

Abstract
Background
Elevated levels of an endogenous Na/K‐ATPase inhibitor marinobufagenin accompany salt‐sensitive hypertension and are implicated in cardiac fibrosis. Immunoneutralization of marinobufagenin reduces blood pressure in Dahl salt‐sensitive (Dahl‐S) rats. The effect of the anti‐marinobufagenin monoclonal antibody on blood pressure, left ventricular (LV) and renal remodeling, and gene expression were investigated in hypertensive Dahl‐S rats.
Methods and Results
Dahl‐S rats were fed high NaCl (8%, HS; n=14) or low NaCl (0.1%, LS; n=14) diets for 8 weeks. Animals were administered control antibody (LS control antibody, LSC; HS control antibody, HSC; n=7 per group) or anti‐marinobufagenin antibody once on week 7 of diet intervention (n=7 per group). Levels of marinobufagenin, LV, and kidney mRNAs and proteins implicated in profibrotic signaling were assessed. Systolic blood pressure was elevated (211±8 versus 133±3 mm Hg, P<0.01), marinobufagenin increased 2‐fold in plasma (P<0.05) and 5‐fold in urine (P<0.01), LV and kidney weights increased, and levels of LV collagen‐1 rose 3.5‐fold in HSC versus LSC. Anti‐marinobufagenin antibody treatment decreased systolic blood pressure by 24 mm Hg (P<0.01) and reduced organ weights and level of LV collagen‐1 (P<0.01) in hypertensive Dahl salt‐sensitive rats with anti‐marinobufagenin antibody versus HSC. The expression of genes related to transforming growth factor‐β–dependent signaling was upregulated in the left ventricles and kidneys in HSC versus LSC groups and became downregulated following administration of anti‐marinobufagenin antibody to hypertensive Dahl‐S rats. Marinobufagenin also activated transforming growth factor‐β signaling in cultured ventricular myocytes from Dahl‐S rats.
Conclusions
Immunoneutralization of heightened marinobufagenin levels in hypertensive Dahl‐S rats resulted in a downregulation of genes implicated in transforming growth factor‐β pathway, which indicates that marinobufagenin is an activator of profibrotic transforming growth factor‐β–dependent signaling in salt‐sensitive hypertension.
Keywords: cardiac hypertrophy, Dahl salt‐sensitive hypertension, marinobufagenin, monoclonal antibody, transforming growth factor
Subject Categories: Cell Signalling/Signal Transduction, Growth Factors/Cytokines, Ion Channels/Membrane Transport, Hypertension, Remodeling
Clinical Perspective
What Is New?
The endogenous steroidal Na/K‐ATPase inhibitor marinobufagenin is a marker of salt‐sensitivity.
In salt‐sensitive hypertension, heightened marinobufagenin levels activate cardiac and renal genes and proteins, implicated in profibrotic transforming growth factor‐β–dependent signaling.
Immunoneutralization of marinobufagenin downregulates genes associated with transforming growth factor‐β–dependent signaling and heart failure, reverses tissue remodeling, and improves function of the heart and kidneys in hypertensive Dahl salt‐sensitive rats.
What Are the Clinical Implications?
The novel finding that marinobufagenin is an upstream activator in profibrotic transforming growth factor‐β signaling implicated in salt‐sensitivity phenomena indicates the significance of further studies of the endogenous inhibitors and regulators of Na/K‐ATPase in salt‐sensitive hypertension in humans.
Immunoneutralization of marinobufagenin can control the exaggerated production of endogenous marinobufagenin, which may be applicable to kidney disease and salt‐sensitive hypertension.
Introduction
Salt sensitivity, ie, elevation of arterial blood pressure (BP) in response to high salt (HS) intake1, 2, 3, 4 involves an endogenous steroidal ligand of Na/K‐ATPase marinobufagenin.5, 6 In mammals, marinobufagenin is primarily synthetized by adrenocortical cells from cholesterol7, 8 and increases in salt‐sensitive hypertension,5, 6 preeclampsia,9, 10 and chronic kidney disease.11 Marinobufagenin participates in regulation of renal Na+ transport, water‐salt homeostasis, and BP9, 12 and activates profibrotic signaling.11, 13, 14, 15, 16 Inhibition of Na/K‐ATPase in renal tubules promotes natriuresis, whereas targeting of Na/K‐ATPase in arterial vascular smooth muscle cells by marinobufagenin leads to an elevation of cytosolic Ca2+ and vasoconstriction via activation of an ionic pathway.17, 18 Excessive production of marinobufagenin is an adaptive response to compensate for the genetically impaired renal Na+ excretion and pressure‐natriuresis mechanisms in salt‐sensitive hypertension.19 An increased marinobufagenin level leads to vasoconstriction,17 which links dietary HS intake and hypertension.5, 6
HS intake in Dahl salt‐sensitive (Dahl‐S) rats is associated with a hypertensive response accompanied by the development of compensatory left ventricular (LV) hypertrophy, followed by the development of decompensated congestive heart failure (HF).20, 21 Salt‐sensitive hypertension in Dahl‐S rats develops as a result of renal Na+ retention, which stimulates marinobufagenin increase in parallel with BP elevation.5, 19, 22 Marinobufagenin induces cardiovascular and renal tissue fibrosis via signaling pathway,13, 14, 15 which is markedly reduced by administration of anti‐marinobufagenin monoclonal antibody (mAb) in the rat model of chronic kidney disease.11, 15 Fibrotic changes in myocardial and renal tissue in hypertensive Dahl‐S rats are accompanied by an activation of transforming growth factor‐β (TGFβ) signaling.20, 23, 24, 25 The association of marinobufagenin and TGFβ was described in normotensive rats26 however, the studies exploring the causative relationship between marinobufagenin and TGFβ in salt‐sensitivity are scant.
Dahl‐S rat strain was bred for a trait of salt sensitivity.27 Although it is known what genes participate in the development of hypertension and cardiovascular remodeling,25, 28, 29, 30, 31 it is not fully understood what signaling mechanism is activated by marinobufagenin via Na/K‐ATPase signal transduction in salt sensitivity. The aim of the present study was to investigate genes involved in marinobufagenin‐induced cardiovascular and renal remodeling in the Dahl‐S rat model of hypertension. We compared the expression of genes in the left ventricle and kidneys in hypertensive Dahl‐S rats given an HS intake with and without anti‐marinobufagenin mAb administration, which provides a tool in the investigation of the effects of marinobufagenin. We hypothesized that: (1) an elevated marinobufagenin level in hypertensive Dahl‐S rats activates cardiovascular and renal TGFβ‐dependent profibrotic signaling, and (2) immunoneutralization of marinobufagenin by an anti‐marinobufagenin mAb will decrease the expression of profibrotic genes, and, thus, diminish tissue fibrosis and improve function of the renal and cardiovascular systems.
Methods
The microarray GEO accession numbers for the data reported in this article are given in the Microarray Data Analysis section. The other data that support the findings of this study are available from the corresponding author upon reasonable request.
Experimental Model
The experimental design of the study was approved by the Animal Care and Use Committee of the Intramural Research Program, National Institute on Aging, National Institutes of Health, and all procedures were performed in accordance with institutional guidelines. Six‐week‐old, male, Dahl‐S rats (SS/JrHsd; Charles River Laboratories), body weight (BW) 218±6 g, were placed on a low salt (LS) diet (n=14; 0.1% NaCl; CAT#TD94268; Harlan Laboratories, Inc) or an HS diet (n=14; 8% NaCl; CAT#TD92012; Harlan Laboratories, Inc) and water ad libitum for 8 weeks.
Seven animals from each dietary salt group were treated once with mouse isotype IgG1 (50 μg IgG/kg BW; CAT#MAB002, clone 11711, R&D Systems, Inc) used as control (LS control [LSC] and HS control [HSC]), or with an anti‐marinobufagenin mAb (clone 3E9) intraperitoneally (50 μg IgG/kg BW) (groups LSAB and HSAB) at week 7 on HS or LS diets.32 Antibody was diluted in sterile 0.9% NaCl to the final concentration 50 μg IgG/mL. In our previous publications, 1 week of the marinobufagenin immunoneutralization exhibited antifibrotic effects in a chronic kidney disease model11 and reduced BP in Dahl‐S rats26 without the adverse effect. Echocardiographic measurements were assessed before and after the antibody treatment (below). Systolic BP was recorded by tail‐cuff plethysmography (IITC Life Science Inc) in conscious animals at baseline and before and after the anti‐marinobufagenin mAb administration. For each time point, 10 to 15 measurements of BP were performed for each rat; the average of at least 5 stable readings was used.
At the end of week 8, rats were placed in metabolic cages for 24 hours for measurement of water consumption and urine output. Animals were deeply anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and euthanized by exsanguination. Blood samples were collected for measurements of plasma marinobufagenin, serum titer of anti‐marinobufagenin mAb (below), and erythrocyte Na/K‐ATPase activity measurement (below). Wet weight of thoracic aortae, hearts, left ventricles, and kidneys were measured and expressed per BW. Tissues were collected for Western blotting analysis and for the estimation of gene expression by real‐time quantitative polymerase chain reaction (qPCR) and microarray analyses (below). The protein and mRNA profiles were assessed in renal medulla because renal tubular cells in the proximal straight tubule and thick ascending limb of Henle's loop are highly responsible for maintaining the salt and water balance of the blood via Na/K‐ATPase, which is a target for marinobufagenin.32, 33, 34
Concentration of urinary Na+ was measured with a Roche‐Hitachi 917 flame photometry (Roche). Urinary creatinine was estimated with a creatinine assay kit (Cayman Chemical). Plasma electrolytes and creatinine were measured by an i‐STAT analyzer (Abbott Laboratories). Fractional excretion of Na+ (FENa) was calculated as FENa=(uNa×pCr×100)/(pNa×uCr), where uNa and pNa are urine Na+ and plasma Na+ concentrations (mmol/L), respectively, uCr and pCr are urine creatinine and plasma creatinine concentrations (mmol/L), respectively, and expressed as percent. Creatinine clearance was calculated as (uCr×uVol)/(pCr×1440), where uCr and pCr are urine creatinine and plasma creatinine concentrations (mmol/L), respectively; uVol is volume of urine in mL produced during 24 hours, or 1440 minutes, and expressed as mL/min.
Isolated Cardiac Myocytes Studies
Ventricular myocytes were isolated from the hearts of 4‐ to 5‐month‐old male Dahl‐S rats (n=6) kept on an LS diet.35 Freshly prepared ventricular myocytes were cultured for 2 to 3 hours on the plates coated with 10 to 20 μg/mL Laminin (Sigma‐Aldrich Inc) in the culture medium M199 (Sigma‐Aldrich Inc).35 After the cells were attached they were washed 2 or 3 times with the culture medium and were treated for 24 hours with vehicle or with physiological doses of marinobufagenin (1 and 10 nmol/L) as described.15, 16 Cells were collected and used for Western blotting analysis (below).
Echocardiography
Evaluation of cardiac function and morphology was performed by transthoracic echocardiography (Sonos 5500, Hewlett‐Packard) before and after control or anti‐marinobufagenin mAb treatment of rats anesthetized by isoflurane (2% in oxygen)36 in a subcohort of Dahl‐S rats (n=5–7 per group). Heart images were obtained in 2‐dimensional mode in the parasternal long axis views. The measurements of LV posterior wall thickness and LV internal diameters were made in systole and diastole (LVIDs and LVIDd). LV percent fractional shortening was calculated as: fractional shortening=[(LVIDd−LVIDs)/LVIDd]×100%. Heart mass was calculated from the 2‐dimensional mode image.
For pulse wave velocity (PWV) measurement ECG leads were placed on 2 front legs and 1 rear leg, and ECG tracing was recorded. Aortic PWV was measured by the transit time method using a 12‐MHz Doppler probe (Sonos 5500) at the transverse aortic arch and the abdominal aorta. The distance (d) between 2 points was measured. The time at the transverse aortic arch (t1) and at the abdominal aorta (t2) was defined as the time from the peak of the ECG P wave to the foot of the velocity upstroke. The transit time (Δt) of the flow wave from the upper thoracic aorta to the lower abdominal aorta was determined as the time difference between 2 measurements (t2−t1). PWV was calculated as d/Δt (m/s). Each measurement of PWV represents the average of 5 independent determinations per rat.
Na/K‐ATPase Measurement
One mL of blood was used for the measurement of erythrocyte Na/K‐ATPase activity as previously reported in detail.32 Total ATPase activity was measured by the production of inorganic phosphate (Pi), and Na/K‐ATPase activity was estimated by the difference between ATPase activity in the presence and in the absence of 5 mmol/L ouabain.
Immunoassays
Plasma and urine samples were extracted on Sep‐Pak C18 cartridges (Waters) and used for the measurement of marinobufagenin concentration by marinobufagenin competitive fluoroimmunoassay, based on a monoclonal murine anti‐marinobufagenin 4G4 antibody, performed as recently described in detail.32 Serum titers of anti‐marinobufagenin mAb were determined via solid‐phase fluoroimmunoassay as previously reported.32 Twenty‐four‐hour excretion of marinobufagenin was calculated as marinobufagenin concentration (pmol/L)×24‐hour volume of urine (L) and expressed as pmol/24‐h. LV tissue level of hydroxyproline as a direct measure of total tissue collagen was estimated using the Sircol soluble collagen assay (Biocolor Ltd) and expressed as μg/g tissue.
Western Blotting Analyses
The LV, renal tissue, and ventricular myocytes were homogenized in radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology, Inc) and pretreated in sample buffer (Thermo Fisher Scientific/Invitrogen) for 5 minutes at 90°C for most proteins detection and for 5 minutes at 37°C for collagen detection. Solubilized proteins (30–40 μg protein per lane) were separated by 4% to 12% Tris‐Glycine polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane (GE Healthcare/Life Sciences). The proteins were visualized using rabbit monoclonal p42/44 mitogen‐activated protein kinase (MAPK) (ERK1/2; 1:500; CAT# 4695), rabbit polyclonal TGFβ‐1 (1:500; CAT# 3709), rabbit monoclonal Mothers Against DPP Homolog (SMAD) 2 (1:1000; CAT# 5339), rabbit polyclonal SMAD4 (1:250; CAT# 9515), rabbit polyclonal MAPK3 (MAPKAPK‐3; 1:250; CAT# 3043) antibodies from Cell Signaling Technology; goat polyclonal collagen‐1 antibody (1:500; CAT# 1310‐01; Southern Biotechnology); rabbit polyclonal protein kinase C (PKC) δ (1:1000; CAT# sc‐937), phospho‐PKCδ‐Ser643/676 (1:1000; CAT# 9376), rabbit polyclonal Friend leukemia integration 1 transcription factor (Fli‐1) antibody (1:100; CAT# sc‐356) antibodies from Santa Cruz Biotechnology, Inc, followed by incubation with peroxidase‐conjugated goat anti‐rabbit antibody (1:10 000; CAT# NA934V; GE Healthcare/Life Sciences), or donkey anti‐goat antiserum (1:1000; CAT# sc‐2020; Santa Cruz, Biotechnology, Inc). Bands were visualized by 1‐ to 15‐minute exposure of nitrocellulose membrane on Premium Blue X‐ray film (Phenix Research Products), and optical density was quantified by the laser densitometry (Kodak Molecular Imaging Software, version 5.0). To normalize levels of proteins against levels of glyceraldehydes‐3‐phosphate dehydrogenase (GAPDH), membranes were stripped and reprobed with a rabbit monoclonal anti‐GAPDH (14C10) antibody (1:1000; CAT# 2118; Cell Signaling Technology).
Total RNA Purification and Real‐Time qPCR
Real‐time qPCR analysis of COL1a2, COL3a1, COL4a1, TGFb1, TGFb2, CTGF1, FN1, MAPK1, SMAD3, and SMAD4 mRNA levels in the left ventricle and kidney was performed by amplification of the resulting cDNAs and normalized to expression of the housekeeping gene (GAPDH) mRNA as an internal standard. In detail, total RNA was purified from the left ventricle (50–60 μg per sample) and renal tissue (50 μg per sample) and using the Qiagen RNeasy Mini Kit (Qiagen Inc). Total RNA samples were then reverse‐transcribed to cDNA using the TaqMan Reverse Transcription Kit (Qiagen Inc). Information for the gene primers and the housekeeping gene GAPDH (Qiagen Inc) used for qPCR is presented in Table 1. qPCR was performed with QuantiFast SYBR Green PCR Kit (Qiagen) in accordance with the manufacturer's protocol with an ABI 7300 Real‐Time PCR System (Life Technologies/Applied Biosystems).
Table 1.
Primers Used for Quantitative Real‐Time Polymerase Chain Reaction Analysis
| Gene | Primer |
|---|---|
| Collagen1a2 rat | Rn_Col1a2_1_SG QuantiTect Primer Assay (Qiagen) |
| Collagen3a1 rat | Rn_Col3a1_1_SG QuantiTect Primer Assay (Qiagen) |
| Collagen4a1 rat | Rn_Col4a1_1_SG QuantiTect Primer Assay (Qiagen) |
| CTGF1 rat | Rn_Ctgf_1_SG QuantiTect Primer Assay (Qiagen) |
| GAPDH rat | Rn_Gapd_1_SG QuantiTect Primer Assay (Qiagen) |
| Fibronectin1 rat | Rn_Fn1_1_SG QuantiTect Primer Assay (Qiagen) |
| MAPK1 rat | Rn_Mapk1_1_SG QuantiTect Primer Assay (Qiagen) |
| SMAD3 rat | Rn_Madh3_1_SG QuantiTect Primer Assay (Qiagen) |
| SMAD4 rat | Rn_Madh4_1_SG QuantiTect Primer Assay (Qiagen) |
| TGFbeta1 rat | Rn_Tgfb1_1_SG QuantiTect Primer Assay (Qiagen) |
| TGFbeta2 rat | Rn_Tgfb2_1_SG QuantiTect Primer Assay (Qiagen) |
Gene expression was analyzed in each sample by the following protocol: activation at 95°C (8 minutes) followed by 40 cycles consisting of a first phase of denaturation at 95°C (10 seconds), and a second phase of annealing/extending at 60°C (30 seconds). Each reaction was performed in triplicate with an inclusion of nontemplate controls in each experiment. A dissociation curve analysis was performed in each experiment to eliminate nonspecific amplification, including primer dimers. The GAPDH C t values were subtracted from the raw sample C t values to obtain the corrected C t. Power conversion (power (2−(correctedCt)) was used to convert corrected Ct to relative RNA quantity. Before microarray analysis, the RNA quality and quantity were checked with an Agilent 2100 Bioanalyzer and RNA nano‐chips.
Microarray Data Analysis
Total RNA was examined by Agilent Bioanalyzer (Agilent) to establish RNA quantity and quality. Biotinylated, amplified cRNA was generated by reverse transcribing 500 ug RNA into cDNA, and incorporating biotin in the process of converting cDNA into cRNA, using the Illumina TotalPrep RNA amplification kit (Cat # AMIL1791; Illumina). The biotinylated cRNA was hybridized to Illumina RatRef‐12 BeadArrays and visualized using a streptavidin‐conjugated Cy3‐labeled fluorescent reporter. Microarray data were analyzed as previously described37, 38 with DIANE 6.0.SUITE on JMP11 platform. Average raw microarray signals on each probe were first subjected to filtering by detection P≤0.02 in at least 1 sample as present and converted to its nature log value to obtain normal distribution form on each sample. These normal distributions were further standardized by Z‐transform to obtain Z‐score normalized gene expression value for statistical analysis. Samples were tested by hierarchical clustering, sample grouped principal component analysis, and sample correlation scatter plots to exclude possible outliners before combining them into different genotyping/treatment groups. Then, independent 1‐way ANOVA on sample groups was utilized to identify highly variance probes within each sample group, ANOVA F test P≤0.05 was used as the data global level variance control. Pairwise Z‐test was employed with the false discovery rate as the multiple comparison correction. Individual genes were selected by employing the following parameters: |Z‐ratio| ≥1.5, Z‐test P≤0.05, and false discovery rate ≤0.3. In addition, the average Z‐score on all comparison values in this gene should be more than 0 to avoid comparing between extremely small amount gene expression groups. Genes meeting the above criteria were considered significantly changed.
The Parametric Analysis of Gene Enrichment (PAGE) algorithm37 was employed for gene set enrichment analysis by using the Z‐ratio of all genes in each sample or each comparison as input against the data set supplied by Gene Ontology Institute39 and pathway gene set of MIT Broad Institute molecular signature database v5.1.40 For each gene set (pathway or gene ontology), the enrichment Z‐scores for each functional grouping were calculated based on the difference of the average Z‐ratio of the individual gene in the gene set to the whole comparison average Z‐ratio normalized by the standard derivation of the Z‐ratio within this gene set to the entire genes on the microarray. For each specific comparison pair, we performed Z‐test of the Z‐ratio values for the genes in the gene set to all individual genes’ Z‐ratio values of the microarray, followed by calculation of a Z‐score aggregation on this gene set to describe the scale change.38 The formula used to calculate the Z‐ratio38 is presented below:
where T is treatment group; C is control group; zscore(T) is the zscore of the treatment group T: {zscore Ti}, i=1,..,nt (nt is the number of samples in the treatment group T); is the mean of the zscores for treatment group T; zscore(C) is the zscore of the control group C: {zscore Ci}, i=1,..,nc (nc is the number of samples in the treatment group C); is the mean of the zscores for control group C; is the standard derivation of the difference between the treatment group zscore(T) average to the control group zscore(C) average.38 In the present study the Z‐ratio was calculated for 2 pairwise comparisons: (1) T=HSC versus C=LSC (n=6 per group), and (2) T=HSAB versus C=HSC (n=6 per group).
Thus, Z‐test–generated values, P≤0.05 with false discovery rate ≤0.3, were used as the cutoff criteria for the selection of significant gene set, and each gene set should have at least 3 genes mapped on the microarray. The gene set could be pathway, gene ontology, or any phenotype terms that were collected by a literature mining process. The significant selected gene set (Pathways or GO ontologies) were selected and further grouped according to their interaction categories. If needed, genes for some specific interested gene set were brought into a commercial pathway analysis suite, such as Ingenuity Pathway Analysis Suite or Pathway Studio Suite, to explore details of gene‐protein interaction networks and disease associations.
Accession Numbers
The microarray GEO accession numbers for the data reported in this article is GSE62376. This is the super series that includes all 5 experiments, each with their own numbers (links are not released).
Statistical Analysis
Results are reported as mean±standard error of the mean. Shapiro–Wilk normality tests (GraphPad Prism software; GraphPad Inc) were conducted for each sample and each variable. All samples passed the normality test (α>0.05). Next, the Bartlett test for equal variances detected that the variances did not differ significantly among the groups. Because our data were consistent with normal distributions with constant variance, the parametric ANOVA was applied for data analysis: 1‐way ANOVA, followed by Newman–Keuls test or t test where applicable (GraphPad Prism software). A 2‐sided P value of <0.05 was considered significant.
Results
Effect of Anti‐Marinobufagenin mAb on Clinical, Physiological, and Biochemical Parameters in Hypertensive Dahl‐S Rats
The physiological parameters assessed in this study are presented in Table 2. Following 8 weeks of HS intake, Dahl‐S rats had lower BW and higher systolic BP compared with the animals on an LS intake. Erythrocyte Na/K‐ATPase activity was lower and plasma marinobufagenin was 2‐fold higher in the HSC versus the LSC group. Urine and water volume, total 24‐hour Na+ excretion, and FENa increased, urine creatinine and creatinine clearance decreased, and plasma creatinine was unchanged in the HSC versus the LSC group. The volumetric ratio of urine to water was higher in the HSC versus the LCS group by t test (Table 2). Hypertensive Dahl‐S rats administered anti‐marinobufagenin mAb during the last week of HS intake (HSAB), exhibited reduced systolic BP (by 24 mm Hg), plasma marinobufagenin (by 33%), plasma creatinine (by 27%), urine volume (by 14%), total Na+ excretion (by 17%), and FENa (by 38%), and increased urine creatinine clearance (1.5‐fold) and erythrocyte Na/K‐ATPase activity (1.7‐fold) compared with nontreated HSC (Table 2). Following 1 week of the intraperitoneal administration of anti‐marinobufagenin mAb, titer of specific IgG in the serum of the treated rats was high and exceeded 1:10 000.
Table 2.
Clinical, Physiological, and Biochemical Parameters in Dahl‐S Rats Following 8 Weeks of a High Salt Diet or a Low Salt Diet With and Without Administration of Monoclonal Anti‐Marinobufagenin 3E9 Antibody
| LSC, n=7 | LSAB, n=7 | HSC, n=7 | HSAB, n=7 | |
|---|---|---|---|---|
| Body weight, g | 408±9 | 409±13 | 366±12* | 373±10 |
| SBP, mm Hg | 133±3 | 134±3 | 211±8* | 187±3*, ‡ |
| HR, beats per min | 417±10 | 390±11 | 423±20 | 407±9 |
| Erythrocyte Na/K‐ATPase, μmol Pi/mL per h | 9.61±0.76 | 9.12±0.29 | 5.81±0.21† | 10.29±0.8‡ 1 |
| Plasma marinobufagenin, nmol/L | 0.49±0.07 | 0.38±0.091 | 0.96±0.08 * | 0.64±0.06‡ |
| Plasma Na+, mmol/L | 140.4±0.9 | 142.0±0.7 | 141.7±0.9 | 143.2±0.5 |
| Plasma K+, mmol/L | 4.8±0.17 | 4.45±0.08 | 4.04±0.08 | 4.15±0.03 |
| Hematocrit, % | 43.2±1.1 | 38.5±0.8 | 37.7±2.0 | 40.3±1.01 |
| Plasma creatinine, μmol/L | 39.79±1.981 | 41.68±1.63 | 42.95±3.57 | 30.95±3.79*, ‡ 1 |
| Urine volume, mL/24 h | 15±2 | 19±4 | 86±4† | 74±6†, § 1 |
| Water volume, mL/24 h | 23±3 | 25±4 | 109±4† | 92±4†, § 1 |
| Urine/water ratio | 0.66±0.05 | 0.76±0.10 | 0.79±0.02∥ | 0.81±0.051 |
| Urine Na+, mmoL/24 h | 0.84±0.06 | 0.75±0.05 | 32.1±3.2* | 26.6±2.2*, ‡ 1 |
| Urine K+, mmol/24 h | 2.20±0.19 | 2.11±0.29 | 2.24±0.12 | 2.08±0.051 |
| Urine creatinine, mmol/L | 11.95±1.63 | 10.38±1.85 | 1.42±0.16† | 1.71±0.12† 1 |
| Creatinine clearance, mL/min | 2.61±0.181 | 2.74±0.18 | 1.94±0.24* | 2.82±0.22*, ‡ 1 |
| FENa, % | 0.14±0.011 | 0.16±0.01 | 8.52±0.86* | 5.30±0.67*, ‡ 1 |
Values are expressed as mean±standard error of the mean at termination (week 8) of the experiment. Dahl‐S indicates Dahl salt‐sensitive; FENa, fractional excretion of Na+; HR, heart rate; LSAB, low salt (LS) with anti‐marinobufagenin antibody (AB); SBP, systolic blood pressure.
*P<0.05, † P<0.01 vs low salt control (LSC).
‡ P<0.05, § P<0.01, effect of anti‐marinobufagenin monoclonal antibody (AB) in high salt (HS) (HSAB) vs high salt control (HSC), by 1‐way ANOVA followed by Newman–Keuls post hoc test, which was used to compare 4 groups defined by 2 factors.
P<0.05 by t test vs LSC (1n=6).
Urinary marinobufagenin excretion was 5‐fold greater in HSC, compared with LSC (HSC, mean marinobufagenin 31.1±3.4 pmol/24 h; LSC, mean marinobufagenin 6.2±0.6 pmol/24 h [P<0.01]), and it was significantly reduced by anti‐marinobufagenin mAb in HSAB versus HSC (HSAB, mean marinobufagenin 18.9±2.9 pmol/24 h; HSC, mean marinobufagenin 31.1±3.4 pmol/24 h [P<0.01]) (Figure 1A). Notably, the anti‐marinobufagenin mAb treatment did not affect urine marinobufagenin level in LSAB compared with nontreated LSC (LSAB, mean marinobufagenin 5.0±0.7 pmol/24 h; LSC, mean marinobufagenin 6.2±0.6 pmol/24 h [P=not significant]).
Figure 1.
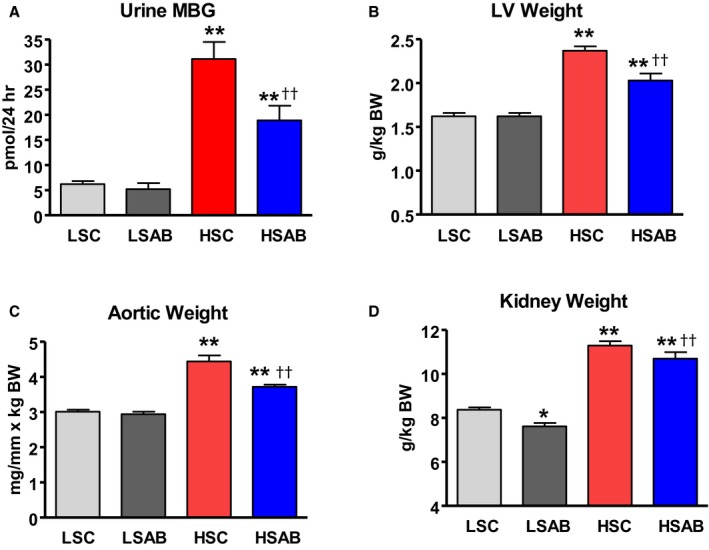
Changes in physiological and biochemical parameters in Dahl salt‐sensitive (Dahl‐S) rats on a high salt (HS) and low salt (LS) diet in the presence and absence of anti‐marinobufagenin antibody (mAb) treatment. A, Marinobufagenin in urine. B through D, Wet weight of left ventricles (LV), aortae, and kidneys correspondently, expressed per body weight (BW) at the end of week 8 of LS and HS diets in Dahl‐S rats treated with control antibody (LS control [LSC] and HS control [HSC]) and with anti‐marinobufagenin mAb (LSAB and HSAB, respectively), n=7 per group. Each bar represents the mean±standard error of the mean. By 1‐way ANOVA followed by Neuman–Keuls test: *P<0.05, **P<0.01, 1 of other groups vs LSC; †† P<0.01, HSAB vs HSC.
Weights of organs, expressed as a wet weight per kg of BW, were significantly higher after 8 weeks on HS diet versus those from the LSC group (HSC, mean LV weight 2.37±0.09 g/kg BW; LSC, mean LV weight 1.66±0.05 g/kg BW [P<0.01]; HSC, mean aortic weight 4.44±0.23 mg/mm×kg BW; LSC, mean aortic weight 3.07±0.08 mg/mm×kg BW [P<0.01]; HSC, mean kidneys weight 11.40±0.23 g/kg BW; LSC, mean kidneys weight 8.53±0.19 g/kg BW [P<0.01], Figure 1B through 1D). LV, aortic, and kidney weights substantially decreased after anti‐marinobufagenin mAb treatment in HSAB compared with nontreated HSC (HSAB, mean LV weight 2.05±0.08 g/kg BW; HSC, mean LV weight 2.37±0.09 g/kg BW [P<0.01]; HSAB, mean aortic weight 3.76±0.08 mg/mm×kg BW; HSC mean aortic weight 4.44±0.23 mg/mm×kg BW [P<0.01]; HSAB, mean kidneys weight 10.72±0.27 g/kg BW; HSC, mean kidneys weight 11.40±0.23 g/kg BW [P<0.01], Figure 1B through 1D). Treatment with anti‐marinobufagenin mAb did not affect LV or aortic weights in LSAB; however, kidney weights were reduced compared with nontreated LSC (LSAB, mean kidneys weight 7.78±0.22 g/kg BW; LSC, mean kidneys weight 8.53±0.19 g/kg BW [P<0.05]) (Figure 1D). Kidney function declined after HS intake (HSC) compared with the LSC group, ie, creatinine clearance was lower and FENa was higher in the HSC group versus the LSC group (Table 2). Treatment of hypertensive Dahl‐S rats (HSAB) with anti‐marinobufagenin mAb significantly increased creatinine clearance (Table 2), indicating the normalization of kidney function after the immunoneutralization of marinobufagenin.
Effect of Anti‐Marinobufagenin mAb on Cardiovascular Remodeling by Echocardiography
Cardiovascular remodeling in Dahl‐S rats on HS diet was accompanied by changes in PWV and in heart structure and function, as determined by echocardiography measurements (Table 3). LV internal diameter in systole and diastole decreased, and LV posterior wall thickness, LV, and heart weights increased in HSC versus LSC. This indicated the beginning of the development of a compensated hypertrophy stage in hypertensive Dahl‐S rats (Table 3). There was a continued decrease in fractional shortening, an increase in LV internal diameter at end systole and end diastole (LVIDd and LVIDs), and an increase in LV posterior wall thickness at diastole in the same group of Dahl‐S rats on HS diet from week 7 to the end of week 8 of HS intake (HSC‐7 and HSC‐8; Table 3). This indicated the initial stage of the development of HF. Treatment of hypertensive Dahl‐S rats with anti‐marinobufagenin mAb in (HSAB‐8) attenuated these changes versus the nontreated group (HSC‐8) (Table 3). Thus, immunoneutralization of marinobufagenin reversed the development of HF in hypertensive Dahl‐S rats versus nontreated hypertensive animals. Heart and LV weight, estimated by echocardiography, significantly increased in vehicle‐treated Dahl‐S rats at week 7 of HS versus LSC and continued to increase during week 8 of HS intake. Anti‐marinobufagenin mAb treatment reversed this increase in heart and LV mass (HSAB‐8 versus HSC‐8 [Table 3]). PWV increased in the HSC group compared with the LSC group and was significantly lower after the immunoneutralization of marinobufagenin in the HSAB group versus the HSC group (Table 3).
Table 3.
Effect of Monoclonal Anti‐Marinobufagenin 3E9 Antibody Administration During the Last Week of Dietary Intervention on Heart Structure and Function in Dahl‐S Rats on High NaCl Intake (by Echocardiography)
| LSC‐7, n=7 | HSC‐7 (Before Treatment), n=6 | HSC‐8 (After Control Antibody Treatment), n=6 | HSAB‐7 (Before Treatment), n=6 | HSAB‐8 (After Anti‐Marinobufagenin mAb Treatment), n=6 | |
|---|---|---|---|---|---|
| FS, % | 51.8±0.5 | 54.5±0.8* | 49.8±0.4§ | 54.8±0.9* | 56.1±1.1¶ |
| LVIDd, mm | 7.45±0.06 | 6.71±0.05† | 7.13±0.03§ | 6.67±0.05† | 6.81±0.09¶ |
| LVIDs, mm | 3.60±0.05 | 3.06±0.06† | 3.58±0.04§ | 3.02±0.06† | 2.99±0.08¶ |
| LVPWd, mm | 1.95±0.05 | 2.94±0.07† | 2.57±0.07*, ‡ | 2.88±0.04† | 2.75±0.06∥ |
| LVW/BW, g/kg | 1.37±0.02 | 1.66±0.04† | 1.84±0.06*, ‡ | 1.61±0.04† | 1.65±0.05∥ |
| HW/BW, g/kg | 3.90±0.12 | 4.95±0.19† | 5.54±0.11*, ‡ | 4.90±0.13† | 5.00±0.12∥ |
| PWV, m/s | 2.6±0.3 | 4.8±0.2* 1 | 6.3±0.7* 1 | 5.3±0.8* | 3.6±0.3∥ |
Values are expressed as mean±standard error of the mean at week 7 of low sodium or high sodium (HS) intake, and at the end of week 8 of HS intake. HSC‐7 and HSC‐8 are the same groups of Dahl salt‐sensitive (Dahl‐S) rats on HS intake before and after control IgG treatment. HSAB‐7 and HSAB‐8 are the same groups of Dahl‐S rats on HS intake before and after anti‐marinobufagenin antibody (mAb) treatment. BW indicates body weight; FS, fractional shortening; HW, heart weight; LVIDd, left ventricular internal diameter at end diastole; LVIDs, left ventricular internal diameter at end systole; LVPWd, left ventricular posterior wall thickness at diastole; LVW, left ventricular weight; PWV, pulse wave velocity (1n=5).
*P<0.05, † P<0.01, effect of HS vs low salt control (LSC)‐7.
‡ P<0.05, § P<0.01, effect of HS intake in vehicle‐treated rats (intragroup: HSC‐8 vs HSC‐7).
∥ P<0.05, ¶ P<0.01, effect of anti‐marinobufagenin mAb at the termination of study vs vehicle‐treated hypertensive animals (intergroup: HSAB‐8 vs HSC‐8), by 1‐way ANOVA followed by Newman–Keuls test, to compare 5 groups defined by 3 factors.
Effect of Anti‐Marinobufagenin Antibody on mRNA and Protein Expression in Renal Medulla and the Left Ventricle
PAGE data analyses of gene expression change as Z‐ratio of mRNA expression in pairs HSC/LSC and HSAB/HSC for the left ventricle and kidneys are presented in Tables 4 and 5. Z‐ratio is calculated from Z‐scores (see Methods for details). Z‐ratio method is used for computing significant fold changes in gene expression between HSC and LSC and between HSAB and HSC. Expression of genes implicated in TGFβ profibrotic signaling was significantly upregulated by HS intake versus LS in both the left ventricle and kidneys (column HSC/LSC). Anti‐marinobufagenin mAb treatment reversed the expression of these genes in the left ventricle and kidney in HSAB versus HSC (Tables 4 and 5; column HSAB/HSC). Sparc, a gene encoding osteonectin, a promoter of calcification and a marker of vascular remodeling,41, 42 was upregulated by HS intake in HSC versus LSC, and downregulated by anti‐marinobufagenin mAb treatment in both the left ventricle (Table 4) and kidneys (Table 5). In addition, gene MGP (matrix Gla protein), whose active form inhibits calcification, was also upregulated in the kidneys of HSC versus LSC (Table 5). Expression of TIMP1, TIMP2, and TIMP3 mRNAs was significantly higher in the renal tissue in HSC versus LSC and reversed with anti‐marinobufagenin mAb treatment (HSAB).
Table 4.
Effect of High Salt Diet and Monoclonal Anti‐Marinobufagenin 3E9 Antibody on Extracellular Matrix and Transcription Factors in the Left Ventricle of Dahl‐S Rats Following 8 Weeks of Dietary Intervention Combined With 1 Week of Anti‐Marinobufagenin mAb Treatment (Pair Analysis of Gene Expression by Microarray)
| Gene | Description | Z‐Ratio | |
|---|---|---|---|
| HSC/LSC | HSAB/HSC | ||
| Ctgf | Connective tissue growth factor | 2.04* | −4.10*, ‡ |
| Tgfb1 | Transforming growth factor‐β1 | 1.72* | −1.82*, ‡ |
| Tgfb2 | Transforming growth factor‐β2 | 2.57† | −3.9†, ‡ |
| MAPK3 | Mitogen‐activated protein kinase 3 | 1.64* | −3.17†, ‡ |
| Sparc | Secreted acidic cysteine rich glycoprotein/osteonectin | 2.08† | −1.64*, ‡ |
Values are expressed as the Z‐ratio of gene expression at termination (week 8) of the experiment. Dahl‐S indicates Dahl salt‐sensitive; AB, marinobufagenin antibody.
*P<0.05, † P<0.01, changes in Z‐ratio within pairs.
P<0.05, high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB)/high salt control (HSC) vs HSC/low salt control (LSC) by unpaired t test; n=6 per group.
Table 5.
Effect of High Salt Diet and Monoclonal Anti‐Marinobufagenin 3E9 Antibody on Extracellular Matrix and Transcription Factors in Kidneys of Dahl‐S Rats Following 8 Weeks of Dietary Intervention Combined With 1 Week of Anti‐Marinobufagenin mAb Treatment (Pair Analysis of Gene Expression by Microarray)
| Gene | Description | Z‐Ratio | |
|---|---|---|---|
| HSC/LSC | HSAB/HSC | ||
| Col1a1 | Collagen, type I, alpha 1 | 4.51† | −4.14†, ‡ |
| Col1a2 | Collagen, type I, alpha 2 | 4.27† | −4.18†, ‡ |
| Col3a1 | Collagen, type III, alpha 1 | 6.17† | −5.16†, ‡ |
| Col4a1 | Collagen, type IV, alpha 1 | 3.48† | −3.17†, ‡ |
| Col5a1 | Collagen, type V, alpha 1 | 3.97† | −2.99†, ‡ |
| Col5a2 | Collagen, type V, alpha 2 | 1.84* | −2.33*, ‡ |
| Col12a1 | Collagen, type XII, alpha 1 | 2.49† | −2.15*, ‡ |
| Pcolce | Procollagen C‐endopeptidase enhancer protein | 3.7† | −2.36*, ‡ |
| Tgfb1 | Transforming growth factor‐β1 | 2.47† | −3.16†, ‡ |
| Tgfb3 | Transforming growth factor‐β3 | 1.78* | −2.3‡ |
| Fn1 | Fibronectin 1 | 6.32† | −3.96*, ‡ |
| Smad1 | Mothers Against DPP Homolog 1 | 1.61* | −0.93 |
| Mmp12 | Matrix metallopeptidase 12 | 1.86* | −2.29*, ‡ |
| Timp1 | Tissue inhibitor of metalloproteinase 1 | 10.72† | −7.50†, ‡ |
| Timp2 | Tissue inhibitor of metalloproteinase 2 | 2.53† | −3.15†, ‡ |
| Timp3 | Tissue inhibitor of metalloproteinase 3 | 2.51* | −4.77*, ‡ |
| MGP | Matrix Gla protein | 6.99† | −6.11†, ‡ |
| Sparc | Secreted acidic cysteine‐rich glycoprotein/osteonectin | 4.62† | −5.57†, ‡ |
Values are expressed as Z‐ratio of gene expression at termination (week 8) of the experiment.
*P<0.05, † P<0.01, changes in Z‐ratio within pairs.
‡P<0.05, hypertensive Dahl salt‐sensitive (Dahl‐S) rats on high salt intake (HS) with anti‐marinobufagenin antibody (AB) (HSAB)/high salt control (HSC) vs HSC/low salt control (LSC) by unpaired t test; n=6 per group.
Abundance of proteins implicated in the TGFβ profibrotic signaling was estimated in the left ventricle by Western blotting (Figure 2) and validated by qPCR analysis (Figure 3) and in the kidneys by Western blotting (Figure 4) and validated by qPCR analysis (Figure 5). The abundance of signaling proteins implicated in the TGFβ pathway, ie, TGFβ‐1 and SMAD4, collagen‐1, and ERK1/2 and MAPK3, were higher in the left ventricle in hypertensive HSC versus LSC and were downregulated to the control levels in HSAB versus HSC (Figure 2). Fli‐1 did not change in HSC, nor in HSAB versus control. Cardiac hydroxyproline level, a direct measure of the total amount of tissue collagen, was higher in the hypertrophied left ventricle in HSC compared with LSC (HSC, mean hydroxyproline level, 130±7 μg/g tissue; LSC, mean hydroxyproline level, 90±5 μg/g tissue [P<0.01]), and significantly decreased after immunoneutralization of marinobufagenin (HSAB, mean hydroxyproline level, 88±8 μg/g tissue; HSC, mean hydroxyproline level, 130±7 μg/g tissue [P<0.01]). The phosphorylated form of PKCδ was detected at a higher amount in HSC versus LSC in the presence of unchanged total PKCδ abundance, and anti‐marinobufagenin mAb reversed phosphorylation of this protein to the control level, making PKCδ less active (Figure 2F).
Figure 2.
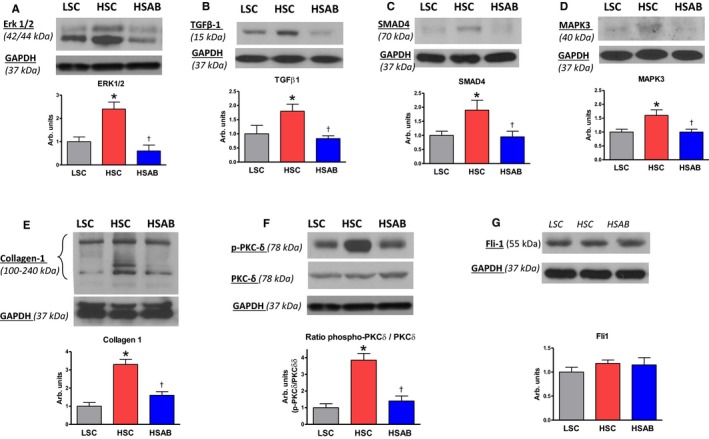
Left ventricle: Effect of a high salt (HS) diet and anti‐marinobufagenin antibody treatment on ERK1/2, transforming growth factor‐β1 (TGFβ‐1), Mothers Against DPP Homolog (SMAD) 4, mitogen‐activated protein kinase 3 (MAPK3), collagen‐1, and protein kinase C (PKC) δ protein abundance by Western blotting analysis in Dahl salt‐sensitive (Dahl‐S) rats. Top panels, Western blotting representative bands; bottom panels, statistical analysis of band density standardized for GAPDH for: (A) total ERK1/2; (B) TGFβ‐1 (total; mature form); (C) total SMAD4; (D) total MAPK3; (E) collagen‐1; (F) the ratio of phosphorylated PKCδ (p‐PKCδ) to total PKCδ protein; and (G) total Friend leukemia integration 1 transcription factor (Fli‐1) in the left ventricles of Dahl‐S rats. Each bar represents the mean±standard error of the mean of at least 3 measurements, n=7 per group. By 1‐way ANOVA followed by Neuman–Keuls post hoc test: *P<0.05, HS control [HSC] vs low salt control [LSC]; † P 0.05, high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB) vs HSC.
Figure 3.
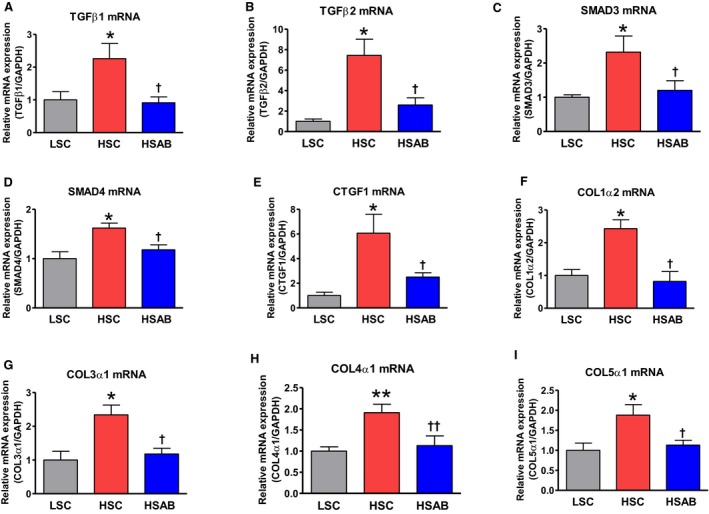
Left ventricle: effect of a high salt (HS) diet and anti‐marinobufagenin antibody treatment on mRNAs in Dahl salt‐sensitive (Dahl‐S) rats. A, Transforming growth factor (TGF) β‐1; (B) TGFβ‐2; (C) Mothers Against DPP Homolog (SMAD) 3; (D) SMAD4; (E) CTGF1; (F) COL1α2; (G) COL3α1; (H) COL4α1; and (I) COL5α1 mRNAs in the left ventricles of Dahl‐S by quantitative polymerase chain reaction analysis. Each bar represents the mean±standard error of the mean of 5 to 6 measurements, n=7 per group. By 1‐way ANOVA followed by Neuman–Keuls post hoc test: *P<0.05, **P<0.01, HS control [HSC] vs low salt control [LSC]; † P<0.05, †† P<0.01, high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB) vs HSC.
Figure 4.
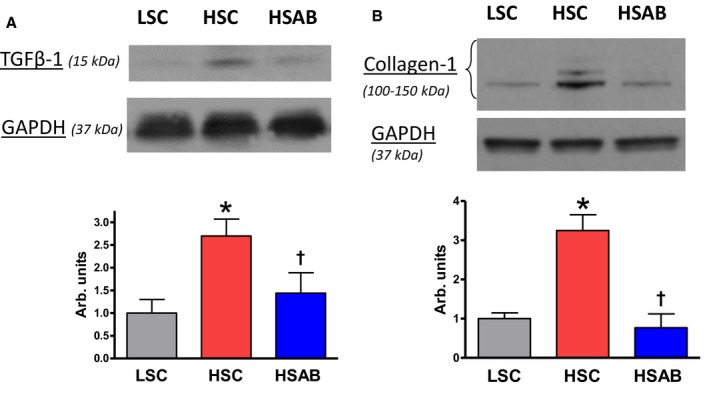
Renal medulla: Effect of a high salt (HS) diet and anti‐marinobufagenin antibody treatment on renal transforming growth factor (TGF) β‐1 (total; mature form) and collagen‐1 protein abundance by Western blotting analysis in Dahl salt‐sensitive (Dahl‐S) rats. Top: Western blotting representative bands. Bottom: Statistical analysis of band density standardized for GAPDH. A, TGFβ‐1; and (B) collagen‐1 in the renal medulla of Dahl‐S rats. Each bar represents the mean±standard error of the mean of at least 3 measurements, n=7 per group. By 1‐way ANOVA followed by Neuman–Keuls post hoc test: *P<0.05, HS control [HSC] vs low salt control [LSC]; † P<0.05, high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB) vs HSC.
Figure 5.
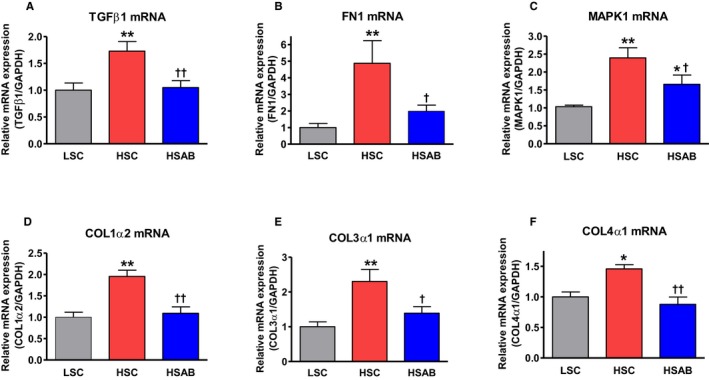
Renal medulla: Effect of a high salt (HS) diet and anti‐marinobufagenin antibody treatment on renal mRNAs. A, Transforming growth factor (TGF) β‐1; (B) fibronectin 1 (FN1); (C) mitogen‐activated protein kinase 1 (MAPK1); (D) COL1α2; (E) COL3α1; and (F) COL4α1 in Dahl salt‐sensitive rats by quantitative polymerase chain reaction analysis. Each bar represents the mean±standard error of the mean of at least 5 measurements, n=7 per group. By 1‐way ANOVA followed by Neuman–Keuls post hoc test: *P<0.05, **P<0.01 one of the other groups vs low salt control [LSC]; † P<0.05, †† P<0.01, high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB) vs HS control (HSC).
The data, assessed via Western blotting in the left ventricle, supported the qPCR results, which demonstrated an upregulation of TGFb1, TGFb2, SMAD3, SMAD4, CTGF, Col1a2, Col3a1, Col4a1, and Col5a1 mRNAs in left ventricles from HSC compared with LSC (Figure 3). These genes were significantly downregulated after anti‐marinobufagenin mAb treatment (HSAB): TGFb1, TGFb2, SMAD3, SMAD4, CTGF, Col1a2, Col3a1, Col4a1, and Col5a1 (Figure 3A through 3I). The microarray data were supported by Western blotting and qPRC analyses.
The abundance of TGFβ‐1 and collagen‐1 proteins, estimated by Western blotting in kidneys, was increased in HSC versus LSC. Anti‐marinobufagenin mAb in vivo reversed the amount of these proteins in the renal tissue (Figure 4). qPCR analysis of renal tissue showed an increase of TGFβ‐1, FN1, MAPK1, Col1a2, Col3a1, and Col4a1 mRNA expression in HSC versus LSC (Figure 5A through 5F). These genes were downregulated after anti‐marinobufagenin mAb treatment in kidneys from HSAB versus HSC (Figure 5).
Heart Failure Network (Microarray Analysis)
The expression of genes related to the Heart Failure Network in the left ventricle, based on PAGE analysis, is presented in Figure 6. Among the genes upregulated during the development of LV hypertrophy and downregulated by anti‐marinobufagenin mAb are the genes related to TGFβ signaling, ie, TGFβ‐1 and TGFβ‐2, CTGF, MAPK3, SCN3B (sodium channel subunit beta‐3), and GRK5 (G protein–coupled receptor kinase 5; related to pathological LV hypertrophy). Some of the genes upregulated in Dahl‐S on an HS diet and related to HF, ie, NPPA (natriuretic peptide A) and NPPB (brain natriuretic peptide), were not affected by immunoneutralization of marinobufagenin, indicating a nondownstream location of these markers in the marinobufagenin‐Na/K‐ATPase profibrotic signaling. The gene expression Heart Failure Network profile shows genes that were upregulated (red) or downregulated (green) in the left ventricle: HSC versus LSC (effect of HS) and HSAB versus HSC (effect of anti‐marinobufagenin mAb) (Figure 6A and 6B).
Figure 6.
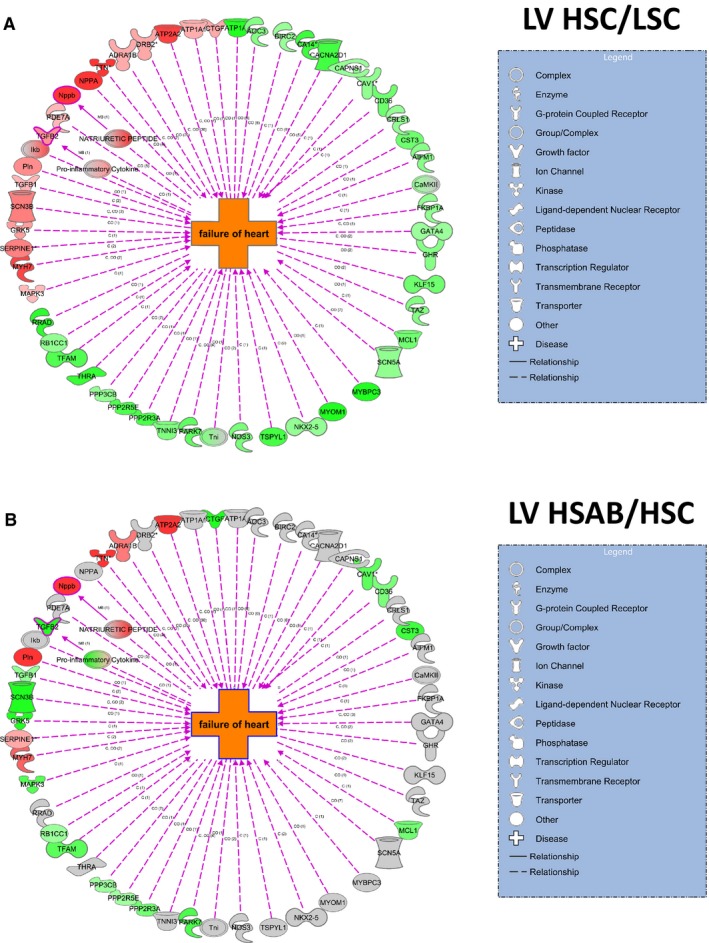
Effect of a high salt (HS) diet and anti‐marinobufagenin antibody treatment on the Heart Failure Network in the left ventricles of Dahl salt‐sensitive rats. Gene expression network profile shows genes in the Heart Failure Network that were upregulated (red) or downregulated (green) in left ventricular (LV) tissue HS control [HSC] vs low salt control [LSC] groups (A), and high salt (HS) with anti‐marinobufagenin antibody (AB) (HSAB) vs HSC groups (B). Green and red indicate significant (P<0.05) changes in gene expression between indicated group pairs; intensity of color indicates the magnitude of Z‐ratio between groups (the darkest color is for the highest Z‐ratio).
Profibrotic Effect of Marinobufagenin in Isolated Ventricular Myocytes
In vitro treatment of cultured ventricular myocytes from control Dahl‐S rats with 1 to 10 nmol/L marinobufagenin for 24 hours increased the abundance of proteins from TGFβ profibrotic signaling (Figure 7A through 7D) including TGFβ‐1, Smad2, Erk1/2, and collagen‐1. Phosphorylation of PKCδ was dose‐dependently stimulated by physiological concentrations of marinobufagenin in LV myocytes (Figure 7E). In addition, marinobufagenin downregulated a negative regulator of collagen‐1 synthesis, Fli‐1 (Figure 7F), which indicates the activation of Fli‐1 profibrotic pathway along with TGFβ signaling by marinobufagenin in the cultured LV myocytes.
Figure 7.
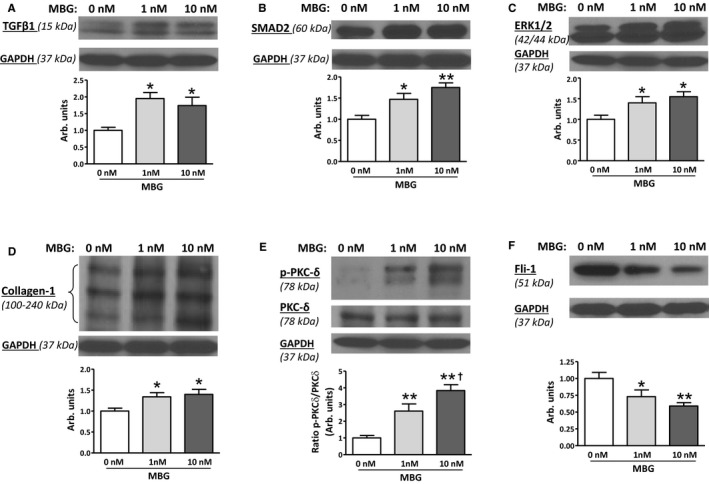
Profibrotic effect of marinobufagenin on cultured left ventricular myocytes of Dahl salt‐sensitive rats kept on a low salt (LS) diet (n=6). Addition of marinobufagenin (1 and 10 nmol/L) for 24 hours to the culture media–activated production of: (A) transforming growth factor (TGF) β‐1 (mature form); (B) Mothers Against DPP Homolog (SMAD) 2; (C) mitogen‐activated protein kinase (MAPK) 42/44; (D) collagen‐1; (E) increased amount of phosphorylated protein kinase C (PKC) δ, and (F) decreased level of Friend leukemia integration 1 transcription factor (Fli‐1). Top panels: representative Western blotting images; bottom panels: statistical analysis of band density for each protein standardized for GAPDH. Each bar represents the mean±standard error of the mean; each protein was tested at least 5 times for each condition. By 1‐way ANOVA followed by Neuman–Keuls post hoc test: *P<0.05, **P<0.01 one of the other groups vs baseline (0 nmol/L marinobufagenin); † P<0.05, 10 nmol/L vs 1 nmol/L marinobufagenin.
Discussion
The present study demonstrates the implication of an endogenous steroidal Na/K‐ATPase inhibitor, marinobufagenin, in the activation of TGFβ‐dependent signaling pathway in Dahl‐S hypertension via Na/K‐ATPase signal transduction. Immunoneutralization of heightened marinobufagenin by anti‐marinobufagenin mAb reversed upregulated profibrotic TGFβ signaling and hypertrophy in cardiovascular and renal tissues from hypertensive Dahl‐S rats. Anti‐marinobufagenin mAb exhibited pronounced cardiac and renal protective effects in hypertensive Dahl‐S rats. In addition, it was demonstrated that marinobufagenin activated TGFβ‐1 signaling in vitro in cultured cardiac ventricular myocytes from Dahl‐S rats.
Upregulated Marinobufagenin Activated TGFβ‐Dependent Tissue Fibrosis via Na/K‐ATPase Cascade
TGFβ‐dependent profibrotic signaling participates in cardiovascular and renal remodeling in hypertensive Dahl‐S rats,24, 25 because administration of anti‐TGFβ mAb to hypertensive Dahl‐S downregulates expression of genes in TGFβ signaling accompanied by antihypertensive and renoprotective effects.43, 44 The mechanism of stimulation of the TGFβ signaling pathway is attributed to several factors, including tissue angiotensin II,45, 46 which stimulates marinobufagenin in vivo and in vitro in a Dahl‐S model, as we previously demonstrated.7 In the present study the immunoneutralization of marinobufagenin restored erythrocyte Na/K‐ATPase activity in hypertensive Dahl‐S rats, indicating that the marinobufagenin‐Na/K‐ATPase complex is another link between angiotensin II and profibrotic signaling, as schematically presented in Figure 8, and proving that anti‐marinobufagenin mAb affects the TGFβ signaling upstream. HS intake was accompanied by a concomitant increase in marinobufagenin and erythrocyte Na/K‐ATPase inhibition compared with those in the LSC group. This observation is in agreement with our previous findings in the Dahl‐S rat model on LS intake.5, 19, 22 Because the immunoneutralization of marinobufagenin in hypertensive Dahl‐S rats resulted in the restoration of Na/K‐ATPase activity and downregulation of genes implicated in TGFβ‐dependent signaling, we conclude that marinobufagenin activates TGFβ profibrotic signaling via Na/K‐ATPase signal transduction (Figure 8). This present observation is supported by previous findings that marinobufagenin and other endogenous Na/K‐ATPase ligands mediate signal transduction via Na/K‐ATPase.47, 48, 49
Figure 8.
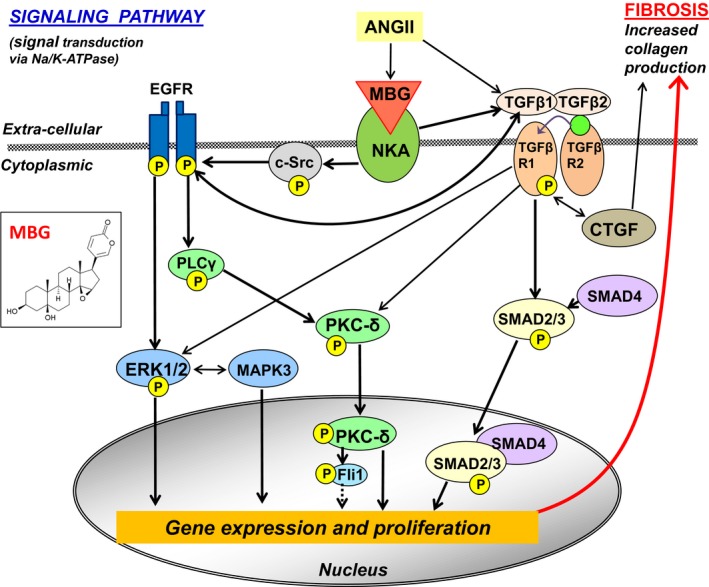
Schematic presentation of signaling (non‐Mothers Against DPP Homolog [SMAD] and SMAD‐dependent) pathways initiated by marinobufagenin via interaction with Na/K‐ATPase. ANGII indicates angiotensin II; c‐Src, proto‐oncogene tyrosine‐protein kinase; EGFR, epidermal growth factor receptor; ERK1/2, mitogen‐activated protein kinase (MAPK); Fli‐1, Friend leukemia integration 1 transcription factor, a negative regulator of collagen‐1 production; NKA, Na/K‐ATPase; PLCγ, phospholipase C gamma; PKCδ, protein kinase C delta; TGFβ, transforming growth factor‐β; TGFβR, TGFβ receptor. The dotted line indicates the release of the procollagen DNA promoter by phosphorylated Fli1; “P” in yellow circle, phosphorylated form of the protein. The chemical structure of marinobufagenin (MBG) is given in the insertion on the left.
TGFβ Profibrotic Signaling: Left Ventricle and Kidney Genes and Proteins Expression in a Dahl‐S Hypertension Model in the Presence of Anti‐Marinobufagenin mAb
In the present study, data on the microarray analysis of the genes implicated in the TGFβ pathway were supported by qPCR and Western blotting analyses. The schematic presentation of the signaling pathway induced by marinobufagenin via inhibition of Na/K‐ATPase is based on our present findings and those previously reported,9, 49, 50, 51, 52, 53, 54 and includes canonical (TGFβ/SMAD‐dependent), noncanonical (Erk1/2 MAPK), and crosstalk between TGFβ and other signaling pathways as schematically presented in Figure 8. The crosstalk of TGFβ and EGFR signaling pathways involves PKCδ, which was phosphorylated (ie, activated) in hypertensive Dahl‐S and dephosphorylated following immunoneutralization of marinobufagenin in the present study (Figure 2). PKCδ was dose‐dependently phosphorylated by marinobufagenin in the cultured cardiomyocytes (Figure 7E). These observations provide an additional indication that marinobufagenin is an important player in profibrotic signaling pathways involved in cardiovascular and renal remodeling in salt‐sensitive hypertension. Previously, we and others have reported the upregulation of PKCδ in LV sarcolemma from hypertensive Dahl‐S rats,55, 56 which indicate the important contribution of PKC in the pathogenesis of heart diseases.57, 58 The PKC family is a desirable target for treatment in the scenario of cardiac disorders including heart hypertrophy and HF. The present observation that the treatment of hypertensive rats with anti‐marinobufagenin mAb was associated with dephosphorylation, ie, deactivation, of PKCδ in LV sarcolemma merits future investigation. In addition, the finding that activated inhibitors of metalloproteinases, tissue inhibitor of metalloproteinases, along with activated TGFβ signaling (Table 5), were reversed by anti‐marinobufagenin mAb, indicates the complex causative relationship between regulators and mediators of fibrosis.
Effect of Anti‐Marinobufagenin mAb on Cardiac Function, Morphology, and PWV in a Dahl‐S Hypertension Model
Higher marinobufagenin levels are associated with increased arterial stiffness, indexed as PWV, and LV mass in clinical studies in patients with normotension59, 60 and patients with borderline hypertension.61 In the current study, the treatment with anti‐marinobufagenin mAb resulted in the improvement of vascular and cardiac function, ie, a decrease in aortic stiffness decrease estimated by reduced PWV, reduction of heart mass, and restoration of cardiac function estimated by echocardiography (Table 3). This restoration was likely caused by the decrease in the activity of TGFβ profibrotic signaling and in the decrease of tissue fibrosis by immunoneutralization of marinobufagenin, because systolic BP was moderately affected and remained high after treatment with anti‐marinobufagenin mAb in the present study. Our present findings are in agreement with previous observations, that passive and active immunoneutralization of heightened marinobufagenin reversed cardiac and renal remodeling in uremic cardiopathy models.11, 13 Anti‐marinobufagenin antibody exhibited an antihypertensive effect in the animal models of preeclampsia32, 62 and salt‐sensitive hypertension32 and reduced vascular fibrosis.63 The vessels from the animals on an HS diet, treated with an anti‐marinobufagenin mAb in vivo, exhibited improved vasorelaxation by sodium nitroprusside ex vivo versus the untreated aortae. This observation demonstrated a compromised endothelium‐independent vasorelaxation caused by a higher aortic wall collagen deposition.63 In the present study, the decreased aortic stiffness after marinobufagenin immunoneutralization is important in the normalization of vascular compliance by anti‐marinobufagenin mAb.
Renoprotective Effect of Anti‐Marinobufagenin mAb in Dahl‐S Hypertension Model
In the present study, the normalization of renal function by anti‐marinobufagenin mAb, estimated by creatinine clearance and FENa (Table 2), was likely attributable to a decrease of renal fibrosis because it was accompanied by a significant decrease in kidney weight, reduced abundance of collagen I, III, and IV, and downregulation of TGFβ profibrotic signaling. These changes in renal medulla were demonstrated by microarray, qPCR, and Western blotting analyses (Figures 4 and 5, Table 5), which is in agreement with our previous observations.11, 13, 15 In addition, kidney levels of FN1 mRNA, a protein, which provide a scaffold for the deposition of interstitial collagens,64 dramatically increased in hypertensive Dahl‐S rats on HS diet, and the anti‐marinobufagenin mAb treatment downregulated FN1 (Table 5). Renal hypertrophy observed in hypertensive rats was mostly caused by the activation of profibrotic TGFβ signaling, resulting in tissue fibrosis and probably the compensatory hyperplasia of the nondamaged nephrons, as suggested in the literature.65 The reduction of renal hypertrophy (Figure 2D) and restoration of renal function, registered as an increase in creatinine clearance in comparison to the HSC (Table 2), by immunoneutralization of marinobufagenin, provides a significant input in the normalization of cardiovascular performance even in the presence of elevated BP. Notably, after the immunoneutralization of marinobufagenin in Dahl‐S rats on an HS diet, total sodium excretion was reduced, which is expected, because marinobufagenin is a natriuretic hormone.5, 19, 22 Thus, the immunoneutralization of the profibrotic and natriuretic factor marinobufagenin manifested a compromised effect on renal function, ie, slightly reduced total 24‐hour sodium and urine excretion, and significantly improved renal function by reversing the profibrotic signaling and hypertrophy.
Profibrotic and natriuretic steroidal inhibitor of Na/K‐ATPase, marinobufagenin, fits the model of salt‐sensitive hypertension,4 playing a dual role as a natriuretic hormone and a profibrotic factor in the scenario of the compromised sodium renal excretion in Dahl‐S rats in comparison to their salt‐resistant counterparts.5, 19, 22 This concept suggests that the nonexcreted sodium ions circulate in the body and add to the plasma volume expansion5, 19, 22 and are also deposited in the skin and muscle interstitial compartments, as was proved in clinical studies and animal models.66, 67 The biological signature of renal surplus osmolyte excretion is a production of endogenous free water, which helps the renal medullar cells to excrete sodium ions and conserve filtrated water by tubular reabsorption.66 Sodium homeostasis is substantially controlled by Na/K‐ATPase, which is regulated by steroidal inhibitors including marinobufagenin. It is unclear whether the interstitial sodium ion concentration and interstitial water are higher in HSAB versus HSC in the present study, because the animals had similar BWs under both conditions. The possible role of marinobufagenin in the water/sodium balance in the interstitial compartments deserves detailed exploration in the Dahl‐S model, which will add a new dimension to the understanding of sodium handling in salt‐sensitive hypertension.
Profibrotic Effect of Marinobufagenin on Cultured LV Myocytes From Dahl‐S Rats
The results of the in vitro 24‐hour treatment of LV myocytes with marinobufagenin proved that physiological, ie, nanomolar concentrations of marinobufagenin, stimulate TGFβ profibrotic signaling in these cells. In addition, marinobufagenin activated a Fli‐1–dependent profibrotic pathway in the LV myocytes (Figure 7). Nuclear factor Fli‐1 is a negative regulator of collagen‐1 biosynthesis. When Fli‐1 is phosphorylated by the phosphorylated PKC‐δ, it releases the procollagen DNA promoter (Figure 8).54, 68 Interestingly, the crosstalk between Fli‐1 and TGFβ profibrotic signaling via PKC‐δ was previously described,68 as schematically presented in Figure 8. Previously, we have demonstrated that the Fli‐1 pathway is also activated by marinobufagenin in vitro in cultured fibroblasts15 and rat vascular smooth muscle cells.26 Our present observation using freshly prepared LV myocytes is in agreement with previous findings15, 26 and indicates that cardiac myocytes also contribute to the production of the extracellular matrix via both Fli‐1 and TGFβ profibrotic signaling. Notably, we did not observe the in vivo activation of Fli‐1 signaling in the present study in hypertensive Dahl‐S rats on HS diet with no changes in the Fli‐1 protein in the left ventricle (Figure 2G), although the Fli‐1 pathway was a predominant profibrotic signal in the animal model of cardiomyopathy.15, 54 We suggest that in the present study in hypertensive Dahl‐S rats with an expedited development of compensated LV hypertrophy, the Fli‐1 profibrotic pathway becomes less pronounced in the presence of abundant TGFβ signaling under the condition of HS intake.
Conclusions
In salt‐sensitive hypertension, heightened endogenous marinobufagenin activates cardiac and renal genes in profibrotic TGFβ‐dependent signaling implicated in the salt‐sensitivity phenomena. Immunoneutralization of marinobufagenin in hypertensive Dahl‐S rats downregulates genes associated with TGFβ‐dependent signaling in the left ventricle and kidneys, reverses heart and renal remodeling, and improves function of the kidneys and cardiovascular system.
Sources of Funding
This work was supported by the Intramural Research Program, National Institute on Aging, National Institutes of Health, USA.
Disclosures
None.
Acknowledgments
The authors are grateful to Anton Bzhelyanskiy, Bruce Ziman, and Khalid Chakir for technical support; Mikayla Hall for editorial assistance; and Christopher H. Morrell for statistical analysis.
(J Am Heart Assoc. 2019;8:e012138 DOI: 10.1161/JAHA.119.012138.)
References
- 1. Folkow B. Physiological aspects of primary hypertension. Physiol Rev. 1982;62:347–504. [DOI] [PubMed] [Google Scholar]
- 2. Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8:II127–II134. [DOI] [PubMed] [Google Scholar]
- 3. Wasserstrom JA, Aistrup GL. Digitalis: new actions for an old drug. Am J Physiol Heart Circ Physiol. 2005;289:H1781–H1793. [DOI] [PubMed] [Google Scholar]
- 4. de Wardener HE, Clarkson EM. Concept of natriuretic hormone. Physiol Rev. 1985;65:658–759. [DOI] [PubMed] [Google Scholar]
- 5. Fedorova OV, Talan MI, Agalakova NI, Lakatta EG, Bagrov AY. Endogenous ligand of alpha(1) sodium pump, marinobufagenin, is a novel mediator of sodium chloride–dependent hypertension. Circulation. 2002;105:1122–1127. [DOI] [PubMed] [Google Scholar]
- 6. Fedorova OV, Lakatta EG, Bagrov AY, Melander O. Plasma level of the endogenous sodium pump ligand marinobufagenin is related to the salt‐sensitivity in men. J Hypertens. 2015;33:534–541; discussion 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fedorova OV, Agalakova NI, Talan MI, Lakatta EG, Bagrov AY. Brain ouabain stimulates peripheral marinobufagenin via angiotensin II signalling in NaCl‐loaded Dahl‐S rats. J Hypertens. 2005;23:1515–1523. [DOI] [PubMed] [Google Scholar]
- 8. Fedorova OV, Zernetkina VI, Shilova VY, Grigorova YN, Juhasz O, Wei W, Marshall CA, Lakatta EG, Bagrov AY. Synthesis of an endogenous steroidal Na pump inhibitor marinobufagenin, implicated in human cardiovascular diseases, is initiated by CYP27A1 via bile acid pathway. Circ Cardiovasc Genet. 2015;8:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev. 2009;61:9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nikitina ER, Mikhailov AV, Nikandrova ES, Frolova EV, Fadeev AV, Shman VV, Shilova VY, Tapilskaya NI, Shapiro JI, Fedorova OV, Bagrov AY. In preeclampsia endogenous cardiotonic steroids induce vascular fibrosis and impair relaxation of umbilical arteries. J Hypertens. 2011;29:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haller ST, Kennedy DJ, Shidyak A, Budny GV, Malhotra D, Fedorova OV, Shapiro JI, Bagrov AY. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am J Hypertens. 2012;25:690–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Strauss M, Smith W, Wei W, Fedorova OV, Schutte AE. Marinobufagenin is related to elevated central and 24‐h systolic blood pressures in young black women: the African‐PREDICT Study. Hypertens Res. 2018;41:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY, Shapiro JI. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension. 2006;47:488–495. [DOI] [PubMed] [Google Scholar]
- 14. Kennedy DJ, Shrestha K, Sheehey B, Li XS, Guggilam A, Wu Y, Finucan M, Gabi A, Medert CM, Westfall K, Borowski A, Fedorova O, Bagrov AY, Tang WH. Elevated plasma marinobufagenin, an endogenous cardiotonic steroid, is associated with right ventricular dysfunction and nitrative stress in heart failure. Circ Heart Fail. 2015;8:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, Smaili S, Periyasamy SM, Hariri IM, Fedorova L, Liu J, Wu L, Kahaleh MB, Xie Z, Malhotra D, Fedorova OV, Kashkin VA, Bagrov AY, Shapiro JI. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension. 2007;49:215–224. [DOI] [PubMed] [Google Scholar]
- 16. Fedorova OV, Emelianov IV, Bagrov KA, Grigorova YN, Wei W, Juhasz O, Frolova EV, Marshall CA, Lakatta EG, Konradi AO, Bagrov AY. Marinobufagenin‐induced vascular fibrosis is a likely target for mineralocorticoid antagonists. J Hypertens. 2015;33:1602–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hamlyn JM, Blaustein MP. Sodium chloride, extracellular fluid volume, and blood pressure regulation. Am J Physiol. 1986;251:F563–F575. [DOI] [PubMed] [Google Scholar]
- 18. Blaustein MP, Ashida T, Goldman WF, Wier WG, Hamlyn JM. Sodium/calcium exchange in vascular smooth muscle: a link between sodium metabolism and hypertension. Ann N Y Acad Sci. 1986;488:199–216. [DOI] [PubMed] [Google Scholar]
- 19. Fedorova OV, Lakatta EG, Bagrov AY. Endogenous Na, K pump ligands are differentially regulated during acute NaCl loading of Dahl rats. Circulation. 2000;102:3009–3014. [DOI] [PubMed] [Google Scholar]
- 20. Inoko M, Kihara Y, Morii I, Fujiwara H, Sasayama S. Transition from compensatory hypertrophy to dilated, failing left ventricles in Dahl salt‐sensitive rats. Am J Physiol. 1994;267:H2471–H2482. [DOI] [PubMed] [Google Scholar]
- 21. Fedorova OV, Talan MI, Agalakova NI, Lakatta EG, Bagrov AY. Coordinated shifts in Na/K‐ATPase isoforms and their endogenous ligands during cardiac hypertrophy and failure in NaCl‐sensitive hypertension. J Hypertens. 2004;22:389–397. [DOI] [PubMed] [Google Scholar]
- 22. Fedorova OV, Kolodkin NI, Agalakova NI, Lakatta EG, Bagrov AY. Marinobufagenin, an endogenous alpha‐1 sodium pump ligand, in hypertensive Dahl salt‐sensitive rats. Hypertension. 2001;37:462–466. [DOI] [PubMed] [Google Scholar]
- 23. Sterzel RB, Luft FC, Gao Y, Schnermann J, Briggs JP, Ganten D, Waldherr R, Schnabel E, Kriz W. Renal disease and the development of hypertension in salt‐sensitive Dahl rats. Kidney Int. 1988;33:1119–1129. [DOI] [PubMed] [Google Scholar]
- 24. Tamaki K, Okuda S, Nakayama M, Yanagida T, Fujishima M. Transforming growth factor‐beta 1 in hypertensive renal injury in Dahl salt‐sensitive rats. J Am Soc Nephrol. 1996;7:2578–2589. [DOI] [PubMed] [Google Scholar]
- 25. Herrera VL, Decano JL, Giordano N, Moran AM, Ruiz‐Opazo N. Aortic and carotid arterial stiffness and epigenetic regulator gene expression changes precede blood pressure rise in stroke‐prone Dahl salt‐sensitive hypertensive rats. PLoS One. 2014;9:e107888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grigorova YN, Wei W, Petrashevskaya N, Zernetkina V, Juhasz O, Fenner R, Gilbert C, Lakatta EG, Shapiro JI, Bagrov AY, Fedorova OV. Dietary sodium restriction reduces arterial stiffness, vascular TGF‐beta‐dependent fibrosis and marinobufagenin in young normotensive rats. Int J Mol Sci. 2018;19:E3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dahl LK, Heine M, Tassinari L. Effects of chronic excess salt ingestion. Evidence that genetic factors play an important role in susceptibility to experimental hypertension. J Exp Med. 1962;115:1173–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moreno C, Williams JM, Lu L, Liang M, Lazar J, Jacob HJ, Cowley AW Jr, Roman RJ. Narrowing a region on rat chromosome 13 that protects against hypertension in Dahl SS‐13BN congenic strains. Am J Physiol Heart Circ Physiol. 2011;300:H1530–H1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flister MJ, Prisco SZ, Sarkis AB, O'Meara CC, Hoffman M, Wendt‐Andrae J, Moreno C, Lazar J, Jacob HJ. Identification of hypertension susceptibility loci on rat chromosome 12. Hypertension. 2012;60:942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chauvet C, Menard A, Xiao C, Aguila B, Blain M, Roy J, Deng AY. Novel genes as primary triggers for polygenic hypertension. J Hypertens. 2012;30:81–86. [DOI] [PubMed] [Google Scholar]
- 31. Yagil C, Hubner N, Monti J, Schulz H, Sapojnikov M, Luft FC, Ganten D, Yagil Y. Identification of hypertension‐related genes through an integrated genomic‐transcriptomic approach. Circ Res. 2005;96:617–625. [DOI] [PubMed] [Google Scholar]
- 32. Fedorova OV, Simbirtsev AS, Kolodkin NI, Kotov AY, Agalakova NI, Kashkin VA, Tapilskaya NI, Bzhelyansky A, Reznik VA, Frolova EV, Nikitina ER, Budny GV, Longo DL, Lakatta EG, Bagrov AY. Monoclonal antibody to an endogenous bufadienolide, marinobufagenin, reverses preeclampsia‐induced Na/K‐ATPase inhibition and lowers blood pressure in NaCl‐sensitive hypertension. J Hypertens. 2008;26:2414–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fedorova OV, Agalakova NI, Morrell CH, Lakatta EG, Bagrov AY. ANP differentially modulates marinobufagenin‐induced sodium pump inhibition in kidney and aorta. Hypertension. 2006;48:1160–1168. [DOI] [PubMed] [Google Scholar]
- 34. Fedorova OV, Kashkin VA, Zakharova IO, Lakatta EG, Bagrov AY. Age‐associated increase in salt sensitivity is accompanied by a shift in the atrial natriuretic peptide modulation of the effect of marinobufagenin on renal and vascular sodium pump. J Hypertens. 2012;30:1817–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korzick DH, Xiao RP, Ziman BD, Koch WJ, Lefkowitz RJ, Lakatta EG. Transgenic manipulation of beta‐adrenergic receptor kinase modifies cardiac myocyte contraction to norepinephrine. Am J Physiol. 1997;272:H590–H596. [DOI] [PubMed] [Google Scholar]
- 36. Ahmet I, Tae HJ, Brines M, Cerami A, Lakatta EG, Talan MI. Chronic administration of small nonerythropoietic peptide sequence of erythropoietin effectively ameliorates the progression of postmyocardial infarction‐dilated cardiomyopathy. J Pharmacol Exp Ther. 2013;345:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. J Mol Diagn. 2003;5:73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gene Ontology Consortium . Member of the Open Biological Ontologies Foundry. The Gene Ontology Consortium is supported by a P41 grant from the National Human Genome Research Institute (NHGRI) [grant U41 HG002273]. Copyright © 1999–2019. Available at: http://geneontology.org/. Accessed February 1, 2013,
- 40. Gene Set Enrichment Analysis . Copyright (c) 2004–2017; Broad Institute, Inc., Massachusetts Institute of Technology, and Regents of the University of California. Available at: http://software.broadinstitute.org/gsea/msigdb/collections.jsp#C2. Accessed February 1, 2013.
- 41. Bassuk JA, Pichler R, Rothmier JD, Pippen J, Gordon K, Meek RL, Bradshaw AD, Lombardi D, Strandjord TP, Reed M, Sage EH, Couser WG, Johnson R. Induction of TGF‐beta1 by the matricellular protein SPARC in a rat model of glomerulonephritis. Kidney Int. 2000;57:117–128. [DOI] [PubMed] [Google Scholar]
- 42. Rivera LB, Bradshaw AD, Brekken RA. The regulatory function of SPARC in vascular biology. Cell Mol Life Sci. 2011;68:3165–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dahly AJ, Hoagland KM, Flasch AK, Jha S, Ledbetter SR, Roman RJ. Antihypertensive effects of chronic anti‐TGF‐beta antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2002;283:R757–R767. [DOI] [PubMed] [Google Scholar]
- 44. Murphy SR, Dahly‐Vernon AJ, Dunn KM, Chen CC, Ledbetter SR, Williams JM, Roman RJ. Renoprotective effects of anti‐TGF‐beta antibody and antihypertensive therapies in Dahl S rats. Am J Physiol Regul Integr Comp Physiol. 2012;303:R57–R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Onozato ML, Tojo A, Kobayashi N, Goto A, Matsuoka H, Fujita T. Dual blockade of aldosterone and angiotensin II additively suppresses TGF‐beta and NADPH oxidase in the hypertensive kidney. Nephrol Dial Transplant. 2007;22:1314–1322. [DOI] [PubMed] [Google Scholar]
- 46. Yoshida J, Yamamoto K, Mano T, Sakata Y, Nishikawa N, Nishio M, Ohtani T, Miwa T, Hori M, Masuyama T. AT1 receptor blocker added to ACE inhibitor provides benefits at advanced stage of hypertensive diastolic heart failure. Hypertension. 2004;43:686–691. [DOI] [PubMed] [Google Scholar]
- 47. Li ZC, Cai T, Tian J, Xie JX, Zhao XC, Liu LJ, Shapiro JI, Xie ZJ. NaKtide, a Na/K‐ATPase‐derived peptide Src inhibitor, antagonizes ouabain‐activated signal transduction in cultured cells. J Biol Chem. 2009;284:21066–21076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xie JX, Shapiro AP, Shapiro JI. The trade‐off between dietary salt and cardiovascular disease; a role for Na/K‐ATPase signaling? Front Endocrinol (Lausanne). 2014;5:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal‐transducing function of Na+/K+‐ATPase. J Biol Chem. 2000;275:27832–27837. [DOI] [PubMed] [Google Scholar]
- 50. Meloche S, Pouyssegur J. The ERK1/2 mitogen‐activated protein kinase pathway as a master regulator of the G1‐ to S‐phase transition. Oncogene. 2007;26:3227–3239. [DOI] [PubMed] [Google Scholar]
- 51. Mishra R, Zhu L, Eckert RL, Simonson MS. TGF‐beta‐regulated collagen type I accumulation: role of Src‐based signals. Am J Physiol Cell Physiol. 2007;292:C1361–C1369. [DOI] [PubMed] [Google Scholar]
- 52. Chen J, Chen JK, Nagai K, Plieth D, Tan M, Lee TC, Threadgill DW, Neilson EG, Harris RC. EGFR signaling promotes TGFbeta‐dependent renal fibrosis. J Am Soc Nephrol. 2012;23:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kotova O, Al‐Khalili L, Talia S, Hooke C, Fedorova OV, Bagrov AY, Chibalin AV. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src‐ and ERK1/2‐dependent mechanism. J Biol Chem. 2006;281:20085–20094. [DOI] [PubMed] [Google Scholar]
- 54. Elkareh J, Periyasamy SM, Shidyak A, Vetteth S, Schroeder J, Raju V, Hariri IM, El‐Okdi N, Gupta S, Fedorova L, Liu J, Fedorova OV, Kahaleh MB, Xie Z, Malhotra D, Watson DK, Bagrov AY, Shapiro JI. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli‐1: implications for uremic cardiomyopathy. Am J Physiol Renal Physiol. 2009;296:F1219–F1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fedorova OV, Talan MI, Agalakova NI, Droy‐Lefaix MT, Lakatta EG, Bagrov AY. Myocardial PKC beta2 and the sensitivity of Na/K‐ATPase to marinobufagenin are reduced by cicletanine in Dahl hypertension. Hypertension. 2003;41:505–511. [DOI] [PubMed] [Google Scholar]
- 56. Koide Y, Tamura K, Suzuki A, Kitamura K, Yokoyama K, Hashimoto T, Hirawa N, Kihara M, Ohno S, Umemura S. Differential induction of protein kinase C isoforms at the cardiac hypertrophy stage and congestive heart failure stage in Dahl salt‐sensitive rats. Hypertens Res. 2003;26:421–426. [DOI] [PubMed] [Google Scholar]
- 57. Ding RQ, Tsao J, Chai H, Mochly‐Rosen D, Zhou W. Therapeutic potential for protein kinase C inhibitor in vascular restenosis. J Cardiovasc Pharmacol Ther. 2011;16:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Newton AC, Antal CE, Steinberg SF. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin Sci (Lond). 2016;130:1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Strauss M, Smith W, Wei W, Bagrov AY, Fedorova OV, Schutte AE. Large artery stiffness is associated with marinobufagenin in young adults: the African‐PREDICT study. J Hypertens. 2018;36:2333–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Strauss M, Smith W, Kruger R, Wei W, Fedorova OV, Schutte AE. Marinobufagenin and left ventricular mass in young adults: the African‐PREDICT study. Eur J Prev Cardiol. 2018;25:1587–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jablonski KL, Fedorova OV, Racine ML, Geolfos CJ, Gates PE, Chonchol M, Fleenor BS, Lakatta EG, Bagrov AY, Seals DR. Dietary sodium restriction and association with urinary marinobufagenin, blood pressure, and aortic stiffness. Clin J Am Soc Nephrol. 2013;8:1952–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fedorova OV, Kolodkin NI, Agalakova NI, Namikas AR, Bzhelyansky A, St‐Louis J, Lakatta EG, Bagrov AY. Antibody to marinobufagenin lowers blood pressure in pregnant rats on a high NaCl intake. J Hypertens. 2005;23:835–842. [DOI] [PubMed] [Google Scholar]
- 63. Grigorova YN, Juhasz O, Zernetkina V, Fishbein KW, Lakatta EG, Fedorova OV, Bagrov AY. Aortic fibrosis, induced by high salt intake in the absence of hypertensive response, is reduced by a monoclonal antibody to marinobufagenin. Am J Hypertens. 2016;29:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vaheri A, Salonen EM, Vartio T. Fibronectin in formation and degradation of the pericellular matrix. Ciba Found Symp. 1985;114:111–126. [DOI] [PubMed] [Google Scholar]
- 65. McCormick CP, Rauch AL, Buckalew VM Jr. Differential effect of dietary salt on renal growth in Dahl salt‐sensitive and salt‐resistant rats. Hypertension. 1989;13:122–127. [DOI] [PubMed] [Google Scholar]
- 66. Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, Lantier L, LaRocque LM, Marton A, Neubert P, Schroder A, Rakova N, Jantsch J, Dikalova AE, Dikalov SI, Harrison DG, Muller DN, Nishiyama A, Rauh M, Harris RC, Luft FC, Wassermann DH, Sands JM, Titze J. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest. 2017;127:1944–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Titze J, Luft FC. Speculations on salt and the genesis of arterial hypertension. Kidney Int. 2017;91:1324–1335. [DOI] [PubMed] [Google Scholar]
- 68. Asano Y, Trojanowska M. Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300/CREB‐binding protein‐associated factor and subsequent acetylation in response to transforming growth factor beta. Mol Cell Biol. 2009;29:1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]