

Summary
Recent studies have linked health fates of children to environmental exposures of their great grandparents. However, few studies have considered whether ancestral exposures influence immune function across generations. Here, we report transgenerational inheritance of altered T cell responses resulting from maternal (F0) exposure to the aryl hydrocarbon receptor ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Since F0 exposure to TCDD has been linked to transgenerational transmission of reproductive problems, we asked whether maternal TCDD exposure also caused transgenerational changes in immune function. F0 exposure caused transgenerational effects on the CD8+ T cell response to influenza virus infection in females but not in males. Outcrosses showed changes were passed through both parental lineages. These data demonstrate that F0 exposure to an aryl hydrocarbon receptor (AHR) agonist causes durable changes to immune responses that can affect subsequent generations. This has broad implications for understanding how the environment of prior generations shapes susceptibility to pathogens and antiviral immunity in later generations.
Subject Areas: Immunology, Environmental Health, Environmental Toxicology, Developmental System Toxicology
Graphical Abstract
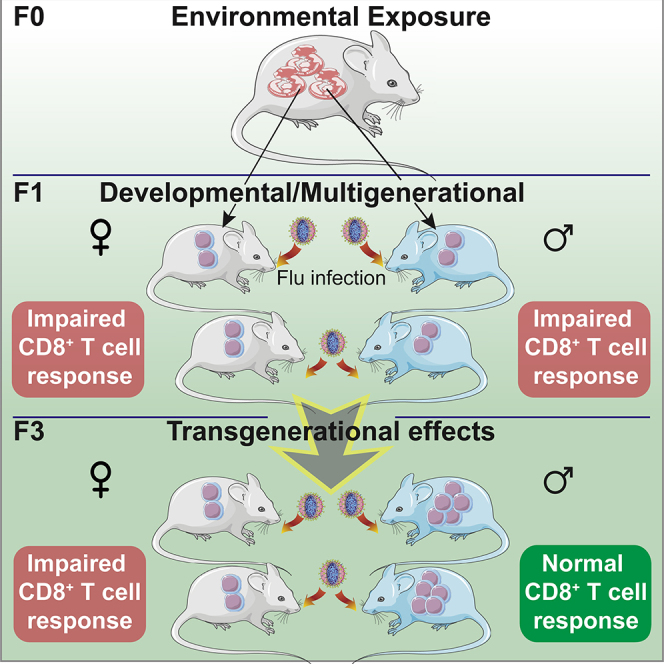
Highlights
-
•
Maternal AHR activation causes transgenerational effects on an immune response
-
•
CD8 T cell response to influenza virus is decreased in male and female F1 offspring
-
•
Transgenerational effects were observed in infected lungs of female F3 offspring
-
•
Both maternal and paternal F1 generations contribute to the immune effects in F3
Immunology; Environmental Health; Environmental Toxicology; Developmental System Toxicology
Introduction
It has become increasingly clear that early life development is a critical and unique window of vulnerability during which environmental exposures influence cellular programming in ways that shape health and disease later in life. There is also growing evidence that maternal stressors and environmental factors affect not just the offspring's health, but also that of subsequent generations as well (Heindel, 2018, Rattan et al., 2018, Rissman and Adli, 2014, Skinner, 2014, Skinner et al., 2010, van Steenwyk et al., 2018, Walker and Gore, 2011). Exposure during pregnancy results in the developing fetus (F1) and its gametes (F2) being exposed. Changes in the F1 and F2 progeny are referred to as developmental and multigenerational effects, respectively. The F3 generation is the first generation with no exposure to the original insult to the pregnant mother; therefore changes in this generation represent transgenerational inheritance (Skinner, 2008). Recent reports of transgenerationally inherited adverse health effects of environmental exposures underscore the importance of this phenomenon to human health and disease (Ferey et al., 2019, Gillette et al., 2018, Klukovich et al., 2019).
The majority of studies demonstrating transgenerational effects of environmental agents have examined their consequences on the reproductive, nervous, or endocrine systems (Babenko et al., 2015, Bohacek et al., 2013, Guerrero-Bosagna and Skinner, 2014, Heindel, 2018, Nilsson and Skinner, 2015, Stegemann and Buchner, 2015). In contrast, research on transgenerational inheritance rarely includes assessment of whether maternal exposures impinge on the function of the immune system. Yet, a properly functioning immune system is fundamentally important to individual and public health. Even slight alterations can reduce defenses against infections or diminish vaccine efficacy (Dallaire et al., 2006, Glynn et al., 2008, Stolevik et al., 2013, Winans et al., 2011). Thus, the consequences of maternal and early life exposures that alter the function of the immune system are broad reaching. Moreover, when it has been examined, developmental exposures to a range of common pollutants, pharmaceuticals, as well as maternal diet have been associated with changes in immune function later in life (Boule and Lawrence, 2016, Dietert, 2009). For instance, higher levels of dioxins and polychlorinated biphenyls (PCBs) in breast milk, cord, and infant blood correlate with increased respiratory tract infections and reduced responses to vaccination (Hochstenbach et al., 2012, Jusko et al., 2016, Miyashita et al., 2011, Stolevik et al., 2013). Animal studies reveal parallel events, showing that maternal and early life exposures durably change immune responses in the offspring (Winans et al., 2011, Boule and Lawrence, 2016). For example, in mouse models, maternal treatment with the dioxin 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) perturbs T cell responses to influenza A virus (IAV) infection in offspring (Boule et al., 2014, Vorderstrasse et al., 2004, Winans et al., 2015). TCDD and other dioxins, as well as PCBs, are persistent organic pollutants. Moreover, many dioxins and PCBs, including TCDD, activate the aryl hydrocarbon receptor (AHR), an environment-sensing transcription factor that plays important, albeit not fully understood, roles in the development and function of the immune system (Rothhammer and Quintana, 2019, Stevens et al., 2009). In addition to pollutants, the AHR has a multitude of ligands ranging from pharmaceuticals to compounds in foods we eat to microbial by-products (Hubbard et al., 2015, Murray et al., 2014, Nguyen and Bradfield, 2008); thus, we are regularly exposed to AHR ligands. Therefore, it is important to understand how exposure to AHR ligands impacts the immune system, including in subsequent generations. TCDD is the most potent and specific AHR agonist and provides a valuable tool to study how developmental activation of the AHR affects the immune system.
In addition to being expressed in cells of the immune system, the AHR is expressed in the reproductive tract, and there have been numerous studies examining the effects of TCDD on the reproductive system (Bruner-Tran et al., 2017, Lew et al., 2011, You et al., 2018). Developmental exposure to TCDD affects both the male and female reproductive systems. For instance, reduced sperm counts in males and higher incidence of endometriosis in females are among the effects observed in F1 offspring (Bruner-Tran et al., 2017, Pilsner et al., 2017). In addition to impacting germ cells and adult F1 offspring, there is also a growing literature base connecting F0 exposure to TCDD with transgenerational effects on both the male and female reproductive systems (Bruner-Tran et al., 2014, Bruner-Tran and Osteen, 2011, Manikkam et al., 2012). Taken together with studies showing that F0 TCDD exposure affects immune responses in F1 offspring (Boule et al., 2014, Boule et al., 2015a, Boule et al., 2015b, Hogaboam et al., 2008, Meyers et al., 2018, Vorderstrasse et al., 2004, Vorderstrasse et al., 2006, Winans et al., 2015), we hypothesized that F0 TCDD exposure also causes transgenerational effects in the immune system.
Influenza A virus (IAV) is a common human pathogen that infects the respiratory system and causes infections that range in severity from moderate to fatal. Every year IAV infections are among the major causes of global morbidity and mortality (Iuliano et al., 2018, Reed et al., 2015). Mouse models of infection with human IAV provide an experimental system with strong relevance to human health, and the murine immune response to IAV closely resembles that of humans (Schmidt and Varga, 2018). In particular, successfully fighting acute primary IAV infection is CD8+ T cell dependent (Topham et al., 1997). The T cell response to IAV occurs in two distinct sites: the lung-draining lymph nodes, such as the mediastinal lymph nodes (MLNs), and the respiratory tract. After encountering IAV in the respiratory tract, dendritic cells (DCs) emigrate to lymph nodes that drain the lung, where they activate naive CD8+ T cells. When they receive the correct signals, virus-specific CD8+ T cells undergo clonal expansion and differentiate into cytotoxic T lymphocytes (CTL), which migrate to the site of infection (i.e., lung), where they directly kill virally infected cells through the release of cytotoxic granules (Kohlmeier and Woodland, 2009).
In this study, we determined whether maternal (F0) exposure to the AHR agonist TCDD causes transgenerational effects on CD8+ T cell responses to IAV infection. Specifically, following maternal exposure to a low, environmentally relevant dose of TCDD (Birnbaum and Tuomisto, 2000, DeVito et al., 1995), we determined the frequency of responding CD8+ T cells in male and female offspring in the F1 and F3 generations, using well-established metrics of their activation, expansion, and differentiation into effector cells. By outcrossing F1 generation mice, we also sought to define whether the transgenerational effects on the immune system are preferentially passed through the maternal or paternal lineage. Delineating which aspects of the CD8+ T cell response to infection are affected across generations furthers our understanding of how environmental AHR ligands cause durable changes to T cell responses and also expands current knowledge of how developmental exposures can impinge on immune responses later in life.
Results
To generate transgenerationally exposed cohorts, F0 dams were administered either vehicle or TCDD orally. The dose of TCDD used in this study does not cause overt toxicity to the dam or pups, nor does it alter immune organ cellularity in naive (unchallenged) offspring (Vorderstrasse et al., 2004, Winans et al., 2015). At maturity, non-sibling F1 offspring were bred to create the F2 generation, and then non-sibling, non-cousin F2 offspring were bred to create the F3 generation (Figure 1A, see Transparent Methods). Consistent with prior reports (Vorderstrasse et al., 2004, Vorderstrasse et al., 2006), maternal treatment with TCDD at this dose and on this schedule did not alter the time to parturition (time from vaginal plug discovery to birth), litter size, or sex ratio in F1 offspring (Figure 1B). Similarly, F0 exposure did not significantly change fertility and duration of pregnancy, as there were no differences in the time between pairing and birth in the F2 or F3 generations, nor were there differences in litter size or sex ratio among the offspring (Figure 1B).
Figure 1.

Breeding Schematic and Reproductive Endpoints
(A) Transgenerational breeding strategy: Pregnant C57/Bl6 dams were treated (p.o.) with either vehicle or TCDD (1 μg/kg) on GD0, 7, 14, and PND2. Separate sires were used for each dam. No culling of litters was performed, and littermates were housed in same-sex groups. Offspring sex was determined, and offspring were randomly assigned to groups for breeding or infection. 15–20 pairs of non-sibling F1 offspring were bred to create the F2 generation. F2 offspring were used to create the F3 generation.
(B) The table summarizes pregnancy metrics, litter size, and sex ratio of offspring from the F1, F2, and F3 generations. Pregnancy metrics: F1 generation, time to parturition, which is defined as the number of days from vaginal plug identification to birth; F2 and F3 generations, the number of days from pairing to giving birth. Data are shown as mean ± SEM. A Student's t test was used to compare TCDD with vehicle groups for each endpoint within each generation.
F0 Exposure to TCDD Has Transgenerational Effects on the CD8+ T cell Response to IAV in the Lungs of Female F3 Offspring
IAV infects epithelial cells in the respiratory tract, and CD8+ T cells are the principle cell type responsible for viral clearance during acute primary infection. In particular, in the lung, virus-specific cytotoxic CD8+ T cells eliminate IAV infected cells (Kohlmeier and Woodland, 2009, McGill and Legge, 2009, Schmidt and Varga, 2018). To assess how F0 AHR activation affects the overall quality of the CD8+ T cell response to IAV across generations, we examined metrics of clonal expansion and effector function in F1 and F3 offspring. At maturity, female mice that were exposed developmentally (F1 generation offspring) or transgenerationally (F3 generation offspring) were infected with IAV. Nine days after infection, which is the peak day of this response (Lawrence et al., 2000, Lawrence et al., 2006), the percentage and number of CD8+ T cells in the lungs of TCDD lineage offspring were significantly reduced in F1 generation offspring, but not in the F3 generation (Table S1). The percentage of viral nucleoprotein (NP)-specific CD8+ T cells was reduced by about 60% in female mice that were developmentally exposed to TCDD (Figure 2A). Similarly, the number of NP+CD8+ T cells was significantly decreased in TCDD lineage F1 offspring compared with infected vehicle F1 offspring (Figure 2B). In the F3 generation, the percentage of NP+CD8+ T cells was not affected by F0 AHR activation (Figure 2C). However, F0 AHR ligand exposure caused a significant decrease in the number of NP+CD8+ T cells in the lungs of F3 female TCDD lineage offspring (Figure 2D). To assess how F0 AHR activation affects the overall magnitude of the CD8+ T cell response to IAV, we also determined the percentage and number of CTL (CD44hiCD62LloCD8+ T cells), a population that includes NP-specific as well as CD8+ T cells specific for other viral peptide epitopes. In both the F1 and F3 generations, the proportion of CD8+ T cells that were CD44hiCD62Llo was not influenced by F0 AHR activation (Figures 2E and 2G). However, the number of CTL was significantly reduced in F1 TCDD lineage offspring (Figure 2F) and 60% reduced in F3 offspring (Figure 2H). These results demonstrate that AHR activation in F0 dams has transgenerational effects on CD8+ T cell clonal expansion during the response to IAV.
Figure 2.

F0 Exposure to TCDD Has Transgenerational Effects on the CD8+ T Response to IAV in the Lung of Female Offspring
At maturity (8–10 weeks of age) 7–9 female mice from each exposure group were infected with IAV, and the CD8+ T cell response in the lung was measured 9 days later.
(A) Flow plots depict the mean percent of NP+CD8+ T cells in vehicle (left) and TCDD (right) F1 offspring.
(B) The graph indicates the mean number of NP+CD8+ T cells in vehicle and TCDD F1 offspring.
(C) Flow plots depict the mean percent of NP+CD8+ T cells in F3 vehicle (left) and TCDD (right) lineage offspring.
(D) The mean number of NP+CD8+ T cells in F3 vehicle and TCDD lineage offspring.
(E) The flow plots display the mean percent of CTL in vehicle (left) and TCDD (right) F1 offspring.
(F) The mean number of CTL in vehicle and TCDD F1 offspring.
(G) Flow plots depict the mean percent of CTL in F3 vehicle (left) and TCDD (right) lineage offspring.
(H) The mean number of CTL in vehicle and TCDD lineage F3 offspring.
(I) The flow plot depicts the mean percent of IFNγ+CD8+ T cells in the lung of F1 offspring.
(J) The mean number of IFNγ+CD8+ T cells in the lung of F1 offspring.
(K) The flow plots show the mean percent of IFNγ+CD8+ T cells in the lung of F3 offspring.
(L) The mean number of IFNγ+CD8+ T cells in the lung of F3 offspring.
(M) The flow plots depict the mean percent of CD107a+CD8+ T cells in the lung of F1 offspring.
(N) The mean number of CD107a+CD8+ T cells in the lung of F1 offspring.
(O) The flow plots depict the mean percent of CD107a+CD8+ T cells in F3 offspring.
(P) The mean number of CD107a+CD8+ T cells in the lung of F3 offspring. Full gating strategy is depicted in Figure S1. NP+CD8+ T cells were derived from the leukocyte gate; all others were generated from the CD8+ T cell gate (Figure S1).
Data are represented as mean ± SEM; * denotes p ≤ 0.05 (Student's t test). The numerical values that correspond to the bar graphs as well as p values for each comparison are listed in Table S1.
A successful CD8+ T cell response to IAV infection not only involves a significant increase in the magnitude of the CD8+ T cell population in the lung, but also indicates that the cells have gained their effector functions. These include secretion of antiviral cytokines and degranulation, which enable CD8+ T cells to kill IAV-infected cells and clear the infection. To assess how F0 TCDD exposure impacts CD8+ T cell effector function, we determined the frequency and number of CD8+ T cells that make the antiviral cytokine IFNγ and that express CD107a, which is an indicator of degranulation. In the F1 generation, IFNγ production by CD8+ T cells in the lung was largely unaffected by F0 AHR activation (Figures 2I and 2J). However, in the F3 offspring, there was a 50% decrease in the number of IFNγ+CD8+ T cells in the lung (Figures 2K and 2L). A similar pattern was observed for CD107a+CD8+ T cells: there were no significant differences in either the percentage or number of CD107a+CD8+ T cells between vehicle and TCDD lineage offspring in the F1 generation (Figures 2M and 2N). However, in the F3 generation, the number, but not proportion, of CD107a+CD8+ T cells was significantly reduced when the AHR was activated in F0 dams (Figure 2P and 2O ). Thus, our findings indicate that F0 exposure to the AHR agonist TCDD has transgenerational effects on both CD8+ T cell effector function and clonal expansion during IAV infection. Interestingly, the transgenerational effects appear more robust with regard to the reduction of clonal expansion than effector function, suggesting that these pathways may be differentially regulated by F0 AHR activation.
F0 Exposure to TCDD Has Developmental but Not Transgenerational Effects on the CD8+ T cell Response to IAV in the Lungs of Male Offspring
In parallel to experiments performed using female mice, we measured the CD8+ T cell response in their male siblings in the F1 and F3 generations. F0 exposure did not significantly modify the percent of NP+CD8+ T cells in the lungs of male F1 or F3 offspring (Figures 3A and 3C, Table S1). The number of NP+CD8+ T cells was reduced by 45% in the F1 generation (Figure 3B) but was not significantly different from vehicle lineage in the F3 generation (Figure 3D). The percentage of CTL in the lungs of TCDD lineage F1 offspring was reduced by approximately 75% compared with vehicle (Figure 3E). Likewise, the size of the CTL population was smaller in TCDD lineage F1 offspring compared with vehicle (Figure 3F). However, in F3 generation male TCDD lineage offspring, neither the percentage nor the number of CTL was affected by F0 exposure to TCDD (Figures 3G and 3H). In the F1 generation, the proportion of CD8+ T cells that produced IFNγ was increased by F0 AHR activation (Figure 3I); however, the number of IFNγ+CD8+ T cells was reduced in F1 TCDD lineage offspring (Figure 3J). In the F3 generation, however, the percentage of IFNγ+CD8+ T cells was not altered by F0 TCDD exposure (Figure 3K). Moreover, similar to the F1 generation, the number of IFNγ+CD8+ T cells was reduced by 60% in TCDD lineage offspring compared with vehicle (Figure 3L). F0 AHR activation had a minimal impact on the CD107a+CD8+ T cell population in lungs of male offspring. Specifically, the proportion of CD8+ T cells that expressed CD107a was not different in either F1 or F3 generation offspring (Figures 3M and 3O). The number of CD107a+CD8+ T cells in F1 generation offspring was reduced by 40% in offspring of TCDD-treated dams (Figure 3N); however, the number of CD107a+CD8+ T cells in TCDD F3 generation offspring was not different from vehicle F3 lineage mice (Figure 3P).
Figure 3.

F0 Exposure to TCDD Has Developmental but Not Transgenerational Effects on the CD8+ T Response to IAV in the Lung of Male TCDD Lineage Offspring
7–8 male mice from each treatment group were infected with IAV, and the CD8+ T cell response in the lung was measured 8 days later.
(A) Flow plots depict the mean percent of NP+CD8+ T cells in vehicle (left) and TCDD (right) F1 offspring.
(B) The mean number of NP+CD8+ T cells in vehicle and TCDD F1 offspring.
(C) Flow plots depict the mean percent of NP+CD8+ T cells in F3 vehicle (left) and TCDD (right) lineage offspring.
(D) The mean number of NP+CD8+ T cells in F3 vehicle and TCDD lineage offspring.
(E) The flow plots display the mean percent of CTL in vehicle (left) and TCDD (right) F1 offspring.
(F) The mean number of CTL in vehicle and TCDD F1 offspring.
(G) Flow plots depict the mean percent of CTL in F3 vehicle (left) and TCDD (right) lineage offspring.
(H) The mean number of CTL in vehicle and TCDD lineage F3 offspring.
(I) The flow plot depicts the mean percent of IFNγ+CD8+ T cells in the lung of F1 offspring.
(J) The mean number of IFNγ+CD8+ T cells in the lung of F1 offspring.
(K) The flow plots show the mean percent of IFNγ+CD8+ T cells in the lung of F3 offspring.
(L) The mean number of IFNγ+CD8+ T cells in the lung of F3 offspring.
(M) The flow plots depict the mean percent of CD107a+CD8+ T cells in the lung of F1 offspring.
(N) The mean number of CD107a+CD8+ T cells in the lung of F1 offspring.
(O) The flow plots depict the mean percent of CD107a+CD8+ T cells in F3 offspring.
(P) The mean number of CD107a+CD8+ T cells in the lung of F3 offspring. Full gating strategy is depicted in Figure S1. NP+CD8+ T cells were derived from the leukocyte gate; all others were generated from the CD8+ cell gate (Figure S1).
Data are shown as mean + SEM; * denotes p ≤ 0.05 (Student's t test). Numerical values for the bar graphs as well as p values that correspond to the comparisons in each panel are shown in Table S1.
In summary, these results indicate that F0 exposure to TCDD affects important aspects of the CD8+ T cell response to IAV in the lungs of both male and female F1 offspring. However, in the F3 generation, F0 exposure had a minimal impact on the CD8+ T cell response to IAV in male offspring. Conversely, F0 exposure caused transgenerational effects on the CD8+ T cell response to infection in the lungs of female F3 offspring, findings that suggest that transgenerational inheritance in the immune system may be sex specific.
In the MLN, F0 AHR Activation Alters the CD8+ T cell Response of Female and Male F1 but Not F3 Generation Offspring
Although the effector functions of CD8+ T cells to IAV infection are predominantly executed in the respiratory tract, their activation begins in the lymph nodes, where CD8+ T cells interact with antigen-presenting cells expressing viral peptides. This stimulates naive CD8+ T cells to clonally expand and differentiate into armed effectors before migrating to the lung. Therefore, we determined whether F0 TCDD exposure also caused transgenerational changes in CD8+ T cell clonal expansion or differentiation in the MLN in response to IAV infection. Across all metrics, maternal exposure reduced the CD8+ T cell response to IAV in the MLN of male and female F1 offspring. Thus, offspring of TCDD-treated dams exhibited significant reductions in the number of NP+CD8+ T cells (Figures 4A and 4E), CTL (Figures 4B and 4F), IFNγ+ (Figures 4C and 4G), and CD107a+CD8+ T cells (Figures 4D and 4H and Table S2). Despite the strong effect that F0 AHR activation had on the CD8+ T cell response in F1 offspring, we did not observe any statistically significant differences in these measures of CD8+ T cell expansion or effector function in the MLN of male or female F3 offspring (Figure 4, Table S2).
Figure 4.

In the MLN, F0 AHR Activation Alters the CD8+ T Cell Response of Female and Male F1 but Not F3 Generation Offspring
F1 and F3 generation offspring were generated as shown in Figure 1A. 7–9 adult F1 and F3 generation offspring from each treatment group were infected with IAV, and MLN cells were harvested on day 8 (males) or 9 (females) post infection. The graphs depict the mean number of cells from vehicle and TCDD offspring found in the MLN on day 8 or 9 post IAV infection.
(A) The number of NP+CD8+ T cells in female F1 (left) and F3 (right) offspring.
(B) The number of CTL in female F1 (left) and F3 (right) offspring.
(C) The number of IFNγ+CD8+ T cells in female F1 (left) and F3 (right) offspring.
(D) The number of CD107a+CD8+ T cells in female F1 (left) and F3 (right) offspring.
(E) The number of NP+CD8+ T cells in male F1 (left) and F3 (right) offspring.
(F) The number of CTL in male F1 (left) and F3 (right) offspring.
(G) The number of IFNγ+CD8+ T cells in male F1 (left) and F3 (right) generation offspring.
(H) The number of CD107a+CD8+ T cells in male F1 (left) and F3 (right) offspring. Black-striped bars, VEH F1; white-striped bars, TCDD F1; black bars, VEH F3; white bars, TCDD F3.
Data are shown as mean +SEM. * denotes p value ≤ 0.05 (Student's t test). Numerical values for all the bar graphs as well as p values for each comparison are shown in Table S2.
Transgenerational Effects on the CD8+ T cell Response to IAV Are Transmitted through Both Parental Lineages
To determine whether the reduced CD8+ T cell responses in the lungs of female F3 generation offspring are preferentially transmitted through the maternal or paternal F1 lineage, we performed outcrosses of F1 mice. The outcrosses generated a group of offspring that had only maternal lineage TCDD exposure and another group of offspring with only paternal lineage TCDD exposure (Figure 5A). As controls, we also carried F1 siblings forward by incrossing F1 males and females from the same F0 exposure groups (i.e., F1 VEH x F1 VEH; F1 TCDD x F1 TCDD). At maturity, we infected F3 offspring from all four crosses and measured CD8+ T cell clonal expansion and effector function. Consistent with our prior observations (Figure 2D), in the F3 generation, the number of virus NP-specific CD8+ T cells was significantly decreased in the incrossed TCDD lineage relative to incrossed vehicle lineage offspring (Figure 5B, black and white bars). Maternal TCDD lineage exposure replicated the significant decrease (Figure 5B, black and light gray bars). Paternal lineage exposure also decreased the number of NP+CD8+ T cells, but the effect was not statistically significant (Figure 5B compare the black and dark gray bars; see also Table S4). TCDD exposure through either the paternal or maternal lineage significantly reduced the number of CTL (Figure 5C). The average number of IFNγ+CD8+ T cells was not significantly different among the outcrossed groups and control; however, the two incrossed groups were significantly different (Figure 5D, Table S4). Similar to viral NP-specific CD8+ T cells, the number of CD107a+CD8+ T cells was significantly reduced in both the TCDD incrossed and maternal lineages, whereas paternal TCDD lineage exposure caused a subtler decrease in the number of CD107a+CD8+ T cells, which did not reach statistical significance (Figure 5E). When considered together, these changes in CD8+ T cell clonal expansion and effector function do not appear to be transmitted solely through a single parental lineage, as F3 effects were observed in both the maternal and paternal TCDD lineage exposures separately. Although both F1 parental lineages affected the CD8+ T cell response to IAV in the F3 generation, these data suggest that exposure through the maternal line had a more potent effect on the CD8+ T cell response than paternal lineage TCDD exposure and that clonal expansion may be more sensitive to the effects of F0 AHR activation than acquisition of CTL effector function.
Figure 5.

Transgenerational Effects on the CD8+ T Cell Response to IAV in the Lung Are Transmitted through Both Parental Lineages
(A) Outcross breeding scheme: F1 offspring were generated as in Figure 1A. A portion of F1 offspring from both vehicle and TCDD treatment groups was used to perform outcrosses. Vehicle males were paired with TCDD females to create the maternal TCDD lineage. For the paternal TCDD lineage, vehicle females were paired with TCDD males. Vehicle x vehicle and TCDD x TCDD incrosses were also made. The resulting F2 offspring were paired to other non-sibling F2 offspring to create the F3 generation. At 8–10 weeks of age, 7–9 male and 7–9 female offspring from different dams were infected with IAV. MLNs and lungs from males were harvested on day 8 post infection and from females on day 9.
(B–E) The CD8+ T cell response in the lung of female F3 outcrossed offspring. The designations to the left of each graph denote the F0 treatment group of the male and female F1 offspring involved in each F1 cross. The graphs depict the mean number of cells found in the lung of F3 female offspring: (B) NP+CD8+ T cells, (C) CTL, (D) IFNγ+CD8+ T cells, and (E) CD107a+CD8+ T cells. Black bars, VEH; white bars, TCDD; light gray bars, maternal TCDD lineage; dark gray bars, paternal TCDD lineage.
Data are shown as mean ± SEM. * denotes p value ≤ 0.05 determined by a two-way ANOVA followed by a Dunnett's test with the control as the VEH x VEH lineage. The numerical values for the bar graphs are listed in Table S3, and the p values generated using two-way ANOVA and Dunnett's test are shown in Table S4. The outcross experiment was performed once.
Developmental AHR Activation Influences the CD8+ T cell Response to Infection via Targeting Hematopoietic Cells
The AHR is widely expressed in hematopoietic and non-hematopoietic cells. To determine whether AHR signaling within the hematopoietic compartment contributes differentially to the effects of developmental exposure in the CD8+ T cell responses of female and male offspring, we used conditional knockout mice that lack Ahr in all hematopoietic cells (Bennett et al., 2018). To create littermates that do and do not express the Ahr in hematopoietic cells, we crossed nulliparous Ahrfx/fx females with Ahrfx/fxVav1cre male mice. The resulting male and female Ahrfx/fx and Vav1creAhrfx/fx offspring of control or TCDD-treated dams were infected with IAV at maturity. Similar to wild-type B6 mice, female Ahrfx/fx F1 offspring that were developmentally exposed to TCDD exhibited reduction in the percentage (Figure 6A) and number (Figure 6B) of NP+CD8+ T cells compared with vehicle-exposed Ahrfx/fx mice. However, when Ahr was excised from hematopoietic cells, maternal exposure did not affect the number of NP+CD8+ T cells in female offspring (Figure 6B). Although there was no difference in the percentage (Figure 6C), infected male Ahrfx/fx offspring of dams treated with TCDD had a statistically significant reduction in the number of NP+CD8+ T cells compared with male Ahrfx/fx offspring of vehicle control dams (Figure 6D). Yet, the number of NP+CD8+ T cells in TCDD exposed Vav1creAhrfx/fx offspring was not significantly different from that of Vav1creAhrfx/fx male offspring of control dams (Figure 6D). Thus, the decreased number of NP+CD8+ T cells in TCDD-exposed F1 offspring requires AHR-mediated signaling in the hematopoietic cells. In addition to measuring the expansion of virus-specific CD8+ T cells, we compared differentiation into CTL. Regardless of maternal exposure, the percentage of CTL was similar in female Ahrfx/fx and Vav1creAhrfx/fx offspring (Figure 6E). Yet, compared with female offspring of control-treated dams, there was a statistically significant reduction in the number of CTL in female Ahrfx/fx mice that were developmentally exposed to TCDD (Figure 6F). Lack of Ahr in hematopoietic cells eliminated this difference in the number of CTL (Figure 6F). In IAV-infected male offspring, there was also no significant difference in the percentage of CTL (Figure 6G), but the number of CTL in TCDD-exposed Ahrfx/fx males was significantly less than in vehicle-exposed Ahrfx/fx offspring (Figure 6H). When Vav1creAhrfx/fx TCDD-exposed offspring were infected, the number of CTL was not significantly different from that of Vav1creAhrfx/fx offspring (Figure 6H). However, similar to NP+CD8+ T cells from Vav1creAhrfx/fx males, the number of CTL was 1.7-fold lower in TCDD-exposed offspring compared with males of vehicle-treated dams means. Thus, when the AHR is triggered during development, it effects hematopoietic cells in a manner that leads to changes in CD8+ T cell responses later in life, although the consequences may be slightly different between sexes.
Figure 6.

Lack of AHR in Hematopoietic Cells Has Differential Effects on CD8+ T Cell Expansion in Female and Male Offspring during IAV Infection
At maturity, 9–11 male or female developmentally exposed Vav1creAhrfx/fx and Ahrfx/fx offspring were infected with IAV. MLN cells were harvested and stained as described in the Transparent Methods section.
(A) The percentage of NP+CD8+ T cells in IAV-infected female vehicle and TCDD-exposed Ahrfx/fx (top row) and Vav1creAhrfx/fx offspring (bottom row). The number on the plots denotes the mean percentage of NP+CD8+ T cells.
(B) The number of NP+CD8+ T cells from vehicle (V) and TCDD (T)-exposed Ahrfx/fx and Vav1creAhrfx/fx offspring on day 9 post IAV infection.
(C) The percentage of NP+CD8+ T cells in male exposed Ahrfx/fx and Vav1creAhrfx/fx offspring on day 9 post IAV infection.
(D) The number of NP+CD8+ T cells from male Ahrfx/fx and Vav1creAhrfx/fx offspring of vehicle and TCDD treated dams.
(E) The percentage of CTL (CD44hiCD62LloCD8+ T cells) in female Ahrfx/fx (top row) and Vav1creAhrfx/fx offspring (bottom row) on day 9 post IAV infection. The number on the plots denotes the mean percentage of CTL.
(F) The number of CD44hiCD62Llo CD8+ T cells in female Ahrfx/fx and Vav1creAhrfx/fx offspring.
(G) The percentage of CTL in male Ahrfx/fx (top row) and Vav1creAhrfx/fx offspring (bottom row). The number on the plots denotes the mean percentage of CTL.
(H) The number of CTL in male Ahrfx/fx and Vav1creAhrfx/fx offspring 9 days after infection. All flow plots are derived from the CD8+ T cell gate (Figure S1).
All data are presented as mean ± SEM. * denotes p value ≤0.05, compared with control offspring with the same genotype (ANOVA followed by Tukey HSD).
Discussion
Recent studies reveal that maternal exposures can cause changes in biological processes that span generations. For example, maternal exposure to endocrine disrupting chemicals causes transgenerational changes in metabolism as well as altered reproductive and nervous system functions (Heindel, 2018, Rattan et al., 2018, Rissman and Adli, 2014, Skinner, 2014, Skinner et al., 2010, van Steenwyk et al., 2018, Walker and Gore, 2011). Other studies have shown that maternal and early life exposures affect immune responses in the F1 generation (Boule and Lawrence, 2016, Winans et al., 2011). Although a recent study indicates that maternal exposure to diesel exhaust particles affects asthma risk across generations (Gregory et al., 2017), no prior studies have directly examined whether maternal exposure to AHR-binding chemicals causes transgenerationally inherited changes in immune responses. The work reported in the present study evaluated whether maternal (F0) exposure affects a key immune defense in a transgenerational manner. We found that F0 exposure caused transgenerational effects on the CD8+ T cell response to influenza A virus infection; however, not all of the effects observed in the F1 generation were transmitted equally to the F3 generation, and the transgenerational effects observed were organ and sex specific.
The major finding of our study was that F0 exposure to TCDD caused transgenerational changes to the CD8+ T cell response to infection. Interestingly, changes in the F3 generation were observed in the infected lung but not in the MLN. This is in contrast to the F1 generation, in which the T cell response to infection is altered in both MLN and lung. Although the site of IAV infection is the respiratory tract, the MLN is the primary site where naive CD8+ T cells become activated, clonally expand, differentiate, and acquire effector functions before migrating to the lung. In addition to recognizing and eliminating infected cells in the lung, newly emigrated CD8+ T cells can also receive additional signals from lung resident DCs. This provides greater stimulation to the CD8+ T cells, prompting them to undergo additional rounds of proliferation and enhancing their cytotoxicity (McGill and Legge, 2009). As such, DC-CD8+ T cell interactions in the lung are important for an optimally effective CD8+ T cell response to IAV (Lawrence et al., 2005, McGill et al., 2008). Thus, a possible explanation for transgenerational differences in the CD8+ T cell response in the lung, but not the MLN, is that there are durable effects of F0 exposure on DC functions that bolster CD8+ T cell responses in the infected lung. A recent report demonstrates that developmental exposure to TCDD alters DC function in adult F1 offspring, including their ability to activate naive IAV-specific CD8+ T cells (Meyers et al., 2018). Other studies provide further evidence that early life exposures influence the function of DCs. For example, offspring of dams sensitized with ovalbumin or diesel exhaust particles (DEPs) are predisposed to airway inflammation later in life, in part due to changes in DCs, and some effects of DEP exposure span generations (Fedulov et al., 2008, Gregory et al., 2017). Thus, changes in DC function potentially carry across generations.
In addition to influencing leukocyte functions, F0 exposure may trigger changes to the programming of lung epithelial cells, and the consequences of this may differ across generations in a manner that influences the quality of the CD8+ T cell response in the infected lung. For example, lung epithelial cells produce thymic stromal lymphopoietin (TSLP), a cytokine that, when produced during an IAV infection, acts on DCs in the lung and enhances the DC-CD8+ T cell interactions necessary for continued expansion of CD8+ T cells (Yadava et al., 2013). Furthermore, activation of the AHR influences Tslp gene expression (Jeong et al., 2018). Therefore, it is possible that the reduced CD8+ T cell response in the lung is due to ineffective DC-CD8+T cell interactions, which arise from changes to the DCs themselves and also from aberrant signaling from the lung epithelium that is programmed by F0 AHR activation.
Another important observation in our study was that the transgenerational effects were sex specific. This was somewhat surprising given that in the F1 generation both male and female offspring of TCDD-treated dams had significantly suppressed CD8+ T cell responses to IAV infection. Yet, in the F3 generation, differences in these same metrics of the CD8+ T cell response to infection were observed in female offspring but not in their male littermates. A simple explanation is that immunomodulatory programming is passed on only maternally. However, results from the outcross experiment indicate that there are consequences of F0 AHR activation in male F1 offspring that contribute to generation-spanning changes in T cell responses. Specifically, paternal lineage TCDD exposure transmitted altered CD8+ T cell responses to female F3 offspring. The difference in the consequences of F0 exposure in the male F1 and F3 generation could indicate that direct (F0, F1) and indirect (F3) exposures have differential effects on programming in the male germ line. AHR activation by TCDD affects the male and female reproductive system (Bruner-Tran et al., 2014, Bruner-Tran and Osteen, 2011, Karman et al., 2012, Manikkam et al., 2012), and the AHR is broadly expressed in many tissues, including immune and non-immune cells. Within the developing hematopoietic system, the Ahr is expressed in fetal liver and thymus and is transcriptionally active. For instance, AHR activation stimulates Cyp1a1 gene expression in fetal liver, hematopoietic stem and progenitor cells, thymus, and also peripheral T cells of neonatal mice that were directly exposed to TCDD (Ahrenhoerster et al., 2014, Majora et al., 2005, Singh et al., 2012, Winans et al., 2015). Yet, the transcriptional activity triggered by early life exposure is transient, as evidenced by the observation that, in adult offspring exposed perinatally, the AHR activity induced by earlier exposure does not persist (Gehrs and Smialowicz, 1997, Ishimaru et al., 2009, Winans et al., 2015). Although the mechanisms by which triggering the AHR lead to generation-spanning alterations in immune function remain to be elucidated, these results suggest that direct exposures (F0, F1) may lead to immunomodulatory effects that are easily discernable but are transient, whereas indirect effects are more subtle. Variations in AHR-mediated changes in gene expression in distinct cell types, as well as in developing male and female fetuses and neonates, may underlie some of the differences observed in our study. Based on a combination of mRNA and protein expression, different cells and tissues express different levels of the Ahr (Esser and Rannug, 2015, Li et al., 1994, Nishihashi et al., 2006). For example, the lung and liver generally express higher levels compared with immune cells (Li et al., 1994). When it has been compared directly, male and female rodents express the Ahr at similar levels (Lee et al., 2015, Nault et al., 2017). A limitation of current knowledge is that, in many studies of Ahr expression and AHR signaling, only one sex has been used. Moreover, in fetal tissues, sex is generally not reported, perhaps owing to technical challenges, such as the need to pool tissue from more than one fetus to obtain sufficient material for assays. Nonetheless, distinct consequences of direct exposure to TCDD in male and female mice have been described (Lee et al., 2015, Pohjanvirta et al., 2012, Prokopec et al., 2015). For instance, Ahr expression levels and induction of known AHR target genes were similar in male and female mice, whereas TCDD treatment elicited changes to the overall transcriptome profile in liver that were different in males and females (Lee et al., 2015, Prokopec et al., 2015). Also, the immune responses of both males and females are similarly influenced by direct exposure to AHR agonists, whereas differences in the magnitude have been observed, although which sex is more sensitive (i.e., showed significant effect at lower dose) depended on the antigen challenge (Mustafa et al., 2008, Sugita-Konishi et al., 2003, Vorderstrasse et al., 2006). Therefore, different levels of Ahr within a particular cell or tissue are not likely sufficient to explain differential responses. Instead, differences reflect a combination of the manner in which AHR signaling converges with other signaling pathways that are specific to cell type and sex and, in the case of immune responses, the specific antigen challenge.
Further support for the idea that the observed consequences of developmental and transgenerational exposures reflect complex interplay comes from our finding that the effects of developmental exposure were not identical in male and female offspring. For instance, in female Vav1creAhrfx/fx offspring, excision of Ahr from hematopoietic cells fully eliminated the effect of maternal TCDD treatment on the response to IAV. In male Vav1creAhrfx/fx mice, the effect appeared blunted, suggesting that in males there may be hematopoietic and non-hematopoietic effects of developmental exposure that contribute to the attenuated CD8+ T cell response. The AHR is expressed widely throughout the body and is highly expressed in barrier organs such as the lung (Esser and Rannug, 2015). Therefore, it is possible that AHR activation in non-hematopoietic cells, such as developing lung epithelial cells, sets off an alternative programming that indirectly affects the CD8+ T cell response to infection in male TCDD-exposed offspring. A related potential explanation is that sex differences in the immune response to infection contribute to the observed differences. Although the overall cellular and molecular defense mechanisms are the same, there are differences in the timing and magnitude of the immune response to IAV in males and females (Vom Steeg and Klein, 2019). For example, during a primary IAV infection, females generally mount adaptive immune responses that are larger in magnitude, yet males have less mortality than females (Klein et al., 2012). However, during secondary challenge with a heterosubtypic virus, females have lower viral titers in their lungs, which is thought to be due to their greater ability to produce antibodies than males (Fink et al., 2018, Lorenzo et al., 2011). The mechanisms that explain sex differences in the response to IAV are not completely understood; however, steroid sex hormones are a contributing factor (Robinson et al., 2011). Given that AHR signaling influences endocrine tissues and endocrine-mediated pathways, it is likely that F0 AHR activation affects myriad different pathways that could potentially influence the programming and function of the immune system.
Although the totality of mechanisms that drive transgenerational inheritance are not fully known, changes in epigenetic regulatory marks are one of the means for conveying changes across generations. Non-coding RNA and histone marks are increasingly recognized as facilitators; however, DNA methylation has been the focus of many studies of transgenerational inheritance (Baxter and Drake, 2019, Blake and Watson, 2016, Boskovic and Rando, 2018, Nilsson and Skinner, 2015). Several pieces of evidence link developmental exposure to the AHR agonist TCDD with altered DNA methylation in transgenerational inheritance of disease phenotypes. For example, in a transgenerational study using rats, developmental exposure to TCDD caused differentially methylated regions (DMRs) in the promoters of sperm DNA isolated from F3 offspring (Manikkam et al., 2012). Developmental exposure to TCDD also altered DNA methylation in the female reproductive tract (Bruner-Tran et al., 2012). A comparison of progesterone receptor (PR) methylation levels in female offspring revealed that developmental TCDD exposure caused durable hypermethylation of the PR gene (Bruner-Tran et al., 2012). Specifically, in F1 offspring developmentally exposed to TCDD, the PR gene was 60% hypermethylated compared with control offspring, and it remained 40% hypermethylated in F3 offspring (Bruner-Tran et al., 2012). Furthermore, in preimplantation embryos treated with TCDD, there was a reduction in gene expression of the imprinted gene H19 that corresponded with an increase in DNA methylation and increased methyltransferase activity (Wu et al., 2004). Another study found that male F1 and F3 rats developmentally exposed to TCDD had altered levels of DNMT3a and DNMT3b in liver tissue (Ma et al., 2015). Hence, there is evidence that developmental exposure to TCDD alters DNA methylation patterns in F1 and F3 offspring. Additionally, separate studies have shown that developmental exposure of mice to TCDD alters DNA methylation patterns in CD8+ T cells in F1 offspring (Winans et al., 2015). DMRs were observed across all genomic features in CD8+ T cells from IAV-infected and from naive mice, suggesting that F0 TCDD exposure influences DNA methylation marks in T cells. In contrast to the liver, changes in overall DNA methylation in CD8+ T cells were not associated with alterations in Dnmt expression levels (Winans et al., 2015). This suggests that, in CD8+ T cells, changes in DNA methylation levels may not be mediated by direct modulation of DNMT expression or that developmental exposure is causing broad changes in the cellular machinery that regulates DNA methylation. Nonetheless, these overall findings indicate that altered DNA methylation likely plays an important role in shaping how exposures in the womb and shortly after birth mold the immune system's function later in life, and potentially across generations.
Few studies of transgenerational inheritance have examined the effects of environmental exposures on the immune system, and no prior studies of developmental exposure to AHR agonists have examined transgenerational effects on immune cells. The work presented here demonstrates that the immune system, like other organ systems, is vulnerable to transgenerational effects caused by environmental exposures. These studies also emphasize the importance of examining both sexes and considering that differences at the site of infection and secondary lymphoid organs may not always be the same. The discovery that maternal exposure to an AHR agonist caused effects on T cells that persisted across generations expands our knowledge of how developmental exposures impact the immune system. Given that early life exposure to environmental chemicals that bind AHR is associated with greater incidence and severity of infectious disease in human populations, the broad implications of these new findings indicate that environmental cues have a tremendous impact on public health, not just in the present but also for future generations.
Limitations of the Study
Although we report novel and exciting findings about transgenerational effects in the immune system, only a single immune challenge was used. This constrains conclusions about sex specificity and whether T cell responses in other peripheral tissues would also be affected across generations. Future studies with additional immune challenges, such as other types of viruses and assessment of antitumor immunity (which also requires CD8+ T cells), will be important extensions of this work.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Brian Rudd (Cornell University) and Dr. Deborah Cory-Slechta (University of Rochester) for thoughtful discussion and critical feedback on our manuscript. We gratefully acknowledge Dr. Sally Thurston (University of Rochester) for her input regarding the overall design of the transgenerational breeding strategy. We are also appreciative of Dr. Timothy Bushnell and the outstanding team at the University of Rochester Flow Cytometry Core. We thank Rachel Garcia for assistance with the graphical abstract. Graphic images are from Servier Medical Art (https://smart.servier.com), licensed under the Creative Commons Attribution 3.0 Unported License Agreement (https://creativecommons.org/licenses/by/3.0/). The authors are supported by grants from the United States National Institutes of Health (R01-ES0004862, R01-ES023260, T32-ES07026, T32-AI007285, T32-HL066988, and P30-ES01247).
Author Contributions
Conceptualization, B.P.L.; Methodology, C.M.P., B.W., L.A.B., and B.P.L.; Investigation, C.M.P., B.W., C.G.B., L.A.B., and C.T.O.; Writing – Original Draft, C.M.P. and L.A.B; Writing – Review & Editing, C.G.B, L.A.B., and B.P.L.; Visualization, C.M.P. and L.A.B.; Project Administration, C.T.O and B.P.L.; Funding Acquisition and Supervision, B.P.L.
Declaration of Interests
The authors declare that they have no actual or potential competing financial or non-financial interests.
Published: October 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.09.014.
Data and Code Availability
All the FCS (flow cytometry standard) files used to generate the data in this paper, as well as FCS files from an independent transgenerational cohort, has been deposited in the NIH FigShare repository (DOI: http://doi.org/10.35092/yhjc.c.4649441)
Supplemental Information
References
- Ahrenhoerster L.S., Tate E.R., Lakatos P.A., Wang X., Laiosa M.D. Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin attenuates capacity of hematopoietic stem cells to undergo lymphocyte differentiation. Toxicol. Appl. Pharmacol. 2014;277:172–182. doi: 10.1016/j.taap.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko O., Kovalchuk I., Metz G.A. Stress-induced perinatal and transgenerational epigenetic programming of brain development and mental health. Neurosci. Biobehav Rev. 2015;48:70–91. doi: 10.1016/j.neubiorev.2014.11.013. [DOI] [PubMed] [Google Scholar]
- Baxter F.A., Drake A.J. Non-genetic inheritance via the male germline in mammals. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019;374:20180118. doi: 10.1098/rstb.2018.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J.A., Singh K.P., Welle S.L., Boule L.A., Lawrence B.P., Gasiewicz T.A. Conditional deletion of Ahr alters gene expression profiles in hematopoietic stem cells. PLoS One. 2018;13:e0206407. doi: 10.1371/journal.pone.0206407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum L.S., Tuomisto J. Non-carcinogenic effects of TCDD in animals. Food Addit. Contam. 2000;17:275–288. doi: 10.1080/026520300283351. [DOI] [PubMed] [Google Scholar]
- Blake G.E., Watson E.D. Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr. Opin. Chem. Biol. 2016;33:101–107. doi: 10.1016/j.cbpa.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Bohacek J., Gapp K., Saab B.J., Mansuy I.M. Transgenerational epigenetic effects on brain functions. Biol. Psychiatry. 2013;73:313–320. doi: 10.1016/j.biopsych.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Boskovic A., Rando O.J. Transgenerational epigenetic inheritance. Annu. Rev. Genet. 2018;52:21–41. doi: 10.1146/annurev-genet-120417-031404. [DOI] [PubMed] [Google Scholar]
- Boule L.A., Burke C.G., Fenton B.M., Thevenet-Morrison K., Jusko T.A., Lawrence B.P. Developmental activation of the AHR increases effector CD4+ T cells and exacerbates symptoms in autoimmune disease-prone Gnaq+/- mice. Toxicol. Sci. 2015;148:555–566. doi: 10.1093/toxsci/kfv203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule L.A., Lawrence B.P. Influences of early life environmental exposures on immune function across the lifespan. In: Esser C., editor. Environmental Influences on the Immune System. Springer-Verlag Wien; 2016. pp. 21–54. [Google Scholar]
- Boule L.A., Winans B., Lambert K., Vorderstrasse B.A., Topham D.J., Pavelka M.S., Jr., Lawrence B.P. Activation of the aryl hydrocarbon receptor during development enhances the pulmonary CD4+ T-cell response to viral infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;309:L305–L313. doi: 10.1152/ajplung.00135.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule L.A., Winans B., Lawrence B.P. Effects of developmental activation of the AhR on CD4+ T cell responses to influenza virus infection in adult mice. Environ. Health Perspect. 2014;122:1201–1208. doi: 10.1289/ehp.1408110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner-Tran K.L., Ding T., Yeoman K.B., Archibong A., Arosh J.A., Osteen K.G. Developmental exposure of mice to dioxin promotes transgenerational testicular inflammation and an increased risk of preterm birth in unexposed mating partners. PLoS One. 2014;9:e105084. doi: 10.1371/journal.pone.0105084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner-Tran K.L., Gnecco J., Ding T., Glore D.R., Pensabene V., Osteen K.G. Exposure to the environmental endocrine disruptor TCDD and human reproductive dysfunction: translating lessons from murine models. Reprod. Toxicol. 2017;68:59–71. doi: 10.1016/j.reprotox.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner-Tran K.L., Osteen K.G. Developmental exposure to TCDD reduces fertility and negatively affects pregnancy outcomes across multiple generations. Reprod. Toxicol. 2011;31:344–350. doi: 10.1016/j.reprotox.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner-Tran K.L., Resuehr D., Ding T., Lucas J.A., Osteen K.G. The Role of endocrine disruptors in the epigenetics of reproductive disease and dysfunction: potential relevance to humans. Curr. Obstet. Gynecol. Rep. 2012;1:116–123. doi: 10.1007/s13669-012-0014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaire F., Dewailly E., Vezina C., Muckle G., Weber J.P., Bruneau S., Ayotte P. Effect of prenatal exposure to polychlorinated biphenyls on incidence of acute respiratory infections in preschool Inuit children. Environ. Health Perspect. 2006;114:1301–1305. doi: 10.1289/ehp.8683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito M.J., Birnbaum L.S., Farland W.H., Gasiewicz T.A. Comparisons of estimated human body burdens of dioxin-like chemicals and TCDD body burdens in experimentally exposed animals. Environ. Health Perspect. 1995;103:820–831. doi: 10.1289/ehp.95103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietert R.R. Developmental immunotoxicology: focus on health risks. Chem. Res. Toxicol. 2009;22:17–23. doi: 10.1021/tx800198m. [DOI] [PubMed] [Google Scholar]
- Esser C., Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Fedulov A.V., Leme A., Yang Z., Dahl M., Lim R., Mariani T.J., Kobzik L. Pulmonary exposure to particles during pregnancy causes increased neonatal asthma susceptibility. Am. J. Respir. Cell Mol. Biol. 2008;38:57–67. doi: 10.1165/rcmb.2007-0124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferey J., Boudoures A.L., Reid M., Drury A., Scheaffer S., Modi Z., Kovacs A., Pietka T., DeBosch B.J., Thompson M.D. Maternal high-fat, high-sucrose diet induces transgenerational cardiac mitochondrial dysfunction independent of maternal mitochondrial inheritance. Am. J. Physiol. Heart Circ. Physiol. 2019;316:H1202–H1210. doi: 10.1152/ajpheart.00013.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink A.L., Engle K., Ursin R.L., Tang W.Y., Klein S.L. Biological sex affects vaccine efficacy and protection against influenza in mice. Proc. Natl. Acad. Sci. U S A. 2018;115:12477–12482. doi: 10.1073/pnas.1805268115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrs B.C., Smialowicz R.J. Alterations in the developing immune system of the F344 rat after perinatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Effects on the fetus and the neonate. Toxicology. 1997;122:219–228. doi: 10.1016/s0300-483x(97)00098-x. [DOI] [PubMed] [Google Scholar]
- Gillette R., Son M.J., Ton L., Gore A.C., Crews D. Passing experiences on to future generations: endocrine disruptors and transgenerational inheritance of epimutations in brain and sperm. Epigenetics. 2018;13:1106–1126. doi: 10.1080/15592294.2018.1543506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn A., Thuvander A., Aune M., Johannisson A., Darnerud P.O., Ronquist G., Cnattingius S. Immune cell counts and risks of respiratory infections among infants exposed pre- and postnatally to organochlorine compounds: a prospective study. Environ. Health. 2008;7:62. doi: 10.1186/1476-069X-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory D.J., Kobzik L., Yang Z., McGuire C.C., Fedulov A.V. Transgenerational transmission of asthma risk after exposure to environmental particles during pregnancy. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;313:L395–L405. doi: 10.1152/ajplung.00035.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Bosagna C., Skinner M.K. Environmentally induced epigenetic transgenerational inheritance of male infertility. Curr. Opin. Genet. Dev. 2014;26:79–88. doi: 10.1016/j.gde.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindel J.J. The developmental basis of disease: update on environmental exposures and animal models. Basic Clin. Pharmacol. Toxicol. 2018;00:1–9. doi: 10.1111/bcpt.13118. [DOI] [PubMed] [Google Scholar]
- Hochstenbach K., van Leeuwen D.M., Gmuender H., Gottschalk R.W., Stolevik S.B., Nygaard U.C., Lovik M., Granum B., Namork E., Meltzer H.M. Toxicogenomic profiles in relation to maternal immunotoxic exposure and immune functionality in newborns. Toxicol. Sci. 2012;129:315–324. doi: 10.1093/toxsci/kfs214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogaboam J.P., Moore A.J., Lawrence B.P. The aryl hydrocarbon receptor affects distinct tissue compartments during ontogeny of the immune system. Toxicol. Sci. 2008;102:160–170. doi: 10.1093/toxsci/kfm283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T.D., Murray I.A., Perdew G.H. Indole and tryptophan metabolism: endogenous and dietary routes to Ah receptor activation. Drug Metab. Dispos. 2015;43:1522–1535. doi: 10.1124/dmd.115.064246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru N., Takagi A., Kohashi M., Yamada A., Arakaki R., Kanno J., Hayashi Y. Neonatal exposure to low-dose 2,3,7,8-tetrachlorodibenzo-p-dioxin causes autoimmunity due to the disruption of T cell tolerance. J. Immunol. 2009;182:6576–6586. doi: 10.4049/jimmunol.0802289. [DOI] [PubMed] [Google Scholar]
- Iuliano A.D., Roguski K.M., Chang H.H., Muscatello D.J., Palekar R., Tempia S., Cohen C., Gran J.M., Schanzer D., Cowling B.J. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet. 2018;391:1285–1300. doi: 10.1016/S0140-6736(17)33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H., Shin J.Y., Kim M.J., Na J., Ju B.G. Activation of aryl hydrocarbon receptor negatively regulates thymic stromal lymphopoietin gene expression via protein kinase Cdelta-p300-NF-kappaB pathway in keratinocytes under inflammatory conditions. J. Invest. Dermatol. 2018;139:1098–1109. doi: 10.1016/j.jid.2018.11.012. [DOI] [PubMed] [Google Scholar]
- Jusko T.A., De Roos A.J., Lee S.Y., Thevenet-Morrison K., Schwartz S.M., Verner M.A., Murinova L.P., Drobna B., Kocan A., Fabisikova A. A birth cohort study of maternal and infant serum PCB-153 and DDE concentrations and responses to infant tuberculosis vaccination. Environ. Health Perspect. 2016;124:813–821. doi: 10.1289/ehp.1510101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman B.N., Basavarajappa M.S., Craig Z.R., Flaws J.A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin activates the aryl hydrocarbon receptor and alters sex steroid hormone secretion without affecting growth of mouse antral follicles in vitro. Toxicol. Appl. Pharmacol. 2012;261:88–96. doi: 10.1016/j.taap.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S.L., Hodgson A., Robinson D.P. Mechanisms of sex disparities in influenza pathogenesis. J. Leukoc. Biol. 2012;92:67–73. doi: 10.1189/jlb.0811427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klukovich R., Nilsson E., Sadler-Riggleman I., Beck D., Xie Y., Yan W., Skinner M.K. Environmental toxicant induced epigenetic transgenerational inheritance of prostate pathology and stromal-epithelial cell epigenome and transcriptome alterations: ancestral origins of prostate disease. Sci. Rep. 2019;9:2209. doi: 10.1038/s41598-019-38741-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier J.E., Woodland D.L. Immunity to respiratory viruses. Annu. Rev. Immunol. 2009;27:61–82. doi: 10.1146/annurev.immunol.021908.132625. [DOI] [PubMed] [Google Scholar]
- Lawrence B.P., Roberts A.D., Neumiller J.J., Cundiff J.A., Woodland D.L. Aryl hydrocarbon receptor activation impairs the priming but not the recall of influenza virus-specific CD8+ T cells in the lung. J. Immunol. 2006;177:5819–5828. doi: 10.4049/jimmunol.177.9.5819. [DOI] [PubMed] [Google Scholar]
- Lawrence B.P., Warren T.K., Luong H. Fewer T lymphocytes and decreased pulmonary influenza virus burden in mice exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) J. Toxicol. Environ. Health A. 2000;61:39–53. doi: 10.1080/00984100050116771. [DOI] [PubMed] [Google Scholar]
- Lawrence C.W., Ream R.M., Braciale T.J. Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J. Immunol. 2005;174:5332–5340. doi: 10.4049/jimmunol.174.9.5332. [DOI] [PubMed] [Google Scholar]
- Lee J., Prokopec S.D., Watson J.D., Sun R.X., Pohjanvirta R., Boutros P.C. Male and female mice show significant differences in hepatic transcriptomic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. BMC Genomics. 2015;16:625. doi: 10.1186/s12864-015-1840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew B.J., Manickam R., Lawrence B.P. Activation of the aryl hydrocarbon receptor during pregnancy in the mouse alters mammary development through direct effects on stromal and epithelial tissues. Biol. Reprod. 2011;84:1094–1102. doi: 10.1095/biolreprod.110.087544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Donat S., Dohr O., Unfried K., Abel J. Ah receptor in different tissues of C57BL/6J and DBA/2J mice: use of competitive polymerase chain reaction to measure Ah-receptor mRNA expression. Arch. Biochem. Biophys. 1994;315:279–284. doi: 10.1006/abbi.1994.1501. [DOI] [PubMed] [Google Scholar]
- Lorenzo M.E., Hodgson A., Robinson D.P., Kaplan J.B., Pekosz A., Klein S.L. Antibody responses and cross protection against lethal influenza A viruses differ between the sexes in C57BL/6 mice. Vaccine. 2011;29:9246–9255. doi: 10.1016/j.vaccine.2011.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Chen X., Liu Y., Xie Q., Sun Y., Chen J., Leng L., Yan H., Zhao B., Tang N. Ancestral TCDD exposure promotes epigenetic transgenerational inheritance of imprinted gene Igf2: methylation status and DNMTs. Toxicol. Appl. Pharmacol. 2015;289:193–202. doi: 10.1016/j.taap.2015.09.024. [DOI] [PubMed] [Google Scholar]
- Majora M., Frericks M., Temchura V., Reichmann G., Esser C. Detection of a novel population of fetal thymocytes characterized by preferential emigration and a TCRgammadelta+ T cell fate after dioxin exposure. Int. Immunopharmacol. 2005;5:1659–1674. doi: 10.1016/j.intimp.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Manikkam M., Tracey R., Guerrero-Bosagna C., Skinner M.K. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One. 2012;7:e46249. doi: 10.1371/journal.pone.0046249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill J., Legge K.L. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J. Immunol. 2009;183:4177–4181. doi: 10.4049/jimmunol.0901109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill J., Van Rooijen N., Legge K.L. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J. Exp. Med. 2008;205:1635–1646. doi: 10.1084/jem.20080314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers J.L., Winans B., Kelsaw E., Murthy A., Gerber S., Lawrence B.P. Environmental cues received during development shape dendritic cell responses later in life. PLoS One. 2018;13:e0207007. doi: 10.1371/journal.pone.0207007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita C., Sasaki S., Saijo Y., Washino N., Okada E., Kobayashi S., Konishi K., Kajiwara J., Todaka T., Kishi R. Effects of prenatal exposure to dioxin-like compounds on allergies and infections during infancy. Environ. Res. 2011;111:551–558. doi: 10.1016/j.envres.2011.01.021. [DOI] [PubMed] [Google Scholar]
- Murray I.A., Patterson A.D., Perdew G.H. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer. 2014;14:801–814. doi: 10.1038/nrc3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa A., Holladay S.D., Goff M., Witonsky S.G., Kerr R., Reilly C.M., Sponenberg D.P., Gogal R.M., Jr. An enhanced postnatal autoimmune profile in 24 week-old C57BL/6 mice developmentally exposed to TCDD. Toxicol. Appl. Pharmacol. 2008;232:51–59. doi: 10.1016/j.taap.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nault R., Fader K.A., Harkema J.R., Zacharewski T. Loss of liver-specific and sexually dimorphic gene expression by aryl hydrocarbon receptor activation in C57BL/6 mice. PLoS One. 2017;12:e0184842. doi: 10.1371/journal.pone.0184842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.P., Bradfield C.A. The search for endogenous activators of the aryl hydrocarbon receptor. Chem. Res. Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson E.E., Skinner M.K. Environmentally induced epigenetic transgenerational inheritance of reproductive disease. Biol. Reprod. 2015;93(6):145. doi: 10.1095/biolreprod.115.134817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihashi H., Kanno Y., Tomuro K., Nakahama T., Inouye Y. Primary structure and organ-specific expression of the rat aryl hydrocarbon receptor repressor gene. Biol. Pharm. Bull. 2006;29:640–647. doi: 10.1248/bpb.29.640. [DOI] [PubMed] [Google Scholar]
- Pilsner J.R., Parker M., Sergeyev O., Suvorov A. Spermatogenesis disruption by dioxins: epigenetic reprograming and windows of susceptibility. Reprod. Toxicol. 2017;69:221–229. doi: 10.1016/j.reprotox.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohjanvirta R., Miettinen H., Sankari S., Hegde N., Linden J. Unexpected gender difference in sensitivity to the acute toxicity of dioxin in mice. Toxicol. Appl. Pharmacol. 2012;262:167–176. doi: 10.1016/j.taap.2012.04.032. [DOI] [PubMed] [Google Scholar]
- Prokopec S.D., Watson J.D., Lee J., Pohjanvirta R., Boutros P.C. Sex-related differences in murine hepatic transcriptional and proteomic responses to TCDD. Toxicol. Appl. Pharmacol. 2015;284:188–196. doi: 10.1016/j.taap.2015.02.012. [DOI] [PubMed] [Google Scholar]
- Rattan S., Brehm E., Gao L., Flaws J.A. Di(2-Ethylhexyl) Phthalate exposure during prenatal development causes adverse transgenerational effects on female fertility in mice. Toxicol. Sci. 2018;163:420–429. doi: 10.1093/toxsci/kfy042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed C., Chaves S.S., Daily Kirley P., Emerson R., Aragon D., Hancock E.B., Butler L., Baumbach J., Hollick G., Bennett N.M. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS One. 2015;10:e0118369. doi: 10.1371/journal.pone.0118369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman E.F., Adli M. Minireview: transgenerational epigenetic inheritance: focus on endocrine disrupting compounds. Endocrinology. 2014;155:2770–2780. doi: 10.1210/en.2014-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D.P., Lorenzo M.E., Jian W., Klein S.L. Elevated 17beta-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. 2011;7:e1002149. doi: 10.1371/journal.ppat.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V., Quintana F.J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019;19:184–197. doi: 10.1038/s41577-019-0125-8. [DOI] [PubMed] [Google Scholar]
- Schmidt M.E., Varga S.M. The CD8 T cell response to respiratory virus infections. Front. Immunol. 2018;9:678. doi: 10.3389/fimmu.2018.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N.P., Singh U.P., Guan H., Nagarkatti P., Nagarkatti M. Prenatal exposure to TCDD triggers significant modulation of microRNA expression profile in the thymus that affects consequent gene expression. PLoS One. 2012;7:e45054. doi: 10.1371/journal.pone.0045054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod. Toxicol. 2008;25:2–6. doi: 10.1016/j.reprotox.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol. Cell Endocrinol. 2014;398:4–12. doi: 10.1016/j.mce.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.K., Manikkam M., Guerrero-Bosagna C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010;21:214–222. doi: 10.1016/j.tem.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemann R., Buchner D.A. Transgenerational inheritance of metabolic disease. Semin. Cell Dev. Biol. 2015;43:131–140. doi: 10.1016/j.semcdb.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens E.A., Mezrich J.D., Bradfield C.A. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolevik S.B., Nygaard U.C., Namork E., Haugen M., Meltzer H.M., Alexander J., Knutsen H.K., Aaberge I., Vainio K., van Loveren H. Prenatal exposure to polychlorinated biphenyls and dioxins from the maternal diet may be associated with immunosuppressive effects that persist into early childhood. Food Chem. Toxicol. 2013;51:165–172. doi: 10.1016/j.fct.2012.09.027. [DOI] [PubMed] [Google Scholar]
- Sugita-Konishi Y., Kobayashi K., Naito H., Miura K., Suzuki Y. Effect of lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on the susceptibility to Listeria infection. Biosci. Biotechnol. Biochem. 2003;67:89–93. doi: 10.1271/bbb.67.89. [DOI] [PubMed] [Google Scholar]
- Topham D.J., Tripp R.A., Doherty P.C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 1997;159:5197–5200. [PubMed] [Google Scholar]
- van Steenwyk G., Roszkowski M., Manuella F., Franklin T.B., Mansuy I.M. Transgenerational inheritance of behavioral and metabolic effects of paternal exposure to traumatic stress in early postnatal life: evidence in the 4th generation. Environ. Epigenet. 2018;4:dvy023. doi: 10.1093/eep/dvy023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vom Steeg L.G., Klein S.L. Sex and sex steroids impact influenza pathogenesis across the life course. Semin. Immunopathol. 2019;41:189–194. doi: 10.1007/s00281-018-0718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorderstrasse B.A., Cundiff J.A., Lawrence B.P. Developmental exposure to the potent aryl hydrocarbon receptor agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin Impairs the cell-mediated immune response to infection with influenza a virus, but enhances elements of innate immunity. J. Immunotoxicol. 2004;1:103–112. doi: 10.1080/15476910490509244. [DOI] [PubMed] [Google Scholar]
- Vorderstrasse B.A., Cundiff J.A., Lawrence B.P. A dose-response study of the effects of prenatal and lactational exposure to TCDD on the immune response to influenza A virus. J. Toxicol. Environ. Health A. 2006;69:445–463. doi: 10.1080/15287390500246985. [DOI] [PubMed] [Google Scholar]
- Walker D.M., Gore A.C. Transgenerational neuroendocrine disruption of reproduction. Nat. Rev. Endocrinol. 2011;7:197–207. doi: 10.1038/nrendo.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winans B., Humble M.C., Lawrence B.P. Environmental toxicants and the developing immune system: a missing link in the global battle against infectious disease? Reprod. Toxicol. 2011;31:327–336. doi: 10.1016/j.reprotox.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winans B., Nagari A., Chae M., Post C.M., Ko C.I., Puga A., Kraus W.L., Lawrence B.P. Linking the aryl hydrocarbon receptor with altered DNA methylation patterns and developmentally induced aberrant antiviral CD8+ T cell responses. J. Immunol. 2015;194:4446–4457. doi: 10.4049/jimmunol.1402044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Ohsako S., Ishimura R., Suzuki J.S., Tohyama C. Exposure of mouse preimplantation embryos to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters the methylation status of imprinted genes H19 and Igf2. Biol. Reprod. 2004;70:1790–1797. doi: 10.1095/biolreprod.103.025387. [DOI] [PubMed] [Google Scholar]
- Yadava K., Sichelstiel A., Luescher I.F., Nicod L.P., Harris N.L., Marsland B.J. TSLP promotes influenza-specific CD8+ T-cell responses by augmenting local inflammatory dendritic cell function. Mucosal Immunol. 2013;6:83–92. doi: 10.1038/mi.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y.A., Mohamed E.A., Rahman M.S., Kwon W.S., Song W.H., Ryu B.Y., Pang M.G. 2,3,7,8-Tetrachlorodibenzo-p-dioxin can alter the sex ratio of embryos with decreased viability of Y spermatozoa in mice. Reprod. Toxicol. 2018;77:130–136. doi: 10.1016/j.reprotox.2018.02.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the FCS (flow cytometry standard) files used to generate the data in this paper, as well as FCS files from an independent transgenerational cohort, has been deposited in the NIH FigShare repository (DOI: http://doi.org/10.35092/yhjc.c.4649441)