DimiG. Of the 1,034 miRNAs, top 30 miRNA candidates predicted by DimiG are given in Table 1. Eighteen miRNAs are supported by the literature or database dbDEMC v2.0 and five miRNAs are used as biomarker for detecting prostate cancer. For example, a meta-analysis shows that the first miRNA miR-939 and the eighth miRNA miR-661 are downregulated and the ninth miRNA miR-637 is upregulated in recurrent prostate cancer (Pashaei et al., 2017). This study also finds that prostate cancer-associated CTNNB1 (Anastas and Moon, 2013) is one hub gene for its interacting targets in a gene network. That is to say, the predicted miRNA genes by DimiG in Table 1 are well consistent with existing knowledge.
Table 1.
The Top 30 Candidate Prostate Cancer-Related miRNAs Predicted by DimiG and Their Support Evidences in Literature
| Rank | MiRNA Name | Reason | Support Evidence |
|---|---|---|---|
| 1 | miR-939 | Downregulated | PMID:28651018 |
| 2 | miR-93 | Overexpressed in prostate cancer, and downregulates capicua levels to promote prostate cancer progression | PMID:26124181 |
| 3 | miR-92a | Regulates tumor suppressor PTEN | PMID:24098737 |
| 4 | miR-874 | Upregulated | dbDEMC v2.0 |
| 5 | miR-766 | Downregulated | dbDEMC v2.0 |
| 6 | miR-765 | Mimics the expression of oncogenic HMGA1 in prostate cancer | PMID:24837491 |
| 7 | miR-744 | Promotes prostate cancer progression by activating Wnt/β-catenin pathway | PMID:28107193 |
| 8 | miR-661 | Downregulated | PMID:28651018 |
| 9 | miR-637 | Upregulated | PMID:28651018 |
| 10 | miR-625 | Upregulated | dbDEMC v2.0 |
| 11 | miR-608 | Target overexpressed FLOT1 in prostate cancer | PMID:28549468 |
| 12 | miR-5196 | – | – |
| 13 | miR-5193 | Downregulates TRIM11 in prostate cancer | PMID:30608062 |
| 14 | miR-504 | Represses overexpressed FOXP1 in prostate cancer | PMID:23022474 |
| 15 | miR-5011 | – | – |
| 16 | miR-491 | Interact with PDGFRA to reduce prostate cancer cell migration | PMID:29312807 |
| 17 | miR-486 | A prostate cancer driver | PMID:29069829 |
| 18 | miR-4739 | Biomarker for prostate cancer | Patent: WO2015190584A1 |
| 19 | miR-4731 | – | – |
| 20 | miR-4728 | Target MST4 involved in prostate cancer progression | PMID:25950472 |
| 21 | miR-4726 | Biomarker for prostate cancer | Patent: WO2015190584A1 |
| 22 | miR-4725 | Biomarker for prostate cancer | Patent: WO2015190584A1 |
| 23 | miR-4723 | Regulate Abl kinases in prostate cancer | PMID:24223753 |
| 24 | miR-4716 | – | – |
| 25 | miR-4667 | Biomarker for prostate cancer | Patent: WO2015190584A1 |
| 26 | miR-4644 | – | – |
| 27 | miR-455 | Target eIF4E as tumor suppressor in prostate cancer | PMID:28350134 |
| 28 | miR-4505 | Biomarker for prostate cancer | Patent: WO2015190584A1 |
| 29 | miR-4498 | – | – |
| 30 | miR-4447 | – | – |
– Means no support evidence.
In another study, the second miRNA miR-93 is frequently overexpressed in prostate cancer and downregulates capicua levels (Choi et al., 2015). In dbDEMC v2.0, three miRNAs miR-874, miR-766, and miR-625 are differentially expressed in prostate cancer. Of them, miR-625 and miR-874 share the same gene target HMGA1 in prediction channel of RAIN database with miR-765. The remaining three miRNAs in the top 10 candidates either regulate prostate cancer-associated genes or activate prostate cancer-associated pathway. We have also noted that of the top 10 miRNAs, only miR-92a and miR-765 are recorded in HMDD v3.0, and the others are not. These results indicate that DimiG can infer novel disease-miRNA associations currently not in the curated databases. Of the remaining 20 miRNAs, eight miRNAs are supported to be associated with prostate cancer by literature and five miRNAs are used as biomarkers for detecting prostate cancer in a filed patent. The results indicate that DimiG is powerful in identifying disease-associated miRNAs.
As shown in Figure 5A, of the 176 prostate cancer-associated miRNAs in HMDD v3.0, 21 are in the top 50 predicted candidates by DimiG. Six of the 15 prostate cancer-associated miRNAs in dbDEMC v2.0 belong to the top 50 candidates predicted by DimiG (Figure S3A). On the prostate cancer-specific dataset constructed from HMDD v3.0, DimiG yields an AUC of 0.724, 0.697, 0.675, and 0.664 on GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively (Figure 5B). Similarly, on the dataset constructed from dbDEMC v2.0, DimiG achieves an AUC of 0.844, 0.836, 0.729, and 0.684 on GTEx, E-MTAB-513, GSE30352, and GSE43520 for prostate cancer, respectively (Figure S3B). The performance is better than that on the dataset constructed from HMDD v3.0 because the extracted disease-miRNA associations are experimentally verified using low-throughput methods and are more reliable. We can observe that more tissues can provide informative clues for predicting prostate cancer-associated miRNAs; even some tissues may be considered not relevant to prostate cancer. The results further demonstrate the power of DimiG.
Figure 5.

Venn Diagram and ROC Curve for Predicting Associated miRNAs for Prostate Cancer, Lung Cancer, and IBD Using DimiG
(A) The overlap between the top 50 predicted miRNAs by DimiG and prostate cancer-associated miRNAs in HMDD v3.0.
(B) ROC curve for predicting prostate cancer associated miRNAs using four tissue expression datasets.
(C) and (D) (C) Venn diagram and (D) ROC for lung cancer.
(E) and (F) (E) Venn diagram and (F) ROC for inflammatory bowel disease (IBD)
We further investigate the predicted miRNA candidates using verified prostate cancer-associated miRNAs from miRCancer and PhenomiR. Of the top 50 predicted miRNAs by DimiG, 10 miRNAs are supported by miRCancer (Figure S4A) and 16 miRNAs are supported by PhenomiR (Figure S5A). On the prostate cancer-specific set derived from miRCancer, as shown in Figure S4B, DimiG yields an AUC of 0.755, 0.743, 0.690, and 0.695 using GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. As demonstrated in Figure S5B, for the prostate cancer-specific set collected from PhenomiR, DimiG achieves an AUC of 0.723, 0.713, 0.645, and 0.653 using GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. DimiG achieves similar results on the two databases as HMDD v3.0.
Lung Cancer
We investigate the prediction ability of DimiG for lung cancer-associated miRNAs. There are 172 such miRNAs recorded in the HMDD v3.0 for lung cancer. As shown in Figure 5C, of the 172 miRNAs, 12 miRNAs are in the top 50 lung cancer-associated miRNAs predicted by DimiG. Then we evaluate the prediction performance on the dataset consisting of 172 lung cancer-associated miRNAs and 172 miRNAs not associated with lung cancer in HMDD v3.0. As shown in Figure 5D, DimiG yields an AUC of 0.733, 0.701, 0.649, and 0.674 for GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively.
We further report the prediction ability on another lung cancer-specific dataset derived from dbDEMC v2.0. In this dataset, there are 16 miRNAs associated with lung cancer and another 16 miRNAs not associated with lung cancer. Of the 16 miRNAs associated with lung cancer, one is in the top 50 predicted miRNAs by DimiG (Figure S3C). As shown in Figure S3D, DimiG achieves an AUC of 0.925, 0.808, 0.806, and 0.869 for GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. The results also indicate that informative clues can be captured in more tissues for predicting lung cancer-associated miRNAs.
According to miRCancer database, 60 miRNAs are associated with lung cancer. Of the 60 miRNAs, three are in the top 50 miRNAs predicted by DimiG (Figure S4C). On the lung cancer-specific set derived from miRCancer (Figure S4D), DimiG yields an AUC of 0.778, 0.740, 0.775, and 0.705 using GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. Based on PhenomiR, 190 miRNAs are associated with lung cancer. Of them, 10 miRNAs are in the top 50 candidates predicted by DimiG (Figure S5C). In addition, as shown in Figure S5D, DimiG obtains an AUC of 0.781, 0.751, 0.757, and 0.754 using the four expression datasets GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. All the above results show that DimiG achieves promising performance for lung cancer.
Inflammatory Bowel Disease
Beside cancer, we also investigate DimiG's prediction ability for another non-cancer disease IBD. In this study, we predicted associated diseases for 1,034 miRNAs, of which 18 miRNAs are deposited for IBD. Seven of the 18 miRNAs are in the top 50 miRNAs predicted by DimiG (Figure 5E). As shown in Figure 5F, DimiG yields an AUC of 0.799, 0.758, 0.756, and 0.673 for GTEx, E-MTAB-513, GSE30352, and GSE43520, respectively. The results show that we can use DimiG for other non-cancer diseases.
Discussion
In this study, we present a semi-supervised multi-label GCN framework to integrate heterogeneous networks of tissue expression profiles, miRNA-PCG interactions, PCG-PCG interactions, and disease-PCG interaction to infer disease-associated miRNAs. The whole pipeline is under the context of interaction network, where disease-miRNA associations are completely not involved in model training. CNN cannot directly process the non-Euclidean domain data, like network data. However, GCN can handle these types of data; they are specially designed to extract abstract features from network data. To prove that, we use T-distributed Stochastic Neighbor Embedding (T-SNE) to map the learned node features in the last hidden layer by the GCN and the original node features from expression profiles in GTEx to 2D space. As shown in Figure S6, the learned node features by GCN have a better shape than original node features. We demonstrate that cross-validating the methods trained on disease-miRNA associations yields an overestimated performance. Our results demonstrate that DimiG outperforms other state-of-the-art methods, which do not require disease-miRNA association information, and two methods trained on disease-miRNA associations.
In this study, we need to set several cutoff values (Figure S1) for PCG-PCG interactions from STRING database, PCG-miRNA interactions from RAIN database, and disease-PCG associations from DISEASES database. All the three databases are developed by the same group, they use similar data quality control, and the confidence values are all scored using the similar pipeline from multiple channels, including experiments, knowledge, text mining, and prediction. Thus the constructed graph should follow GCN's assumption that it is a simple one-modal graph, in which all nodes are of the same type (all nodes are genes) and all edges have the same semantic meaning (Kipf and Welling, 2017).
As shown in Figure S7, for the low cutoff values of STRING with 300 and RAIN with 0.10, DimiG yields much lower performance with AUC 0.653, it is because lower cutoff may introduce more false-positive interactions. For cutoff value of STRING greater than 400 and cutoff value of RAIN greater than 0.15, DimiG yields similar AUCs. DimiG yields the best AUC 0.754 at cutoff values 400 and 0.20 for STRING and RAIN, respectively, but these higher cutoff values will lead to fewer miRNAs. Thus, we use cutoff value 400 of STRING and 0.15 of RAIN in this study, and DimiG yields an AUC 0f 0.748 and is capable of finding more miRNAs, which is just a little lower than 0.753 with cutoff value 400 and 0.20 for STRING and RAIN databases, respectively, as the trade-off. We also evaluate the impact of confidence thresholds 1.5 and 2.5 for disease-PCG associations on the performance of DimiG. As shown in Figure S8, DimiG yields an AUC of 0.719 and 0.742 for thresholds 1.5 and 2.5, respectively; both are lower than 0.748 when using threshold 2. The possible reason is that lower confidence threshold 1.5 introduces more false-positives for model training and higher threshold 2.5 makes the number of training samples much fewer, which are both not good for training machine learning model. In this study, we use PCG-PCG interactions from STRING v10 instead of STRING v11, because both RAIN and DISEASES databases are based on STRING v10. Some gene identifiers are changed between STRING v10 and v11. We directly use PCG-PCG interactions from STRING v11 for DimiG, only 5,092 PCGs are kept for constructing the interaction network, and DimiG achieves a lower AUC of 0.718.
There are other computational models with reported cross-validation AUC over 0.8, in which disease-miRNA associations are involved in model training. We demonstrate that cross-validation could report overoptimistic performance of the methods and could not generalize well to unseen disease-miRNA associations. Disease-miRNA associations can be incorporated into model training; however, randomly dividing the disease-miRNA pairs into the training and test sets for cross-validation could be biased. To better evaluate the performance of one model, a strictly independent test set should be at least constructed; e.g., the model is trained on all data published up to a specific year and predictions are evaluated on data published after that, or the model is trained on data in the older version of database and evaluated on those new added disease-miRNA associations in the updated database. In addition, DimiG predicts associated miRNAs for 248 diseases in one model. Many previous methods formulate the disease-miRNA prediction as binary classification problems, and they require constructing negative disease-miRNA associations, which may introduce false-negatives into model training.
Limitation of the Study
In this study, we used only expression profiles across tissues as node features. Some studies have revealed that functional domain information can assist identifying disease-associated miRNAs (Yang et al., 2018). In future work, we can combine the gene ontology (GO) information and expression values across tissues into the node features, or ensemble the two GCN models trained on each representation, which is expected to further improve the prediction performance of DimiG.
DimiG does not require disease-miRNA associations for model training, but it requires the miRNA-PCG interactions to construct the graph. Each miRNA must have at least one interacting PCG; all nodes, including miRNAs and PCGs, need be present during the training. Thus the trained node embedding can be propagated to miRNAs and further used for inferring miRNA-associated diseases. This precondition makes us to discard some miRNAs, and DimiG cannot infer associated diseases for these miRNAs without interacting PCGs. In addition, more and more novel miRNAs are being discovered, and their interactions with PCGs may not be readily available. Thus the trained models cannot be generalized to miRNAs not in the graph. Luckily, some recent GCN models have tried to solve this issue, which are trained on a set of nodes and generalized to any augmentation of the graph (Hamilton et al., 2017). This inductive GCN model can be applied for novel miRNAs not in the graph in our future study.
Conclusion
In this study, we present a semi-supervised multi-label learning framework DimiG to integrate interaction data for inferring miRNA-associated diseases. DimiG does not use any disease-miRNA associations for model training. This new approach achieves promising performance and outperforms other baseline methods not trained on disease-miRNA associations with a large margin on our benchmark dataset. We observe that cross-validation performance of methods trained on known disease-miRNA associations could be overestimated and could not reflect their actual abilities for inferring new disease-miRNA associations. Our results demonstrate that the tissue expression profiles can provide informative signals for inferring disease-miRNA associations. We expect DimiG to be used to discover novel miRNA biomarkers for diseases and that the framework can be extended to other tasks based on network data, e.g., functional annotations of proteins.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (No. 2018YFC0910500), the National Natural Science Foundation of China (No. 61903248, 61725302, 61671288, 91530321, 61603161), and the Science and Technology Commission of Shanghai Municipality (No. 16ZR1448700, 16JC1404300, 17JC1403500).
Author Contributions
Conceptualization, X.P. and H.-B.S.; Methodology: X.P.; Writing – Original Draft, X.P.; Writing – Review & Editing, X.P. and H.-B.S.; Funding Acquisition, H.-B.S.
Declaration of Interests
The authors declare no competing interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.013.
Contributor Information
Xiaoyong Pan, Email: xypan172436@gmail.com.
Hong-Bin Shen, Email: hbshen@sjtu.edu.cn.
Data and Code Availability
All the data and code are available at https://github.com/xypan1232/DimiG or http://www.csbio.sjtu.edu.cn/bioinf/DimiG.
Supplemental Information
References
- Alipanahi B., Delong A., Weirauch M.T., Frey B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015;33:831–838. doi: 10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- Anastas J.N., Moon R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer. 2013;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Angermueller C., Parnamaa T., Parts L., Stegle O. Deep learning for computational biology. Mol. Syst. Biol. 2016;12:878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antanaviciute A., Daly C., Crinnion L.A., Markham A.F., Watson C.M., Bonthron D.T., Carr I.M. GeneTIER: prioritization of candidate disease genes using tissue-specific gene expression profiles. Bioinformatics. 2015;31:2728–2735. doi: 10.1093/bioinformatics/btv196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M.A., Davis S.J., Liu P.Y., Pan X.Q., Williams A.M., Iczkowski K.A., Gallagher S.T., Bishop K., Regner K.R., Liu Y. Tissue-specific microRNA expression patterns in four types of kidney disease. J. Am. Soc. Nephrol. 2017;28:2985–2992. doi: 10.1681/ASN.2016121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Chen X., Huang L. LRSSLMDA: laplacian regularized sparse subspace learning for MiRNA-Disease Association prediction. PLoS Comput. Biol. 2017;13:e1005912. doi: 10.1371/journal.pcbi.1005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Gong Y., Zhang D.H., You Z.H., Li Z.W. DRMDA: deep representations-based miRNA-disease association prediction. J. Cell Mol. Med. 2018;22:472–485. doi: 10.1111/jcmm.13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Xie D., Wang L., Zhao Q., You Z.H., Liu H. BNPMDA: bipartite network projection for MiRNA-disease association prediction. Bioinformatics. 2018;34:3178–3186. doi: 10.1093/bioinformatics/bty333. [DOI] [PubMed] [Google Scholar]
- Chen X., Xie D., Zhao Q., You Z.H. MicroRNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2019;20:515–539. doi: 10.1093/bib/bbx130. [DOI] [PubMed] [Google Scholar]
- Chen X., Yin J., Qu J., Huang L. MDHGI: matrix decomposition and heterogeneous graph inference for miRNA-disease association prediction. PLoS Comput. Biol. 2018;14:e1006418. doi: 10.1371/journal.pcbi.1006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Liu M.X., Yan G.Y. RWRMDA: predicting novel human microRNA-disease associations. Mol. Biosyst. 2012;8:2792–2798. doi: 10.1039/c2mb25180a. [DOI] [PubMed] [Google Scholar]
- Ching T., Himmelstein D.S., Beaulieu-Jones B.K., Kalinin A.A., Do B.T., Way G.P., Ferrero E., Agapow P.M., Zietz M., Hoffman M.M. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface. 2018;15 doi: 10.1098/rsif.2017.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi N., Park J., Lee J.S., Yoe J., Park G.Y., Kim E., Jeon H., Cho Y.M., Roh T.Y., Lee Y. miR-93/miR-106b/miR-375-CIC-CRABP1: a novel regulatory axis in prostate cancer progression. Oncotarget. 2015;6:23533–23547. doi: 10.18632/oncotarget.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defferrard M., Bresson X., Vandergheynst P. Convolutional neural networks on graphs with fast localized spectral filtering. J. Univers. Comput. Sci. 2016;19:3844–3852. [Google Scholar]
- Friedman R.C., Farh K.K.H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene C.S., Krishnan A., Wong A.K., Ricciotti E., Zelaya R.A., Himmelstein D.S., Zhang R., Hartmann B.M., Zaslavsky E., Sealfon S.C. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015;47:569–576. doi: 10.1038/ng.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton, W.l., Ying, R., and Leskovec, J. (2017). Inductive Representation Learning on Large Graphs. 31st Conference on Neural Information Processing Systems (NIPS 2017).
- Jia L., Zhang Z., Wang L., Jiang W.M., Zhao M.B. Adaptive neighborhood propagation by joint L2,1-norm regularized sparse coding for representation and classification. IEEE 16th International Conference on Data Mining (ICDM) 2016:201–210. [Google Scholar]
- Jiang Q.H., Hao Y.Y., Wang G.H., Juan L.R., Zhang T.J., Teng M.X., Liu Y.L., Wang Y.D. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Biol. 2010;4(Suppl 1):S2. doi: 10.1186/1752-0509-4-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipf, T.N., and Welling, M. (2017). Semi-Supervised Classification with Graph Convolutional Networks. 5th International Conference on Learning Representations (ICLR-17).
- Lan W., Wang J., Li M., Liu J., Wu F.X., Pan Y. Predicting MicroRNA-disease associations based on improved MicroRNA and disease similarities. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018;15:1774–1782. doi: 10.1109/TCBB.2016.2586190. [DOI] [PubMed] [Google Scholar]
- Lecun Y., Bottou L., Bengio Y., Haffner P. Gradient-based learning applied to document recognition. Proc. IEEE. 1998;86:2278–2324. [Google Scholar]
- Lehtinen S., Lees J., Bahler J., Shawe-Taylor J., Orengo C. Gene function prediction from functional association networks using kernel partial least squares regression. PLoS One. 2015;10:e0134668. doi: 10.1371/journal.pone.0134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J., Thomas J., Salvatore M., Phillips R., Lo E., Shad S., Hasz R., Walters G., Garcia F., Young N. The genotype-tissue expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig N., Leidinger P., Becker K., Backes C., Fehlmann T., Pallasch C., Rheinheimer S., Meder B., Stahler C., Meese E. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–3877. doi: 10.1093/nar/gkw116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mork S., Pletscher-Frankild S., Palleja Caro A., Gorodkin J., Jensen L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics. 2014;30:392–397. doi: 10.1093/bioinformatics/btt677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.Y., Shen H.B. OUGENE: a disease associated over-expressed and under-expressed gene database. Sci. Bull. 2016;61:752–754. [Google Scholar]
- Pan X., Shen H.B. RNA-protein binding motifs mining with a new hybrid deep learning based cross-domain knowledge integration approach. BMC Bioinformatics. 2017;18:136. doi: 10.1186/s12859-017-1561-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Shen H.B. Predicting RNA-protein binding sites and motifs through combining local and global deep convolutional neural networks. Bioinformatics. 2018;34:3427–3436. doi: 10.1093/bioinformatics/bty364. [DOI] [PubMed] [Google Scholar]
- Pan X., Rijnbeek P., Yan J., Shen H.-B. Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics. 2018;19:511. doi: 10.1186/s12864-018-4889-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Jensen L.J., Gorodkin J. Inferring disease-associated long non-coding RNAs using genome-wide tissue expression profiles. Bioinformatics. 2019;35:1494–1502. doi: 10.1093/bioinformatics/bty859. [DOI] [PubMed] [Google Scholar]
- Park Y., Marcotte E.M. Flaws in evaluation schemes for pair-input computational predictions. Nat. Methods. 2012;9:1134–1136. doi: 10.1038/nmeth.2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pashaei E., Pashaei E., Ahmady M., Ozen M., Aydin N. Meta-analysis of miRNA expression profiles for prostate cancer recurrence following radical prostatectomy. PLoS One. 2017;12:e0179543. doi: 10.1371/journal.pone.0179543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinero J., Bravo A., Queralt-Rosinach N., Gutierrez-Sacristan A., Deu-Pons J., Centeno E., Garcia-Garcia J., Sanz F., Furlong L.I. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletscher-Frankild S., Palleja A., Tsafou K., Binder J.X., Jensen L.J. DISEASES: text mining and data integration of disease-gene associations. Methods. 2015;74:83–89. doi: 10.1016/j.ymeth.2014.11.020. [DOI] [PubMed] [Google Scholar]
- Tang C., Zhou H., Zheng X., Zhang Y., Sha X. Dual Laplacian regularized matrix completion for microRNA-disease associations prediction. RNA Biol. 2019;16:601–611. doi: 10.1080/15476286.2019.1570811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J.W., Wang Y. Cost-sensitive label propagation for semi-supervised face recognition. IEEE Trans. Inf. Foren. Sec. 2019;14:1729–1743. [Google Scholar]
- Xuan P., Dong Y., Guo Y., Zhang T., Liu Y. Dual convolutional neural network based method for predicting disease-related miRNAs. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19123732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan P., Han K., Guo Y., Li J., Li X., Zhong Y., Zhang Z., Ding J. Prediction of potential disease-associated microRNAs based on random walk. Bioinformatics. 2015;31:1805–1815. doi: 10.1093/bioinformatics/btv039. [DOI] [PubMed] [Google Scholar]
- Xuan P., Shen T., Wang X., Zhang T., Zhang W. Inferring disease-associated microRNAs in heterogeneous networks with node attributes. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018 doi: 10.1109/TCBB.2018.2872574. [DOI] [PubMed] [Google Scholar]
- Yang Y., Fu X., Qu W., Xiao Y., Shen H.B. MiRGOFS: a GO-based functional similarity measurement for miRNAs, with applications to the prediction of miRNA subcellular localization and miRNA-disease association. Bioinformatics. 2018;34:3547–3556. doi: 10.1093/bioinformatics/bty343. [DOI] [PubMed] [Google Scholar]
- You Z.H., Huang Z.A., Zhu Z., Yan G.Y., Li Z.W., Wen Z., Chen X. PBMDA: a novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017;13:e1005455. doi: 10.1371/journal.pcbi.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Li F.Z., Jia L., Qin J., Zhang L., Yan S.C. Robust adaptive embedded label propagation with weight learning for inductive classification. IEEE Trans. Neur. Net. Lear. 2018;29:3388–3403. doi: 10.1109/TNNLS.2017.2727526. [DOI] [PubMed] [Google Scholar]
- Zhou J., Troyanskaya O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015;12:931. doi: 10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitnik M., Agrawal M., Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018;34:i457–i466. doi: 10.1093/bioinformatics/bty294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoidi O., Tefas A., Nikolaidis N., Pitas I. Positive and negative label propagations. IEEE Trans. Circ. Syst. Vid. 2018;28:342–355. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data and code are available at https://github.com/xypan1232/DimiG or http://www.csbio.sjtu.edu.cn/bioinf/DimiG.