

Abstract
Background
Mutations in the POT1 gene explain abnormally long telomeres and multiple tumors including cardiac angiosarcomas (CAS). However, the link between long telomeres and tumorigenesis is poorly understood.
Methods and Results
Here, we have studied the somatic landscape of 3 different angiosarcoma patients with mutations in the POT1 gene to further investigate this tumorigenesis process. In addition, the genetic landscape of 7 CAS patients without mutations in the POT1 gene has been studied. Patients with CAS and nonfunctional POT1 did not repress ATR (ataxia telangiectasia RAD3‐related)–dependent DNA damage signaling and showed a constitutive increase of cell cycle arrest and somatic activating mutations in the VEGF (vascular endothelial growth factor)/angiogenesis pathway (KDR gene). The same observation was made in POT1 mutation carriers with tumors different from CAS and also in CAS patients without mutations in the POT1 gene but with mutations in other genes involved in DNA damage signaling.
Conclusions
Inhibition of POT1 function and damage‐response malfunction activated DNA damage signaling and increased cell cycle arrest as well as interfered with apoptosis, which would permit acquisition of somatic mutations in the VEGF/angiogenesis pathway that drives tumor formation. Therapies based on the inhibition of damage signaling in asymptomatic carriers may diminish defects on cell cycle arrest and thus prevent the apoptosis deregulation that leads to the acquisition of driver mutations.
Keywords: cardiac angiosarcoma, cell cycle arrest, damage response, POT1, VEGF/angiogenesis pathway
Subject Categories: Angiogenesis, Basic Science Research, Mechanisms, Pathophysiology, Genetics
Clinical Perspective
What Is New?
In this study we describe how mutations in the POT1 gene, which explain long telomeres, correlate with cell cycle arrest increase in angiosarcoma patients.
The same increase was observed in other cardiac angiosarcoma patients even without mutations in the POT1 gene but in the damage response signaling.
This malfunction would bypass the apoptosis mechanism and would trigger the acquisition of somatic activating mutations in the angiogenesis pathway.
What Are the Clinical Implications?
Our results suggest that the use of angiogenesis inhibitors might regulate the tumor progression; however, targeting ATM/ATR (ataxia telangiectasia mutated/RAD3‐related) activity would rescue the cell cycle control and would prevent the acquisition of somatic driver mutations in patients affected with angiosarcoma tumors and asymptomatic patients carrying POT1 mutations.
Introduction
The Li‐Fraumeni syndrome is an autosomal dominant syndrome representing a genetic predisposition to a wide spectrum of tumors and is typically linked to mutations of the TP53 tumor suppressor gene.1 Li‐Fraumeni‐like families have a similar clinical presentation, but Li‐Fraumeni‐like syndrome is less frequently associated with mutations in the TP53 gene. Recently, we studied different Li‐Fraumeni‐like families with multiple tumors including various cases of cardiac angiosarcoma (CAS), which is the most common and most aggressive type of primary malignant neoplasm of the heart in adults.2 Patients affected with CAS are generally diagnosed at advanced stages with very poor prognosis and short survival rates (5‐year survival rate of 14%).3 The genetic landscape that determines the tumorigenic process of angiosarcomas (AS) is poorly understood and not well established.4, 5 Previous studies by our group uncovered a deleterious missense mutation in the POT1 gene (c.349C>T [p.Arg117Cys], pathogenic, Li‐Fraumeni‐like syndrome/CAS, autosomal dominant)6 causing AS in 4 families (3 in cardiac tissue and 1 in breast).6 Germline mutations in the POT1 gene have also been related with the development of other familial cancer types.7, 8, 9, 10, 11, 12 POT1 is a component of the so‐called shelterin complex, which is involved in telomere elongation in germline and stem cells (Figure 1A).13 In normal conditions the shelterin complex protects telomere cap ends in somatic cells by preventing access of the telomerase to chromosome ends.14 The shelterin complex also masks single‐stranded telomeres from the DNA damage response, thereby preventing the activation of ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia RAD3‐related) to avoid cell cycle arrest through POT1 and TPP1 (Figure 1B).15 TPP1 (which is also called ACD gene) anchors the telomere by POT1 and TRF1 (telomeric repeat binding factor 1) proteins. When telomeres are critically short, the shelterin complex does not prevent activation of the ATM and ATR response, which can drive the cell to senescence and apoptosis (Figure 1C).
Figure 1.
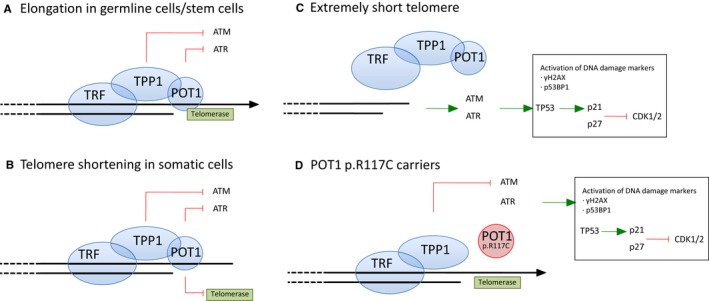
Telomere biology and damage signaling. A, Elongation in germline/stem cells. The shelterin complex mediates telomere elongation by recruiting telomerase. Shelterin also represses the DNA damage response by preventing the activation of ATM and ATR through TPP1 and POT1 proteins, respectively. TPP1 is anchored to the chromosome by POT1 and TRF (telomeric repeat binding factor 1), which is another component of the shelterin complex. B, Telomere shortening in somatic cells. Somatic divisions entail telomere shortening due to the inhibition of telomerase recruitment mediated by the POT1 protein. C, Extremely short telomere. The shelterin complex cannot bind critically short telomeres. DNA damage response ATM/ATR activates the TP53/p21 cascade to inhibit CDK1/2. DNA damage markers such as γH2AX and TP53BP1 bind the short telomeres. DNA damage signaling mediates senescence, cell cycle arrest, and apoptosis. D,POT1 p.Arg117Cys (p.R117C) mutation carriers. The POT1 p.R117C mutation prevents POT1 from binding to TPP1 and from forming the OB‐fold to bind single‐strand DNA, which prevents POT1 from repressing ATR signaling. Telomere damage response is activated in POT1 p.R117C mutation carriers.
Our previous studies demonstrated that cardiac angiosarcoma patients carrying the POT1 p.Arg117Cys mutation showed abnormally long telomeres due to the lack of repression of telomerase, which led to increased fragility and damage.6 Other described POT1 mutations associated with risk of developing familial glioma and familial melanoma tumors also led to abnormally long and fragile telomeres.7 However, the link between telomere instability and tumorigenesis is not well understood. In addition, studies of the biological pathways involved in the progression of angiosarcomas are very scarce. Recently, next‐generation sequencing studies uncovered somatic alterations in the VEGF/PLCG1 pathway in cardiac angiosarcoma16, 17 and driver mutations in the PI3K/AKT/mTOR and MEK pathways in angiosarcomas other than cardiac,18, 19, 20, 21 but these studies did not distinguish between constitutive and somatic mutations and did not clarify the genetics and underlying mechanisms.
In order to understand how telomere instability links the angiosarcoma process, in the present work we studied somatic events in different angiosarcoma patients carrying the POT1 p.Arg117Cys mutation described in Calvete et al6: 2 patients (F1 and F3) affected with CAS and 1 patient (F2) with 2 tumors, a breast AS and a papillary thyroid tumor. In addition, we studied the genetic and molecular somatic landscape that drives tumor progression in 7 patients with sporadic CAS tumors (NT1‐NT3 and T1 to T4) who did not carry mutations in the POT1 gene.
Methods
The data, methods used in the analysis, and materials used to conduct the research are available to any researcher for purposes of reproducing the results or replicating the procedure.
Ethics Statement
Institutional Review Board approval was obtained; the ethics committee of the CNIO, the Institute of Health Carlos III, and SaludMadrid (Autonomous Community of Madrid) approved this study (CS9679). Informed consent was received from participants before inclusion in the study.
Patients
Formalin‐fixed paraffin‐embedded (FFPE) tissue samples from 10 patients were selected. The 3 familial angiosarcoma individuals carrying the POT1 p.Arg117Cys mutation were selected from Calvete et al.6 Seven FFPE tissue samples from individuals affected with sporadic CAS not carrying mutations in the POT1 gene were also selected: 3 FFPE tissue sections contained normal (N) and tumor (T) tissue (NT1, NT2 and NT3), and the other 4 FFPE sections contained only tumor tissue (T1 to T4).
Samples
Genomic DNA was extracted from FFPE tissue samples and from fixed tissue slides (microdissection) using the DNeasy Blood & Tissue Kit (Cat No. 69504, Qiagen, Hilden, Germany) following the manufacturer's instructions. Histology of hematoxylin and eosin–stained sections of the tissues was assessed by a pathologist (M.M.).
Immunohistochemistry
FFPE blocks were cut into 5‐μm‐thick sections for immunohistochemistry (IHC) studies. The cell cycle in normal tissue was tested with anti‐p21 (WAF1) from Merck (Darmstadt, Germany; Ref MABE325), anti‐p27 (57/Kip1) from BD Biosciences (Franklin Lakes, NJ; Ref 610242), and anti–phosphohistone γH2AX (Ser139) from Millipore (Burlington, MA; Ref 05‐636) antibodies. Activation of the VEGF‐angiogenesis pathway was assessed with 2 different antibodies against phosphorylated (activated) proteins: anti–phospho‐p44/42‐MAPK (ERK1/2) and anti–phospho‐S6 ribosomal protein (Ser235/236) from Cell Signaling Technology (Danvers, MA; Refs 9101 and 2211, respectively). Both the absence of staining and excess nonspecific staining were considered as negative staining. Staining was considered separately in normal (N) and tumor (T) tissues. IHC controls were performed by using normal tissue from biopsies of healthy individuals.
Whole Exome Sequencing and Bioinformatics Pipelines
Exomes from selected tissues were captured and enriched using the SureSelect Human All Exon Kit (78 Mb) (Agilent Technologies, Santa Clara, CA). Enriched samples were paired‐end sequenced on an Illumina Genome Analyzer II sequencing platform using 2 lanes per sample and generating 101 base‐pair long reads. FASTQ files of short reads were aligned using the BWA algorithm to the GRCh37/hg19 reference genome22; 96.11% (ranging from 93.34% to 98.74%) of the reads aligned in the reference genomes (effectiveness). GATK‐based variant calling was performed for aligned reads considering DP (Read Depth) values of >30 and Quality‐by‐Depth scores for a variant confidence of >1.00. A total of 92.28% (ranging from 91.70% to 92.16%) of the variants that were well aligned and annotated passed the quality and coverage filters. Strict filtering for only well‐defined variants by quality controls, and those not included in repeat regions were included to prevent false positives. Tumor variants (<10% alternative variant allele frequency) and those with low coverage (<6×) were discarded.
Only quality‐filtered variants affecting coding sequences of canonical transcripts (nonsynonymous, essential splice site, frame shift or gain/loss of stops) were taken into account. Variant type annotation, population statistics, disease‐specific sequence databases, and in silico predictive algorithms were according to AMCG standards and guidelines.23 Variants with a minor allele frequency of <0.01 and <0.05 (dbSNP130, HapMap, or 1000 Genomes) were considered for stringent and relaxed filtering, respectively. Their potential damaging effect was assessed using the VEP24 script software package (including Sift, Polyphen and Condel damage predictors). Stringent filtering only considered the variants annotated as pathogenic by all damage predictors. Whole‐exome sequencing data have been deposited in the ArrayExpress public database under accession number E‐MTAB‐7999 (available at https://www.ebi.ac.uk/fg/annotare/).
Variants found in DNA blood samples and variants found in common between paired normal/tumor tissues were considered as constitutional. Variants found only in tumor tissue were considered somatic variants. Constitutional variants were validated in DNA from blood and normal tissue samples, and somatic variants were validated in DNA from tissue samples (tumor) by Sanger sequencing.
Pathway Enrichment Analyses
Two different software packages were used for independent assessments of the gene set analyses. Data were analyzed with Qiagen's Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City, CA www.qiagen.com/ingenuity) and ConsensusPathDB (available at http://cpdb.molgen.mpg.de/).25 Overrepresentation analysis of the gene set list was performed with a minimum overlap of 4 genes with the pathway database set size and a P‐value cutoff of 0.001. Induced network module analysis without intermediate nodes and considering high‐confidence binary protein and genetic/gene regulatory interactions were also evaluated.
Trusight Tumor 170 Panel and IBM Watson Study
Resequencing of tumor DNA from FFPE tissue samples was performed with TruSight Tumor 170 from Illumina (San Diego, CA), a novel sequencing platform that predicts the most probable variant causing the pathology and provides suggestions for translating the data to the clinic. The sequencing of the hybrid capture was run on the HiSeq 2500 System. The captured gene content in the TruSight Tumor 170 Assay, data sheet, and specifications are available at https://www.illumina.com. Low variant quality of <20 and low depth (<100 for variant calls and <250 for reference call filters) were considered. Variants were supported with >7 reads and filtered by frequency (minor allele frequency <0.05). The copy number variation call was calculated for the fold‐change results for each gene. Variants were classified and analyzed later by the IBM Watson for Genomics platform, which searches electronic medical databases to find information that may be relevant to a particular genomic sequence (available at https://www.ibm.com/watson/).
Results
Angiosarcoma Patients Carrying the POT1 p.Arg117Cys Mutation
Constitutional Effect in Normal Tissue of POT1 p.Arg117Cys Mutation Carriers
In normal conditions the POT1 protein represses downstream activation of the DNA damage response at telomeres in somatic cells (Figure 1B).15 The POT1 p.Arg117Cys protein shows a reduced capacity to bind telomeres and TPP16 and may affect the regulation of the damage response. Damage response activation leads to cell cycle arrest, replicative senescence, and apoptosis (Figure 1C). In order to decipher the putative effect of POT1 malfunction, patient tissues were stained with anti‐P‐γH2AX (DNA damage marker) and anti‐p21 and anti‐p27 antibodies (inhibitors of CDK1/2 to arrest the cell cycle). IHC studies were carried out in normal (N) tissues of patients from the families carrying the constitutional POT1 p.Arg117Cys mutation studied in Calvete et al6: 2 patients (F1 and F3) affected with CAS and 1 patient (F2) with 1 breast AS and 1 papillary thyroid tumor.
Increased IHC staining with anti‐p21, ‐p27, and ‐P‐γH2AX antibodies was observed in N tissues of all studied patients in comparison with the corresponding N tissues from healthy non–mutation carriers (Table 1 and Figure 2A). Therefore, the reduced binding to telomeres and to TPP1 by the POT1 p.Arg117Cys protein correlates with activation of damage response signaling mediated by p21 and p27 (Figure 1D).
Table 1.
Total Cases and Number of Variants Found in the Whole Exome Sequencing
| Individual | Pathology | Tissue | Variant Calling | Filtered Variants | Somatic Variantsa | Constitutional Variantsb | |
|---|---|---|---|---|---|---|---|
| POT1 p.Arg117Cys carriers | F1 | CAS | T | 1095 | 46 | NA+ | |
| N | |||||||
| F2 | Papillary thyroid | T | 100 506 | 1294 | 5 | NA+ | |
| N | 100 371 | 1281 | |||||
| Breast AS | T | 93 496 | 1276 | 13 | NA+ | ||
| Without mutations in the POT1 gene | NT1 | Sporadic CAS | T | 102 560 | 1266 | 62 | 1032 |
| N | 111 686 | 1333 | |||||
| NT2 | Sporadic CAS | T | 100 120 | 1231 | 36 | 1101 | |
| N | 141 248 | 1239 | |||||
| NT3 | Sporadic CAS | T | 97 233 | 704 | 37 | 1180 | |
| N | 113 358 | 762 |
| Individual | Pathology | Tissue | Variant Calling | Filtered Variants | Stringent Filtering | |
|---|---|---|---|---|---|---|
| Only T tissue availablec | T1 | Sporadic CAS | T | 93 374 | 1181 | 315 |
| T2 | Sporadic CAS | T | 95 492 | 1274 | 403 | |
| T3 | Sporadic CAS | T | 104 545 | 1328 | 443 | |
| T4 | Sporadic CAS | T | 93 859 | 1233 | 361 |
CAS indicates cardiac angiosarcoma; N, normal tissue; NA+, not applicable (POT1 p.Arg117Cys carriers from Calvete et al [2015]); T, tumor tissue.
Variants found in tumor tissue that were not found in normal tissue.
Variants found in normal tissue that were not found in tumor tissue.
Individuals with only T tissue sequenced.
Figure 2.
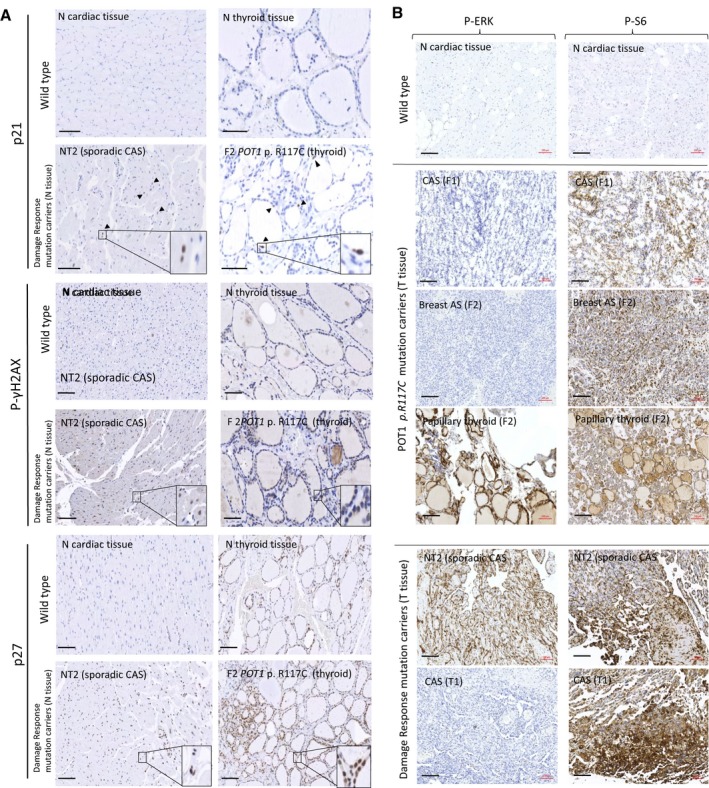
Immunohistochemical staining. A, Tissue stress was tested in normal tissue of carriers of the POT1 p.Arg117Cys (p.R117C) mutation and carriers of mutations in the damage response‐signaling pathway (sporadic CAS) in comparison with the corresponding normal tissue without mutations (wild type). Above: Wild‐type cardiac and thyroid tissues from healthy controls without mutations. Below: normal tissue of individual NT2 (sporadic CAS individual with constitutional mutations in the ATR,ATM and TP53BP genes) and normal tissue of individual F2 (papillary thyroid tumor with POT1 p.R117C mutation) as representative examples (see Table 2 for all studied individuals). Increased cell cycle arrest was observed in the normal tissue of both patients. Black arrowheads show some of the stained nuclei. Detailed fields (10×) are also shown. Scale bar (in black): 100 μm. B, IHC staining with anti–P‐ERK and anti–P‐S6 antibodies in tumor tissues. Tumor tissues from carriers (F1 and F2) and noncarriers (T1 and NT2) of the POT1 p.R117C mutation compared with a normal tissue section (negative staining) are shown as examples (see Table 2 for all studied individuals). Three tumors from the 2 patients (F1 and F2) carrying the POT1 p.R117C mutation are shown: both angiosarcomas (CAS tumor tissue from F1 and breast AS from patient F2) only showed immunoreactivity with anti–P‐S6 antibody, while the papillary thyroid tumor (patient F2) also showed immunoreactivity with anti–P‐ERK antibody. Two staining patterns were observed in sporadic CAS patients without mutations in the POT1 gene: tissue from patient T1 only showed immunoreactivity with anti–P‐S6, whereas tissue from patient NT2 (sporadic CAS) was stained with both anti–P‐S6 and anti–P‐ERK antibodies. Scale bar (in black): 100 μm. AS indicates angiosarcoma; CAS, cardiac angiosarcoma; N, normal tissue; T, tumor tissue.
Somatic Events in POT1 p.Arg117Cys Mutation Carriers
To evaluate possible somatic events in affected individuals carrying the POT1 p.Arg117Cys mutation that might lead to the formation of AS, the exomes of normal (N) and tumor (T) tissues of the F1 and F2 individuals were sequenced. Variants found in T tissue but not in N tissue were considered to be somatic (Table S1). In patient F1 (CAS), 46 filtered somatic variants were found in T but not in N cardiac tissue (Table 2) including an in‐frame deletion in the KDR gene (p.Asn704del), which encodes VEGF receptor 2 (VEGFR2), which belongs to the VEGF‐angiogenesis signaling pathway (Figure 3A and 3B).
Table 2.
Immunohistochemistry Staining Results for the Studied Cases
| Mutation | Sample | Pathology | Tissue | N Tissue | Mutations in VEGF‐Angiogenesis Pathway | T Tissue | |||
|---|---|---|---|---|---|---|---|---|---|
| γH2AX | p21 | p27 | P‐ERK | P‐S6a | |||||
| POT1 | F1 | CAS | Cardiac (N+T) | + | + | + | KDR | − | + |
| p.Arg117Cys | F2 | Breast AS | Breast T | NA | NA | NA | PI3K | − | + |
| Thyroid (N +T) | + | + | + | BRAF | + | + | |||
| DR genes | NT2 | CAS | Cardiac (N+T) | + | + | + | VEGF2/RAS‐MAPK | + | + |
| T1 | CAS | Cardiac (T) | NA | NA | NA | Akt‐PI3K | − | + | |
| T3 | CAS | Cardiac (T) | NA | NA | NA | RAS‐MAPK/Akt‐PI3K | + | + | |
| T4 | CAS | Cardiac (T) | NA | NA | NA | VEGFA/Akt‐PI3K | + | + | |
+ indicates overexpression; −, no expression; CAS, cardiac angiosarcoma; DR, damage response; N, normal; NA, tissue not available; T, tumor.
Positive staining in lining epithelium and tissue.
Figure 3.
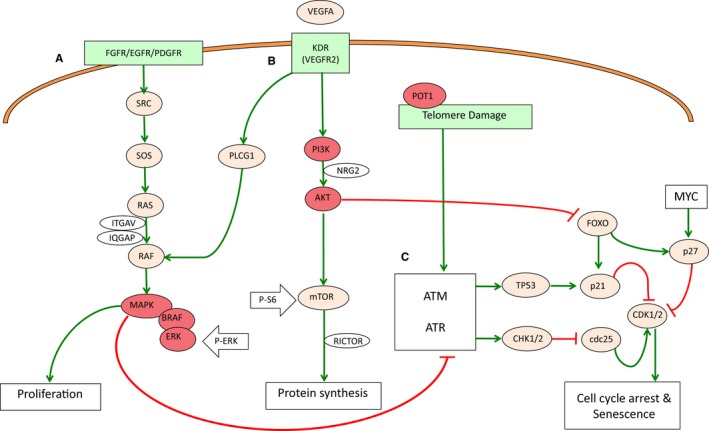
VEGF (vascular endothelial growth factor)‐angiogenesis signaling and cell cycle regulation pathways. VEGF signaling is a growth factor pathway to stimulate vasculogenesis and angiogenesis. A, MAPK/ERK signaling regulates cell proliferation. B, AKT/PI3K signaling is related to protein synthesis and cell cycle signaling. Locations of anti–P‐ERK and anti–P‐S6 used in IHC studies are also shown in the pathway (white arrows). C, Damage‐signaling pathway. AKT/PI3K signaling inhibits apoptosis, senescence, and cell cycle arrest through inhibition of FOXO, which in turn positively regulates p21 and p27 activity.
Regarding the F2 individual, only 13 filtered somatic variants were found in the T tissue of the breast AS (Table S1). Two of them were variants annotated in the genes PLCG1 (p.Leu752Val) and PIK3CA (p.Arg88Gln) belonging to the VEGF‐angiogenesis pathway and previously described to be involved in different angiosarcomas and primary breast cancer.26 Another 5 somatic variants were found in the papillary thyroid T tissue from the same individual (Table S1). Three of these belonged to the VEGF‐angiogenesis pathway (PIK3R6 [p.Arg59Lys], RASSF1 [p.Arg227His], and BRAF [p.Val600Glu]). Overall, 32/52 (62%) of the somatic missenses were C:G>T:A changes (Table S1). Genes with mutations are shown in the angiogenesis pathway depicted in Figure 3.
Finally, only cardiac tumor tissue from patient F3 (CAS) was sequenced (Table 2). A total of 297 filtered variants were found in the tumor tissue of this patient (Table S2), including another mutation in the KDR gene (p.Thr771Arg). Overall, 60% of the somatic variants found in patient F3 were annotated as C:G>T:A changes (Table S2).
Thus, somatic mutations in the VEGF signaling pathway were found in the tumor tissues of all affected individuals carrying the POT1 p.Arg117Cys germline mutation. Interestingly, both studied CAS patients had mutations in KDR, which activates VEGF signaling to regulate angiogenesis by the MAPK/ERK and AKT/PI3K pathways (Figure 3A and 3B). MAPK/ERK regulates proliferation activity, while AKT/PI3K promotes protein synthesis. Interestingly, both molecular activities regulate the cell cycle by inhibiting senescence promotion (Figure 3C). In order to test the putative effects of the mutations found in the VEGF angiogenesis pathway, we studied the MAPK/ERK and AKT/PI3K molecular pathways by IHC with antibodies against the activated (phosphorylated) forms of ERK and S6, respectively (Figure 3A and 3B). All angiosarcomas (CAS and the breast AS tumors) did not stain with anti–P‐ERK but were positively stained (>70%) with the anti–P‐S6 antibody. However, papillary thyroid tumor tissue stained with both antibodies (Table 1 and Figure 2B).
In summary, somatic activating mutations of the VEGF‐angiogenesis pathway were found in all studied tumor tissues of POT1‐mutated patients independently of the tumor type (cardiac, breast, and thyroid). Somatic mutations in the KDR gene were found in both CAS patients (F1 and F3). In addition, positive P‐S6 staining was observed in all angiosarcomas with somatic mutations in the KDR (CAS) and PI3K (breast AS) genes (AKT/PI3K pathway). The papillary tumor (F2), which had a mutation in the MAPK/ERK signaling pathway (BRAF gene), was stained with both anti–P‐ERK and anti‐PS6 antibodies (Figures 2B and 3).
Sporadic Cardiac Angiosarcoma Patients Without Mutations in the POT1 Gene
To assess the whole genetic landscape of CAS tumors, another 7 patients with sporadic CAS tumors who were not carrying mutations in the POT1 gene were studied. Normal (N) and tumor (T) cardiac tissues were available from 3 sporadic CAS individuals (NT1, NT2, and NT3), whereas only tumor tissue was available from the other 4 CAS individuals (T1 to T4). We found 1032, 1101, and 1180 constitutional variants in cardiac tissue for the NT1, NT2, and NT3 CAS individuals, respectively; 62, 36, and 37 somatic variants were found for individuals NT1, NT2, and NT3, respectively (Table 2).
Regarding the 4 tumor samples of which only T tissue was available, no distinction could be made between constitutional and somatic variants. We found 1181, 1274, 1328, and 1233 variants in the T tissue of patients T1, T2, T3, and T4, respectively (Table 2).
To further delineate the genetic landscape of sporadic CAS tumors, the genes encompassing filtered variants were grouped into 2 different pathway enrichment analyses. The first set included genes with constitutional (found in both N and T tissues from cases NT1, NT2, and NT3) and all genes with variants from the other 4 CAS individuals with only the tumor tissue sequenced (constitutional or somatic) (2501 unique genes in total); the second set included the genes with somatic variants (only present in T tissue) from cases with N and T tissue (NT1, NT2, and NT3) and again all genes with variants from the other 4 CAS individuals with only the sequenced T tissue cases (T1 to T4) (1522 unique genes in total).
Constitutional Events in Normal Tissue in Sporadic CAS
This study revealed that the most represented pathway and the pathway with the major number of genes were the “Sustainability of p53 pathway” (genes ATM, TP53, and RFWD2) (P value 0.003) and the “Repair modulation pathway” (genes ATR, ATM, TP53, RFWD2, SIRT7, BRCA2, CDK8, UBE2D1, WRN, PMS2, BRIP1, TP53BP2, and APC2) (P value 0.0022), respectively (Table S3). Mutations in genes from these pathways were found in all 7 sporadic CAS individuals (Figure 4). These genes belong to the damage response signaling pathway and may deregulate the cell cycle in the same manner as previously observed for the POT1 mutation carriers. Thus, N cardiac tissue from the sporadic CAS individuals (NT series) was also stained with anti–P‐γH2AX, anti‐p21, and anti‐p27 antibodies. Positive staining was also observed in N tissue of sporadic CAS individuals (Table 1), which correlated with cell cycle deregulation as observed in familial angiosarcomas (POT1 p.Arg117Cys mutation carriers). Therefore, the familial angiosarcomas (POT1 p.Arg117Cys carriers) and sporadic CAS patients behaved in a similar way regarding cell cycle arrest regulation.
Figure 4.

Genes with mutations in the studied CAS individuals. Genes are distributed considering the gene ontology among telomere instability and damage response or VEGF (vascular endothelial growth factor)‐angiogenesis signaling pathways. Exomes from normal/tumor tissues of 3 patients (F1, F2, and F3) carrying the constitutional POT1 p.Arg117Cys mutation (orange) and 3 sporadic CAS individuals not carrying mutations in the POT1 gene (NT1, NT2, and NT3) were sequenced. Constitutional mutations refer to variants found in blood samples and found in common in both N and T tissues (red). Mutations found only in tumor tissue were considered somatic (purple). Only tumor tissue from another 4 sporadic CAS individuals (T1 to T4) was also sequenced. A distinction between first event and somatic variants could not be made for the mutations found in these individuals (blue). Less stringent frequency filtering corresponds to a minor allele frequency <0.05 (light purple) instead of <0.01 (light blue). AS indicates angiosarcoma; CAS, cardiac angiosarcoma; N, normal tissue; P. thyroid, papillary thyroid tumor; T, tumor tissue.
Somatic Events in Tumor Tissue in Sporadic CAS
A second pathway enrichment analysis was performed with the gene set including the somatic variants found in T tissues of sporadic CAS individuals (see above). This enrichment analysis revealed that the most represented pathway was the “gf‐hypoxia and angiogenesis” pathway (Biocarta: 16.7%) (P value 0.00557). The pathway with the major number of affected genes was the “Signaling by VEGF” pathway (Reactome, 24 genes) (P value 0.00101) (Table S3). Both enrichment analyses corresponded with the VEGF‐angiogenesis pathway. Genes with mutations are shown in the angiogenesis pathway of Figure 3. Mutations in genes from these pathways were found in all 7 sporadic CAS individuals (Figure 4). Activation of the MAPK/ERK and AKT‐PI3K pathways was studied by IHC with anti–P‐ERK and anti–P‐S6 antibodies, respectively.
Tumors of all studied sporadic CAS individuals (NT and T series) stained positive with anti‐PS6 antibodies, which demonstrates that somatic mutations were activating the VEGF‐angiogenesis pathway (Table 1). Especially intense staining was also observed in the endothelial lining of blood vessels. However, not all tissues from individuals affected with sporadic CAS stained with anti‐PERK antibody. Tissue of individual T1, who only had mutations in the AKT‐PI3K signaling pathway (Table 1), did not stain with anti–P‐ERK. The tumors with mutations in the 2 molecular signaling pathways (NT1, NT2, and T4) also stained positive with the 2 antibodies (anti–P‐ERK and anti–P‐S6) (Table 1). Interestingly, the individual with mutations only in the MAPK/ERK signaling pathway (T3) also stained positive with both antibodies (Table 1). Stained tissues from patients T1 and NT2 are shown in Figure 2B as representative examples.
In summary, all studied individuals with CAS (either familial or sporadic) had mutations (either constitutional or somatic) in normal tissue affecting damage response signaling (Figure 4). IHC with anti‐p21 and anti‐p27 antibodies confirmed cell cycle arrest deregulation in N tissue that leads to constitutional cell cycle arrest and cessation of cell division (Figure 2A and Table 1). In addition, somatic activating mutations in the VEGF‐angiogenesis pathway were found in tumor tissue of familial and sporadic angiosarcomas, independently of the presence of POT1 mutations (Figures 2B and 4; Table 1).
Sequencing Replication With the Truseq170 Panel and IBM Watson for Genomics Platform
A sequencing replication was performed for 2 previously sequenced CAS patients without mutations in the POT1 gene. The Truseq170 panel was run for tumor tissue of the T1 and T4 individuals and analyzed with the IBM Watson for Genomics platform (version 33.148), which is a novel sequencing platform that predicts the most probable variant causing the pathology and provides suggestions for translating the data to the clinic. The Watson for Genomics pipeline revealed 15 variants of unknown significance, 2 alterations without proposed therapies, and only 1 actionable alteration for patient T1 (Table S4). Three variants of unknown significance, 3 alterations with no proposed therapies, and another 3 actionable alterations were described for patient T4 (Table S5). Interestingly, a not previously detected copy number gain was found for the KIT gene in the tumor tissue of patient T1. The gained region is involved in tumor cell proliferation, angiogenesis, and metastatic disease. Only 1 actionable pathway was found in common for both CAS patients. The variants found in the TP53 gene were highlighted as actionable alterations, as previously found in the WES study (TP53 p.Arg175His and TP53 p.Val143Met for patients T1 and T4, respectively). No Food and Drug Administration–approved therapies for angiosarcoma were recommended for these 2 patients, but 3 therapies with clinical trials were observed in common for both patients: AZD1775 (NCT numbers NCT02576444 and NCT018227384), Transferrin Receptor‐Targeted Liposomal p53 (NCT02354547), and Modified Vaccinia Virus Ankara Vaccine (NCT02432963). The 3 therapies for the alterations common to the 2 CAS patients target TP53 to restore cell cycle regulation.
Discussion
Telomere Instability in POT1 Mutation Carriers Increases Cell Cycle Arrest in Constitutional Tissue and Increases Acquisition of Somatic Mutations in the Angiogenesis Pathway
The single‐strand DNA response in telomeres is inactivated by the shelterin complex (Figure 1A and 1B).15 We previously observed that the POT1 p.Arg117Cys mutation prevented the POT1 protein from binding to TPP1 and forming the OB‐fold to bind singles‐strand DNA, which led to abnormally long telomeres.6 Here, we observed that abnormal telomere length found in our patients correlates with a genomic instability scenario that, in consequence, activates DNA damage‐signaling (γH2AX‐positive staining). Individuals carrying the POT1 p.Arg117Cys mutation overexpressed the p21 and p27 proteins in constitutional tissue (Table 1), which correlates with prevention of the repression of ATR signaling and leads to cell cycle arrest (Figure 1D). This increased senescence in nontumor tissue would result in reduced cell cycling and cessation of cell division. A similar mechanism was proposed to explain how short telomeres can lead to vascular senescence and diminished proliferative capacity that involved exhaustion of cell pools in mice.27, 28 In addition, tumor tissue of POT1 p.Arg117Cys mutation carriers was studied in order to evaluate the involvement of constitutional cell cycle arrest in tumor progression. On average, 61% of the somatic variants found in these patients were C:G>T:A changes (Tables S1 and S2). At this time we cannot rule out a correlation between this observed bias and a specific mutation signature in these tumors. Megquier et al29 have established an association with somatic deamination of cytosine to thymine in a large series of angiosarcomas (different from cardiac), which is in agreement with our observation. Somatic mutations in the VEGF‐angiogenesis pathway were found in tumor tissue of all individuals (Figure 4). Interestingly, the F1 and F3 individuals (CAS) also had a somatic mutation in the KDR gene, which is involved in VEGF/PLCG1 activation and was described altered in 2 recently studied sporadic CAS cases.16, 17 In the F2 patient the recurrent somatic mutations PIK3CA p.Arg88Gln and BRAF p.Val600Glu were found in the breast AS and the papillary thyroid tumor, respectively. Twenty‐five percent of all breast cancers have somatic mutations in the PIK3CA gene. Specifically, the PIK3CA p.Arg88Gln mutation was described in a primary breast cancer.26 The somatic mutation BRAF p.Val600Glu is the most common genetic change in papillary thyroid cancers (35.8%).30 In addition, the BRAF p.Val600Glu and PIK3CA p.Arg88Gln mutations have been described as activators of the angiogenic response.31, 32 The somatic mutation BRAF p.Val600Glu has also been reported in gliomas33 and melanomas,34 where constitutional mutations in the POT1 gene were also described.10, 11, 12 In summary, individuals with nonfunctional POT1 did not repress damage signaling (ATR) and showed a constitutive increase of p21 and p27 expression, which is in agreement with cell cycle arrest. In addition, somatic activating mutations in the angiogenesis pathway were acquired. Interestingly, activating somatic mutations in the KDR gene were found in both studied patients affected with CAS and carrying a POT1 germline mutation (F1 and F3). This was also observed in other tissues and tumors different from cardiac angiosarcomas with mutations in the POT1 gene (thyroid and breast from patient F2), demonstrating that not only in cardiac tissue is there a strong correlation between constitutional senescence mediated by POT1 malfunction and the acquisition of somatic mutations in the angiogenesis pathway.
Sporadic Cardiac Angiosarcomas Without POT1 Mutations
Although in 4 of the 10 patients studied no discrimination between germline and somatic mutations could be made, mutations in the pathways that were affected by the mutation in the POT1 gene at both the constitutional and the somatic level were also found in these patients.
Studied CAS individuals without constitutional mutations in the POT1 gene presented mutations in genes that were involved in damage response signaling (ATM‐ATR‐TP53) (Figure 4). IHC studies with anti‐p21 and anti‐p27 antibodies confirmed the damage response activation in all sporadic CAS individuals (Table 1 and Figure 2). Therefore, our results suggest a relation between telomere instability (familial CAS) and altered damage signaling (sporadic CAS) on 1 hand, and increased cell cycle arrest leading to the cessation of cell division on the other. Our observation is in agreement with previous studies in which overexpression of TP53 was detected by immunohistochemistry in 49% of angiosarcoma patients.19 Somatic activating mutations in the VEGF‐angiogenesis pathway were also found in tumor tissue of all sporadic CAS individuals (Figures 2 and 4). Similar studies performed in angiosarcomas also uncovered somatic driver mutations in the VEGF/PLCG1 angiogenesis pathway.18, 20, 21
Constitutional Cell Cycle Arrest May Fuel the Acquisition of Somatic Mutations in the Angiogenesis Pathway in Angiosarcomas
Activation of damage signaling in both familial (POT1 mutations carriers) and sporadic angiosarcomas (ATM‐ATR‐TP53 mutation carriers) induced constitutional senescence and reduced cell division (Figure 2). Our results strongly suggest a correlation between constitutional cell cycle arrest and the acquired somatic mutations in the VEGF‐angiogenesis pathway that drive angiosarcoma formation. Moreover, increased cell cycle arrest due to POT1 malfunction also uncovered somatic angiogenesis activation in tumors other than cardiac angiosarcomas. Damage response signaling and the VEGF‐signaling pathway are mutually regulated (Figure 3). Recently, telomere biology and the PI3K pathway (angiogenesis) were also shown to be functionally connected, and phosphorylation activity of the PI3K/AKT pathway was demonstrated to affect telomere stability in vitro.35 In vitro studies with stem cells from Pot1a knockout mice with increased telomere dysfunction also suggested a correlation with increased proliferation.36 Our results indicate that the observed cell cycle deregulation may interfere with apoptosis. This bypass of apoptosis would permit the acquisition of somatic mutations. In addition, cells carrying somatic mutations in genes involved in attenuating cell cycle arrest, which depletes progenitor stem cells in nontumor tissues, may undergo positive selection. A bypass of apoptosis was also suggested in studies in which POT1 was inactivated in vitro, and induced genomic instability enabled cancer cells to acquire additional mutations and conferred aggressive behavior. Attenuation of the damage response was suggested to allow tumor cells to bypass the proliferation defect imposed by POT1 inhibition.36 Therefore, under senescence conditions, activating somatic mutations in the angiogenesis pathway would acquire an important role to replenish the depleted tissue. However, the mutations found in the angiogenesis pathway would contribute to angiosarcoma formation and progression.
Clinical Significance of the Identified Mechanism
Activation of the VEGF‐angiogenesis pathway was found in all studied individuals with AS (familial and sporadic). However, our IHC studies revealed 2 different staining patterns that correlated with the location of the somatic mutations (Figure 2). Tumors with mutations in the AKT‐PI3K signaling pathway (F1, F2, F3, and T1) were only positively stained with the anti‐P‐S6 antibody (Table 1 and Figure 4). The individuals with mutations only in the MAPK/ERK signaling pathway (NT1, NT2 and the papillary thyroid tumor of F2) or in both molecular signaling pathways (T3 and T4) were positively stained with both antibodies (Table 1). Therefore, different somatic alterations regarding increased angiogenesis may occur in response to senescence. These results give important clues regarding the diagnosis and classification of angiosarcomas.
Our results also have an important clinical relevance regarding treatment and translational research. Inhibition of angiogenesis may be useful to stop tumor progression.37 However, angiogenesis inhibition would mitigate the effect of the driving somatic mutations but would not revert cell cycle arrest or the suggested bypass of apoptosis and would therefore not curtail the acquisition of new somatic mutations. Treatment with PI3K inhibitors of patient‐derived xenografts also showed increased telomeric DNA damage.35 Our results suggest that further studies regarding ATM/ATR, damage signaling and cell cycle inhibition activity might lead to recovering cell cycle control and preventing the acquisition of somatic mutations, including in asymptomatic patients carrying POT1 mutations. Regarding this issue, the second sequencing experiment with the IBM Watson platform also pointed to the damage‐signaling pathway as a therapeutic target. Actionable variants in the TP53 gene were highlighted in both studied CAS individuals (T1 and T4). Therefore, although somatic driver variants were found to occur in the angiogenesis pathway, only damage response signaling was found actionable for both studied angiosarcomas (Tables S4 and S5).
In summary, our current results demonstrate that inhibition of POT1 gene function and damage response malfunction both activate ATR‐dependent DNA damage signaling, which increases cell cycle arrest that would diminish cell proliferation in constitutional tissue, and that triggers somatic activating mutations in the angiogenesis pathway in angiosarcomas. Interestingly, our results and the 2 previously studied CAS patients16, 17 suggest a strong correlation between constitutional mutations in the POT1 gene, somatic activating mutations in the KDR gene, and CAS development. Importantly, the same mechanism was observed in tumor types different from cardiac tumors for patients carrying POT1 mutations and long telomeres (Figure 2). The significance of this mechanism needs to be further evaluated, and it is conceivable that POT1 mutations lead to the same acquired somatic alterations in other tissues and tumor types such as glioma, melanoma, or colorectal cancer.7 Therefore, mutations found in the POT1 gene and other genes involved in DNA damage‐response signaling (ATR/ATM and TP53) in the studied cardiac angiosarcomas correlate with constitutional cell cycle arrest, which would deplete the progenitor cells and trigger tissue stress. This tissue stress would give rise to a bypass of the apoptotic regulation, which permits the acquisition of multiple somatic events. In all studied CAS cases (patients with familial CAS carrying the POT1 mutation and patients with sporadic CAS), somatic activating mutations were found in the angiogenesis pathway, which drives tumor formation. At a translational level, inhibition of angiogenesis might be useful to halt tumor progression. However, inhibition of angiogenesis would not reverse cell cycle arrest or the suggested bypass of apoptosis in constitutional asymptomatic tissue. Instead, the use of ATM/ATR activity inhibitors might restore cell cycle control and prevent the acquisition of somatic mutations.
Sources of Funding
Benitez's laboratory is partially funded by Centro de Investigación (CIBERER), Horizon2020 BRIDGES project, and by the Spanish Ministry of Health supported by Federación Española de Enfermedades Raras (FEDER) funds (PI16/00440). Garcia‐Pavia's group is partially supported by the Instituto de Salud Carlos III (ISCIII) (grants CB16/11/00432 and PI14/0967) and by the Spanish Ministry of Economy and Competitiveness (grant SAF2015‐71863‐REDT). Garcia‐Pavia's and Crespo‐Leiro's groups are supported by FEDER funds. Urioste's laboratory is funded by Spanish Ministry of Health supported by FEDER fund (PI14/00459).
Disclosures
None.
Supporting information
Table S1. Filtered Somatic Variants Found in the WES of POT1 p.Arg117Cys Carriers, 1 CAS and 1 Breast AS With Papillary Thyroid Cancer
Table S2. Filtered Variants Found in the WES of Patient F3 Affected With CAS (POT1 p.R117C Carrier)
Table S3. Mutations Found in the WES for Chromosome/Telomere Instability and the VEGF‐Angiogenesis Pathway in Sporadic CAS Sequenced Individuals (Non–POT1 Mutation Carriers)
Table S4. Actionable Variants Found in the Sequencing Experiment With IBM Watson for Genomics Platform for Patient T1
Table S5. Actionable Variants Found in the Sequencing Experiment With IBM Watson for Genomics Platform for Patient T4
Acknowledgments
We are grateful to Dr Manuel Serrano from the Tumour Suppression Group (CNIO) for critical revision of the manuscript. We want to acknowledge the patients and the BioBank of the Complejo Hospitalario Universitario de Santiago (CHUS) (PT17/0015/0002), integrated into the Spanish National Biobanks Network, for their collaboration and Centro Nacional de Análisis Genómico (CNAG) for the technical support with WES data.
(J Am Heart Assoc. 2019;8:e012875 DOI: 10.1161/JAHA.119.012875.)
Contributor Information
Oriol Calvete, Email: ocalvete@cnio.es.
Javier Benitez, Email: jbenitez@cnio.es.
References
- 1. Malkin D. Li‐Fraumeni syndrome. Genes Cancer. 2011;2:475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Butany J, Nair V, Naseemuddin A, Nair GM, Catton C, Yau T. Cardiac tumours: diagnosis and management. Lancet Oncol. 2005;6:219–228. [DOI] [PubMed] [Google Scholar]
- 3. Patel SD, Peterson A, Bartczak A, Lee S, Chojnowski S, Gajewski P, Loukas M. Primary cardiac angiosarcoma—a review. Med Sci Monit. 2014;20:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Casha AR, Davidson LA, Roberts P, Nair RU. Familial angiosarcoma of the heart. J Thorac Cardiovasc Surg. 2002;124:392–394. [DOI] [PubMed] [Google Scholar]
- 5. Keeling IM, Ploner F, Rigler B. Familial cardiac angiosarcoma. Ann Thorac Surg. 2006;82:1576. [DOI] [PubMed] [Google Scholar]
- 6. Calvete O, Martinez P, Garcia‐Pavia P, Benitez‐Buelga C, Paumard‐Hernández B, Fernandez V, Dominguez F, Salas C, Romero‐Laorden N, Garcia‐Donas J, Carrillo J, Perona R, Triviño JC, Andrés R, Cano JM, Rivera B, Alonso‐Pulpon L, Setien F, Esteller M, Rodriguez‐Perales S, Bougeard G, Frebourg T, Urioste M, Blasco MA, Benítez J. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53‐negative Li‐Fraumeni–like families. Nat Commun. 2015;6:8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calvete O, Garcia‐Pavia P, Domínguez F, Bougeard G, Kunze K, Braeuninger A, Teule A, Lasa A, Ramón y Cajal T, Llort G, Fernández V, Lázaro C, Urioste M, Benitez J. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur J Hum Genet. 2017;25:1278–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Speedy HE, Kinnersley B, Chubb D, Broderick P, Law PJ, Litchfield K, Jayne S, Dyer MJS, Dearden C, Follows GA, Catovsky D, Houlston RS. Germline mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood. 2016;128:2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chubb D, Broderick P, Dobbins SE, Frampton M, Kinnersley B, Penegar S, Price A, Ma YP, Sherborne AL, Palles C, Timofeeva MN, Bishop DT, Dunlop MG, Tomlinson I, Houlston RS. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. 2016;7:11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bainbridge MN, Armstrong GN, Gramatges MM, Bertuch AA, Jhangiani SN, Doddapaneni H, Lewis L, Tombrello J, Tsavachidis S, Liu Y, Jalali A, Plon SE, Lau CC, Parsons DW, Claus EB, Barnholtz‐Sloan J, Il'yasova D, Schildkraut J, Ali‐Osman F, Sadetzki S, Johansen C, Houlston RS, Jenkins RB, Lachance D, Olson SH, Bernstein JL, Merrell RT, Wrensch MR, Walsh KM, Davis FG, Lai R, Shete S, Aldape K, Amos CI, Thompson PA, Muzny DM, Gibbs RA, Melin BS, Bondy ML. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2014;107:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robles‐Espinoza CD, Harland M, Ramsay AJ, Aoude LG, Quesada V, Ding Z, Pooley KA, Pritchard AL, Tiffen JC, Petljak M, Palmer JM, Symmons J, Johansson P, Stark MS, Gartside MG, Snowden H, Montgomery GW, Martin NG, Liu JZ, Choi J, Makowski M, Brown KM, Dunning AM, Keane TM, López‐Otín C, Gruis NA, Hayward NK, Bishop DT, Newton‐Bishop JA, Adams DJ. POT1 loss‐of‐function variants predispose to familial melanoma. Nat Genet. 2014;46:478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shi J, Yang XR, Ballew B, Rotunno M, Calista D, Fargnoli MC, Ghiorzo P, Bressac‐de Paillerets B, Nagore E, Avril MF, Caporaso NE, McMaster ML, Cullen M, Wang Z, Zhang X, Bruno W, Pastorino L, Queirolo P, Banuls‐Roca J, Garcia‐Casado Z, Vaysse A, Mohamdi H, Riazalhosseini Y, Foglio M, Jouenne F, Hua X, Hyland PL, Yin J, Vallabhaneni H, Chai W, Minghetti P, Pellegrini C, Ravichandran S, Eggermont A, Lathrop M, Peris K, Scarra GB, Landi G, Savage SA, Sampson JN, He J, Yeager M, Goldin LR, Demenais F, Chanock SJ, Tucker MA , Goldstein AM, Liu Y, Landi MT. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46:482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. 2017;18:175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martínez P, Blasco MA. Telomere‐driven diseases and telomere‐targeting therapies. J Cell Biol. 2017;216:875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. [DOI] [PubMed] [Google Scholar]
- 16. Kunze K, Spieker T, Gamerdinger U, Nau K, Berger J, Dreyer T, Sindermann JR, Hoffmeier A, Gattenlöhner S, Bräuninger A. A recurrent activating PLCG1 mutation in cardiac angiosarcomas increases apoptosis resistance and invasiveness of endothelial cells. Cancer Res. 2014;74:6173–6183. [DOI] [PubMed] [Google Scholar]
- 17. Zhrebker L, Cherni I, Gross LM, Hinshelwood MM, Reese M, Aldrich J, Guileyardo JM, Roberts WC, Craig D, Von Hoff DD, Mennel RG, Carpten JD. Case report: whole exome sequencing of primary cardiac angiosarcoma highlights potential for targeted therapies. BMC Cancer. 2017;17:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lahat G, Dhuka AR, Hallevi H, Xiao L, Zou C, Smith KD, Phung TL, Pollock RE, Benjamin R, Hunt KK, Lazar AJ, Lev D. Angiosarcoma: clinical and molecular insights. Ann Surg. 2010;251:1098–1106. [DOI] [PubMed] [Google Scholar]
- 19. Italiano A, Chen CL, Thomas R, Breen M, Bonnet F, Sevenet N, Longy M, Maki RG, Coindre JM, Antonescu CR. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: a pattern distinct from other sarcomas with complex genomics. Cancer. 2012;118:5878–5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Antonescu CR, Yoshida A, Guo T, Chang NE, Zhang L, Agaram NP, Qin LX, Brennan MF, Singer S, Maki RG. KDR activating mutations in human angiosarcomas are sensitive to specific kinase inhibitors. Cancer Res. 2009;69:7175–7179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Behjati S, Tarpey PS, Sheldon H, Martincorena I, Van Loo P, Gundem G, Wedge DC, Ramakrishna M, Cooke SL, Pillay N, Vollan HKM, Papaemmanuil E, Koss H, Bunney TD, Hardy C, Joseph OR, Martin S, Mudie L, Butler A, Teague JW, Patil M, Steers G, Cao Y, Gumbs C, Ingram D, Lazar AJ, Little L, Mahadeshwar H, Protopopov A, Al Sannaa GA, Seth S, Song X, Tang J, Zhang J, Ravi V, Torres KE, Khatri B, Halai D, Roxanis I, Baumhoer D, Tirabosco R, Amary MF, Boshoff C, McDermott U, Katan M, Stratton MR, Futreal PA, Flanagan AM, Harris A, Campbell PJ. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat Genet. 2014;46:376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li H, Durbin R. Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics. 2010;26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, Peters BA, Velculescu VE, Park BH. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–775. [DOI] [PubMed] [Google Scholar]
- 27. Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006;72:9–17. [DOI] [PubMed] [Google Scholar]
- 28. Wong LSM, Oeseburg H, De Boer RA, Van Gilst WH, Van Veldhuisen DJ, Van Der Harst P. Telomere biology in cardiovascular disease: the TERC‐/‐ mouse as a model for heart failure and ageing. Cardiovasc Res. 2009;81:244–252. [DOI] [PubMed] [Google Scholar]
- 29. Megquier K, Turner‐Maier J, Swofford R, Kim J‐H, Sarver AL, Wang C, Sakthikumar S, Johnson J, Koltookian M, Lewellen M, Scott MC, Graef AJ, Borst L, Tonomura N, Alfoldi J, Painter C, Thomas R, Karlsson EK, Breen M, Modiano JF, Elvers I, Lindblad‐Toh K. Genomic analysis reveals shared genes and pathways in human and canine angiosarcoma. bioRxiv Available at: https://www.biorxiv.org/content/biorxiv/early/2019/03/15/570879.full.pdf. Accessed March 8, 2019. [DOI] [PMC free article] [PubMed]
- 30. Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC‐RAS‐BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63:1454–1457. [PubMed] [Google Scholar]
- 31. Bottos A, Martini M, Di Nicolantonio F, Comunanza V, Maione F, Minassi A, Appendino G, Bussolino F, Bardelli A. Targeting oncogenic serine/threonine‐protein kinase BRAF in cancer cells inhibits angiogenesis and abrogates hypoxia. Proc Natl Acad Sci USA. 2012;109:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pratilas CA, Xing F, Solit DB. Targeting oncogenic BRAF in human cancer. Curr Top Microbiol Immunol. 2012;355:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Penman CL, Faulkner C, Lowis SP, Kurian KM. Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low‐grade gliomas. Front Oncol. 2015;5:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlino MS, Long GV, Kefford RF, Rizos H. Targeting oncogenic BRAF and aberrant MAPK activation in the treatment of cutaneous melanoma. Crit Rev Oncol Hematol. 2015;96:385–398. [DOI] [PubMed] [Google Scholar]
- 35. Méndez‐Pertuz M, Martínez P, Blanco‐Aparicio C, Gómez‐Casero E, Belen García A, Martínez‐Torrecuadrada J, Palafox M, Cortés J, Serra V, Pastor J, Blasco MA. Modulation of telomere protection by the PI3K/AKT pathway. Nat Commun. 2017;8:1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pinzaru AM, Hom RA, Beal A, Phillips AF, Ni E, Cardozo T, Nair N, Choi J, Wuttke DS, Sfeir A, Denchi EL. Telomere replication stress induced by POT1 inactivation accelerates tumorigenesis. Cell Rep. 2016;15:2170–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maj E, Papiernik D, Wietrzyk J. Antiangiogenic cancer treatment: the great discovery and greater complexity (review). Int J Oncol. 2016;49:1773–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Filtered Somatic Variants Found in the WES of POT1 p.Arg117Cys Carriers, 1 CAS and 1 Breast AS With Papillary Thyroid Cancer
Table S2. Filtered Variants Found in the WES of Patient F3 Affected With CAS (POT1 p.R117C Carrier)
Table S3. Mutations Found in the WES for Chromosome/Telomere Instability and the VEGF‐Angiogenesis Pathway in Sporadic CAS Sequenced Individuals (Non–POT1 Mutation Carriers)
Table S4. Actionable Variants Found in the Sequencing Experiment With IBM Watson for Genomics Platform for Patient T1
Table S5. Actionable Variants Found in the Sequencing Experiment With IBM Watson for Genomics Platform for Patient T4