

Abstract
Interstitial lung diseases encompass a large number of entities, which are characterised by a small number of partially overlapping fibrosing injury patterns, either alone or in combination. Thus, the presently applied morphological diagnostic criteria do not reliably discriminate different interstitial lung diseases. We therefore analysed critical regulatory pathways and signalling molecules involved in pulmonary remodelling with regard to their diagnostic suitability. Using laser‐microdissection and microarray techniques, we examined the expression patterns of 45 tissue‐remodelling associated target genes in remodelled and non‐remodelled tissue samples from patients with idiopathic pulmonary fibrosis/usual interstitial pneumonia (IPF/UIP), non‐specific interstitial pneumonia (NSIP), organising pneumonia (OP) and alveolar fibroelastosis (AFE), as well as controls (81 patients in total). We found a shared usage of pivotal pathways in AFE, NSIP, OP and UIP, but also individual molecular traits, which set the fibrosing injury patterns apart from each other and correlate well with their specific morphological aspects. Comparison of the aberrant gene expression patterns demonstrated that (1) molecular profiling in fibrosing lung diseases is feasible, (2) pulmonary injury patterns can be discriminated with very high confidence on a molecular level (86–100% specificity) using individual gene subsets and (3) these findings can be adapted as suitable diagnostic adjuncts.
Keywords: interstitial lung diseases, idiopathic interstitial pneumonia, usual interstitial pneumonia, non‐specific interstitial pneumonia, organising pneumonia, alveolar fibroelastosis
Introduction
Interstitial lung disease (ILD; also referred to as diffuse parenchymal lung disease) is an umbrella term for a diverse and complex group of over 300 non‐neoplastic pulmonary diseases 1, 2. The earliest description of ILD dates from the 19th century, when Ludwig von Buhl, a Munich‐based pathologist, coined the phrase ‘chronic interstitial pneumonia’ in 1872 3. For the next century, there was little progress in understanding and classifying ILD, until – in the late 1960s – Carrington and Liebow started categorising different ILDs on the basis of their histopathological presentation 4, 5, 6, 7.
Currently, ILD includes – among others – entities with lung involvement by primarily extra pulmonary (e.g. autoimmune disorders as rheumatoid arthritis) or granulomatous diseases (e.g. sarcoidosis) 8, 9, 10, 11, as well as the group of idiopathic interstitial pneumonias (IIP) with no recognisable underlying cause 1. Although the exact cause of most ILD is not known, current studies have linked both genetic and environmental factors to their genesis, particularly in idiopathic pulmonary fibrosis (IPF) 12, 13. However, the exact mechanisms promoting pulmonary remodelling are still incompletely understood. All ILDs are defined and should be diagnosed by a synoptic, interdisciplinary evaluation of clinical, radiological and morphological criteria 14. Unfortunately, from a diagnostic point of view, lung injury patterns are per se not pathognomonic for any given entity 15. There are a number of reasons for the limited specificity of conventional morphology in ILD diagnosis, especially in transbronchial biopsies (TBB): (1) a restricted number of injury patterns accounts for a large number of clinical entities, whose features (2) show significant overlap and (3) a heterogeneous distribution, often with a peripheral accentuation in the lung parenchyma. Thus, even with a multidisciplinary approach, surgical lung biopsies would often be warranted for the correct diagnosis, but are rarely performed due to their high complication rates 16, 17, 18, 19.
Because specific treatment options have recently become available for some ILD, the correct pattern identification and integrated diagnoses have become more important 12, 19, 20, 21. A better understanding of the molecular pathogenesis and evolution of the lung's response to injury might reveal new diagnostic criteria for a better separation of injury patterns among ILD, especially usual interstitial pneumonia (UIP), non‐specific interstitial pneumonia (NSIP), organising pneumonia (OP) and alveolar fibroelastosis (AFE) (Figure 1) 22 as well as help define new therapeutic targets.
Figure 1.
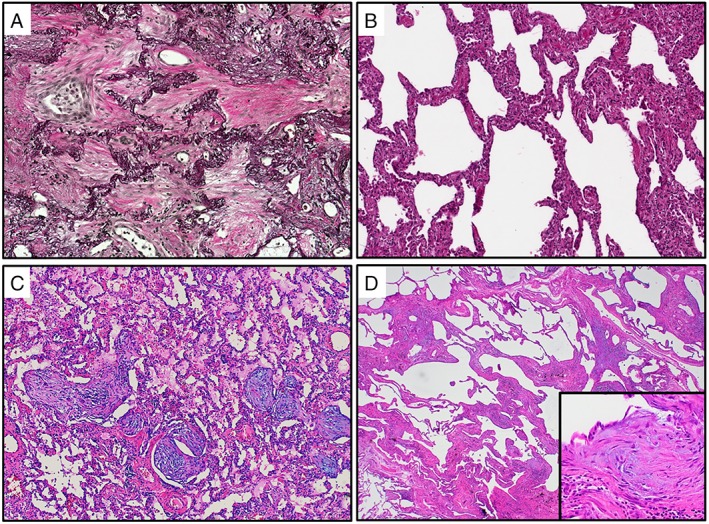
Histological patterns of fibrosing pulmonary injury patterns. (A) AFE (Van Gieson's stain) is characterised by prominent elastosis (black) of the remnant alveolar walls, while the former alveoli are filled up with collagen (red). Note the interspersed fibroblasts and macrophages embedded in the ECM (original magnification ×200). (B) NSIP (H&E stain), characterised by a diffuse, temporally homogeneous and chronic interstitial inflammation with scant fibrotic broadening of the alveolar walls (original magnification ×100). (C) OP (H&E stain) with intra‐alveolar proliferation of anastomosing mesenchymal plugs, rich in (myo)fibroblasts protruding into the alveoli (original magnification ×200). (D) UIP (H&E stain), with marked distortion of the pulmonary architecture: note the discontinuous, patchy and temporally heterogeneous interstitial fibrosis with prominent fibroblastic foci (insert), consisting of activated myofibroblasts aligned in parallel (original magnification ×20/×200).
The UIP pattern is rather specific – but not pathognomonic – for the most important entity of IIP, so‐called IPF. IPF is defined as ‘a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause’ with a 5‐year survival rate of only about 30% 23. The morphological hallmark of the UIP pattern is spatially and temporally heterogeneous interstitial fibrosis driven by so‐called fibroblastic foci, with secondary honeycomb changes, resulting in massive architectural distortion. Antifibrotic pharmaceuticals, for example, nintedanib and pirfenidone have recently become available for IPF and show promising initial results in slowing disease progression and prolonging patient survival 12. In this context, first studies have been put forward that use machine learning classifiers to distinguish UIP from other forms of ILD, so far with moderate to good accuracy based on transcriptomics data from pooled lung biopsy samples 24, 25.
The NSIP pattern can occur in its pure form as an idiopathic clinical entity, but often manifests as a secondary pulmonary reaction pattern 26, 27, 28. Unlike IPF, NSIP shows homogeneous and mild interstitial fibrosis, which may be accompanied by chronic interstitial inflammation. Patients afflicted do not harbour the marked architectural disturbance seen in UIP and show a significantly less aggressive clinical course 29.
Another, frequently observed, reaction to injury pattern is OP, which, again, can occur as an idiopathic variant or as the sequela of trauma, infection or toxic lung damage 30, 31, 32. OP is a focal lesion and the sites of remodelling are the small airways, with continuous granulation tissue plugs, protruding into bronchioli and alveoli. The impairment of pulmonary architecture and function is usually only mild 32.
Another, recently defined, injury pattern is AFE, also referred to as pleuroparenchymal fibroelastosis in some clinical settings 22, 33, 34. Again, AFE can manifest idiopathically, or in the context of other pulmonary injuries. Histologically, fully developed AFE shows fibrotic remodelling featuring intra‐alveolar collagenous obliteration with inconspicuous inflammation and a dominant elastosis in the adjacent, former alveolar walls. Patient survival is stage‐dependent, as progression tends to be relentless and, aside from lung transplantation, there is no effective treatment available 22.
All these morphological patterns are the sequelae of a disturbed response to injury and share an abnormal interaction between the epithelium and the underlying mesenchyme, resulting in aberrant activation of myofibroblasts as a common feature 22, 32, 35.
From a purely functional point of view, we and others have demonstrated complex dysregulation of a large number of mediators in ILD, governing functional areas such as fibrin digestion, macrophage and (myo)fibroblast recruitment, vascular remodelling and epithelial–mesenchymal transition 22, 36. Of these, deregulation of fibrosis‐associated genes involving the pivotal TGF‐β cascade, such as TGF‐β receptors, bone morphogenetic proteins (BMPs) and ECM remodelling enzymes such as matrix metalloproteinases (MMP) or lysyl oxidase (LOX), is one of the joint molecular hallmarks 22, 32. In addition, modulators of regulatory T‐cells, like forkhead‐box‐protein‐3 (FOXP3), thrombocyte regulating thrombospondin (THBS1) or inflammatory associated cytokines such as CCL5, were found to be specifically regulated in ILD 22, 32, 37.
The aims of the current study were (1) to identify gene expression profiles and molecular motifs specific for different injury patterns, (2) to elucidate the contribution of disparate differentiation pathways in development and the extent of fibrotic remodelling and (3) to analyse the discriminatory power of gene analysis for conventional diagnostics. For this purpose, we performed compartment‐specific analyses of distinct injury patterns in well‐defined ILD cases.
Materials and methods
Specimens and study groups
All lung explants sampled during allogeneic lung transplantation at Hannover Medical School (ca. 120–140 procedures annually as of 2018), were evaluated for the present study. As lung specimens can show a variety of histological changes as sequelae of trauma, inflammation and infection, we extensively sampled the selected, well fixed lungs to make sure of characteristic and uniform forms of pulmonary injury patterns – or lack thereof – in the individual patient groups (see below). Of the cases available we included 81 patients/donors in the present study, of which we analysed a total of ~300 compartment‐specific samples (see below).
A total of 20 lungs were explanted because of IPF with UIP pattern histology (4 females, age at transplantation: 59.4 years (arithmetic mean [AM], SD 8.9). A total of 15 patients had undergone transplantation due to idiopathic lung disease with a dominant fibrosing NSIP pattern (11 females, age at transplantation: 46.7 years [AM, SD 15.4]), 14 were diagnosed with AFE (age at transplantation 48.1 years [AM, SD 17.4, 12 females]). In addition to the explants, we selected 15 lung resection specimens from patients with pronounced OP (8 females, age at sampling: 48.2 years [AM, SD 16.6]). Here, underlying diseases that led to diagnostic resection included cystic fibrosis, bronchiectasis, chronic obstructive pulmonary disease (COPD) and non‐specific reaction after surgical trauma as well as cryptogenic OP (COP). All the specimens were inflated with formalin and fixed overnight before being sampled and embedded in paraffin (formalin‐fixed paraffin‐embedded [FFPE]) 37. In all groups, we selected cases on the basis of representative histomorphology, as well as consistency. As a reference, we selected another 17 samples originating from morphologically inconspicuous downsizing tissue of allografts, which were sampled prior to implantation 37.
The FFPE samples were retrieved from the archives of the Institute of Pathology of Hannover Medical School and handled anonymously, following the requirements of the local ethics committee (Ethics Committee vote no. 3381‐2016).
Immunohistochemistry
Serial slides were immunohistochemically stained for different cell surface markers to enable leukocyte sub differentiation, as well as Ki‐67 and smooth muscle actin (SMA) for fibroblast proliferation, following a standard ABC protocol (antibodies listed in Tables 1 and 2) 38. The staining results observed in different compartments and histological patterns were scored semi‐quantitatively in at least three locations of the lungs, ranging from no apparent reaction (score 0), positivity in less than 30% (score 1), positivity in 30% and more/less than 60% (score 2) and positivity in 60% of cells or more (score 3) 37. In AFE specimens, we scored the stroma of the fibroelastic remodelled parenchyma. In UIP specimens, we separately scored fibroblastic foci, the epithelium directly adjacent to them as well as the scarred interstitium. In NSIP, we scored the alveolar epithelium and the interstitium, in OP the stroma of the mesenchymal plugs as well as the epithelium of the adjacent alveoli. The inflammatory cells in AFE, NSIP, OP and UIP were quantitated by counting the number of positively marked cells per high‐power field (HPF; positivity for CD3, CD20, CD68 and mast‐cell tryptase; 15 HPF per specimen). Pairwise comparisons between groups were performed using Student's t‐test.
Table 1.
Immunohistochemical quantification of inflammatory cells
| Detection system | DAB Zytomed HRP kit | |||||
|---|---|---|---|---|---|---|
| Measured parameter | Positive cells per HPF (AM ± SD) | |||||
| Antibody | CD3 | CD20 | CD68 | FOXP3 | Mast cell tryptase+ | |
| Company | Dako | Dako | Dako | Zytomed | Leica | |
| Group | Dilution | 1:200 | 1:50 | 1:100 | 1:100 | 1:50 |
| UIP | Peribronchial | 66.4 ± 17.3*** | 82.3 ± 25.7*** | 12.4 ± 4.6*** | 6.9 ± 4.0* | 0.0 ± 0.0 |
| Fibroblastic foci | 2.3 ± 2.8 | 1.1 ± 1.8 | 13.3 ± 4.6*** | 0.5 ± 1.1 | 0.0 ± 0.0 | |
| Remodelled interstitium | 12.0 ± 6.2*** | 10.9 ± 4.2*** | 57.3 ± 10.8*** | 0.8 ± 1.4 | 3.0 ± 0.8*** | |
| NSIP | Peribronchial | 54.5 ± 12.7*** | 54.5 ± 12.7*** | 9.3 ± 2.6*** | 8.9 ± 6.1* | 0.1 ± 3.0 |
| Remodelled interstitium | 11.0 ± 3.9*** | 5.3 ± 3.0** | 11.7 ± 4.6*** | 4.9 ± 4.0** | 2.1 ± 1.2*** | |
| OP | Peribronchial | 62.4 ± 21.7*** | 77.1 ± 10.1*** | 10.8 ± 3.4*** | 8.9 ± 6.3* | 0.0 ± 0.0 |
| OP lesion | 1.9 ± 3.4 | 4.1 ± 5.2 | 12.6 ± 5.1*** | 0.8 ± 1.4 | 1.0 ± 0.9 | |
| AFE | Peribronchial | 99.6 ± 16.5*** | 118.4 ± 8.2*** | 37.1 ± 6.4*** | 12.9 ± 7.2 | 0.0 ± 0.0 |
| AFE lesion | 40.8 ± 16.2*** | 50.3 ± 17*** | 42.3 ± 19.6*** | 6.4 ± 6.7* | 0.8 ± 0.9 | |
| Controls | Peribronchial | 14.9 ± 7.0 | 24.5 ± 9.0 | 4.5 ± 1.9 | 3.9 ± 3.7 | 0.0 ± 0.0 |
| Interstitium | 3.9 ± 2.1 | 2.3 ± 1.5 | 4.1 ± 2.4 | 1.4 ± 2.0 | 0.4 ± 0.7 | |
A Student's t‐test of absolute cell counts was performed on all diseased tissues against the respective control tissue (‘AFE lesion’, ‘OP lesion’ and ‘fibroblastic foci’ were compared against the interstitium of controls). Further pairwise comparisons are included in supplementary material, Figure S1.
p < 0.05; significant difference against control.
p < 0.01; significant difference against control.
p < 0.001; significant difference against control.
Table 2.
Quantification of proliferating and apoptotic cells
| Method | TUNEL assay | Ki‐67 & Smooth‐muscle actin (SMA) immunohistochemical double staining | ||
|---|---|---|---|---|
| Detection System | ApopTag plus peroxidase in‐situ apoptosis detection kit | DAB Zytomed HRP kit | ||
| Associated biological function | Apoptosis | Proliferation | ||
| Group | Measured parameter | Positive cells per HPF (AM ± SD) | Positive cell ratio compared to controls | Fraction (0–100%) of positive cells per HPF (AM ± SD) |
| UIP | Peribronchial | 0.80 ± 0.78** | 12.0 | 4.7 ± 0.3 |
| Fibroblastic foci | 0.07 ± 0.26 | 0.5 | 9.3 ± 0.5* | |
| Remodelled interstitium | 0.80 ± 0.78** | 6.0 | 5.3 ± 0.5 | |
| NSIP | Peribronchial | 0.13 ± 0.35* | 2.0 | 8.0 ± 0.6 |
| Remodelled interstitium | 0.53 ± 0.74 | 4.0 | 3.4 ± 1.5 | |
| OP | Peribronchial | 0.53 ± 0.74* | 8.0 | 7.3 ± 0.6 |
| OP lesion | 0.33 ± 0.49 | 2.5 | 4.4 ± 6.6* | |
| AFE | Peribronchial | 0.00 ± 0.00 | 0.0 | 9.3 ± 0.6 |
| AFE lesion | 0.80 ± 0.78** | 12.0 | 1.3 ± 2.0 | |
| Controls | Peribronchial | 0.07 ± 0.26 | – | 3.3 ± 0.4 |
| Interstitium | 0.13 ± 0.35 | – | 3.3 ± 0.3 | |
A Student's t‐test of absolute cell counts was performed on all diseased tissues against the respective control tissue (‘AFE lesion’, ‘OP lesion’ and ‘fibroblastic foci’ were compared against the interstitium of controls).
p < 0.05; significant difference against control.
p < 0.01; significant difference against control.
For negative controls, the primary antibody was replaced by BSA 37, 38.
Immunofluorescence double‐staining
To assess the rate of proliferating (myo)fibroblastic cells in different compartments of ILD entities, we performed immunofluorescence double‐staining for Ki‐67 and SMA, essentially as previously described 39.
TUNEL assay
To analyse the rate and role of apoptosis in the remodelling process in ILD, we estimated the content of fragmented DNA in different compartments of ILD entities, using an apoptosis detection kit (ApopTag plus peroxidase in situ apoptosis detection kit, Millipore, Temecula, CA, USA), following the manufacturer's protocol 38. Pairwise comparisons between groups were performed using Student's t‐test.
Compartment‐specific analysis of ILD entities
As the aim of the present study was to identify discrete gene expression profiles and molecular motifs in the respective (sub)compartments of fibrosing ILD, we used laser‐assisted micro‐dissection to generate expression profiles from the different pathologically affected lung compartments. The micro‐dissected compartments encompassed ECM, (myo)fibroblasts, smooth muscle cells, small pre‐ and post‐capillary vessels, capillaries, remnant epithelium and infiltrating leukocytes at different ratios. The morphologically naïve controls consisted of the regular alveolar septae composed of the alveolar epithelium, interstitium, capillaries, small bronchioli and pre‐ and post‐capillary vessels.
Laser‐assisted micro‐dissection and RNA extraction
FFPE 5 μm thick tissue sections were mounted on a poly‐l‐lysin‐coated membrane attached to a metal frame. After routine deparaffinisation and haemalum staining, the CellCut Plus System (MMI Molecular Machines & Industries AG, Eching, Germany) was used for laser‐assisted micro‐dissection of target compartments: we isolated sections of lung parenchyma that showed characteristic changes of AFE, NSIP, OP and AFE (excluding large vessels and airways), respectively, using a no‐touch technique, essentially as described 37. In the explanted lungs, the respective compartments were sampled from at least four different locations in both lungs 37. In wedge biopsies, we also sampled the micro‐dissected material from four different areas within the specimens. The micro‐dissected samples were morphologically chosen to adequately reflect the average degree of cellularity/inflammation and fibrosis in that subgroup 37. In the UIP group, we also collected small bronchi with the adjacent stroma and areas of lung parenchyma that showed no delimitable interstitial fibrosis.
In the reference specimens, we harvested non remodelled lung parenchyma – including small pre‐/post‐capillary vessels and small airways/bronchioles (<0.1 mm in diameter) – but excluding larger arterial and venous vessels and bronchi.
Approximately 8500 cells were collected from serial sections from each compartment in every group. The micro‐dissected tissue was subsequently suspended in a proteinase K digestion buffer by placing the buffer directly in the adhesive cap. After overnight digestion, RNA was isolated using phenol‐chloroform extraction and precipitation following our established procedure 37.
cDNA synthesis
Complementary DNA of each sample was generated from 1 μg of RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA) and following the manufacturer's protocol 37, 40.
Low‐density arrays
Forty‐five target genes as well as three reference genes were selected for our custom‐made TaqMan Low‐Density Array (LDA); genes were included on the basis of our previous and refined analyses of non‐neoplastic remodelling in human lungs and incorporate biomarkers of inflammation, (myo)fibroblast activation, ECM deposition and modification, and so on. (LDA: Applied Biosystems, Foster City, CA, USA; genes and abbreviations are listed in supplementary material, Table S1 in alphabetical order) 22, 40, 41. An amplicon size below 100 bp was one of the criteria for target genes, enabling reliable gene expression analysis by LDA in FFPE samples, essentially as described 41. The primer sets were spotted eight‐fold (8 × 48) into a 384‐well plate, thus allowing the synchronous analysis of eight samples per PCR run. The TaqMan LDAs were performed as single runs on a 7900HT Fast Real‐Time PCR system and recorded by the 7900HT SDS 2.3 software (Applied Biosystems). For negative controls, cDNA was replaced by water 37, 40.
Statistical analysis
All statistical analyses were performed in the computational language R 42. To generally assess the relative expression of the selected target genes in the different groups and compartments, C T values were calculated by normalisation to the mean expression of three endogenous control genes (GAPDH, GUSB, POLR2A) and converted into values. Prior to normalisation, missing values were imputed with the maximum of all observed C T values rounded up to the next whole‐number, that is, 45. Subsequently, we identified significant differences in gene expression between groups via the Mann–Whitney U test (for pairwise comparisons) and the Kruskal–Wallis test (for multiple group comparisons) followed by calculation of false discovery rates (FDRs). We considered FDRs significant according to the following levels of confidence: FDR < 0.05 (*), FDR < 0.01 (**) and FDR < 0.001 (***). Because of the lack of standardised reference values for the measured genes, the median of each gene in the control group was used as a level for defining ‘high’ levels of the respective variable.
Classification of entities via gene expression
In order to predict different ILD entities based on the expression of our target genes, we trained models for the discrimination of (1) all occurring entities (healthy, AFE, NSIP, OP, UIP), (2) healthy from diseased entities, (3) AFE from other diseased entities as well as (4) the discrimination of NSIP, OP and UIP from each other. For modelling purposes, we randomly split the samples into training and blinded test sets (30 and 70% of samples, respectively). The training sets were then used as input for various machine learning algorithms (R package caret 43 version 6.0‐81; algorithms used: lda, rf, mlpML, nnet, multinom, svmLinear and svmRadial). Tuning parameters and intervals were left at default settings. Leave‐one‐out cross validation was used for resampling and estimating the preliminary model performance. The resulting models were subsequently ranked based on the performance against their test set. For each possible number of genes that can be used for classification, the most suitable gene subset was identified using an exact leaps and bounds algorithm aimed at optimising the Tau‐squared coefficient (R package subselect 44) or using the random forest importance metric in case of random forest models. Gene subset selection was limited to significantly regulated genes. Also, genes with highly correlated expression were removed in a stepwise manner prior to subset selection (correlation ≥0.9).
Functional analysis
In order to complement our molecular data with predictions on the activation/inhibition of specific physiological functions, data were analysed via a comparison analysis of biological functions from the Ingenuity Pathway Analysis tool (IPA, Qiagen Inc., Venlo, Netherlands) 45. As input, we used the gene expression data from each sample relative to the respective median gene expression in the controls as separate observations. The function activity prediction of IPA was executed with the default settings, thus using information from both in vivo and in vitro experiments. The resulting z‐scores give a quantitative estimate of how biological functions are effectively regulated by the observed differences in gene expression. Biological pathways linked to fibrosis were then selected and significant differences in the regulation of these functions estimated via pairwise Mann–Whitney U tests and Kruskal–Wallis tests (for multi‐group comparison of AFE, NSIP, OP and UIP only). Here, we considered P values significant according to the following levels of confidence: p < 0.05 (*), p < 0.01 (**) and p < 0.001 (***).
Results
Immunohistochemical characterisation of AFE, NSIP, OP and UIP
The immunohistochemical characterisation of the inflammatory and myofibroblastic cells is summarised in Tables 1 and 2, with additional details in supplementary material, Figure S1. Focusing on the remodelled interstitium in UIP and NSIP, the fibroblastic foci of UIP as well as the intra‐alveolar lesions in OP and AFE, two inflammatory reaction patterns can be discriminated: on the one hand OP and fibroblastic foci as low‐grade inflammatory lesions and on the other hand AFE and the remodelled interstitium in NSIP and UIP as inflammatory rich lesions. Among the densely inflamed lesions AFE stands out, as the absolute numbers of T‐cells, B‐cells and macrophages – with the exception of the remodelled interstitium in UIP – are significantly higher than in all other lesions/compartments, not only within the remodelled but also the peribronchial areas. However, it has to be taken into account that we analysed not yet fully evolved end‐stage AFE lesions with (still) ongoing inflammation; sampling fully developed end‐stage lesions would show a lower inflammatory reaction. The remodelled interstitial areas in UIP and NSIP showed comparable levels of T‐cells, but UIP revealed significantly higher infiltrates of B‐cells and especially macrophages and significantly fewer regulatory T‐cells than NSIP.
In all lesions, with the exception of AFE, macrophages made up the predominant component of the inflammatory infiltrate. In contrast to T‐ and B‐cells, the majority of the macrophage population resided not in the peribronchial areas but within the fibrotic lesions themselves. In addition, macrophages accumulated in (remnant) alveoli in AFE, NSIP, OP and UIP specimens, as well as in the peribronchial areas.
CD3+ T‐cells were found to be concentrated mainly in circumscribed peribronchial aggregates in AFE, NSIP, OP and UIP and the remodelled areas of AFE, less frequently in the remodelled interstitium of UIP and NSIP and only sparsely in OP and fibroblastic foci. The majority of FOXP3+ regulatory T‐cells were located peribronchially in all entities – albeit in far lesser quantities when compared to other leukocyte types. Only AFE and NSIP showed significant levels of regulatory T‐cells intra‐lesionally.
In specimens from AFE, NSIP, OP and UIP, CD20+ B‐cells were located mainly in the (remnant) peribronchial‐associated lymphoid tissue. In the remodelled compartments of UIP, NSIP and OP comparably low levels could be shown; here, only AFE revealed significant numbers of B‐cells.
Mast cell tryptase+ (MCT+) mast cells were quite scarce and evenly dispersed overall in the remodelled/scarred interstitium with slightly higher values in UIP and NSIP.
As expected, Ki‐67 and SMA double staining of UIP specimens (Table 2) demonstrated a prominent proliferation of SMA‐positive myofibroblasts in the fibroblastic foci and moderate proliferation of myofibroblasts in the remodelled interstitium. In NSIP, double staining showed modest positivity in the mildly remodelled interstitium, whereas in OP patients pronounced proliferation of myofibroblasts was found in the intra‐alveolar mesenchymal plugs that are characteristic of this injury pattern. In AFE lesions, the proliferation of myofibroblasts was limited to the former alveolar walls, but was not found in the former alveolar spaces. Ki‐67 positivity in the alveoli adjacent to AFE lesions was mainly due to proliferation of macrophages.
Assessment of cell apoptosis
According to TUNEL assays (Table 2), there were only infrequent apoptotic mesenchymal cells in the scarred interstitium and peribronchially in UIP lungs. Similarly, only few apoptotic mesenchymal cells were found in the interstitium of NSIP and in the intra‐alveolar mesenchymal plugs of OP specimens. In AFE lesions apoptosis was found to be significantly higher (12‐fold) compared to controls.
Fibrosis‐related gene expression analysis in ILD
Comparison of gene expression profiles among UIP, NSIP, OP and AFE
The expression of 45 remodelling‐related candidate genes was analysed in patients with AFE, NSIP, OP and UIP. As illustrated in Figure 2, the expression of four out of all analysed genes was higher in all ILD entities as compared to controls. Remarkably, all entities shared a significantly higher expression of collagen 3 (COL3A1), matrix metalloproteinases 2 and 14 (MMP2 and MMP14) and stromal cell derived factor 1 α (CXCL12).
Figure 2.
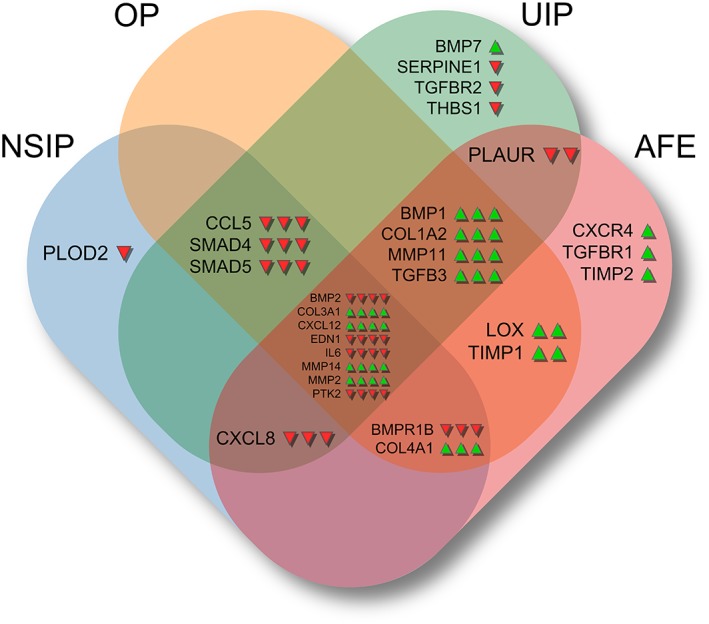
Significantly regulated genes in fibrosing pulmonary injury patterns as compared to controls. Significant up‐ or down‐regulation of genes compared to healthy controls is indicated by arrows (Mann–Whitney U test, FDR < 0.05). The mid‐section shows genes that are significantly regulated in all examined entities. For each intersection area, the arrows indicating a significant regulation correspond to the entities of that intersection in left‐to‐right orientation, that is, NSIP > OP > UIP > AFE.
The control specimens we selected as references showed significantly stronger expression of BMP 2 (BMP2), endothelin 1 (EDN1), IL‐6 (IL6) and protein tyrosine kinase 2 (PTK2) when compared to the ILD groups.
Entity‐specific gene expression patterns in ILD
Generally speaking, ILDs share strong similarities but also exhibit marked differences in regard to gene expression profiles (Figure 2). In comparison to controls and to one another, NSIP is characterised by down‐regulation of PLOD2 while BMP1, COL1A2, MMP11 and TGFB3 are only up‐regulated in AFE, OP and UIP. The latter is characterised by up‐regulation of BMP7 and down‐regulation of SERPINE1, TGFBR2 and THBS while, in contrast to other ILDs, the genes BMPR1B and COL4A1 are not significantly regulated. AFE is characterised by up‐regulation of CXCR4, TGFBR2 and TIMP2 while CCL5, SMAD4 and SMAD5 are only regulated in NSIP, OP and UIP. OP samples showed no exclusive gene regulation. However, the gene CXCL8 is significantly down‐regulated in all ILDs except OP.
Entity‐specific gene expression patterns in ILDs relative to each other
When the gene expression levels of the ILDs shown above are scaled towards one another the relative differences of the gene expression profiles are enhanced. In this context, up‐ or down‐regulation always compares to the mean of all ILD samples. Overall, it can be seen that ILDs are characterised by specific gene expression patterns (Figure 3). For convenience these patterns are emphasised using colour coding in the background of the respective histograms. The genes forming these patterns can be subdivided into four groups dependent on the pattern‐forming characteristics. The first group consists of MMP13, BMP7 and BMPR1B. In AFE, NSIP and OP these genes are down‐regulated, with the exception of BMPR1B in AFE, which is not regulated here and MMP13 in OP which is up‐regulated. In contrast this group of genes is up‐regulated in UIP and BMPR1B especially is a defining factor for the identification of UIP.
Figure 3.
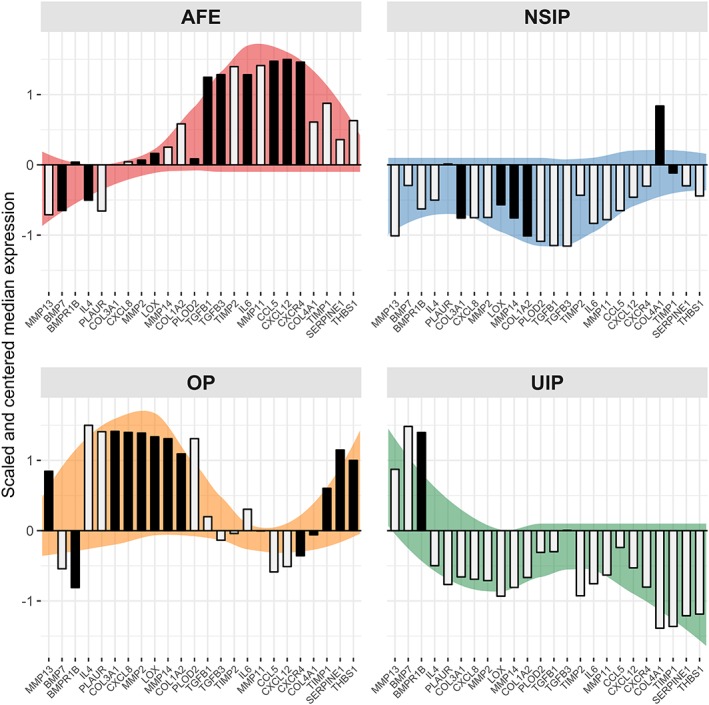
Relative expression profiles of genes significantly regulated between fibrosing pulmonary injury patterns. When comparing the pulmonary injury patterns addressed in this manuscript directly, 24 out of 45 examined genes are significantly regulated (Kruskal–Wallis test, FDR < 0.05). Differences between AFE, NSIP, OP and UIP are emphasised via gene‐wise centring and scaling of expression values (a scaled expression value of zero represents the mean of all four groups). Coloured backgrounds highlight the observed expression patterns and serve no analytical purpose. Darkened bars point out genes that have been used in the creation of the most accurate predictive model for a given entity.
The next group is IL4, PLAU, COL3A1, CXCL8, MMP2, LOX, MMP14, COL1A2 and PLOD2; these genes were found to be slightly up‐regulated in AFE and strongly up‐regulated in OP, but down‐regulated in NSIP and UIP.
The third group consists of TGFB1, TGFB3, TIMP2, IL6, MMP11, CCL5, CXCL12 and CXCR4. This group of genes is strongly up‐regulated in AFE, while mostly down‐regulated in NSIP, OP and UIP.
The last group of genes consists of COL4A1, TIMP1, SERPINE1 and THBS1. They are all up‐regulated in AFE and even more in OP with the exception of COL4A1. In NSIP only COL4A1 is up‐regulated and in UIP all genes in this group are strongly down‐regulated.
Gene expression analysis in remodelled and non‐remodelled areas in UIP lungs
Subsequently, we compared the gene expression motifs between two different compartments from lungs with a prominent UIP pattern to control samples: first, areas of alveolar parenchyma without apparent morphological changes and second, areas with prominent fibroblastic foci and interstitial fibrosis.
Remodelled and non‐remodelled areas shared significant up‐regulation of COL1A2, COL3A1, MMP2 and MMP14 when compared to controls (see supplementary material, Table S2). Also, significantly differentially regulated compared to controls, that is, down‐regulated in remodelled and up‐regulated in non‐remodelled areas, are the genes BMP2, CCL5, CXCL8, SERPINE1 and THBS1. The non‐remodelled areas showed prominent mRNA up‐regulation of COL4A1, IL17A, LOX, TGFB1, TGFB2, TIMP1 and TIMP2.
The remodelled areas displayed a significant increase of mRNA expression of BMP1, BMP7, CXCL12, MMP11 and TGFB3 whereas expression of EDN1, IL6, PLAUR, PTK2, SMAD4, SMAD5 and TGFBR2 was decreased.
Discrimination analysis of ILD entities by molecular markers
In order to discriminate ILD entities from healthy samples and each other based on molecular motifs, we created a set of binary classifiers using the gene expression data presented above following a ‘divide‐and‐conquer’‐like approach (see supplementary material, Figures [Link], [Link], [Link] and Appendix S1). This modelling strategy was devised since the best performing model for multiclass classification of all healthy and diseased samples yielded only about 65% precision against a randomly chosen, common test set of 55 samples (see supplementary material, Table S3) using the expression data of 8 genes. The binary classifiers in turn yield individual precision values ranging from 75 up to 100% (see supplementary material, Table S3). Our modelling approach uses the five best performing binary classifiers in a decision tree that subsequently aims to discriminate (1) healthy from diseased samples, (2) AFE from other disease entities and, lastly,(3) NSIP, OP and UIP from each other. This allows for the optimisation of overall precision and especially class‐wise specificity since unambiguous and contradictory predictions can be rejected (‘unclassifiable samples’). Following this strategy, 43 out of 55 samples from the common test set can be classified with a final precision of 95% (Table 3 and see supplementary material, Table S3).
Table 3.
Classification results
| True class | Predicted class | Unclassifiable samples | Class‐wise specificity (%) | Class‐wise sensitivity (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Control | AFE | NSIP | OP | UIP | |||||
| Control | 11 | 100 | 100 | (100) | |||||
| AFE | 7 | 1 | 1 | 100 | 88 | (78) | |||
| NSIP | 6 | 5 | 100 | 100 | (55) | ||||
| OP | 6 | 1 | 3 | 86 | 86 | (70) | |||
| UIP | 11 | 3 | 92 | 100 | (79) | ||||
Predictions made by the decision tree of binary models using a test set of 55 samples. Class‐wise sensitivity values in parentheses take unclassifiable samples into account as false negative predictions. The overall precision for classifiable samples is 95%.
Gene function interactions in ILD entities
In order to analyse the biological functions underlying the ILD entities we used the IPA tool to calculate activation z‐scores for these functions from our gene expression data (Figure 4). Each sample was treated as an individual observation and scores were computed relative to healthy controls (equivalent to a z‐score of zero). With regard to functions that are presumed to be involved in fibrotic remodelling we found the following biological functions and pathways: ‘migration of mononuclear and endothelial precursor cells’, ‘cellular adhesion’, ‘general cellular proliferation and mesenchymal precursor proliferation’, ‘inflammation’, aberrant angiogenesis’ and ‘pulmonary development’.
Figure 4.
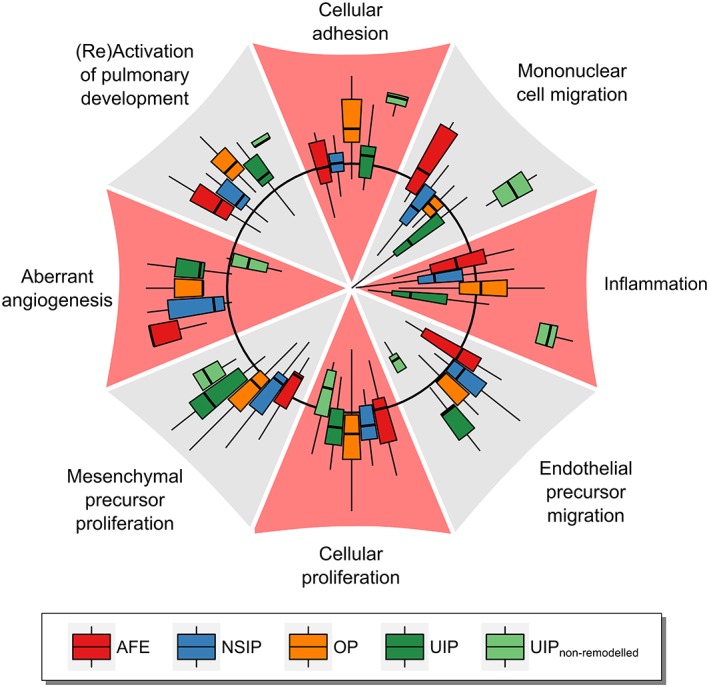
Activation of biological functions in fibrosing pulmonary injury patterns according to IPA. The activation of biological functions in pulmonary injury patterns compared to healthy controls was predicted for each sample using the IPA tool. The black circle represents an activation z‐score of zero (no regulation), while the outer areas indicate positive z‐scores (up‐regulation) and the inner areas negative z‐scores (down‐regulation). The displayed z‐scores range from −3.32 up to 3.85. Outliers are not shown, but are included in the calculation of quartile ranges.
In general, all four ILD entities feature up‐regulation of ‘pulmonary development’ and ‘aberrant angiogenesis’. In AFE samples, activation of ‘mononuclear cell migration’ and ‘aberrant angiogenesis’ is predicted whereas ‘mesenchymal precursor proliferation’ is decreased. OP is characterised by activated ‘cellular adhesion’, ‘aberrant angiogenesis’, ‘mesenchymal precursor proliferation’ and ‘mitosis’. Remodelled areas in UIP featured an increase in ‘endothelial precursor migration’ and ‘mesenchymal precursor proliferation’ as well as a decrease in ‘mononuclear cell migration’ and ‘inflammation’. In contrast, activation scores of functions from non‐remodelled parenchyma in patients with UIP were drastically different from other ILDs. Here, ‘cellular adhesion’ and ‘mononuclear cell migration’ were strongly activated, whereas ‘endothelial cell migration’ and ‘aberrant angiogenesis’ were decreased. NSIP samples involved only moderate up‐regulation of ‘endothelial precursor migration’ and moderate down‐regulation of ‘mononuclear cell migration’. ‘Mesenchymal precursor proliferation’ and ‘cellular adhesion’ mostly clustered around the baseline.
Discussion
Although the first effective medical treatments for IPF have recently been introduced, the current state of affairs is still sobering 46, 47, 48: ILDs remain a very complex group of diseases, with serious diagnostic challenges 24, 49. Further progress regarding the development and use of specific therapies hinges on accurate diagnoses and separation of clinical entities 22. This is considerably hampered by the overlap of morphological patterns – in histopathology and radiology alike, the heterogeneous spatial and chronological distribution of morphological changes, as well as the inter observer variability of morphological assessment 50, 51. As many cases cannot be satisfyingly classified even by application of a multidisciplinary approach using the currently established criteria, new diagnostic approaches are needed.
To address the heterogeneous nature of remodelling in ILD, we previously started to combine morphological and molecular studies of different injury patterns to identify disease‐specific molecular motifs 22, 40, 52. Thereby, we showed that (1) molecular subtyping of specific pulmonary injury patterns seen in the context of lung and stem cell transplantation can be reliably performed, (2) stepwise development of injury corresponds to predictable and compartment‐specific changes of the pulmonary microenvironment and (3) morphologically identical injury patterns seen in different clinical settings, such as AFE or obliterative airway remodelling (OAR), also correspond on the molecular level: ‘AFE is AFE and OAR is OAR’ and (4) morphologically inconspicuous tissue samples from areas adjacent to aforementioned lesions already show characteristic molecular alterations 22, 32, 37, 40.
The goals of this paper were to (1) identify discrete gene expression profiles and molecular motifs in ILD, (2) amalgamate conventional morphology, gene expression and cellular functions and (3) use gene expression motifs in a diagnostic perspective to discriminate among different prevalent and relevant pulmonary injury patterns and disease entities.
The current American Thoracic Society/European Respiratory Society (ATS/ERS) guidelines for the diagnosis of ILD 15 – and IPF in particular –have put a strong emphasis on high resolution computed tomography (HRCT) findings in the corresponding clinical context. This is understandable, as the most characteristic features of UIP, such as fibroblastic foci and architectural distortion, are usually not accessible by TBB due to their heterogeneous distribution and/or peripheral localisation. Moreover, the rather high complication rates with mortality in elective surgical biopsies of ILD patients ranging between 1.7 and 3.9%, and up to 16% in non‐elective procedures 53, discourage their use if the patient's CT findings are ‘probable’ or ‘indeterminate’ regarding an UIP pattern.
Therefore, using molecular methods to gain additional information even from TBB without specific morphological changes would significantly enhance their diagnostic value. In this context, we have previously shown that (1) scoring gene expression levels of the TGF‐β axis in biopsies from lung transplanted patients allows for reliable detection and separation of patients with specific sub‐forms of (2) airway fibrosis/allograft dysfunction and, of note, (3) is also predictive for outcome/prognosis (Risk Assessment of Chronic Lung Allograft Dysfunction (CLAD) [RACA] score) 22, 37, 40. Most importantly, we showed that even morphologically inconspicuous lung parenchyma, adjacent to different foci of fibrosis and inflammation, already bears characteristic ‘molecular lesions’ 37, 40, 52. As a variety of pulmonary injury patterns arise in transplanted and non‐transplanted patients alike, morphology in conjunction with molecular motifs may well also form a diagnostic adjunct for the correct classification of ILD.
To this end, we analysed compartment‐specific expression patterns of remodelling‐associated genes to identify the most relevant molecular changes and thereby predict the biological functions underlying the respective ILD entities 40 using the established IPA tool. Aberrant regulations were also interpreted in the context of the degree of histological and immunohistochemical remodelling (Figure 5).
Figure 5.
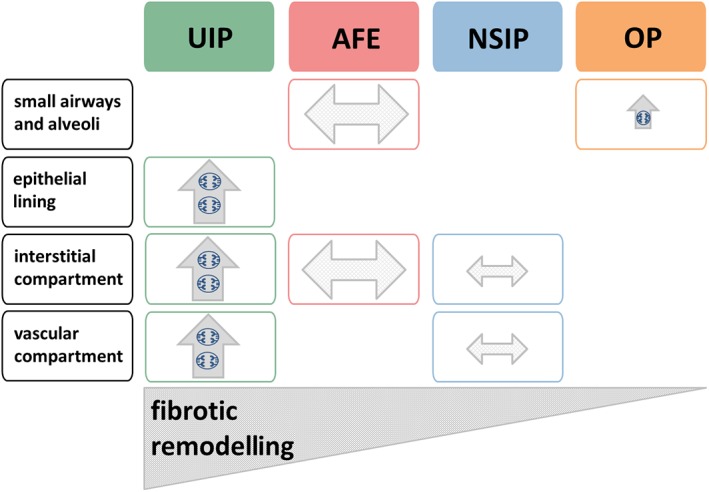
Compartment‐specific synopsis of remodelling in fibrosing pulmonary injury patterns. UIP, AFE, NSIP and OP graded according to the degree of pulmonary architectural distortion in the respective, fully developed injury pattern. Ongoing remodelling in the involved compartments (y‐axis) is indicated by an upward arrow (↑), while end‐stage remodelling is indicated by a horizontal arrow (↔). Relative size of the arrows indicates the extent of remodelling in a given compartment.
Not surprisingly, we found a certain level of molecular similarity among these entities, mainly regarding genes regulating fibrogenesis: AFE, NSIP, OP and UIP share up‐regulation of CXCL12 54, which recruits CXCR4 positive fibrocyte progenitors and T‐cells 55, classical fibrosis associated collagen 3A1 and matrix remodelling MMP2 and 14 – which are in principle responsible for ECM degradation and clearance of fibrosis 56, 57. This is supported by immunohistochemistry, which shows an overall increase in inflammation (Table 1).
The least fibrotic remodelling is seen in OP with only focal epithelial injury and a subsequent granulation tissue‐like remodelling of the terminal air ducts and alveoli. On a functional level, predicted by IPA, OP lesions themselves are characterised by pronounced myofibroblast activation and the alveolar parenchyma adjacent to these by inflammatory processes (Table 1). Concurrently, no decrease in inflammation is predicted in contrast to other ILDs (Figure 4), while expression of IL‐4 is consistently increased (Figure 3). Regarding fibrogenesis, OP shares similarities with AFE and UIP which is indicative of fibrotic consolidation based on the expression TGF‐β‐dependent cascades via TGFB3 together with collagens 1A2 and 3A1, but in the absence of expression of TIMP2 (Figure 2). Although our OP lung specimens came from patients with quite different underlying diseases and conditions – bearing in mind that OP represents about the most unspecific pulmonary reaction to injury – and were resected rather than followed up, it appears plausible that the usually dramatic response of OP to anti‐inflammatory treatment can be explained to some extent by this dominant inflammatory microenvironment 58.
NSIP also shows only a rather limited degree of remodelling with a mild, homogeneous widening of the interstitial compartment by a limited proliferation of fibroblasts with matrix deposition as well as capillary remodelling. On a cellular level NSIP is dominated by CD3+/FOXP3+ regulatory T‐cells. Regarding molecular signals of fibrogenesis, NSIP shows a gene expression pattern common to other injury patterns examined, except aberrant expression of basal membrane collagen 4A1, which is similar to AFE but differs from OP and UIP (Figure 2).
Generally speaking, AFE presents a severe impairment of the pulmonary architecture. Here, airspaces are rendered non‐functional by an initial influx of monocyte‐derived macrophages and subsequent fibrotic obliteration by myofibroblasts producing collagens 1A2, 3A1 and 4A1. Concurrently, epithelia undergo apoptosis and the former alveolar wall is remodelled by a deposition of elastic fibres 22, 33. Overall, molecular signals in AFE (Figure 2) indicate a rather early and florid fibrotic microenvironment which is functionally predicted to be dominated by infiltration, but not proliferation, of fibroblasts and their precursors (Figure 4), with a distinctive up‐regulation of CXCR4, the complementary receptor to the universally up‐regulated CXCL12 59. This molecular phenotype (1) is consistent with the fact that our not (yet) fully end‐stage AFE lesion showed interspersed inflammatory cells and (2) leads to mononuclear cell migration/recruitment 60, 61 reflected by the interstitial and intra‐lesional inflammatory and fibrocyte infiltrate, setting AFE apart from NSIP, OP and UIP specimens. Additionally, AFE is characterised by a pronounced expression of members of the TGF‐beta signalling family, as well as remodelling associated Inhibitors (TIMP1 and 2) of MMPs in addition to collagens 1A2, 3A1 and 4A1, indicating active fibrotic remodelling.
The most severe distortion of the pulmonary architecture occurs in UIP. Here, a heterogeneous pattern of intermixed obliterated and patent airspaces predominates, the latter featuring a multifocal bronchiolar metaplasia of the epithelia 62. The interstitium reveals a massive proliferation of myofibroblasts with prominent matrix deposition, partially organised in circumscribed groups, referred to as fibroblastic foci; adjacent to these, aberrant (neo) angiogenesis is a characteristic finding (Figure 4). Functionally, UIP is mainly characterised by a proliferation of mesenchymal and endothelial precursor cells, as well as endothelial precursor migration, a function involved in sprouting angiogenesis, despite the average down‐regulation of angiogenetic factors like CXCL8. However, both pro and anti‐angiogenetic factors are known to be expressed in fibroblastic foci in UIP lungs that is dependent on the degree and localisation of angiogenesis within the remodelled areas 63, 64. Whereas fibroblastic foci are only sparsely inflamed lesions with a predominance of matrix‐producing myofibroblasts, the remodelled interstitial areas show a predominance of macrophages with only intermediate values of T‐cells. However, the low activation scores (z‐scores) for mononuclear cell migration predicted by IPA argue for a rather inactive compartment with regard to attraction of new leucocytes.
Molecularly, the most prominent difference between UIP and the other injury patterns is up‐regulation of BMP7. This BMP is involved in progressive tissue remodelling as it shows a limited capability to reverse fibrosis and clear collagen deposition. Up‐regulation of the secreted endopeptidases BMP1 and MMP11, as well as the membrane‐based MMP14, which in turn activates MMP2, correlates well with the continuous and progressive remodelling and (sub)total loss of the original pulmonary architecture that is so characteristic for UIP pattern IPF cases 56, 65, 66, 67. The prominent expression of the key mediators for lung remodelling in the rather characteristic fibroblastic foci underlines their importance as the focus of pulmonary remodelling. This is accompanied by a slight increase in apoptosis in the interstitium of the remodelled areas. The prominent expression of TGF‐β‐dependent cascades via TGFB3 together with collagens 1A2 and 3A1 and the absence of expression of TIMP1 and 2 indicate active consolidation of the fibrotic foci as the main function. In addition, it should be noted that non‐remodelled areas within the same lung already present a gene expression profile indicating activation of all cellular functions analysed (Figure 4). The actual predictive value for UIP remains to be determined and will be the subject of future studies.
The use of objective data generated by molecular analysis and machine learning approaches is thought to be the next step in modern diagnostics. This is especially needed since inter‐observer variability in ILD diagnostics based on histology alone is often challenging 50, 51. So far, first studies have shown their high value for the analysis of TBB in IPF to discriminate UIP from other ILD patterns, using untargeted microarray approaches 25, 68. Here, we performed a more conservative approach based on the analysis of fibrosis‐related genes using a low density microarray system in micro‐dissected tissue samples 40. We identified a subset of 24 genes that is sufficient to discriminate between AFE, NSIP, OP and UIP on a molecular level with 95% confidence. Generating the sample sizes needed to build robust and accurate models can be challenging depending on the required materials and diseases. Nonetheless, we employed a rather sophisticated modelling approach with only about one third of the analysed mRNA samples being used for training purposes and about two thirds for model validation, in order to avoid over fitting. The large size of the validation set ensures that the accuracy values of our models are of high confidence. Employing the models created in this work in a stepwise decision tree allowed for identifying and discarding ambiguous predictions, thus reducing false positives and yielding a high overall accuracy of 95% for a common test set of 55 samples. Considering that smaller training sets are generally accompanied by a decrease in model accuracy, our ‘divide‐and‐conquer’‐like approach therefore seems to be well suited for working with small sample sizes. However, ambiguous predictions (i.e. unclassifiable samples) could be considered as false‐negatives. In this case our approach would possibly also reduce class‐wise sensitivity.
As shown here, the molecular discrimination of ILD entities in diseased tissue is feasible and very accurate. However, in order to be relevant for diagnostic approaches, non‐invasive methods for material acquisition, such as bronchoalveolar lavage (BAL), would be favourable. Notably, all of the 24 significantly regulated genes we used for the classification of ILD entities are detectable in BAL at the transcript level 69. Also, the majority of these genes (15/24) have also been shown to be detectable at the protein level in BAL 70, 71. Therefore, the future transfer of this work's findings to BAL appears promising. In addition, our analysis suggests that drugs such as pirfenidone and nintedanib, which act on collagens 1 and 3, are candidates for treatment of not only UIP but also OP and even AFE, since collagens 1A2 and 3A1 are dominantly overexpressed in these entities. However, their application might be of limited use in NSIP, since here collagen 1A2 is not significantly overexpressed.
In conclusion, we have demonstrated that (1) molecular profiling in different forms of ILD is feasible, particularly in regard to (2) comprehensive discrimination of pulmonary injury patterns, and that (3) disease entities appear to be defined by characteristic functional profiles:
OP: prominent inflammation and minute changes to the original pulmonary architecture with mainly granulation‐like tissue in the alveoli.
NSIP: discrete fibrotic remodelling and mild disturbance of the pulmonary architecture due to aberrant expression of basal membrane components.
AFE: severe fibrotic remodelling in the former alveoli and disturbance of the pulmonary architecture, likely due to fibrocyte invasion.
UIP: severe destruction of the original lung architecture, predominantly in the interstitium, with increase in sprouting angiogenesis and fibroblast proliferation.
Future studies should aim to translate our compartment‐specific findings to the analysis of BAL samples, which are more easily obtainable, and assess the potential effects of novel anti‐fibrotic drugs on the molecular microenvironment of different ILD entities.
Author contributions statement
DJ, FL, MK and HS designed the study. HS, TW, NI, SJ, GW, AH collected data. PB MK, HS, DJ, TW and FL performed data analysis and interpretation. All authors provided literature research. HS, DJ, MK and LN generated the figures. DJ, FL, HS, SJ, GW and MK wrote the manuscript. FL and MK contributed equally and share last authorship. All authors had final approval of the submitted and published versions.
Abbreviations
See supplementary material, Table S4.
Supporting information
Figure S1. Statistical analysis of the immunohistochemical quantification of inflammatory cells
Figure S2. Schema of designed modelling strategy involving a decision tree of binary classifiers
Figure S3. Performance of designed modelling strategy
Figure S4. Possible prediction outcomes of designed modelling strategy
Table S1. Low density array – target and reference genes
Table S2. Gene expression data
Table S3. Classification results
Table S4. Abbreviations
Appendix S1. 7z‐Archive (7‐Zip) containing the best‐performing binary classifiers trained in this work as serialised objects for use with the R programming language
Acknowledgements
The authors would like to thank Johanna Rische, Regina Engelhardt, Anette Müller‐Brechlin, Christina Petzold, Enrico Calvino Iglesias, Tim Klein, Svea Schwarze and Allison Seidel for their excellent technical assistance and Michael Morgan and Gareth Griffiths for editing the manuscript. This project received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (consolidator grant XHaLe no. 19281137 to Danny Jonigk); Mark Kuehnel and Lavinia Neubert were financed via a research grant from the German Research Foundation (Deutsche Forschungsgemeinschaft [DFG], SFB 738/3, project B09 to Danny Jonigk and Florian Laenger); additional funding was received from the German Federal Ministry of Economics and Technology (Bundesministerium für Wirtschaft und Technologie [BMWI]) via the Central Innovation Programme for small and medium‐sized enterprises (SMEs) to Danny Jonigk and Mark Kuehnel (no. ZF4549001CR8).
No conflicts of interest were declared.
References
- 1. Travis WD, Costabel U, Hansell DM, et al An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188: 733–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee EY. Interstitial lung disease in infants: new classification system, imaging technique, clinical presentation and imaging findings. Pediatr Radiol 2013; 43: 3–13. [DOI] [PubMed] [Google Scholar]
- 3. Homolka J. Idiopathic pulmonary fibrosis: a historical review. CMAJ 1987; 137: 1003–1005. [PMC free article] [PubMed] [Google Scholar]
- 4. Liebow AA, Steer A, Billingsley JG. Desquamative interstitial pneumonia. Am J Med 1965; 39: 369–404. [DOI] [PubMed] [Google Scholar]
- 5. Carrington CB, Liebow AA. Limited forms of angiitis and granulomatosis of Wegener's type. Am J Med 1966; 41: 497–527. [DOI] [PubMed] [Google Scholar]
- 6. Liebow AA, Carrington CB. Hypersensitivity reactions involving the lung. Trans Stud Coll Physicians Phila 1966; 34: 47–70. [PubMed] [Google Scholar]
- 7. Carrington CB, Addington WW, Goff AM, et al Chronic eosinophilic pneumonia. N Engl J Med 1969; 280: 787–798. [DOI] [PubMed] [Google Scholar]
- 8. Spagnolo P, Lee JC, Sverzellati N, et al The lung in rheumatoid arthritis – focus on interstitial lung disease. Arthritis Rheumatol 2018; 70: 1544–1554. [DOI] [PubMed] [Google Scholar]
- 9. Rossi G, Cavazza A, Colby TV. Pathology of sarcoidosis. Clin Rev Allergy Immunol 2015; 49: 36–44. [DOI] [PubMed] [Google Scholar]
- 10. Cottin V. Idiopathic interstitial pneumonias with connective tissue diseases features: a review. Respirology 2016; 21: 245–258. [DOI] [PubMed] [Google Scholar]
- 11. Ferri C, Manfredi A, Sebastiani M, et al Interstitial pneumonia with autoimmune features and undifferentiated connective tissue disease. Autoimmun Rev 2016; 15: 61–70. [DOI] [PubMed] [Google Scholar]
- 12. Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018; 378: 1811–1823. [DOI] [PubMed] [Google Scholar]
- 13. Kaur A, Mathai SK, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med 2017; 4: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Larsen BT, Smith ML, Elicker BM, et al Diagnostic approach to advanced fibrotic interstitial lung disease: bringing together clinical, radiologic, and histologic clues. Arch Pathol Lab Med 2017; 141: 901–915. [DOI] [PubMed] [Google Scholar]
- 15. Raghu G, Collard HR, Egan JJ, et al An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011; 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lettieri CJ, Veerappan GR, Helman DL, et al Outcomes and safety of surgical lung biopsy for interstitial lung disease. Chest 2005; 127: 1600–1605. [DOI] [PubMed] [Google Scholar]
- 17. Kreider ME, Hansen‐Flaschen J, Ahmad NN, et al Complications of video‐assisted thoracoscopic lung biopsy in patients with interstitial lung disease. Ann Thorac Surg 2007; 83: 1140–1144. [DOI] [PubMed] [Google Scholar]
- 18. Blackhall V, Asif M, Renieri A, et al The role of surgical lung biopsy in the management of interstitial lung disease: experience from a single institution in the UK. Interact Cardiovasc Thorac Surg 2013; 17: 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutchinson JP, McKeever TM, Fogarty AW, et al Surgical lung biopsy for the diagnosis of interstitial lung disease in England: 1997–2008. Eur Respir J 2016; 48: 1453–1461. [DOI] [PubMed] [Google Scholar]
- 20. Richeldi L, du Bois RM, Raghu G, et al Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- 21. Hutchinson J, Fogarty A, Hubbard R, et al Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J 2015; 46: 795–806. [DOI] [PubMed] [Google Scholar]
- 22. Jonigk D, Rath B, Borchert P, et al Comparative analysis of morphological and molecular motifs in bronchiolitis obliterans and alveolar fibroelastosis after lung and stem cell transplantation. J Pathol Clin Res 2016; 3: 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hutchinson JP, McKeever TM, Fogarty AW, et al Increasing global mortality from idiopathic pulmonary fibrosis in the twenty‐first century. Ann Am Thorac Soc 2014; 11: 1176–1185. [DOI] [PubMed] [Google Scholar]
- 24. Raghu G, Remy‐Jardin M, Myers JL, et al Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018; 198: e44–e68. [DOI] [PubMed] [Google Scholar]
- 25. Kim SY, Diggans J, Pankratz D, et al Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high‐dimensional transcriptional data. Lancet Respir Med 2015; 3: 473–482. [DOI] [PubMed] [Google Scholar]
- 26. Xu W, Xiao Y, Liu H, et al Nonspecific interstitial pneumonia: clinical associations and outcomes. BMC Pulm Med 2014; 14: 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Belloli EA, Beckford R, Hadley R, et al Idiopathic non‐specific interstitial pneumonia. Respirology 2016; 21: 259–268. [DOI] [PubMed] [Google Scholar]
- 28. Kono M, Nakamura Y, Yoshimura K, et al Nonspecific interstitial pneumonia preceding diagnosis of collagen vascular disease. Respir Med 2016; 117: 40–47. [DOI] [PubMed] [Google Scholar]
- 29. Nunes H, Schubel K, Piver D, et al Nonspecific interstitial pneumonia: survival is influenced by the underlying cause. Eur Respir J 2015; 45: 746–755. [DOI] [PubMed] [Google Scholar]
- 30. Feinstein MB, DeSouza SA, Moreira AL, et al A comparison of the pathological, clinical and radiographical, features of cryptogenic organising pneumonia, acute fibrinous and organising pneumonia and granulomatous organising pneumonia. J Clin Pathol 2015; 68: 441–447. [DOI] [PubMed] [Google Scholar]
- 31. Baque‐Juston M, Pellegrin A, Leroy S, et al Organizing pneumonia: what is it? A conceptual approach and pictorial review. Diagn Interv Imaging 2014; 95: 771–777. [DOI] [PubMed] [Google Scholar]
- 32. Izykowski N, Kuehnel M, Hussein K, et al Organizing pneumonia in mice and men. J Transl Med 2016; 14: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khiroya R, Macaluso C, Montero MA, et al Pleuroparenchymal fibroelastosis: A review of histopathologic features and the relationship between histologic parameters and survival. Am J Surg Pathol 2017; 41: 1683–1689. [DOI] [PubMed] [Google Scholar]
- 34. Rosenbaum JN, Butt YM, Johnson KA, et al Pleuroparenchymal fibroelastosis: a pattern of chronic lung injury. Hum Pathol 2015; 46: 137–146. [DOI] [PubMed] [Google Scholar]
- 35. Selman M, Pardo A. Revealing the pathogenic and aging‐related mechanisms of the enigmatic idiopathic pulmonary fibrosis. An integral model. Am J Respir Crit Care Med 2014; 189: 1161–1172. [DOI] [PubMed] [Google Scholar]
- 36. von der Thüsen JH, Hansell DM, Tominaga M, et al Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol 2011; 24: 1633–1639. [DOI] [PubMed] [Google Scholar]
- 37. Jonigk D, Merk M, Hussein K, et al Obliterative airway remodeling: molecular evidence for shared pathways in transplanted and native lungs. Am J Pathol 2011; 178: 599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jonigk D, Golpon H, Bockmeyer CL, et al Plexiform lesions in pulmonary arterial hypertension: composition, architecture, and microenvironment. Am J Pathol 2011; 179: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jonigk D, Lehmann U, Stuht S, et al Recipient‐derived neoangiogenesis of arterioles and lymphatics in quilty lesions of cardiac allografts. Transplantation 2007; 84: 1335–1342. [DOI] [PubMed] [Google Scholar]
- 40. Jonigk D, Izykowski N, Rische J, et al Molecular profiling in lung biopsies of human pulmonary allografts to predict chronic lung allograft dysfunction. Am J Pathol 2015; 185: 3178–3188. [DOI] [PubMed] [Google Scholar]
- 41. Bockmeyer CL, Jonigk D, Kreipe H, et al MicroRNA profiling using RNA from microdissected immunostained tissue. Methods Mol Biol 2011; 755: 85–94. [DOI] [PubMed] [Google Scholar]
- 42. R Core Team . R: A Language and environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- 43. Kuhn M, Wing J, Weston S, et al. caret: Classification and Regression Training. 2017
- 44. Cerdeira JO, Silva PD, Cadimo J, et al subselect: Selecting Variable Subsets. R package version 0.13. 2017.
- 45. Krämer A, Green J, Pollard J, et al Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014; 30: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Raghu G, Selman M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am J Respir Crit Care Med 2015; 191: 252–254. [DOI] [PubMed] [Google Scholar]
- 47. Hayton C, Chaudhuri N. Current treatments in the management of idiopathic pulmonary fibrosis: pirfenidone and nintedanib. Clin Med Insights Ther 2017; 9: 1179559X1771912. [Google Scholar]
- 48. Margaritopoulos GA, Vasarmidi E, Antoniou KM. Pirfenidone in the treatment of idiopathic pulmonary fibrosis: an evidence‐based review of its place in therapy. Core Evid 2016; 11: 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wells AU, Costabel U, Poletti V, et al Challenges in IPF diagnosis, current management and future perspectives. Sarcoidosis Vasc Diffuse Lung Dis 2015; 32: 28–35. [PubMed] [Google Scholar]
- 50. Mäkelä K, Hodgson U, Piilonen A, et al Analysis of the histologic features associated with Interobserver variation in idiopathic pulmonary fibrosis. Am J Surg Pathol 2018; 42: 672–678. [DOI] [PubMed] [Google Scholar]
- 51. Hashisako M, Tanaka T, Terasaki Y, et al Interobserver agreement of usual interstitial pneumonia diagnosis correlated with patient outcome. Arch Pathol Lab Med 2016; 140: 1375–1382. [DOI] [PubMed] [Google Scholar]
- 52. Jonigk D, Theophile K, Hussein K, et al Obliterative airway remodelling in transplanted and non‐transplanted lungs. Virchows Arch 2010; 457: 369–380. [DOI] [PubMed] [Google Scholar]
- 53. Marjański T, Halman J, Taniewska S, et al Open lung biopsy performed in idiopathic pulmonary fibrosis is a safe procedure. Kardiochirurgia Torakochirurgia Pol 2017; 14: 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu J, Mora A, Shim H, et al Role of the SDF‐1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol 2007; 37: 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Molon B, Gri G, Bettella M, et al T cell costimulation by chemokine receptors. Nat Immunol 2005; 6: 465–471. [DOI] [PubMed] [Google Scholar]
- 56. Pardo A, Cabrera S, Maldonado M, et al Role of matrix metalloproteinases in the pathogenesis of idiopathic pulmonary fibrosis. Respir Res 2016; 17: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giannandrea M, Parks WC. Diverse functions of matrix metalloproteinases during fibrosis. Dis Model Mech 2014; 7: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Epler GR, Colby TV, McLoud TC, et al Bronchiolitis obliterans organizing pneumonia. N Engl J Med 1985; 312: 152–158. [DOI] [PubMed] [Google Scholar]
- 59. Nagasawa T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J Mol Med 2014; 92: 433–439. [DOI] [PubMed] [Google Scholar]
- 60. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015; 348: 74–80. [DOI] [PubMed] [Google Scholar]
- 61. Zheng H, Fu G, Dai T, et al Migration of endothelial progenitor cells mediated by stromal cell‐derived factor‐1α/cxcr4 via Pi3k/akt/enos signal transduction pathway. J Cardiovasc Pharmacol 2007; 50: 274–280. [DOI] [PubMed] [Google Scholar]
- 62. Smith M, Dalurzo M, Panse P, et al Usual interstitial pneumonia‐pattern fibrosis in surgical lung biopsies. Clinical, radiological and histopathological clues to aetiology. J Clin Pathol 2013; 66: 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Strieter RM, Gomperts BN, Keane MP. The role of CXC chemokines in pulmonary fibrosis. J Clin Invest 2007; 117: 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Barratt S, Millar A. Vascular remodelling in the pathogenesis of idiopathic pulmonary fibrosis. QJM 2014; 107: 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pardo A, Selman M. Matrix metalloproteases in aberrant fibrotic tissue remodeling. Proc Am Thorac Soc 2006; 3: 383–388. [DOI] [PubMed] [Google Scholar]
- 66. Craig VJ, Zhang L, Hagood JS, et al Matrix metalloproteinases as therapeutic targets for idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 2015; 53: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Selman M, Ruiz V, Cabrera S, et al TIMP‐1, ‐2, ‐3, and ‐4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am J Physiol Cell Mol Physiol 2000; 279: L562–L574. [DOI] [PubMed] [Google Scholar]
- 68. Raghu G, Flaherty KR, Lederer DJ, et al Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med 2019; 7: 487–496. [DOI] [PubMed] [Google Scholar]
- 69. Lee J, Arisi I, Puxeddu E, et al Bronchoalveolar lavage (BAL) cells in idiopathic pulmonary fibrosis express a complex pro‐inflammatory, pro‐repair, angiogenic activation pattern, likely associated with macrophage iron accumulation. PLoS One 2018; 13: e0194803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Foster MW, Morrison LD, Todd JL, et al Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res 2015; 14: 1238–1249. [DOI] [PubMed] [Google Scholar]
- 71. DeBoer EM, Wagner BD, Popler J, et al Novel application of aptamer proteomic analysis in cystic fibrosis bronchoalveolar lavage fluid. Proteomics Clin Appl 2019; 13: e1800085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Statistical analysis of the immunohistochemical quantification of inflammatory cells
Figure S2. Schema of designed modelling strategy involving a decision tree of binary classifiers
Figure S3. Performance of designed modelling strategy
Figure S4. Possible prediction outcomes of designed modelling strategy
Table S1. Low density array – target and reference genes
Table S2. Gene expression data
Table S3. Classification results
Table S4. Abbreviations
Appendix S1. 7z‐Archive (7‐Zip) containing the best‐performing binary classifiers trained in this work as serialised objects for use with the R programming language