

Abstract
Objective
To evaluate the efficacy and safety of oral levosimendan in patients with amyotrophic lateral sclerosis (ALS). This phase II, randomised, double-blind, placebo-controlled, crossover, three-period study with 6 months open-label follow-up enrolled adults with ALS and sitting slow vital capacity (SVC) 60%–90 % of predicted from 11 sites in four countries.
Methods
Patients received levosimendan 1 mg daily, 1 mg two times a day or placebo during three 14-day crossover periods and levosimendan 1–2 mg daily during open-label follow-up. Primary endpoint was sitting SVC; secondary endpoints included supine SVC, ALS Functional Rating Scale-Revised (ALSFRS-R), tolerability and safety.
Results
Of 66 patients randomised, 59 contributed to the double-blind results and 50 entered open-label follow-up. Sitting SVC was not significantly different between the treatments. In post hoc analysis using period-wise baselines, supine SVC favoured levosimendan over placebo, estimated mean differences from baseline being −3.62% on placebo, +0.77% on levosimendan 1 mg daily (p=0.018) and +2.38% on 1 mg two times a day (p=0.001). Headache occurred in 16.7% of patients during levosimendan 1 mg daily (p=0.030), 28.6% during 1 mg two times a day (p=0.002) and 3.3% during placebo. The respective frequencies for increased heart rate were 5.1% (p=0.337), 18.5% (p=0.018) and 1.7%. No significant differences between the treatments were seen for other adverse events.
Conclusions
Levosimendan did not achieve the primary endpoint of improving sitting SVC in ALS. Headache and increased heart rate were increased on levosimendan, although it was otherwise well tolerated. A phase III study to evaluate the longer term effects of oral levosimendan in ALS is ongoing.
Keywords: levosimendan, SVC, respiratory function, amyotrophic lateral sclerosis
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease of upper and lower motor neurons resulting in progressive weakness, with death occurring from respiratory failure commonly as a result of diaphragmatic weakness typically within 3–4 years. None of the current therapies offer substantial clinical benefit for patients with ALS, riluzole having a modest effect on survival and edaravone on the rate of functional decline.1 Despite the beneficial effects of non-invasive ventilation,2 3 poor respiratory function is a major source of disability, fatigue, morbidity and mortality in ALS, and there is an urgent need for an effective therapy to improve symptoms associated with the respiratory decline.
Levosimendan binds selectively to troponin C sensitising cardiac and skeletal muscles to calcium,4 and currently, an intravenous formulation of levosimendan is indicated for the treatment of acute worsening of severe heart failure. The mean elimination half-lives of levosimendan and its two active metabolites, OR-1855 and OR-1896, are about 1 and 60 hours, respectively. Due to its having similar free plasma concentrations to levosimendan, OR-1896 is believed to contribute to therapeutic efficacy during prolonged treatment (Orion Pharma, data on file). Levosimendan does not increase consumption of ATP or oxygen5 and does not cross the blood–brain barrier.6
There is a compelling scientific rationale for the development of an oral formulation of levosimendan for symptomatic treatment of ALS. Two experimental studies have shown that in diaphragm muscle fibres obtained from rats and humans, levosimendan improves submaximal force generation of diaphragm (both slow and fast muscle fibres) by about 15%–25%.7 8 In addition, levosimendan has been reported to improve neuromechanical efficiency of human diaphragm function by 21% in healthy people.9 Further support is provided by the development programmes of other calcium sensitisers, tirasemtiv and reldesemtiv. In a phase IIb study, tirasemtiv showed a positive effect on sitting slow vital capacity (SVC), but not on the primary efficacy endpoint ALS Functional Rating Scale-Revised (ALSFRS-R).10
We therefore carried out a phase two multicentre, randomised, double-blind, placebo-controlled trial of oral levosimendan in ALS.
Methods
Study design and participants
This was a randomised, double-blind, placebo-controlled, crossover, three-period study with 6 months open-label follow-up. Each treatment period lasted for 2 weeks separated by 19–23 day washout periods (online supplementary figure 1). There were 11 sites from the UK, Germany, Ireland and the Netherlands. The main inclusion criteria were diagnosis of definite, probable or laboratory-supported probable ALS according to El Escorial revised criteria11; sitting SVC 60%–90% of predicted for age, height and gender and disease duration from symptom onset of 12–48 months. The main exclusion criteria were other causes of neuromuscular weakness, diagnosis of another neurodegenerative disease, assisted ventilation or gastrostomy within 3 months and history of significant cardiac disease or cardiac events. All entry criteria are listed in the online supplementary table 1.
jnnp-2018-320288supp002.pdf (209.2KB, pdf)
jnnp-2018-320288supp004.pdf (59.4KB, pdf)
All participants provided written informed consent for the study. The study was conducted according to the Declaration of Helsinki and in compliance with Good Clinical Practice. An independent board monitored safety throughout the study. EudraCT number: 2014-004567-21; ClinicalTrials.gov identifier: NCT02487407.
Study medications, randomisation and dosing
Levosimendan 1 mg capsules and identical placebo capsules were used. Patients were randomised to three crossover periods using six possible treatment sequences according to William’s design,12 equal allocation ratio and central randomisation.
Dosing was twice daily. During the 1 mg daily period, levosimendan 1 mg was taken in the morning and placebo in the evening. During the 2 mg daily period, dosing was 1 mg two times a day and during the placebo period, placebo capsules were taken two times a day.
During open-label follow-up, all patients were started on 1 mg levosimendan taken in the morning for 2 weeks, after which the dose was increased to 1 mg two times a day if tolerated. The dose could be decreased back to 1 mg daily or discontinued if required for any reason.
Efficacy assessments
The primary endpoint was SVC (% of predicted normal) measured in the sitting position. Secondary endpoints included SVC measured supine, sniff nasal pressure (SNP), ALSFRS-R, overnight oxygen saturation (SpO2), hand grip strength, submaximal hand grip strength endurance, Clinical Global Impression of Change (CGI-C) assessed by patients and investigators, visual analogue scale (VAS) assessing fatigue and quality of life (QoL) scales (five-level EuroQoL five dimension (EQ-5D-5L) and Short Form-36 (SF-36)). Non-invasive ventilation support, permanent continuous ventilator dependence, tracheostomy and survival were recorded. During the 6-months open-label follow-up period, sitting and supine SVC, SNP, ALSFRS-R, EQ-5D-5L and SF-36 were assessed (see online supplementary file and online supplementary table 2).
jnnp-2018-320288supp007.pdf (36.2KB, pdf)
jnnp-2018-320288supp005.pdf (49.3KB, pdf)
Safety assessments
Safety was assessed by physical examination, vital signs, laboratory tests, 12-lead ECG, 24 hours Holter-ECG and recording of adverse events (AEs).
Other assessments
Plasma samples for levosimendan, OR-1855, OR-1896 and riluzole concentrations were collected in the morning before study treatment intake on days 1 and 14 of each treatment period. DNA samples were collected for pharmacogenomic assessments and acetylation status, based on polymorphism in the N-acetyltransferase enzyme affecting the metabolism of OR-1855 to OR-1896. All study assessments are presented in the online supplementary table 2.
Statistical analyses
Sample size estimation was based on additional results from the BENEFIT-ALS trial.10 SVC was assumed to decline during each double-blind crossover period by 4% for placebo and 1% for levosimendan, with a common SD of 9% and within subject correlation of 0.70. A sample size of 54 provides over 80% power at 5% significance.
Intention-to-treat analysis without imputation for missing data was used throughout reporting. Appropriate descriptive statistics, frequency tables and plots were used to summarise all data.
The primary efficacy endpoint was change in sitting SVC, comparing baseline (period 1 day 1) and day 14 predose assessments. Due to a significant period effect, changes from period-wise baselines (period 1 day 1, period 2 day 1 and period 3 day 1, respectively) were analysed as primary comparisons post hoc. Analysis of co-variance, appropriate to crossover design, was used for primary analysis. The statistical model included treatment dose, baseline SVC, treatment sequence and period as fixed effects and subject and site as random effects. All pairwise comparisons were performed using Tukey’s adjustment. Slope of decline in SVC was evaluated including all data from both double-blind crossover and open-label follow-up using a random slope and intercept model.
Secondary efficacy endpoints were primarily evaluated at the end of the double-blind crossover. A two-sided 0.05 significance level was allocated for the analyses of secondary variables. The secondary efficacy variables were analysed using the same statistical principles as the primary variable. Respiratory function and quality of life endpoints were evaluated during the open-label follow-up using descriptive statistics only.
Levosimendan, OR-1855, OR-1896 and riluzole concentrations were summarised and plotted using descriptive statistics. Acetylation status was determined from all subjects participating in the study. Acetylation status was summarised by treatments and the effects of acetylation status on levosimendan, metabolites, SVC, SNP and ambulatory heart rate (HR) were evaluated using analysis of variance (ANOVA) appropriate for the crossover design. Potential effects of levosimendan treatment on riluzole concentration were evaluated using ANOVA.
Safety data including AEs, vital signs, laboratory results, 12-lead ECG and 24 hours Holter-ECG were displayed by treatment and study part (crossover and open label).
Results
Study population
Of the 66 patients randomised, 71.2% were male, 92.4% white European and 83.3% had spinal-onset disease. Median disease duration from symptom onset was 21.2 months (table 1).
Table 1.
Baseline demographics and characteristics
| Variable | Total N=66 |
| Age, years | |
| Median | 56.5 |
| Range | 36–75 |
| Sex, n (%) | |
| Male | 47 (71.2) |
| Female | 19 (28.8) |
| Weight, kg | |
| Mean (SD) | 76.7 (15.9) |
| BMI, kg/m2 | |
| Mean (SD) | 25.6 (4.0) |
| Race, n (%) | |
| Caucasian | 61 (92.4) |
| Asian | 3 (4.5) |
| Black | 1 (1.5) |
| Other | 1 (1.5) |
| Disease duration from symptom onset, months | |
| Median | 21.2 |
| Range | 12–48 |
| Sitting SVC % of predicted | |
| Mean (SD) | 75.3 (9.1) |
| Supine SVC % of predicted | |
| Mean (SD) | 73.3 (14.0) |
| ALSFRS-R | |
| Mean (SD) | 36.7 (5.4) |
| Site of disease onset, n (%) | |
| Spinal | 55 (83.3) |
| Bulbar | 11 (16.7) |
| Treated with riluzole, n (%) | 66 (100) |
ALSFRS-R, Amyotrophic Lateral Sclerosis Functional Rating Scale-Revised; BMI, body mass index; SVC, slow vital capacity.
Study medication exposure
During the crossover, double-blind part of the study, 59 patients received levosimendan 1 mg daily, 59 levosimendan 1 mg two times a day and 58 placebo.
In the open-label follow-up part of the study, 50 people received levosimendan, 44 of them increasing the dose from 1 mg to 2 mg daily at 2 weeks (figure 1). The mean duration of the treatment for all study participants was 147.9 days (range 6–195 days) during the open-label follow-up.
Figure 1.
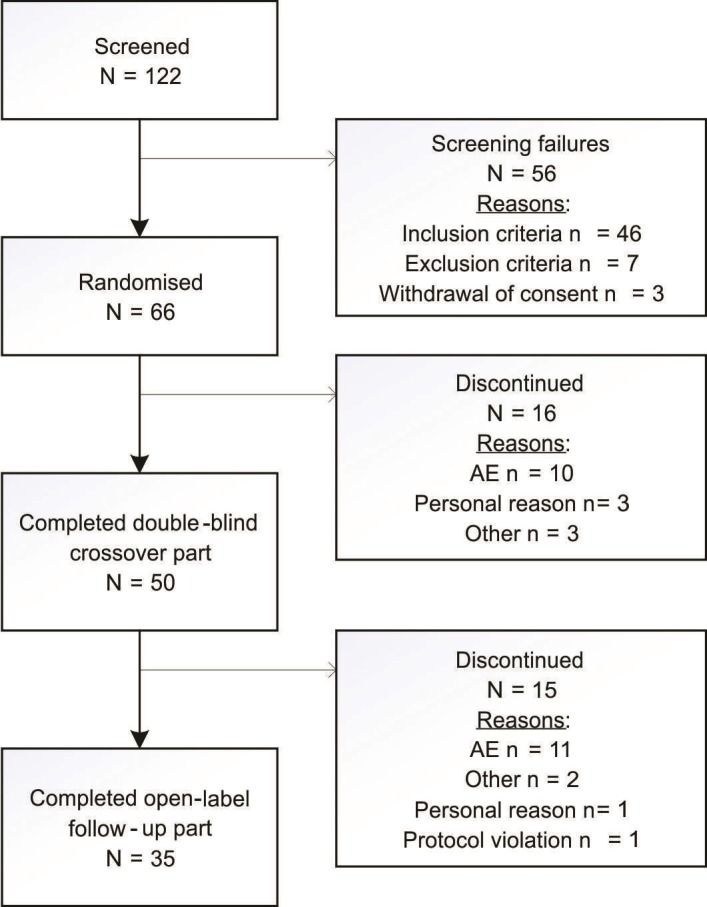
Disposition of subjects. AE, adverse event.
Efficacy in the crossover, double-blind part of the study
We initially designed the study under the assumption that patients would not deteriorate significantly during the first 3 months and therefore that the baseline of the first treatment period would be appropriate to use as baseline for all treatment periods. However, due to a significant period effect (p<0.0001) with period-wise baseline affected by previous treatment period, the data did not allow appropriate interpretation of efficacy using the period 1, day 1 baseline for all periods. We therefore performed a post hoc period-wise analysis with the period-specific baseline used for each period (eg, period 2, day 1 baseline for period 2).
Sitting SVC
Using the original baseline definition, estimated mean differences from baseline in sitting SVC were −2.01% for placebo, −2.99% for levosimendan 1 mg daily (p=0.67 vs placebo) and −2.25% for levosimendan 2 mg daily (p=0.98 vs placebo). Using period-wise baselines (post hoc), the differences between levosimendan and placebo remained similar (data not shown).
Supine SVC
There is no formula to predict the effects of age, height and gender on supine SVC, and expected values were therefore calculated using the formula for sitting SVC. Using the original baseline definition, mean differences from baseline were −4.51% for placebo, −2.57% for levosimendan 1 mg daily (p=0.35 vs placebo) and −1.97% points for levosimendan 2 mg daily (p=0.17 vs placebo). Using period-wise baselines (post hoc), mean differences from baseline were −3.62% for placebo, +0.77 for levosimendan 1 mg daily (p=0.018 vs placebo) and +2.38% points for levosimendan 2 mg daily (p<0.001 vs placebo). Supine SVC results using the period-wise baselines (post hoc) are presented for each period separately and all the periods combined in figure 2 and the results with the primary analyses are shown for each period separately in the online supplementary figure 2.
Figure 2.

Slow vital capacity (SVC) (per cent of predicted normal) measured in the supine position. Change from period-wise baselines (period 1 day 1, period 2 day 1 and period 3 day 1, respectively; post hoc analysis) in per cent predicted SVC is shown for all three 14-day crossover treatment periods combined (left) and each period separately (right).
jnnp-2018-320288supp003.pdf (66.3KB, pdf)
Patients with bulbar onset had lower baseline supine SVC than patients with spinal onset. In the subgroup analyses, changes from baseline were different depending on onset site (p=0.008 for interaction), the treatment effect being larger in patients with bulbar onset (−9.02 for placebo, +4.06 levosimendan 1 mg daily (p=0.003 vs placebo) and +5.06, levosimendan 2 mg daily (p=0.001 vs placebo). Numerically, greater treatment effects were seen in patients who had baseline supine SVC below the median value of 75.0% points (p=0.291 for interaction): −7.00 for placebo, −0.02 levosimendan 1 mg daily (p=0.049 vs placebo) and +1.14, levosimendan 2 mg daily (p=0.016 vs placebo).
ALSFRS-R
There was no significant difference in the ALSFRS-R total or respiratory scores. Estimated mean differences from baseline were −0.82 for placebo, −0.46 for levosimendan 1 mg daily (p=0.49 vs placebo) and −0.37 for levosimendan 2 mg daily (p=0.34 vs placebo). Estimated mean differences from baseline in respiratory domain scores were −0.22 for placebo, +0.04 for levosimendan 1 mg daily (p=0.13 vs placebo) and +0.05 for levosimendan 2 mg daily (p=0.12 vs placebo).
Other efficacy endpoints
There were no trends seen between placebo and levosimendan in SNP, VAS of fatigue, overnight SpO2, hand grip assessments, CGI-C or in QoL scales
Key efficacy parameters during the 6 months open-label part of the study are presented in the online supplementary file.
Tolerability and safety
In the double-blind crossover part of the study, AEs were reported by 71% during levosimendan 1 mg daily, 85% levosimendan 2 mg daily and 53% of patients on placebo. Most of the AEs were mild, with severe AEs reported in isolated cases only. The most commonly reported AEs during the crossover part were headache, fall and increased HR (terms ‘HR increased’, ‘tachycardia’ and ‘sinus tachycardia’ combined) (table 2). Of the 50 patients continuing to open-label follow-up, AEs were reported for 42 (84%), with the most commonly reported events being fall (28%), dysphagia (12%), respiratory failure (12%), headache (10%) and nasopharyngitis (8%).
Table 2.
Most common adverse events during the study
| Preferred term | Double-blind cross-over | Open-label follow-up | ||
| Levosimendan 1 mg (N=59) |
Levosimendan 1 mg two times a day (N=59) |
Placebo (N=58) |
Levosimendan 1–2 mg (N=50) |
|
| Participants (%) | ||||
| Headache | 10 (16.9)* | 17 (28.8)† | 2 (3.4) | 5 (10.0) |
| Fall | 9 (15.3) | 9 (15.3) | 5 (8.6) | 14 (28.0) |
| Heart rate increased‡ | 3 (5.1)§ | 11 (18.6)¶ | 1 (1.7) | 1 (2.0) |
| Nasopharyngitis | 4 (6.8) | 4 (6.8) | 3 (5.2) | 4 (8.0) |
| Cough | 6 (10.2) | – | 1 (1.7) | 3 (6.0) |
| Contusion | 2 (3.4) | 4 (5.1) | 1 (1.7) | – |
| Nausea | 4 (6.8) | 1 (1.7) | 1 (1.7) | 3 (6.0) |
| Diarrhoea | 1 (1.7) | 3 (5.1) | 1 (1.7) | 3 (6.0) |
| Constipation | 2 (3.4) | 2 (3.4) | 3 (5.2) | 1 (2.0) |
| Oxygen saturation decreased** | 3 (5.1) | 4 (6.8) | – | 1 (2.0) |
*P=0.030.
†p=0.002.
‡Preferred terms ‘heart rate increased’, ‘tachycardia’ and ‘sinus tachycardia’ combined.
§P=0.337.
¶P=0.018.
**Decreased oxygen saturation was reported in a total of two patients from two centres.
During the double-blind crossover part of the study, 13 of the 66 patients discontinued the study due to an AE, the most common reason being increase in HR fulfilling a predefined study treatment stopping rule (one during levosimendan 1 mg daily, eight during 2 mg daily and one during placebo). Based on the 24 hours Holter-ECG, mean changes from baseline in mean HR were 5.2 beats per minute (bpm) during levosimendan 1 mg daily, 10.7 bpm during 2 mg daily and −0.4 bpm during placebo. Other AEs leading to discontinuation of the study during the crossover periods were headache (n=1) and atrial fibrillation and ECG QT prolonged (n=1) during levosimendan 2 mg daily, and bradycardia and cardiac arrest (n=1) during placebo. During the open-label follow-up, six subjects discontinued the study due to AEs, five of them being serious adverse events (SAEs) (pulmonary embolism, aspiration pneumonia, respiratory failure, acute myocardial infarction and dysphagia) and one non-serious AE (HR increased).
During the crossover part of the study, SAEs were reported in four (7%) patients both during levosimendan 1 mg daily and placebo and in two (3%) patients during levosimendan 2 mg daily. Nineteen patients (38%) reported SAEs during open-label follow-up. The most commonly reported SAE terms were respiratory failure, dysphagia and ALS. Two of the SAEs were assessed as related to the study treatment by the investigator: bradycardia and cardiac arrest during placebo in the crossover part and acute myocardial infarction during the open-label follow-up. Five patients died during the study (one during the crossover and four during the open-label part of the study); in all cases, the death was assessed as not related to the study treatment by the investigator. Two patients died of ALS and one each of ‘pneumonia aspiration, ‘pneumonia bacterial, myocardial infarction and pneumothorax’ and ‘respiratory failure’.
Mean changes from baseline in supine systolic blood pressure were numerically greater after levosimendan (from −4.0 to −8.6 mm Hg) than after placebo (from 1.9 to −0.1 mm Hg). Changes in diastolic blood pressure were similar to those in systolic blood pressure. No differences were seen in mean orthostatic test results between treatments.
Levosimendan, OR-1855, OR-1896 and riluzole concentrations
Slow acetylators had higher OR-1855 concentrations, but lower OR-1896 concentrations than rapid acetylators (online supplementary table 3). Samples taken after the washout period did not indicate any carryover effects. Acetylation status had no effect on supine SVC effect observed (interaction p=0.667), the apparent treatment effect being similar in slow (2 mg daily 2.05%, 1 mg daily 1.79% and placebo −3.22%) and fast (2 mg daily 2.57%, 1 mg daily −0.55% and placebo −4.45%) acetylators.
jnnp-2018-320288supp006.pdf (37.8KB, pdf)
There were no differences in plasma concentrations of riluzole between day 1, day 14 or between the treatment periods (data not shown).
Discussion
This was the first study with oral levosimendan in patients with ALS. The study was designed to find treatment differences within a short period of time with three crossover periods during double-blind comparison. Fourteen-day treatment periods were selected based on earlier studies reporting that a single oral 1 mg dose of levosimendan was able to increase cardiac output in patients with severe heart failure by 22% (p<0.005; Orion Pharma data on file) and that a short levosimendan infusion improved diaphragm function in healthy people.9 It was expected, however, that most of the outcome measures such as ALSFRS-R13 and QoL scales would not show any differences within 14-day periods.
In contrast to our primary assumptions, there was a clear period effect (but no carryover effect) seen in the crossover part of the study, and therefore the efficacy results were analysed period-wise, using period-specific baselines (post hoc analyses). Although the primary endpoint of sitting SVC did not show significant differences between the treatments, supine SVC in the post hoc analyses indicated a dose-related treatment effect favouring levosimendan against placebo (4.39% points difference between placebo and levosimendan 1 mg and 6.00% points difference between placebo and 2 mg). Similar results were seen during all three periods, effectively replicating the findings two further times, thus strengthening our confidence in the results (figure 2). Diaphragmatic performance is reduced more dramatically by lying than by sitting, and the first clinical signs of respiratory insufficiency often appear during sleeping or when lying down.14 15 Seeing differences between the treatments in supine but not sitting SVC is supported by the finding that in patients with ALS, supine vital capacity is a more sensitive measure of diaphragmatic strength than that measured in the upright position, suggesting that upright vital capacity might not reveal abnormalities becoming noticeable in the supine position.16 There is also evidence that supine vital capacity is a better predictor of survival than upright vital capacity,17 18 and among several different respiratory measures, it has been reported to correlate most closely with diaphragmatic weakness, especially in patients with vital capacity <75% of predicted.16 Treatment effects seen in supine SVC between levosimendan and placebo treatments in patients with SVC <75% predicted at baseline are in line with this expectation. Although no significant changes were seen, both the ALSFRS-R total score and respiratory domain subscore numerically favoured levosimendan, supporting the supine SVC results. However, one needs to be cautious in interpreting ALSFRS-R results as the absolute differences were small and the treatment duration was short. Apart from supine SVC, other secondary efficacy endpoints did not show any difference between treatments.
All 50 patients completing the crossover part of the study continued to the open-label follow-up. Mean supine SVC at study baseline was 73.3% points and declined to 61.8% points at the end of the 6 months open-label follow-up. In turn, mean ALSFRS-R total score was 36.7 points at baseline and declined to 28.9 points at the end of open-label follow-up. During the entire 9-months study, the mean decline in supine SVC and ALSFRS-R total score were 1.93% points/month and 1.03 points/month, respectively. Mean declines were smaller over the last 6 months when all patients were on levosimendan. Based on a previous retrospective analysis, slowing the rate of decline of SVC by 1.5% point monthly (from −4.23 to −2.73 and to −1.23) corresponds to a reduction of about 20% in the risk for respiratory events or death.19 In light of this analysis, our results are encouraging, but no conclusions can be drawn since our uncontrolled data from the open-label follow-up are not directly comparable due to differences in study design and duration, and the position in which SVC was measured.
In patients with rapid acetylator status more active metabolite OR-1896 of levosimendan is formed20 and the active metabolite levels were higher in rapid than slow acetylators in this study. Despite this, there was no difference in the change of supine SVC. This is in line with the earlier findings in patients with heart failure, in whom haemodynamic effects were similar in rapid and slow acetylators.20
Levosimendan was generally well tolerated. Headache and increase in HR were more common during levosimendan treatment than placebo showing a dose-dependent increase in frequency. Headache, most probably due to vasodilatation by levosimendan, was usually short lasting and did not result in discontinuation of the study except for one patient with a medical history of migraine. In most cases, the increase in HR was reported as an AE based on a predefined study treatment stopping rule (increase in mean HR of over 15 bpm from baseline in the 24 hours Holter recording) and was not due to subjective symptoms of tachycardia. Most of the other frequently reported AEs, such as falls, nasopharyngitis, dysphagia and respiratory failure, were not considered to be related to the study treatment, reflecting the signs and symptoms of ALS. Supraventricular and ventricular tachyarrhythmias were reported in a low number of patients and were evenly distributed across the treatment arms. AEs such as dizziness or somnolence were reported as isolated events only and no AEs such as ataxia, agitation, confusion or delirium were reported in the study. All deaths and most other SAEs were assessed as not related to the study treatment by the investigator, indicating incidental events expected to be related to ALS.
In summary, we found no evidence that levosimendan improves SVC in the sitting position. Levosimendan treatment was well tolerated but with a dose-dependent increase in headache and tachycardia compared with placebo. In the light of the post hoc analysis indicating a possible dose-dependent treatment effect of levosimendan on the supine SVC compared with placebo, a phase III study is ongoing to evaluate longer term effects of oral levosimendan in ALS (NCT03505021) (online supplementary video).
jnnp-2018-320288supp001.mp4 (37.8MB, mp4)
Acknowledgments
We thank patients and their caregivers for participation in the study and Sanna Valkonen (Orion Corporation) for editorial support. Pamela Shaw is supported as an NIHR Senior Investigator. This trial was supported by the NIHR Clinical Research Facility and the NIHR Biomedical Research Centre at the University of Sheffield and Sheffield Teaching Hospitals NHS Foundation Trust, and the NIHR Maudsley Biomedical Research Centre.
Footnotes
Contributors: AAC contributed to acquisition of data. AAC, VA and MK contributed to the study concept and design, analysis and interpretation of the data. AAC, VA, TS and MK contributed to manuscript preparation. PJS, PNL and TS contributed to study planning. PJS, PNL, LHvdB, OH and ACL contributed to study execution and manuscript critique. TS contributed to statistical analysis. AA and VA are responsible for the overall content.
Funding: The study was funded by Orion Corporation Orion Pharma.
Competing interests: VVA and TS are employees of Orion Corporation. Other authors have no conflicts to disclose.
Patient consent for publication: Not required.
Ethics approval: The study was approved by local Ethical committees: The Medical Ethical Review Committee of the Netherlands (15/425); Central Ethics Committee/Ethics Committee of the University of ULM, Germany (170/15); Ethics (Medical Research) Committee, Beaumont Hospital, Ireland (15/46); London Bridge Ethics Committee, UK (15/LO/0684). The study was conducted according to principles of the Declaration of Helsinki and in compliance with Good Clinical Practice. All participants provided written informed consent for the study. An independent board monitored safety throughout the study. The study was registered in EudraCT number: 2014-004567-21; ClinicalTrials.gov identifier: NCT02487407.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: The data sets of this study are available from the corresponding author on reasonable request.
References
- 1. van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet 2017;390:2084–98. 10.1016/S0140-6736(17)31287-4 [DOI] [PubMed] [Google Scholar]
- 2. Berlowitz DJ, Howard ME, Fiore JF, et al. Identifying who will benefit from non-invasive ventilation in amyotrophic lateral sclerosis/motor neurone disease in a clinical cohort. J Neurol Neurosurg Psychiatry 2016;87:280–6. 10.1136/jnnp-2014-310055 [DOI] [PubMed] [Google Scholar]
- 3. Chiò A, Calvo A, Moglia C, et al. Non-invasive ventilation in amyotrophic lateral sclerosis: a 10 year population based study. J Neurol Neurosurg Psychiatry 2012;83:377–81. 10.1136/jnnp-2011-300472 [DOI] [PubMed] [Google Scholar]
- 4. Haikala H, Levijoki J, Lindén IB. Troponin C-mediated calcium sensitization by levosimendan accelerates the proportional development of isometric tension. J Mol Cell Cardiol 1995;27:2155–65. 10.1016/S0022-2828(95)91371-8 [DOI] [PubMed] [Google Scholar]
- 5. Deschodt-Arsac V, Calmettes G, Raffard G, et al. Absence of mitochondrial activation during levosimendan inotropic action in perfused paced guinea pig hearts as demonstrated by modular control analysis. Am J Physiol Regul Integr Comp Physiol 2010;299:R786–R792. 10.1152/ajpregu.00184.2010 [DOI] [PubMed] [Google Scholar]
- 6. Antila S, Huuskonen H, Nevalainen T, et al. Site dependent bioavailability and metabolism of levosimendan in dogs. Eur J Pharm Sci 1999;9:85–91. 10.1016/S0928-0987(99)00048-2 [DOI] [PubMed] [Google Scholar]
- 7. van Hees HWH, Andrade Acuña G, Linkels M, et al. Levosimendan improves calcium sensitivity of diaphragm muscle fibres from a rat model of heart failure. Br J Pharmacol 2011;162:566–73. 10.1111/j.1476-5381.2010.01048.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Hees HWH, Dekhuijzen PNR, Heunks LMA. Levosimendan enhances force generation of diaphragm muscle from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2009;179:41–7. 10.1164/rccm.200805-732OC [DOI] [PubMed] [Google Scholar]
- 9. Doorduin J, Sinderby CA, Beck J, et al. The calcium sensitizer levosimendan improves human diaphragm function. Am J Respir Crit Care Med 2012;185:90–5. 10.1164/rccm.201107-1268OC [DOI] [PubMed] [Google Scholar]
- 10. Shefner JM, Wolff AA, Meng L, et al. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:426–35. 10.3109/21678421.2016.1148169 [DOI] [PubMed] [Google Scholar]
- 11. Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–9. 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 12. Williams EJ. Experimental designs balanced for the estimation of residual effects of treatments. Aust J Chem 1949;2:149–68. 10.1071/CH9490149 [DOI] [Google Scholar]
- 13. Rooney J, Burke T, Vajda A, et al. What does the ALSFRS-R really measure? A longitudinal and survival analysis of functional dimension subscores in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2017;88:381–5. 10.1136/jnnp-2016-314661 [DOI] [PubMed] [Google Scholar]
- 14. Wade OL, Gilson JC. The effect of posture on diaphragmatic movement and vital capacity in normal subjects with a note on spirometry as an aid in determining radiological chest volumes. Thorax 1951;6:103–26. 10.1136/thx.6.2.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Loh L, Goldman M, Davis JN. The assessment of diaphragm function. Medicine 1977;56:165–9. 10.1097/00005792-197703000-00006 [DOI] [PubMed] [Google Scholar]
- 16. Lechtzin N, Wiener CM, Shade DM, et al. Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest 2002;121:436–42. 10.1378/chest.121.2.436 [DOI] [PubMed] [Google Scholar]
- 17. Baumann F, Henderson RD, Morrison SC, et al. Use of respiratory function tests to predict survival in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2010;11:194–202. 10.3109/17482960902991773 [DOI] [PubMed] [Google Scholar]
- 18. Schmidt EP, Drachman DB, Wiener CM, et al. Pulmonary predictors of survival in amyotrophic lateral sclerosis: use in clinical trial design. Muscle Nerve 2006;33:127–32. 10.1002/mus.20450 [DOI] [PubMed] [Google Scholar]
- 19. Andrews JA, Meng L, Kulke SF, et al. Association between decline in slow vital capacity and respiratory insufficiency, use of assisted ventilation, tracheostomy, or death in patients with amyotrophic lateral sclerosis. JAMA Neurol 2018;75:58–64. 10.1001/jamaneurol.2017.3339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kivikko M, Sundberg S, Karlsson MO, et al. Acetylation status does not affect levosimendan's hemodynamic effects in heart failure patients. Scand Cardiovasc J 2011;45:86–90. 10.3109/14017431.2010.540762 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jnnp-2018-320288supp002.pdf (209.2KB, pdf)
jnnp-2018-320288supp004.pdf (59.4KB, pdf)
jnnp-2018-320288supp007.pdf (36.2KB, pdf)
jnnp-2018-320288supp005.pdf (49.3KB, pdf)
jnnp-2018-320288supp003.pdf (66.3KB, pdf)
jnnp-2018-320288supp006.pdf (37.8KB, pdf)
jnnp-2018-320288supp001.mp4 (37.8MB, mp4)