

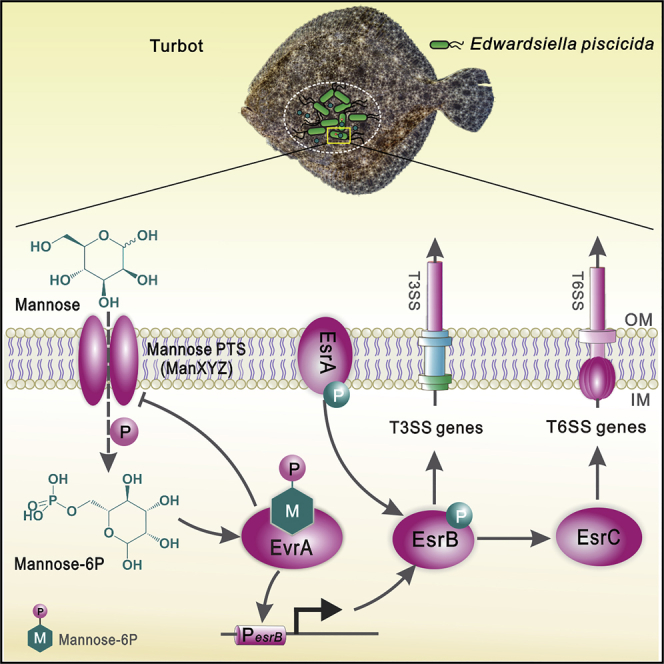
Summary
Bacterial pathogens are thought to activate expression of virulence genes upon detection of host-associated cues, but identification of the nature of such signals has proved difficult. We generated a genome-scale defined transposon mutant library in Edwardsiella piscicida, an important fish pathogen, to quantify the fitness of insertion mutants for intracellular growth in macrophages and in turbot (Scophthalmus maximus). These screens identified EvrA, a transcription activator that induces expression of esrB, a key virulence regulator. EvrA is directly bound and activated by mannose-6-phosphate (man-6P) derived from actively imported mannose. Mutants lacking EvrA or expressing an EvrA unable to bind man-6P were similarly attenuated in turbot. Exogenously added mannose promoted E. piscicida virulence, and high levels of mannose were detected in fish tissue. Together, these observations reveal that binding of a host-derived sugar to a transcription factor can facilitate pathogen sensing of the host environment and trigger virulence programs.
Subject Areas: Pathogenic Organism, Microbiology, Fish Culture
Graphical Abstract
Highlights
-
•
An E. piscicida defined mutant library is generated and analyzed in vitro and in vivo
-
•
EvrA is a key transcriptional activator of the known virulence regulator esrB
-
•
EvrA is directly bound and activated by mannose-6-phosphate from imported mannose
-
•
Extracellular mannose augments E. piscicida virulence in an evrA-dependent manner
Pathogenic Organism; Microbiology; Fish Culture
Introduction
Edwardsiella piscicida (formerly included in Edwardsiella tarda) is a Gram-negative facultative intracellular bacterial pathogen that causes edwardsiellosis, a serious systemic infectious disease that afflicts more than 20 species of freshwater and marine fish (Abayneh et al., 2013, Shao et al., 2015, Wang et al., 2009, Yang et al., 2012). This organism is also an opportunistic pathogen of humans, where it can cause gastroenteritis or wound infections and occasionally septicemia (Leung et al., 2012). All close relatives of E. piscicida, including E. tarda, E. hoshinae, E. ictaluri, and E. anguillarum, also infect farmed fish (Shao et al., 2015). As a result, edwardsiellosis causes severe economic losses in the aquaculture industry worldwide (Park et al., 2012). Moreover, these pathogens are increasingly becoming resistant to multiple antibiotics (Wang et al., 2009), limiting treatment options for the aquaculture industry and highlighting the need for the development of new prevention strategies, including vaccines (Park et al., 2012).
E. piscicida is thought to initiate infection by attaching to the epithelia of its principal host entry sites, the gastrointestinal tract or gills. Subsequently, the organism can survive and proliferate within host cells, particularly phagocytes (Leung et al., 2012), evading innate immune defenses, before causing hemorrhagic septicemia. Like phylogenetically related Enterobacteriaceae bacteria Salmonella spp., E. piscicida pathogenicity depends on both its type III secretion system (T3SS) and type VI secretion system (T6SS) as well as their distinct sets of effectors in animal models of infection (Chen et al., 2017, Liu et al., 2017, Srinivasa Rao et al., 2004, Zheng and Leung, 2007). Expression of these virulence-associated secretion systems requires a two-component system, EsrA-EsrB, and an AraC family transcriptional regulator EsrC in Edwardsiella bacteria (Rogge and Thune, 2011, Zheng et al., 2005). However, there is little knowledge of the environmental factors that trigger activation of these virulence-associated secretion systems or of non-T3/T6SS E. piscicida gene products required for fitness during infection or in aquatic environments.
Transposon-insertion site sequencing (TIS) is a potent high-throughput approach for determining the genetic requirements for bacterial fitness in distinct conditions (Chao et al., 2016, Price et al., 2018). Usually, highly saturated transposon mutant libraries are created so that TIS-based screens can provide high-resolution maps of the fitness contributions of individual loci and domains (Chao et al., 2016). However, less complex libraries, e.g., arrayed libraries containing mutants with a single insertion in a known genomic location, can also be useful, particularly when experimental bottlenecks are limiting (Abel et al., 2015, Fu et al., 2013), such as in some animal models of infection. Defined (or arrayed) mutant libraries, which usually contain one or two insertions per gene, have been created for several pathogens and model organisms, e.g., Pseudomonas aeruginosa and Vibrio cholerae (Cameron et al., 2008, Jacobs et al., 2003, Liberati et al., 2006), and have proved to be of value for screens where bottlenecks constrain the number of mutants that can screened (Fu et al., 2013). Moreover, such defined libraries serve as valuable resources because they often consist of collections of insertion mutants in almost all non-essential loci for an organism of interest.
Here, we created a comprehensive defined transposon mutant library in E. piscicida EIB202, a highly pathogenic isolate derived from a moribund turbot (Scophthalmus maximus) (Wang et al., 2009). We used pooled subsets of this library to analyze the fitness consequences of >7,000 insertion mutants during growth in media, in phagocytes and in vivo. An additional screen of the insertion mutants that had reduced fitness in turbot led to the identification of EvrA (ETAE_ 2071, Edwardsiella virulence regulator A), a transcription factor that directly activates expression of esrB, thereby leading to increased T3/T6SS expression (Liu et al., 2017, Zheng et al., 2005). Mannose imported into E. piscicida as mannose-6-phosphate (man-6P) binds to EvrA, promoting its activation of esrB expression. Moreover, mannose is present in host tissue and elevates E. piscicida virulence in fish. Thus, mannose appears to serve as a host-derived cue that activates a genetic circuit facilitating pathogenicity.
Results
Identification of Genes Important for Pathogen Growth in Fish Using a Defined Transposon Insertion Mutant Library
To facilitate genome-scale studies of the fish pathogen E. piscicida (formerly included in E. tarda) (Wang et al., 2009, Abayneh et al., 2013, Shao et al., 2015), we created a library of transposon mutants, where the site of each insertion was determined. MKGR, a derivative of the mariner transposon Himar1 (Rubin et al., 1999), was engineered for these studies (Figure 1A). Mutants generated by MKGR insertion should be resistant to gentamicin (Gm) and exhibit mCherry fluorescence, and a subset of mutants, with insertions downstream of active promoters, will be resistant to Km and exhibit GFP fluorescence; this expectation was confirmed experimentally (Figures S1A and 1B).
Figure 1.

Utilization of a Defined E. piscicida Transposon Insertion Mutant Library to Characterize Requirements for Pathogen Growth in Different Environments
(A) Schematic of the MKGR transposon and workflow overview for defined mutant library generation and subsequent TIS analysis.
(B–D) Scatterplots of input (LB grown) and output abundance of transposon insertion mutants after growth in DMEM (B), J774A.1 macrophages (C), and turbot fish (D). Genes with under-representation in the outputs (“significantly depleted”), based on a cutoff of Log2(Output/Input) (fold change, FC) ≤ −2.0 and p < 0.05, are highlighted in blue triangles; the FC of T3SS (pink squares) and T6SS (cyan diamonds) genes are also shown.
(E) Venn diagram depicting conditionally depleted genes from the three conditions tested. There were no depleted genes in common across all conditions.
The MKGR transposon was delivered by conjugation into E. piscicida EIB202 (ΔP), an otherwise wild-type (WT) and fully virulent strain cured of the endogenous R plasmid pEIB202 encoding genes resisting to various antibiotics, including chloramphenicol (Cm) (Figures 1A and S2A) (Wang et al., 2009). Individual insertion mutants were manually picked into 96-well plates. The insertion sites of mutants were sequenced and mapped to the EIB202 genome (Figures 1A and S2B–S2E, Tables S1 and S2). A total of 2,806 of the 3,599 predicted coding genes were disrupted with an average of approximately five insertions per gene (Table S1). The 78.0% ORF coverage (2,806/3,599) in the E. piscicida defined mutant library is similar to that reported for defined libraries created in other pathogens (Cameron et al., 2008, Gallagher et al., 2007, Gallagher et al., 2013).
To overcome experimental limitations present with very-high-density transposon libraries, e.g., infection bottlenecks (Chao et al., 2016, Fu et al., 2013), a subset library composed of 7,299 randomly selected mutants, including one or two distinct insertions for each disrupted protein coding gene and intergenic region, was assembled from the set of 20,346 unique insertion mutants (Tables S1, S2, S3, S4, S5, S6, and S7). We compared the fitness consequences of the insertion mutations present in this library after growth in Dulbecco's Modified Eagle's medium (DMEM), murine macrophage-like J774A.1 cells, where E. piscicida grows intracellularly (Chen et al., 2017, Liu et al., 2017, Okuda et al., 2009), and in turbot, a natural E. piscicida host (Figures 1B–1D) (Wang et al., 2009). The library was grown in LB medium, the source of the “input” for TIS analyses, before inoculation into each condition. Mutant bacteria recovered from DMEM, J774A.1 cells, or turbot livers (the most robustly colonized tissue [Yang et al., 2017]), were used as “outputs” for TIS analyses. In each condition, correlation coefficients of three biological replicates were high (Figure S3), suggesting that these experiments were not severely compromised by infection bottlenecks or other factors that might stochastically limit library complexity.
There was no overlap in the genes categorized as conditionally depleted (fold change [FC] cutoff = 4, p < 0.05) after growth in DMEM and J774A.1 cells (Figure 1E) (Tables S8 and S9). Genes encoding the T3SS (e.g., eseB and esaM) (Liu et al., 2017, Zheng et al., 2005) and T6SS (e.g., evpC and evpI) (Zheng and Leung, 2007) were not found to be required for growth in DMEM (Figure 1B), even though this medium is known to promote the transcription of T3SS genes (Liu et al., 2017), illustrating the difference between the genetic requirements for growth and the transcriptional reprogramming that may occur in different environments. In fact, insertions in several key activators of T3SS gene expression including esrC, esrA, and esrB, displayed slightly enhanced fitness (FC > 1) in DMEM (Figure 1B), presumably due to reduced metabolic costs associated with production of the T3SS in these mutants (Figure S4A). In contrast to growth in DMEM, E. piscicida growth in J774A.1 cells was dependent on several T3SS genes (Figures 1C and 1E, Table S9), revealing the importance of the pathogen's T3SS for macrophage infection. However, T6SS genes did not contribute to the pathogen's fitness within J774.A1 cells (Figure 1C). This observation was confirmed using turbot-derived macrophages and E. piscicida strains containing single deletions of evpP, evpC, or evpI, critical T6SS structural genes (Zheng and Leung, 2007); these deletion mutants grew as robustly inside turbot macrophages as the WT strain (Figures S5A and S5B). Thus, the T6SS, which is important for E. piscicida growth in turbot (Yang et al., 2017), may primarily promote extracellular growth of the pathogen in vivo.
More genes (258) (Table S10) were categorized as conditionally depleted after growth in turbot than in DMEM or macrophages (Figure 1E), consistent with the idea that the pathogen must rely on a broader array of genes to confront the diverse and changing challenges present in fish tissues. Nearly all of E. piscicida's genes associated with T3SS (29 of 34) and T6SS (16 of 16) (Figure 1D) were conditionally depleted in turbot, confirming the importance of these pathogenesis-linked secretion systems (Srinivasa Rao et al., 2004, Yang et al., 2017, Zheng and Leung, 2007). A genome-scale comparison highlighted the importance of four gene clusters, encoding LPS (region 1, including waaG, waaQ, waaL, waaF, waaC, walW, walR, wabH, wabK [ETAE_0073–0082]), and NADH dehydrogenase (region 3, including nuoM, nuoJ, nuoI, nuoF, nuoD, and nuoA), in addition to the T3SS (region 2) and the T6SS (region 4), as particularly important for E. piscicida growth in turbot (Figure S4B) (Table S11). Region 3 genes were also important for growth in J774A.1 cells, suggesting that the pathogen relies on oxidative phosphorylation for growth inside macrophages as well as in fish. Notably, the largest number (56) of conditionally depleted genes in turbot were of unknown function (Figure S4A); future studies defining the functions of these genes will reveal new aspects of pathogen physiology enabling growth in vivo.
Validation of Conditional Depleted Genes
Specific genes from the above-mentioned four regions of interests were chosen (Figure S4B) (waaQ, walW, wabK, esrA, esrB, eseB, esaM, nuoM, nuoA, nuoI, evpP, evpC, and evpI) for validation using in-frame deletion mutants. In these experiments, E. piscicida ΔP (WT(ΔP)) was mixed 1:1 with each of these mutants and inoculated into LB, DMEM, J774A.1 cells, or turbot in competition assays. In LB, none of the mutants exhibited growth defects, whereas in DMEM, the mutants with insertions in LPS synthesis genes (waaQ, walW, and wabK) were significantly outcompeted by the WT (Figure 2A), mirroring the findings from the screen. Similarly, in the competition experiments in J774A.1 cells and turbot, all insertion mutants that were classified as conditionally depleted in the screens exhibited significant defects in the competition assays (Figures 2B and 2C). Furthermore, the competitive indices found with the waaQ, esrB, eseB, nuoM, evpP, and evpI deletion mutants were similar in J774A.1 cells and turbot primary macrophages (Figure S5B). Thus, the observations from the competition assays strongly correlate with TIS screens. Moreover, there was also an excellent correlation in the fitness measures calculated from the competition and TIS assays (R2 = 0.753, Figure 2D). This correlation was calculated using data presented in Figures 2A–2C along with similar data obtained with 16 additional mutants containing in-frame deletions in genes that covered a range of FC values calculated in the TIS screens. The strong correlation over a large range of FC values derived from the TIS and competition experiments with deletion mutants suggests that the genome-scale datasets presented in Figure 1 and Tables S8–S10 constitute a robust resource for E. piscicida studies.
Figure 2.

Validation of TIS Studies with Competitive Assays in DMEM, Macrophages, and Turbot
(A–C) Selected in-frame deletion mutants were competed 1:1 versus WT(Δp) in LB and DMEM (A), J774A.1 (B), and turbot fish (C). Competitive indices are shown, and the data presented are mean ± SD from three to nine replicates. *p < 0.05, ***p < 0.001 based on ANOVA followed by Bonferroni's multiple-comparison post-test to compare the data with the values from the WT/WTΔp competitions.
(D) Correlation of FC values derived from competition experiments with deletion mutants and from the TIS screen (panels A–C). Each point represents the FC value and standard error (SE) for one gene in both screens. In total, 28 deletion mutants were tested in DMEM, J774A.1, and turbot. A linear regression analysis was used to determine the correlation.
Identification and Characterization of evrA, an In Vivo Virulence Regulator
To identify mutants with defective activation of E. piscicida's T3SS, we individually screened the 258 insertion mutants found to have growth defects in turbot (Table S10) along with 34 mutants displaying auto-aggregation defects when grown in DMEM (Table S12) for their capacities to enter into and proliferate within J774A.1 cells. Although fewer insertion mutants (34 versus 67) showed deficiencies in intracellular growth from this screen as compared with the initial TIS analysis in J774A.1 cells (Tables S9 and S12), most of the mutants that answered this secondary screen (24/34) contained insertions in T3SS-related genes (Table S12). Several of the other mutants had insertions in genes implicated in metabolic processes. One of these genes, ETAE_3493, encodes a homologue of glnA (glutamine synthetase), which is known to modulate production of the E. piscicida T3SS and to be required for E. piscicida pathogenicity (Guan et al., 2018, Yang et al., 2017). We focused our work on another mutant, which contained an insertion in ETAE_ 2071 (hereafter referred to as EvrA for Edwardsiella virulence regulator A) because this gene had not previously been linked to the pathogen's expression of its T3SS or virulence. Since EvrA bears similarity to the DeoR family of transcriptional regulators, which modulate sugar and nucleotide metabolism in diverse bacteria (Figure S6) (Gaigalat et al., 2007, Ishikawa et al., 2002), we speculated that it could provide insight into the metabolic control of expression of E. piscicida's T3SS. In the initial TIS turbot screen, the evrA insertion mutant had an ∼8-fold reduced abundance (FC = 0.13, p < 0.001) (Figure 1D) and the evrA deletion mutant exhibited a competitive defect versus WT(ΔP) in turbot and in J774A.1 cells and turbot macrophages (Figures 2 and S5B), but not in LB or DMEM (Figure 2A). Similarly, assayed on its own, the evrA deletion mutant exhibited reduced invasion of and/or proliferation within J774A.1 cells and caused less cytotoxicity as well, and both these defects were complementable (Figures S5C and S5D). Moreover, evrA transcript abundance was elevated in turbot relative to DMEM (Figure 3A). Together, these observations suggested that evrA may be an in vivo-induced regulator of E. piscicida virulence.
Figure 3.

EvrA Regulates E. piscicida Virulence
(A) evrA transcript levels in WT E. piscicida grown in DMEM (12 h), HeLa cells (8 h.p.i.), J774A.1 cells (6 h.p.i.), and turbot liver (8 d.p.i.), relative to that in the bacteria grown in LB for 12 h. gyrB was used as an internal housekeeping control. n = 3, *, p < 0.05; ***, p < 0.001 as compared with LB (arbitrarily set as 1) and DMEM based on Student's t test.
(B) Auto-aggregation and extracellular protein (ECP) profiles in the indicated strains.
(C) Expression of eseB in turbot and associated CFU burdens; the indicated strains harboring PeseB-luc reporter plasmids were inoculated into turbot and luminescence and bacterial burden was measured at 8 d.p.i. ***, p < 0.001 based on ANOVA analysis of the relative fluorescence units (RFU) and the bacterial burden (n = 3).
(D) Comparison of transcriptomes of WT and ΔevrA. The log2 of the ratio of the abundances of each transcript in ΔevrA versus WT (M) was plotted against the average log2 of the abundance of that transcript in both strains. T3SS, T6SS, and PTS-related genes are highlighted.
(E) qRT-PCR analysis of the transcript levels of indicated T3/T6SS genes in ΔevrA and ΔesrB strains bearing evrA or esrB expressing plasmids driven by their native promoters or a constitutive Plac promoter for esrB, relative to that in WT. gyrB was used as a control. n = 3, *p < 0.05, ***p < 0.001 based on Student's t test.
(F) EMSA of EvrA binding to PesrB. Purified EvrA was added to 20 ng of PesrB or mutant (PesrB Mut1 and PesrB Mut2) Cy5-labeled probes. B, bound DNA; U, unbound DNA.
(G) DNase I footprinting analysis of EvrA binding to a site in the esrB promoter (shown in the dashed box). Electropherograms show a DNase I digestion of the PesrB probe after incubation with 0 or 200 nM of EvrA. The corresponding nucleotide sequence (198 bp 5′ of the translational start codon) protected by EvrA is indicated below. The mutant PesrB motifs used for the EMSA in (C) are shown.
We next investigated if EvrA promotes expression of the pathogen's T3SS and T6SS. E. piscicida aggregates due to the production of EseB, a T3SS apparatus protein, whose expression is directly activated by EsrB, a critical activator of the pathogen's T3SS and T6SS (Gao et al., 2015, Liu et al., 2017, Yin et al., 2018). The evrA deletion mutant did not auto-aggregate (Figure 3B) and produced reduced amounts of T3SS and T6SS proteins in cell lysates (Figure S5E) and in cell supernatants (Figures 3B and S5E) as determined by western blot analysis. Reintroduction of evrA into ΔevrA fully complemented the auto-aggregation and T3/T6SS production defects (Figures 3B and S5E), demonstrating that EvrA augments expression of E. piscicida's T3/T6SS.
In vivo bioluminescence imaging was used to investigate T3SS expression during E. piscicida infection of turbot (Yin et al., 2018). A luciferase reporter of PeseB expression was introduced into a neutral position on the chromosome of WT, ΔevrA and ΔevrA complemented strains, and these strains were inoculated intraperitoneally into turbot. By 8 days post infection (d.p.i.), when luciferase activity was detected in the WT and complemented strains, PeseB-luc activity was not detectable in the ΔevrA background (Figure 3C). Moreover, there was ∼10–16x fewer ΔevrA than WT or the complemented strain CFU recovered from infected fish at this time point (Figure 3C). Together, these observations suggest that EvrA contributes to E. picicida growth in the host by activation of T3SS and T6SS genes.
EvrA Binds Directly to the esrB Promoter to Activate Virulence Gene Expression
To further elucidate how EvrA modulates E. piscicida growth in vivo, we used RNA sequencing (RNA-seq) to define the EvrA regulon by comparing the transcriptomes of the WT and ΔevrA strains. Transcripts of 166 genes were significantly decreased (log2FC < −1 and p < 0.05) and 78 were increased (log2FC > 1 and p < 0.05) in ΔevrA compared with the WT (Figure 3D and Table S13). Many genes in the T3/T6SS gene clusters had lower transcript levels in the evrA mutant, consistent with the idea that their expression is activated by EvrA (Figure 3D and Table S13). qRT-PCR assays corroborated that transcript levels of established T3SS regulatory genes (esrA, esrB, and esrC), T3SS structural genes (eseB and esaM), and T6SS gene evpP were all reduced in the absence of evrA but restored in the complemented strain, ΔevrA + pUTt-evrA (Figure 3E). Levels of these six transcripts were even lower in the ΔesrB mutant, but their levels were not reduced further in an ΔevrAΔesrB mutant, suggesting that evrA acts upstream of and in the same pathway as esrB. Introduction of esrB driven by its native promoter into the double mutant only partially restored transcript levels, whereas introduction of esrB driven by the unrelated lac promoter fully restored transcript amounts to WT or greater levels (Figure 3E). Collectively, these findings support the idea that EvrA promotes esrB expression.
Electrophoretic mobility shifts assays (EMSAs) using purified EvrA were carried out to begin to test whether EvrA directly regulates esrB expression. EvrA bound to a DNA probe that included the upstream region of the esrB gene (Figure 3F). The binding site of EvrA in the esrB promoter region was defined with a DNase I footprint assay performed on a DNA fragment that encompassed the entire intergenic region between esrB and ETAE_0887, the adjacent upstream gene. EvrA protected a region (5′-TTTATCCAAAAT-3′) bearing an AT-rich palindrome structure found 198 bp upstream of the esrB start codon (Figure 3G); this AT-rich sequence is similar to the known binding sites for other DeoR family proteins (Gaigalat et al., 2007). Substitution of the AT nucleotides with GC (PesrB Mut1) but not the replacement of CC with AA (PesrB Mut2) abolished the capacity of EvrA to bind to this fragment (Figure 3F), demonstrating that EvrA binds to a distinct site in the esrB promoter. These observations are consistent with the idea that EvrA modulates E. piscicida virulence gene expression by directly activating EsrB transcription.
Mannose Stimulates evrA-Dependent Virulence Gene Activation
DeoR family proteins often modulate sugar utilization (Figure S6) (Anantharaman and Aravind, 2006, Gaigalat et al., 2007, Ishikawa et al., 2002), and the RNA-seq experiment revealed changes in transcript levels of several sugar transport/utilization genes (e.g., ptsH and manXYZ) in the evrA mutant (Figure 3D). After testing various sugars, we found that supplementation of DMEM with mannose, a C-2 epimer of glucose, selectively induced PesrB-luxAB reporter expression, even though bacterial growth was similar in all of the fermentable carbohydrates screened (Figures 4A and S7A). Activation of esrB promoter activity during growth in mannose required evrA (Figures 4A and S7B), suggesting that mannose promotes evrA-dependent induction of esrB transcription. Consistent with this idea, we found that growth in mannose augmented evrA-dependent production of T3/T6SS proteins (Figure 4B).
Figure 4.

Mannose Promotes EvrA-Dependent Virulence Gene Expression
(A) Chromosomal PesrB-luxAB reporter activity in the indicated strains grown for 12 h in DMEM medium supplemented with the 5 mg/mL of indicated sugars. Data shown are the mean ± SEM of results for triplicate assays. *p < 0.05; ***, p < 0.001 based on Student's t test.
(B) ECP profiles (upper panel) and western blot (lower panel) analysis of T3SS protein EseB expression in cell lysates (WCP) of the indicated strains in the presence of 5 mg/mL glucose (glu) or mannose (man). DnaK was used as the loading control. The numbers correspond to densitometry measurements.
(C) ChIP-qPCR analysis of the relative enrichment in PesrB DNA molecules bound to EvrA from cells grown in glucose (glu) or mannose (man). The results are normalized to the control gene gyrB as well as to ChIPs from ΔevrA + pUTt-Flag cells. ***p < 0.001, t test. NS, not significant.
(D) EMSA of the binding of EvrA to PesrB in the presence of various mannose derivatives. Purified EvrA was mixed with mannose-6-phosphate (man-6P), mannose-1-phosphate (man-1P), GDP-man, or man and then added to 20 ng of Cy5-labeled PesrB probe. B, bound DNA; U, unbound DNA.
Chromatin immunoprecipitation (ChIP)-qPCR analyses revealed that EvrA binding to PesrB was greater in cells grown in mannose than in glucose (Figures 4C and S7C), suggesting that mannose can regulate EvrA DNA-binding activity. However, EMSA analysis showed that addition of mannose to EvrA did not modify its binding to the esrB promoter region (Figure S7D). We hypothesized that EvrA may be directly responsive to a mannose-derived metabolite instead of the native sugar, as bacterial import systems such as the phosphotransferase system (PTS) couple sugar import to modifications such as phosphorylation. Accordingly, we found that mannose-6-phosphate (man-6P), but not mannose-1-phosphate (man-1P) or GDP-mannose, enhanced EvrA binding to the esrB promoter (Figures 4D and S8A–S8D). Electrospray ionization mass spectrometry revealed that purified EvrA forms a folding-dependent complex with man-6P, strongly suggesting that EvrA can directly bind man-6P (Figures 5A and S9).
Figure 5.

Mannose-6-Phosphate (man-6P) Binding to EvrA Enhances esrB Expression
(A) Electrospray ionization mass spectrometry of native and denatured EvrA C-domain complex with man-6P.
(B) Structural model of EvrA interacting with an in silico-docked man-6P based on homology alignment to the D-ribose-5-phosphate isomerase RpiA (PDB_ID: 1LK7) (Ishikawa et al., 2002). The ligand M6P and residues involved in the interaction, as well as R178 were highlighted as sticks with C atoms colored in yellow and cyan (P, orange; N, blue; O, red), respectively.
(C) EMSA of binding of WT or mutant EvrA to PesrB in the presence of man-6P. Purified EvrA or its variants mixed with man-6P and 20 ng of Cy5-labeled PesrB probe were added to the EMSA reactions. B, bound DNA; U, unbound DNA.
(D) Chromosomal PesrB-luxAB reporter activity in the indicated strains grown for 12 h in DMEM medium supplemented with glucose or mannose. Data shown are the mean ± SEM of results for triplicate assays. *p < 0.05; ***, p < 0.001 based on Student's t test. NS, not significant.
(E) Western blot analysis of T3SS protein EseB expression in cell lysates of the indicated strains in the presence of glucose or mannose. DnaK was used as the loading control.
EvrA shares secondary structure with Pyrococcus horikoshii d-ribose-5-phosphate-isomerase (RpiA) (Figure S6F). We performed homology modeling using the known crystal structure of RpiA bound to its ligand (PDB 1LK7) (Ishikawa et al., 2002), to predict how EvrA binds man-6P (Figure 5B). The modeling suggests that EvrA binding to man-6P is dominated by ionic interactions between the phosphate group of the sugar and the sidechain of R221, which protrudes into the binding pocket. The main chain nitrogen atoms of S96 and T97 also likely participate in ligand coordination through hydrogen bonds. Besides R221, two additional arginines were targeted for mutagenesis: R178 from the DeoRC domain, which is predicted to be dispensable for ligand binding, and R7 from the unmodeled HTH domain, which is likely critical for EvrA promoter recognition (Figures 5B, S6D, and S6E).
Biophysical characterization of man-6P-EvrA binding by isothermal titration calorimetry indicated that the complex forms at a micromolar Kd (22.5 μM) with a 1:1 stoichiometry, comparable with other known DeoR-ligand interactions (Figure S8A) (Ishikawa et al., 2002). Alanine substitutions at R221, but not the predicted DNA-contacting R7 or the neutral R178, substantially eliminated man-6P binding to EvrA (Figures S8E–S8G), supporting the role of the R221 in coordinating ligand binding (Figure 5B). The binding studies were corroborated with EMSA-based binding analyses. Man-6P stimulated binding of WT and R178A EvrA to PesrB (Figure 5C) but did not modify binding of the R7A or R221A forms of the protein, presumably because of the loss of their DNA or ligand recognition capacities, respectively (Figures 5C and S8H). The reasons why EvrAR221A bound the PesrB probe with lower apparent affinity than the WT protein (Figure S8H) are not known, but it is possible that the WT protein may co-purify with bound man-6P. Next, we expressed the mutant EvrA proteins in E. piscicida (Figure S10A) to test their function in cells. Only E. piscicida strains expressing EvrA or EvrAR178A led to auto-aggregation (Figure S10B) and exhibited mannose augmentation of esrB expression and EseB production (Figures 5D, 5E, and S10C). Thus, EvrA's capacity to bind man-6P and DNA appear to be critical for the protein to promote virulence gene expression.
We speculated that EvrA might also play a role in mannose uptake because the transcriptomic data suggested that EvrA modestly represses manX (FC ∼ 2) (Figure 3D and Table S13), a component of the mannose-specific PTS, which imports mannose into the cell (Erni et al., 1987). Growth of WT E. piscicida in mannose augmented evrA expression but decreased manX expression, suggestive of negative feedback (Figure S7E). Consistent with the RNA-seq data, the ΔevrA mutant had higher levels of manX transcripts than the WT grown in mannose as well as in glucose, suggesting that EvrA represses manX expression (Figure S7E). Since EvrA binds to the manX promoter region via an AT-rich palindrome (Figure S7F), these findings suggest that EvrA directly represses the manXYZ operon. Collectively, these observations are consistent with the idea that EvrA directly represses manX expression, potentially creating a negative feedback loop dampening mannose-induced, evrA-dependent induction of esrB expression (Figure S4C).
Mannose Augments E. piscicida Virulence in an evrA-Dependent Manner
Finally, we investigated the roles of evrA, manX, and mannose in E. piscicida virulence in turbot. As shown previously, the ΔesrB, ΔT3SS, and ΔT6SS mutant strains were highly attenuated and ∼90% of fish remained alive at 14 d.p.i (Figure 6A) (Yin et al., 2018). The ΔevrA and ΔmanX mutants were also attenuated with a median survival time of over 14 and 7 days, respectively (Figure 6A). More than 50% of fish survived infection with the ΔevrA mutant, and although no animals survived infection with the ΔmanX mutant, these animals survived longer than animals infected with the WT strain (p = 0.0038, Figure 6A). These observations are congruent with the diminished in vivo fitness of both the evrA (FC = 0.13) and manX (FC = 0.24) transposon mutants observed in the TIS screen (Table S10) and demonstrate that evrA and manX contribute to E. piscicida virulence.
Figure 6.

Mannose Promotes E. piscicida pathogenicity in an EvrA-Dependent Manner
(A) Survival curves of turbot challenged with the indicated strains. Phosphate-buffered saline (PBS, pH 7.4) supplemented with 5 mg/mL glucose and mannose was used as a control. The bacterial strains were suspended in PBS with or without 5 mg/mL glucose or mannose and injected into each turbot at a dose of 2.0 × 104 CFU/fish (n = 30 fish/group). Kaplan-Meier survival analysis with a log rank test is shown. **, p < 0.001; NS, not significant, p > 0.05.
(B) In vivo PeseB-luc plasmid reporter activity and associated liver CFU burden at 8 d.p.i. from fish inoculated with the indicated strains. ***, p < 0.001 based on ANOVA analysis of the relative fluorescence units (RFU) and the bacterial burden (n = 3).
(C) Survival curves of turbot challenged with the indicated strains. PBS supplemented with 5 mg/mL mannose was used as a control; otherwise these data were acquired and analyzed identically to that in (A).
(D) Mannose (blue) and man-6P/man-1P (red) levels in extracts from turbot intestines or livers before (filled) or after (open) infection with WT E. piscicida (left) and in WT E. piscicida grown in DMEM supplemented with glucose or mannose (right). Mean ± SE from five fish or three bacterial samples are shown; there was no detectable mannose in the intestine of turbot or in E. piscicida, which is indicated by *, below detection limit. p Values are calculated based on unpaired two-tailed Student's t test.
(E) Schematic of E. piscicida mannose responsive virulence gene regulatory circuit.
Co-inoculation of the WT strain with mannose (5 mg/mL), but not glucose (5 mg/mL), accelerated the mortality of the fish (p = 0.0045, Figure 6A), consistent with the prior observation in carp that elevated tissue mannose, and man-6P levels are correlated with lethal E. tarda infection Guo et al., 2014. In contrast, co-inoculation of the ΔevrA or ΔmanX strains in mannose or glucose did not alter the kinetics of fish survival (p > 0.05, Figure 6A). Mannose supplementation also led to increased expression of eseB in vivo and greater E. piscicida proliferation (Figure 6B). In this assay, strains expressing the evrA mutants that were incapable of mannose-stimulated esrB expression in culture (EvrAR221A and EvrAR7A) phenocopied the evrA deletion mutant (Figure 6B). Similarly, strains expressing these non-functional EvrA mutants were as attenuated in vivo as the strain lacking evrA (Figure 6C); in contrast, the strain expressing the mannose-response evrA mutant (EvrAR178A) killed fish with similar kinetics as the WT.
The observation that EvrAR221A phenocopied the ΔevrA strain, even in the absence of mannose supplementation, suggests that during infection, the EvrA ligand is present and detected by EvrA. Mannose and mannose phosphates (man-6P and man-1P) were found at micromolar levels in the intestines and livers of uninfected turbot (Figure 6D). Consistent with their micromolar presence in vivo, man-6P/man-1P accumulated to micromolar levels in E. piscicida grown in mannose-supplemented cultures, suggesting that the ligand of EvrA (but not its precursor) can naturally accumulate in the intracellular bacterial space (Figure 6D). Furthermore, levels of mannose and man-6P/man-1P were greater in livers from infected fish than those in naive fish, suggesting that E. piscicida growth in vivo may stimulate host production of this sugar. Moreover, esrB expression could be efficiently activated by mannose concentrations detected in vivo (20–200 μM, Figure S11). These findings are consistent with the idea that mannose stimulation of EvrA-dependent induction of virulence gene expression can promote E. piscicida pathogenicity during infection and that exposure to host mannose can prime E. piscicida's virulence.
Discussion
E. piscicida and closely related Edwardsiella species are important fish pathogens that inflict great damage on the aquaculture industry globally. These facultative intracellular organisms are particularly compelling for pathogenesis studies because their virulence depends on both T3SS and T6SS (Srinivasa Rao et al., 2004, Yang et al., 2017, Zheng and Leung, 2007). The defined E. piscicida transposon mutant library (Tables S1 and S2) and the resulting genome-scale datasets representing the genetic requirements for the pathogen's growth in DMEM, J774A.1 cells, and fish (Tables S8–S10, Figure S4C) presented here should provide a valuable resource for future analyses of this pathogen's virulence and functional genomics. There was a remarkable congruence in the observations derived from the three TIS screens and in studies using 28 deletions mutants (Figure 2), including in the magnitudes of the calculated fitness defects for both insertion and deletion mutants, suggesting that the findings from the screens are robust. Besides confirming the importance of the E. piscicida T3/T6SS for turbot growth, our findings delineated many metabolic pathways that the organism depends on to proliferate during infection (Figure S4C) and revealed a new positive regulator of virulence, EvrA.
The genes found to facilitate E. piscicida fitness in the DMEM, J774A.1, and turbot screens were largely distinct, reflecting the manifold differences in these conditions. The genes enabling robust growth in J774A.1 cells and in DMEM did not overlap, and only 28/67 genes facilitating growth in these murine macrophage-like cells were scored as important for fitness in turbot (Figures 1E and S4C). Furthermore, there were nearly 4x as many genes required for fitness in turbot than in J774A.1 cells, illustrating the more diverse demands imposed by an intact host versus the intracellular milieu. The 230 genes facilitating growth in turbot but not J774A.1 cells enable both extracellular and intracellular growth in vivo; furthermore, the pathogen may proliferate within more than one cell type in vivo (Hu et al., 2019). Entry into and growth within J774A.1 cells and turbot macrophages required E. piscicida's T3SS but not its T6SS, even though both secretion systems are required in vivo. Thus, the organism may rely on its T6SS primarily for extracellular growth in fish where it may facilitate competition with tissue-resident microbiota to support pathogen colonization (Anderson et al., 2017, Fu et al., 2018, Zhao et al., 2018).
The utility of the defined mutant library is underscored by the secondary screen that led to the rapid identification of EvrA, a new regulator of E. piscicida virulence. EvrA acts upstream of the master virulence regulator EsrB to influence the expression of both the T3SS and T6SS, while also likely coordinating a negative feedback loop with its ligand, man-6P, to fine-tune its activity (Figure 6E). We found that EvrA is specifically activated by man-6P, the cytosolic form of mannose imported by the PTS, and that mannose is found in the tissues of E. piscicida's host. Our discovery of the EvrA-man-6P regulatory axis suggests that specific carbohydrates may be co-opted as signaling intermediaries between host and microbe in addition to their known roles as substrates for metabolism (Bäumler and Sperandio, 2016, Olive and Sassetti, 2016). For example, availability of fucose in the mammalian intestine acts as a spatial cue for virulence regulation in enterohemorrhagic Escherichia coli (Pacheco et al., 2012). Additionally, the conservation of EvrA in other virulent microbes, including the close relative Salmonella enterica (Figure S6), suggests that directly coupling detection of host sugars to virulence regulation may be a common theme in bacterial pathogenesis.
We posit that mannose detection by E. piscicida can be used both to regulate its virulence and to support its metabolism, although the exact contribution of host mannose to bacterial growth in vivo remains to be determined. In the proposed model, EvrA serves as a “metabolic switch” that links availability of a specific nutrient to activation of the virulence program (Figure 6E). Although it is not clear whether mannose is present outside the host, upregulation of host-specific colonization factors such as the T3SS would suggest it may serve as a host niche-specific signal. Future analysis of additional transposon libraries in E. piscicida strains lacking EvrA will enable additional understanding of the pathogen's metabolic priorities and whether other sugars may play complementary roles to mannose in virulence regulation. This work deepens our understanding of how bacterial pathogens can unite sugar availability and virulence regulation and establishes a framework for future studies that employ high-throughput genetics to dissect the metabolic cross talk between pathogen and host.
Limitations of the Study
The principal limitation of the study is that we were unable to obtain structural data that confirmed direct binding of man-6P to EvrA. Crystallization of the EvrA-man-6P complex proved extremely difficult because purified EvrA is prone to precipitation. Co-expression of protein tags, chaperones, or protein truncations may circumvent these issues and facilitate the future structural and biochemical analysis of EvrA and EvrA-man-6P complex.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by grants from the National Key Research and Development Program of China (2018YFD0900504 to Q.W.), National Natural Science Foundation of China (31430090 to Y.Z., 31602200 to X.L.), Ministry of Agriculture of China (CARS-47), and the Science and Technology Commission of Shandong and Shanghai Municipality (2017CXGC0103 and 17391902000). B.S. is supported by the Natural Sciences and Engineering Council of Canada (PGSD3-487259-2016). M.K.W. is supported by the Howard Hughes Medical Institute (HHMI) (CC30430) and the National Institutes of Health (RO1-AI-042347).
Author Contributions
Conceptualization, L.W. and Q.W.; Investigation, L.W., H.Q., K.Y., G.Y., R.M., J.M., C.Y., J.Y., Y.M., J.X., and X.L.; Writing – Original Draft, L.W. and B.S.; Writing – Review & Editing, Y.Z., Q.W., and M.K.W.; Funding Acquisition, Q.W., B.S., and M.K.W. All authors edited and agreed on the final version.
Declaration of Interests
The authors declare no competing interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.028.
Supplemental Information
References
- Abayneh T., Colquhoun D.J., Sørum H. Edwardsiella piscicida sp. nov., a novel species pathogenic to fish. J. Appl. Microbiol. 2013;114:644–654. doi: 10.1111/jam.12080. [DOI] [PubMed] [Google Scholar]
- Abel S., Abel zur Wiesch P., Chang H.H., Davis B.M., Lipsitch M., Waldor M.K. Sequence tag-based analysis of microbial population dynamics. Nat. Methods. 2015;12:223–226. doi: 10.1038/nmeth.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V., Aravind L. Diversification of catalytic activities and ligand interactions in the protein fold shared by the sugar isomerases, eIF2B, DeoR transcription factors Acyl-CoA transferases and methenyltetrahydrofolate synthetase. J. Mol. Biol. 2006;356:823–842. doi: 10.1016/j.jmb.2005.11.031. [DOI] [PubMed] [Google Scholar]
- Anderson M.C., Vonaesch P., Saffarian A., Marteyn B.S., Sansonetti P.J. Shigella sonnei encodes a functional T6SS used for interbacterial competition and niche occupancy. Cell Host Microbe. 2017;21:769–776. doi: 10.1016/j.chom.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Bäumler A.J., Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535:85–93. doi: 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D.E., Urbach J.M., Mekalanos J.J. A defined transposon mutant library and its use in identifying motility genes in Vibrio cholerae. Proc. Natl. Acad. Sci. U S A. 2008;105:8736–8741. doi: 10.1073/pnas.0803281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao M.C., Abel S., Davis B.M., Waldor M.K. The design and analysis of transposon insertion sequencing experiments. Nat. Rev. Microbiol. 2016;14:119–128. doi: 10.1038/nrmicro.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Yang D.H., Han F.J., Tan J.C., Zhang L.Z., Xiao J.F., Zhang Y.X., Liu Q. The bacterial T6SS effector EvpP prevents NLRP3 inflammasome activation by inhibiting the Ca2+-dependent MAPK-Jnk pathway. Cell Host Microbe. 2017;21:47–58. doi: 10.1016/j.chom.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Erni B., Zanolari B., Kocher H.P. The mannose permease of Escherichia coli consists of three different proteins. J. Biol. Chem. 1987;262:5238–5247. [PubMed] [Google Scholar]
- Fu Y., Waldor M.K., Mekalanos J.J. Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe. 2013;14:652–663. doi: 10.1016/j.chom.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Ho B.T., Mekalanos J.J. Tracking Vibrio cholerae cell-cell interactions during infection reveals bacterial population dynamics within intestinal microenvironments. Cell Host Microbe. 2018;23:274–281. doi: 10.1016/j.chom.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaigalat L., Schlüter J.P., Hartmann M., Mormann S., Tauch A., Pühler A., Kalinowski J. The DeoR-type transcriptional regulator SugR acts as a repressor for genes encoding the phosphoenolpyruvate:sugar phosphotransferase system (PTS) in Corynebacterium glutamicum. BMC Mol. Biol. 2007;8:104. doi: 10.1186/1471-2199-8-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher L.A., Ramage E., Jacobs M.A., Kaul R., Brittnacher M., Manoil C. A comprehensive transposon mutant library of Francisella novicida, a bioweapon surrogate. Proc. Natl. Acad. Sci. U S A. 2007;104:1009–1014. doi: 10.1073/pnas.0606713104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher L.A., Ramage E., Patrapuvich R., Weiss E., Brittnacher M., Manoil C. Sequence-defined transposon mutant library of Burkholderia thailandensis. MBio. 2013;4:e604–e613. doi: 10.1128/mBio.00604-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z.P., Nie P., Lu J.F., Liu L.Y., Xiao T.Y., Liu W., Liu J.S., Xie H.X. Type III secretion system translocon component EseB forms filaments on and mediates autoaggregation of and biofilm formation by Edwardsiella tarda. Appl. Environ. Microbiol. 2015;81:6078–6087. doi: 10.1128/AEM.01254-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y., Yin K.Y., Zhou M., Yang M., Zhang Y.X., Liu X.H., Wang Q.Y. EsrB negatively regulates expression of the glutamine synthetase GlnA in the fish pathogen Edwardsiella piscicida. FEMS Microbiol. Lett. 2018;365:fny007. doi: 10.1093/femsle/fny007. [DOI] [PubMed] [Google Scholar]
- Guo C., Huang X.Y., Yang M.J., Wang S., Ren S.T., Li H., Peng X.X. GC/MS-basedmetabolomics approach to identify biomarkers differentiating survivals from death in cruciancarps infected by Edwardsiella tarda. Fish Shellfish Immunol. 2014;39:215–222. doi: 10.1016/j.fsi.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Hu T., Zhang L., Wang W., Yang D., Xiao J., Zhang Y., Liu X., Liu Q. Edwardsiella piscicida enters nonphagocytic cells via a macropinocytosis-involved hybrid mechanism. J. Bacteriol. 2019;201 doi: 10.1128/JB.00548-18. e00548–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K., Matsui I., Payan F., Cambillau C., Ishida H., Kawarabayasi Y., Kikuchi H., Roussel A. A hyperthermostable D-ribose-5-phosphate isomerase from Pyrococcus horikoshii characterization and three-dimensional structure. Structure. 2002;10:877–886. doi: 10.1016/s0969-2126(02)00779-7. [DOI] [PubMed] [Google Scholar]
- Jacobs M.A., Alwood A., Thaipisuttikul I., Spencer D., Haugen E., Ernst S., Will O., Kaul R., Raymond C., Levy R. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U S A. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K.Y., Siame B.A., Tenkink B.J., Noort R.J., Mok Y. Edwardsiella tarda – virulence mechanisms of an emerging gastroenteritis pathogen. Microbes Infect. 2012;14:26–34. doi: 10.1016/j.micinf.2011.08.005. [DOI] [PubMed] [Google Scholar]
- Liberati N.T., Urbach J.M., Miyata S., Lee D.G., Drenkard E., Wu G., Villanueva J., Wei T., Ausubel F.M. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci. U S A. 2006;103:2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zhao L.Y., Yang M.J., Yin K.Y., Zhou X.H., Leung K.Y., Liu Q., Zhang Y.X., Wang Q.Y. Transcriptomic dissection of the horizontally acquired response regulator EsrB reveals its global regulatory roles in the physiological adaptation and activation of T3SS and the cognate effector repertoire in Edwardsiella piscicida during infection toward turbot. Virulence. 2017;8:1355–1377. doi: 10.1080/21505594.2017.1323157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda J., Kiriyama M., Suzaki E., Kataoka K., Nishibuchi M., Nakai T. Characterization of proteins secreted from a type III secretion system of Edwardsiella tarda and their roles in macrophage infection. Dis. Aquat. Organ. 2009;84:115–121. doi: 10.3354/dao02033. [DOI] [PubMed] [Google Scholar]
- Olive A.J., Sassetti C.M. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat. Rev. Microbiol. 2016;14:221–234. doi: 10.1038/nrmicro.2016.12. [DOI] [PubMed] [Google Scholar]
- Pacheco A.R., Curtis M.M., Ritchie J.M., Munera D., Waldor M.K., Moreira C.G., Sperandio V. Fucose sensing regulates bacterial intestinal colonization. Nature. 2012;492:113–117. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.B., Aoki T., Jung T.S. Pathogenesis of and strategies for preventing Edwardsiella tarda infection in fish. Vet. Res. 2012;43:67. doi: 10.1186/1297-9716-43-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Wetmore K.M., Waters R.J., Callaghan M., Ray J., Liu H., Kuehl J.V., Melnyk R.A., Lamson J.S., Suh Y. Mutant phenotypes for thousands of bacterial genes of unknown function. Nature. 2018;557:503–509. doi: 10.1038/s41586-018-0124-0. [DOI] [PubMed] [Google Scholar]
- Rogge M.L., Thune R.L. Regulation of the Edwardsiella ictaluri type III secretion system by pH and phosphate concentration through EsrA, EsrB, and EsrC. Appl. Environ. Microbiol. 2011;77:4293–4302. doi: 10.1128/AEM.00195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E.J., Akerley B.J., Novik V.N., Lampe D.J., Husson R.N., Mekalanos J.J. In vivo transposition of mariner-based elements in enteric bacteria and mycobacteria. Proc. Natl. Acad. Sci. U S A. 1999;96:1645–1650. doi: 10.1073/pnas.96.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S., Lai Q.L., Liu Q., Wu H.Z., Xiao J.F., Shao Z.Z., Wang Q.Y., Zhang Y.X. Phylogenomics characterization of a highly virulent Edwardsiella strain ET080813T encoding two distinct T3SS and three T6SS gene clusters: propose a novel species as Edwardsiella anguillarum sp. nov. Syst. Appl. Microbiol. 2015;38:36–47. doi: 10.1016/j.syapm.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Srinivasa Rao P.S., Yamada Y., Tan Y.P., Leung K.Y. Use of proteomics to identify novel virulence determinants that are required for Edwardsiella tarda pathogenesis. Mol. Microbiol. 2004;53:573–586. doi: 10.1111/j.1365-2958.2004.04123.x. [DOI] [PubMed] [Google Scholar]
- Wang Q.Y., Yang M.J., Xiao J.F., Wu H.Z., Wang X., Lv Y.Z., Xu L.L., Zheng H.J., Wang S.Y., Zhao G.P. Genome sequence of the versatile fish pathogen Edwardsiella tarda provides insights into its adaptation to broad host ranges and intracellular niches. PLoS One. 2009;4:e7646. doi: 10.1371/journal.pone.0007646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.J., Lv Y.Z., Xiao J.F., Wu H.Z., Zheng H.J., Liu Q., Zhang Y.X., Wang Q.Y. Edwardsiella comparative phylogenomics reveal the new intra/inter-species taxonomic relationships, virulence evolution and niche adaptation mechanisms. PLoS One. 2012;7:e36987. doi: 10.1371/journal.pone.0036987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G.H., Billings G., Hubbard T.P., Park J.S., Leung K.Y., Liu Q., Davis B.M., Zhang Y.X., Wang Q.Y., Waldor M.K. Time-resolved transposon insertion sequencing reveals genome-wide fitness dynamics during infection. MBio. 2017;8:e1517–e1581. doi: 10.1128/mBio.01581-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin K.Y., Guan Y.P., Ma R.Q., Wei L.F., Liu B., Liu X.H., Zhou X.S., Ma Y., Zhang Y.X., Waldor M.K., Wang Q.Y. Critical role for a promoter discriminator in RpoS control of virulence in Edwardsiella piscicida. PLoS Pathog. 2018;14:e1007272. doi: 10.1371/journal.ppat.1007272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Caro F., Robins W., Mekalanos J.J. Antagonism toward the intestinal microbiota and its effect on Vibrio cholerae virulence. Science. 2018;359:210–213. doi: 10.1126/science.aap8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Leung K.Y. Dissection of a type VI secretion system in Edwardsiella tarda. Mol. Microbiol. 2007;66:1192–1206. doi: 10.1111/j.1365-2958.2007.05993.x. [DOI] [PubMed] [Google Scholar]
- Zheng J., Tung S.L., Leung K.Y. Regulation of a type III and a putative secretion system in Edwardsiella tarda by EsrC is under the control of a two-component system, EsrA-EsrB. Infect. Immun. 2005;73:4127–4137. doi: 10.1128/IAI.73.7.4127-4137.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.