

Abstract
Background
Heart failure (HF) is one of the most significant causes of morbidity and mortality for the cardiovascular risk population. We found previously that extracellular HSP70 (heat shock protein) is an important trigger in cardiac hypertrophy and fibrosis, which are associated with the development of heart dysfunction. However, the potential role of HSP70 in response to HF and whether it could be a target for the therapy of HF remain unknown.
Methods and Results
An HF mouse model was generated by a single IP injection of doxorubicin at a dose of 15 mg/kg. Ten days later, these mice were treated with an HSP70 neutralizing antibody for 5 times. We observed that doxorubicin treatment increased circulating HSP70 and expression of HSP70 in myocardium and promoted its extracellular release in the heart. Blocking extracellular HSP70 activity by its antibody significantly ameliorated doxorubicin‐induced left ventricular dilation and dysfunction, which was accompanied by a significant inhibition of cardiac fibrosis. The cardioprotective effect of the anti‐HSP70 antibody was largely attributed to its ability to promote the resolution of myocardial inflammation, as evidenced by its suppression of the toll‐like receptor 2–associated signaling cascade and modulation of the intracellular distribution of the p50 and p65 subunits of nuclear factor‐κB.
Conclusions
Extracellular HSP70 serves as a noninfectious inflammatory factor in the development of HF, and blocking extracellular HSP70 activity may provide potential therapeutic benefits for the treatment of HF.
Keywords: cardiac dysfunction, cardiac remodeling, damage‐associated molecular patterns, nuclear factor‐κB, toll‐like receptor
Subject Categories: Heart Failure, Inflammatory Heart Disease
Clinical Perspective
What Is New?
Elevated extracellular HSP70 (heat shock protein 70) level correlates positively with the development and progression of cardiac dysfunction and remodeling, suggesting that extracellular HSP70 is a potential therapeutic target for the treatment of heart failure.
Extracellular HSP70 promotes heart failure through activating toll‐like receptor 2–p38–nuclear factor‐κB–mediated noninfective myocardial inflammation.
Blocking extracellular HSP70 activity not only reverses doxorubicin‐induced persistent inflammation in myocardial tissue but also significantly attenuates cardiac dysfunction and remodeling.
What Are the Clinical Implications?
The current study reveals a new mechanistic insight in the involvement of extracellular HSP70 in the pathogenesis of heart failure and indicates the possibility to use extracellular HSP70 as a new drug target for the treatment of noninfective inflammation‐mediated cardiac dysfunction and remodeling.
Systemic or local administration of HSP70 neutralizing antibody may be the newest prospect for the management of heart failure.
Introduction
Heart failure (HF) is the final stage of a variety of cardiovascular disorders and the leading cause of hospitalization for people aged >65 years.1 Although the current treatments have largely improved the short‐term prognosis of patients with HF, there are still no convincing or effective prevention strategies to avoid repeated and prolonged rehospitalization.2 Numerous studies have documented that persistent or accumulated noninfectious myocardial inflammation participates in the development and progression of HF3, 4 and can be caused by a wide variety of insults, including ischemia, hypoxia, hemodynamic overloading, and chemotherapeutic agents.5 The host responds to these noninfectious insults and intricately recognizes damage‐associated molecular patterns (DAMPs), which are released from damaged cardiac tissues, through an interaction with pattern recognition receptors on myocardial immune or parenchyma cells.6, 7 Indeed, endogenous DAMPs, including high‐mobility group box 1, HSPs (heat shock proteins), and S100A8/A9, trigger the inflammatory injury process of HF.8, 9, 10 Hence, inhibiting the extracellular function of the DAMPs may be of great therapeutic benefit for the prevention of HF.
The HSP70 family is the highly conserved and best studied class of HSPs. HSP70 (used herein to denote HSP70A1A) is the main member of the 70‐kDa HSP family, and its expression is dramatically induced in response to a variety of stress stimuli, including ischemia, hyperthermia, irradiation, oxidative stress, and inflammation.11, 12 Elevated serum level of HSP70 has been detected in patients with acute myocardial infarction, and it is negatively correlated with left ventricular (LV) ejection fraction.13 An inhibitory antibody to heat shock cognate protein 70 was able to attenuate ischemia‐induced cardiac dysfunction.14 Indeed, we reported previously that intracellular and extracellular HSP70 play different roles in the regulation of cardiac remodeling in response to hypertension. The extracellular HSP70, released from damaged cardiomyocytes, can function as a DAMP to trigger myocardial inflammation and is a potential therapeutic target in the treatment of cardiac hypertrophy and fibrosis.15 However, the precise role and mechanism of HSP70 in the development of HF is largely unknown.
Doxorubicin has long been used as a powerful antitumor agent in the fight against many types of cancer. Its dose‐dependent and irreversible cardiotoxic effect is the most serious drawback to its use.16 We and others have shown that mice or rats treated with 10 to 30 mg/kg doxorubicin developed HF.17, 18 Although the precise mechanisms responsible for the cardiotoxicity of doxorubicin have not been fully elucidated, it is widely accepted that doxorubicin‐induced cardiac injury is mainly caused by increased necrotic or apoptotic loss of cardiomyocytes,19 which could eventually lead to the passive release of various DAMPs from cardiomyocytes.20 Our preliminary data showed that circulating HSP70 level is increased in doxorubicin‐treated mice in comparison with control mice. We, thus, speculated that extracellular HSP70 might act as a DAMP and be involved in the pathogenesis of doxorubicin‐induced HF. Indeed, our present study indicates that blocking extracellular HSP70 activity with a neutralizing antibody not only attenuates doxorubicin‐induced cardiac dysfunction and remodeling but also remarkably reverses persistent inflammation in myocardial tissue. This is largely caused by inhibition of the intranuclear nuclear factor (NF)‐κB in a toll‐like receptor 2 (TLR2)–dependent manner, suggesting that the antagonism of the extracellular HSP70 has a therapeutic potential against HF.
Methods
The data, analytic methods, and materials that support the findings of current study will be available to other researchers on reasonable request.
Animal Model and Treatments
Male C57BL/6J mice (aged 8 weeks) were obtained from Vital River Laboratory Animal Technology (Beijing, China). All mice were housed in a facility with a 12‐hour/12‐hour light/dark cycle and given free access to water and standard rodent chow. All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals, prepared and approved by the Animal Care and Use Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College. HF was induced in 10‐week‐old male C57BL/6J mice by a single IP injection of doxorubicin at a dose of 15 mg/kg. The mice were treated with anti‐HSP70 antibody (Thermo; MA3‐009), isotype‐matched IgG antibodies, or saline on days 10, 14, 18, 25, and 32 after doxorubicin injection (n=60 for saline or doxorubicin group; n=40 for HSP70 antibody or IgG antibody group). The dosing and timing of antibodies were determined on the basis of our previous studies and pilot experiment.15 The antibodies were dissolved in sterile saline and administered by tail vein injection. The first dose was 200 mg/kg, and the remaining doses were 100 mg/kg. Six mice per group were euthanized by an overdose of sodium pentobarbital injection (IP, 100 mg/kg), and the hearts were harvested and sliced into several sections on day 1, 3, 7, 11, 13, 17, 24, and 36 for the indicated analyses, as described below.
Echocardiographic Measurements
Six hours before doxorubicin injection (day 0) and on day 1, 3, 7, 11, 13, 17, 24, and 36 after doxorubicin treatment, mice were anesthetized with sodium pentobarbital (IP, 45 mg/kg), and echocardiography was performed with the Visual Sonics Vevo 770 system (VisualSonics, Canada) using a 30‐MHz image transducer. The heart rate was controlled at ≈500 beats per minute to acquire measurements under physiologically relevant conditions. After a good‐quality 2‐dimensional image was obtained, M‐mode images of the LV were recorded, showing the motion of the myocardial walls to assess contractile patterns, ventricular wall thickness, and chamber size. End‐diastolic LV internal diameter, end‐systolic LV internal diameter, and end‐diastolic LV anterior wall were measured. Cardiac function was evaluated individually by the LV fractional shortening.
ELISA Assays
HSP70 in serum was detected, as described previously.15 Briefly, a high‐binding ELISA plate was coated overnight with monoclonal anti‐HSP70 (Enzo; ADI‐SPA‐810) in carbonate buffer. Samples or recombinant mouse HSP70 protein (Abcam; ab113187) was added and incubated for 2 hours, followed by 3 washes in PBS with Tween 20. A rabbit polyclonal anti‐HSP70 (Enzo; ADI‐SPA‐812) was used as a detection antibody and incubated for 2 hours, followed by 5 washes. Finally, 3,3′,5,5′‐Tetramethylbenzidine substrate reagent (BD Biosciences) was added, and the absorbance was read by a microplate reader at 450 nm. Lipoxin A4 levels or NF‐κBp65 activity in heart tissue homogenates was examined by using an ELISA kit from Neogen or Cell Signaling, following the manufacturers’ instructions.
Histological Analysis and Confocal Microscopy
The hearts were harvested and fixed with 4% paraformaldehyde and embedded in paraffin for histopathological examination. Heart sections (4 μm) were stained with hematoxylin‐eosin or Masson's trichrome blue for evaluation of cardiac morphological features, inflammation, and fibrosis, which were blindly assessed by an experienced pathologist (K.L.). Frozen heart sections (6 μm) were fixed and permeabilized for 10 minutes in acetone and 10 minutes in 0.2% Triton X‐100. Nonspecific binding was blocked by incubation with 3% BSA for 45 minutes at room temperature, followed by incubation with indicated primary antibodies overnight at 4°C. After washing twice with PBS, the sections were incubated with Alex488‐ or Alex647‐conjugated secondary antibodies in the dark for 1 hour at 37°C. The nuclei were stained with 4′,6‐diamidino‐2‐phenylindole for 40 minutes at room temperature. The colocalization of HSP70 and α‐actin as well as the recruitment and infiltration of M1 (F4/80+CD86+) and M2 (F4/80+CD206+)type macrophages were examined using a confocal microscope (Leica Microsystems) and analyzed with the Leica confocal software.
Flow Cytometry Analysis
Single‐cell suspensions were prepared from murine hearts, as previously described, with minor modifications.21 Briefly, hearts rinsed with cold PBS were minced into ∼1‐mm pieces, resuspended in digestion solution containing collagenase IV (2 μg/mL) and DNase (50 μg/mL), and gently agitated at 37 °C for 60 minutes. After a red blood cell lysis and sequential filtration through 200‐μm filters, digested cells were resuspended in PBS and incubated with fluorescein isothiocyanate– or phycoerythrin‐conjugated monoclonal antibody against F4/80 or Gr‐1 to analyze the macrophage and neutrophil populations in the heart. Isotype‐matched monoclonal antibodies were used in the control samples. The percentage of apoptotic neutrophils was assessed by staining with annexin V‐APC and propidium iodide. At least 20 000 stained cells were collected and analyzed using a flow cytometer (BD Biosciences).
Cell Culture and Treatments
Rat myocardial H9C2 cells were purchased from the American Type Culture Collection and cultured in Dulbecco's modified eagle media supplemented with 10% fetal bovine serum and 100 μg/mL penicillin/streptomycin. Tlr2 small interfering RNA, Tlr4 small interfering RNA, and control small interfering RNA were produced by GenePharma (Suzhou, China) and transfected using Lipofectamine RNA interference MAX Transfection Reagent (Life Technologies), according to the manufacturer's instructions. For detection of the NF‐κBp65 activity and intracellular distribution of the p50 and p65 subunits of NF‐κB, H9C2 cells were preincubated with neutralizing antibodies to TLR2 (R&D; MAB1530), TLR4 (BioLegend; 117608), and HSP70 (Thermo; MA3‐009) as well as isotype‐matched IgG for 3 hours, followed by treatment with recombinant HSP70 (100 ng/mL; Enzo; ADI‐ESP‐502) for an additional 60 minutes.
Western Blotting
Protein samples extracted from heart tissues or cultured cells were subjected to Western blot analysis, as described previously.17 The nuclear and cytoplasmic extractions of the cell samples were performed using the NE‐PER Nuclear and Cytoplasmic Extraction Kit (Pierce, Rockford, IL) following the manufacturer's instructions. Antibodies against GAPDH or proliferating cell nuclear antigen (PCNA) and tubulin served as controls for normalization of nuclear and cytosolic fractions, respectively. Specific antibody binding was visualized and quantified by an electrochemiluminescence system (GE Healthcare).
Statistical Analysis
All results are represented as the mean±SEM. Two‐group comparisons were made by Student t test, as appropriate, whereas multiple comparisons among ≥3 groups were performed using 1‐way ANOVA, which was conducted first across all investigated groups in measurements of myocardial function during echocardiography analysis, in which Shapiro‐Wilk test was performed for normality, and there was no evidence of deviation from normality for all variables. Thereafter, post hoc pairwise tests were performed with assessment of statistical significance after Bonferroni correction of P values. P<0.05 was considered statistically significant. All statistics were analyzed using SPSS 17.0 software.
Results
Doxorubicin Increases HSP70 Levels in Serum and Myocardium
We have recently reported that pressure overload induces anomalous expression and distribution of HSP70 in the serum and heart tissue in abdominal aortic constriction mice. To identify the roles and mechanism of cardiac HSP70 in response to doxorubicin treatment, we examined the circulating levels and cardiac expression profile of HSP70 at different time points, after doxorubicin intraperitoneal injection. Significantly increased HSP70 levels were observed not only in serum but also in the heart, which was observed from day 1 after doxorubicin injection. In addition, the peak level of HSP70 expression occurred on day 11, and high levels were maintained until the end of the experiment (Figure 1A and 1B). To further determine whether the distribution of HSP70 was altered or not, the LV sections on day 11 after doxorubicin injection were analyzed using confocal microscopy. As shown in Figure 1C, mice treated with doxorubicin showed a significant up‐regulation of HSP70 expression and accumulation of HSP70 on the membrane of cardiomyocytes. These data suggest that elevated extracellular HSP70 expression is associated in the doxorubicin‐mediated myocardial damage.
Figure 1.
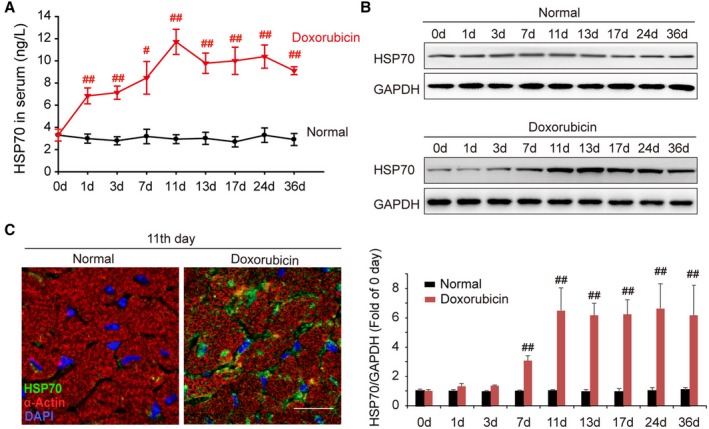
Doxorubicin elevates the level of HSP (heat shock protein) 70 in serum and myocardium. A, Time course of the content of HSP70 in mouse serum after intraperitoneal administration of doxorubicin. Data are the mean±SEM of 3 assays (n=6–8/group). # P<0.05, ## P<0.01, compared with normal mice. B, At the indicated intervals, the expression of HSP70 in the heart was analyzed by Western blot assay. Representative immunoblots and the ratio of HSP70/GAPDH are presented. Data are the mean±SEM of 3 assays. ## P<0.01, compared with normal mice before doxorubicin treatment. C, Localization of HSP70 in cardiac tissue was detected by confocal microscopy on day 11 after the treatment with doxorubicin (green, HSP70; red, α‐actin; blue, nuclei; bar=20 μm). DAPI indicates 4′,6‐diamidino‐2‐phenylindole.
Functional Antagonism of Extracellular HSP70 Attenuates Doxorubicin‐Induced Cardiac Remodeling and Dysfunction
To clarify whether enhanced extracellular HSP70 expression was the main cause of doxorubicin‐induced myocardial remodeling and dysfunction, an anti‐HSP70 neutralizing antibody was used on days 10, 14, 18, 25, and 32 after doxorubicin injection to block extracellular HSP70 activity in mice with HF. Thirty‐six days after the initial doxorubicin injection, the mice exhibited significant LV dilation and myocardium loss, as indicated by hematoxylin‐eosin staining of global heart and the reduced ratio of heart weight/tibia length, whereas the anti‐Hsp70 antibody–treated mice showed milder cardiac remodeling (Figure 2A and 2B). In addition, we found that the doxorubicin‐induced cardiotoxic injury caused persistent myocardial inflammation and fibrosis, whereas targeting the extracellular HSP70 activity conspicuously attenuated the recruitment and infiltration of inflammatory cells as well as the interstitial and perivascular accumulation of collagen in myocardium (Figure 2C and 2D). Activated cardiac myofibroblasts secret procollagen and cytokines and are integral to the pathogenesis of cardiac fibrosis. To evaluate the presence of activated myofibroblasts, immunoblotting of α‐smooth muscle actin and collagen‐I in myocardial tissue was performed at different time points after the doxorubicin or HSP70 antibody treatment. As shown in Figure 2E, doxorubicin significantly up‐regulated the expression of α‐smooth muscle actin and collagen‐I in the myocardium, which could be reversed by HSP70 antibody treatment. Cardiac function and remolding were further dynamically evaluated by echocardiography on day 0, 1, 3, 7, 11, 13, 17, 24, and 36 after doxorubicin treatment. More important, HSP70 antibody treatment exhibited significant functional and constructional improvement of HF (Figure 2F), as demonstrated by the increased LV fraction shortening (Figure 2G), as well as reduced end‐diastolic and end‐systolic LV internal diameter accompanied by an enhanced LV anterior wall thickness at the end diastole (Figure 2H). Together, these data indicate that the elevated extracellular HSP70 is a potential therapeutic target for doxorubicin‐induced myocardial damage and blocking the extracellular HSP70 activity provides beneficial effects for the treatment of HF.
Figure 2.
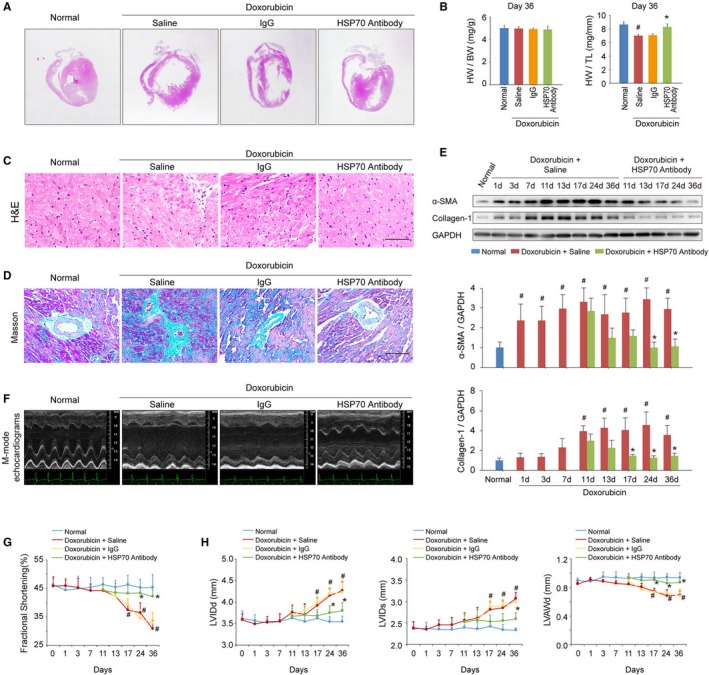
Blocking extracellular HSP (heat shock protein) 70 activity attenuates doxorubicin‐induced cardiac remodeling and dysfunction. A, Blocking extracellular HSP70 with a neutralizing antibody attenuated doxorubicin‐induced ventricular dilatation (global heart section). B, The ratios of heart weight/body weight (HW/BW) and HW/tibia length (HW/TL) are presented (n=6–8/group). # P<0.05, compared with normal mice; *P<0.05, compared with doxorubicin‐treated mice. C, Blocking extracellular HSP70 inhibited the recruitment of inflammatory cells (bar=50 μm), as indicated by hematoxylin‐eosin (H&E) staining of the heart sections. D, Representative photomicrographs of Masson's trichrome staining of the heart sections for cardiac fibrosis evaluation (bar=50 μm). E, Expression of α‐smooth muscle actin (α‐SMA) and collagen I was assayed by Western blot analysis. The expression ratio of the indicated protein to GAPDH from 3 independent experiments is presented. # P<0.05, compared with normal mice; *P<0.05, compared with doxorubicin‐treated mice. F, Representative images of left ventricular (LV) M‐mode echocardiograms (n=6–8/group). Cardiac functional (G) and structural (H) parameters were measured by echocardiography analysis. Data are mean±SEM (n=6–8/group). # P<0.05, compared with normal mice; *P<0.05, compared with doxorubicin‐treated mice. LVAWd indicates LV diastolic anterior wall thickness; LVIDd, end‐diastolic LV internal diameter; LVIDs, end‐systolic LV internal diameter.
Blockage of Extracellular HSP70 Promotes the Resolution of Doxorubicin‐Initiated Myocardial Inflammation
To investigate the regulatory effects of targeting extracellular HSP70 on the immune microenvironment in doxorubicin‐treated heart, expression levels of interleukin‐6, transforming growth factor‐β1, interleukin‐17A, and interleukin‐10 were evaluated at different time points after doxorubicin and/or HSP70 antibody treatments. We observed that doxorubicin challenge dramatically enhanced the expression of proinflammatory cytokines in the myocardium, including interleukin‐6, transforming growth factor‐β1, and interleukin‐17A, and inhibited the expression of the anti‐inflammatory cytokine interleukin‐10 (Figure 3A). However, continuous administration of HSP70 antibody significantly reduced the production of interleukin‐6, transforming growth factor‐β1, and interleukin‐17A (Figure 3B through 3D) but increased the release of interleukin‐10 (Figure 3E). Moreover, HSP70 antibody treatment markedly inhibited the expression of inducible NO synthase and cyclooxygenase 2 but elevated the expression of B‐cell lymphoma 3 in doxorubicin‐treated mice compared with saline or IgG treatments (Figure 3F), suggesting that there is continuous inflammation in the hearts of doxorubicin‐induced HF mice, which can be efficiently reversed by antagonism of the extracellular HSP70 activity.
Figure 3.
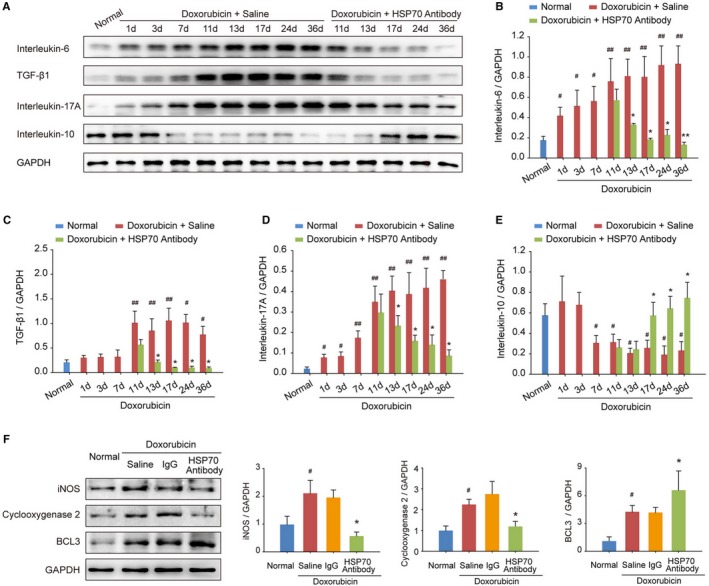
Blocking extracellular HSP (heat shock protein) 70 restricts doxorubicin‐induced myocardial inflammation. A through E, At the indicated time points, the protein levels of interleukin‐6, transforming growth factor (TGF)‐β1, interleukin‐17A, and interleukin‐10 in heart tissue homogenates were detected by Western blot analysis. In a time‐dependent manner, HSP70 neutralizing antibody treatment inhibited the increase in the levels of interleukin‐6 (B), TGF‐β1 (C), and interleukin‐17A (D) but upregulated the level of interleukin‐10 (E) in the myocardium. Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. Data are the mean±SEM of 3 assays. # P<0.05, ## P<0.01, compared with normal mice; *P<0.05, **P<0.01, compared with doxorubicin‐treated mice. F, The expression of the proinflammatory proteins inducible NO synthase (iNOS) and cyclooxygenase 2 as well as the anti‐inflammatory protein B‐cell lymphoma 3 (BCL3) in heart tissue was examined by Western blot analysis at the experimental end point. Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. Data are the mean±SEM of 3 assays. # P<0.05, compared with normal mice; *P<0.05, compared with doxorubicin‐treated mice.
Recent studies have shown that the accumulation of interleukin‐1α in the initial phase of inflammation is correlated with the infiltration of neutrophils, whereas enhanced interleukin‐1β is associated with subsequent migration of macrophages during the development of a sterile inflammatory response.22 Indeed, treatment with doxorubicin resulted in a transient increase of interleukin‐1α and a gradual increase of interleukin‐1β in the myocardium, and HSP70 antibody treatment resulted in a significant decrease in interleukin‐1α production as well as an intense increase in interleukin‐1β levels at the later stage of doxorubicin‐induced inflammatory injury (Figure 4A). Flow cytometry analysis and quantitative results also confirmed that blockade of extracellular HSP70 not only decreased doxorubicin‐enhanced content (Figure 4B) and apoptosis (Figure 4C) of neutrophils, but also further increased the recruitment of macrophages (Figure 4B) in myocardial tissues. As an endogenously produced lipid mediator, lipoxin A4 plays a critical role in promoting inflammation resolution by increasing macrophage infiltration and the phagocytosis of apoptotic polymorphonuclear neutrophils (PMNs). There were no apparent alterations in the production of lipoxin A4 in myocardium challenged with doxorubicin, whereas HSP70 antibody treatment dramatically increased the lipoxin A4 levels at day 7 after the first administration; and an accelerating increase was observed until the experimental end point (Figure 4D). Furthermore, we performed immunostaining for M1‐ or M2‐type macrophages to more precisely define the immune microenvironment in myocardium and observed that doxorubicin‐induced cardiac injury caused recruitment of a large number of M1‐type macrophages (F4/80+CD86+) in heart tissues. Blocking extracellular HSP70 could further increase the total content of macrophages, among which M2‐type macrophages (F4/80+CD206+) were predominantly observed (Figure 4E). Together, these data provide further evidence that blocking the extracellular HSP70 activity contributes to the resolution of inflammation in the hearts of doxorubicin‐treated mice.
Figure 4.
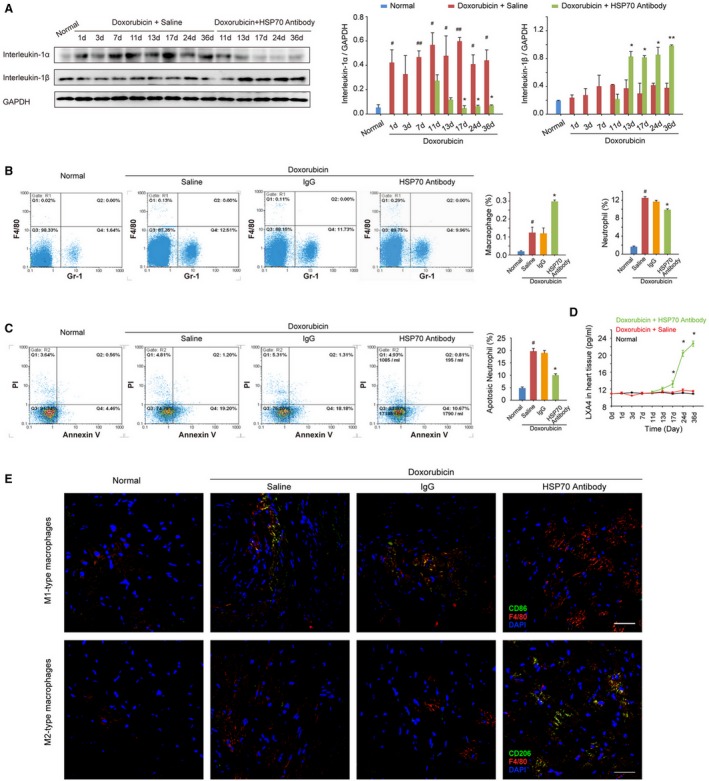
Antagonism of extracellular HSP (heat shock protein) 70 promotes resolution of inflammation in the heart tissue of doxorubicin‐treated mice. A, The time‐dependent expressions of interleukin‐1α and interleukin‐1β in the mouse myocardium after administration of doxorubicin and/or HSP70 antibody. Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. B, The heart single‐cell suspensions were prepared, and the Gr‐1–positive neutrophils and F4/80‐positive macrophages were determined by flow cytometry analysis. Representative scattergrams and the percentage of neutrophils and macrophages are presented. C, The apoptosis of neutrophils was determined by the annexin V and propidium iodide (PI) staining, followed by flow cytometry. Representative scattergrams and percentage of apoptotic neutrophils are presented. D, At the indicated time points, the contents of lipoxin A4 (LXA4) in heart tissue homogenates were detected by ELISA analysis. All data are the mean±SEM of 3 assays. # P<0.05, ## P<0.01, compared with normal mice; *P<0.05, **P<0.01, compared with doxorubicin‐treated mice. E, Recruitment and infiltration of M1‐type macrophages (F4/80+CD86+) and M2‐type macrophages (F4/80+CD206+) in myocardium were detected by confocal microscopy analysis on day 36 after the treatment with doxorubicin (bar=50 μm).
Extracellular HSP70 Activates NF‐κB in a TLR2‐Dependent Manner
Our previous study demonstrates that TLR2 and TLR4 play distinct roles in the progression of doxorubicin‐induced cardiac dysfunction.17 We, thus, hypothesized that TLR2 or TLR4 signaling might also be involved in extracellular HSP70‐mediated myocardial inflammation injury and examined whether blocking extracellular HSP70 affects TLR2/4‐mediated downstream signaling. As shown in Figure 5A, functional antagonism of extracellular HSP70 by an anti‐HSP70 antibody remarkably attenuated doxorubicin‐enhanced expression of MyD88, a universal adapter protein for proinflammatory signals triggered by both TLR2 and TLR4, but did not inhibit the HSP70 expression in myocardium (Figure 5B). Furthermore, targeting extracellular HSP70 attenuated the activation of both p38 and NF‐κB (Figure 5C), indicating the suppression of MyD88‐dependent TLR2/4 downstream signaling. However, HSP70 antibody had no marked effect on MyD88‐independent TLR4 signaling, as indicated by the unchanged expression of toll like receptor adaptor molecule 1 and phosphorylation of interferon regulatory factor 3 (Figure 5D), implying that the TLR2‐associated signaling pathway may play a more vital role in mediating the proinflammatory effect of extracellular HSP70. To evaluate this hypothesis, we performed additional in vitro analyses to examine the downstream signals of TLRs in Tlr2‐ or Tlr4‐silenced H9C2 cells on HSP70 stimulation; and we found that genetic depletion of Tlr2 but not Tlr4 could remarkably reverse HSP70‐induced activation of p38 and NF‐κB (Figure 5E). Furthermore, we exposed HSP70, TLR2, or TLR4 neutralizing antibody preincubated H9C2 cells to a recombinant HSP70 protein and found that HSP70 significantly stimulated the phosphorylation of NF‐κBp65, which could be abolished by HSP70 antibody and TLR2 antibody but not TLR4 antibody (Figure 5F). In addition, the nucleoplasmic translocation of NF‐κBp65 and NF‐κBp50 was detected to verify the activation of NF‐κB. We observed greater induction of p65 nuclear translocation and p50 cytoplasm translocation with HSP70 treatment, which could be distinctly reversed by TLR2 antibody but not TLR4 antibody or isotype IgG (Figure 5G). Taken together, these data suggest that TLR2 plays a crucial role in the extracellular HSP70‐induced inflammatory effects in the myocardium of HF mice.
Figure 5.
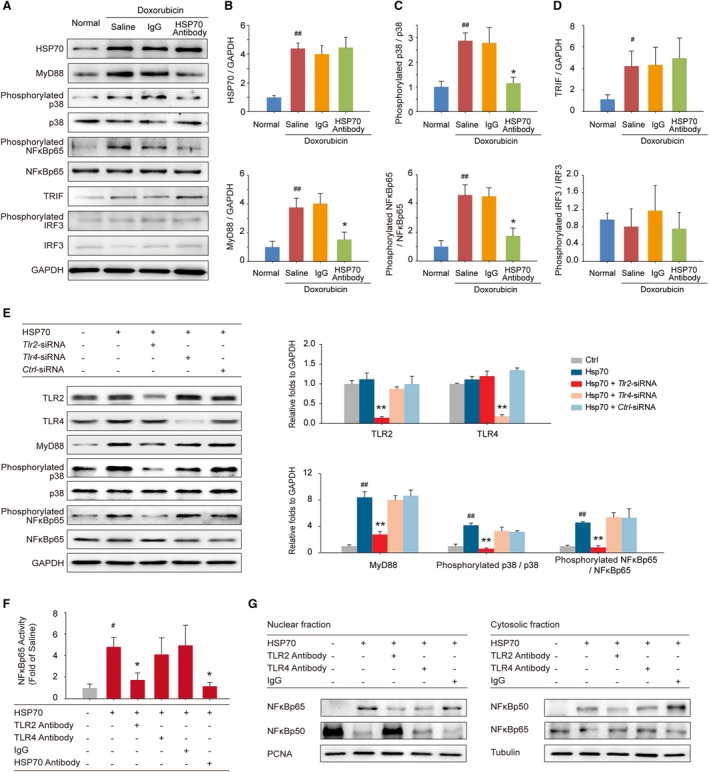
Extracellular HSP (heat shock protein) 70 activates nuclear factor (NF)‐κBp65 and modulates the distribution of the NF‐κB p50 and p65 subunits in a toll‐like receptor 2 (TLR2)–dependent manner. A, Expressions of HSP70, MyD88, phosphorylated p38, p38, phosphorylated NF‐κBp65, NF‐κBp65, toll like receptor adaptor molecule 1 (TRIF), phosphorylated interferon regulatory factor 3 (IRF3), and IRF3 in the mouse myocardium were detected by Western blot analysis. Neutralizing extracellular HSP70 inhibited the MyD88–p38–NF‐κB pathway (B and C) but did not affect the TRIF‐IRF3 pathway (D). Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. # P<0.05, ## P<0.01, compared with normal mice; *P<0.05, **P<0.01, compared with doxorubicin‐treated mice. E, Tlr2–small interfering RNA (siRNA)–, Tlr4‐siRNA–, or Ctrl‐siRNA–transfected H9C2 cells were treated with 100 ng/mL HSP70 recombinant protein for 24 hours; and the TLR2, TLR4, MyD88, phosphorylated p38, p38, phosphorylated NF‐κBp65, and NF‐κBp65 were detected by Western blot analysis. Representative immunoblots and the ratio of the indicated protein to GAPDH are presented. ## P<0.01, compared with untreated cells; **P<0.01, compared with HSP70‐treated cells. F and G, TLR2 antibody, TLR4 antibody, or IgG preincubated H9C2 cells were treated with HSP70 recombinant protein. F, The phosphorylation of NF‐κBp65 in cell homogenates was detected by ELISA analysis. # P<0.05, compared with untreated cells; *P<0.05, compared with HSP70‐treated cells. G, Nuclear and cytosolic extracts were subjected to Western blot analysis with antibodies to the NF‐κB subunits p50 and p65. PCNA and tubulin were used as internal controls for the nuclear and cytosolic fractions, respectively. Representative immunoblots of 3 independent assays are presented.
Discussion
Accumulated studies have documented that the resolution of inflammation is a highly coordinated and active process that is essential for the maintenance of tissue homeostasis.23, 24 However, if the host is unable to neutralize the short‐term injurious agent and/or there is a failure for endogenous proresolving mediators to invoke resolution, chronic inflammation may become prolonged and result in varying degrees of tissue damage and remodeling.25 In our study, we observed that blockage of extracellular HSP70 inhibited cardiac inflammation and promoted its resolution in doxorubicin‐induced HF by switching the cardiac immune microenvironment to an anti‐inflammatory state, as shown by the lower production of interleukin‐6, transforming growth factor‐β1, and interleukin‐17A, as well as higher level of interleukin‐10, which indicates that extracellular HSP70 could be a key factor obstructing the resolution of myocardial inflammation at the early stage of acute injury‐evoked HF.
During the early inflammatory phase, PMNs, acting as the first responders and the hallmark of acute inflammation, are first recruited to the wound site and ingest microorganisms or particles.26 However, after completion of their tasks, an adequate and rapid elimination of neutrophils by infiltrated macrophages is a prerequisite and key histological event for resolving inflammation and restoring tissue function.27 In our current study, we detected the local infiltration of PMNs and macrophages as well as apoptotic PMNs in the heart tissue for the confirmation of the progress of inflammation resolution. As expected, blocking the extracellular HSP70 activity decreased the content of apoptotic PMNs but enhanced the content of macrophages. The study by Rider et al suggests that interleukin‐1α, released from dying cells, initiates sterile inflammation by inducing recruitment of neutrophils, whereas interleukin‐1β promotes the recruitment and retention of macrophages.22 Indeed, our findings confirmed that the dynamic changes in the levels of interleukin‐1α and interleukin‐1β are consistent with the histological and immunological features of the doxorubicin‐induced myocardial injury and imply that the extracellular HSP70 may be a pathological alarm signal for HF. Although we did not address whether the extracellular HSP70 represents a useful diagnostic marker for patients with HF, investigating this possibility may provide valuable information and is worthy of further investigation.
Extracellular HSP70 has been shown to colocalize with multiple membrane receptors (ie, TLRs), among which TLR2 and TLR4 are the most likely functional candidates.28 Indeed, TLR2‐ or TLR4‐knockout mice have been demonstrated to reduce infarct size, improve cardiac function, and suppress myocardial inflammation.29, 30, 31 However, our previous study demonstrated that an adjuvant application of TLR4 agonist could observably promote the resolution of myocardial inflammation in hypertrophic heart through shifting the suppressive immune response toward a type 1 helper T cell type immune response.32 The apparently contradictory effects of TLR4 activity indicate that there is a “double‐edged sword” role of TLR4 in the regulation of myocardial inflammation and targeting TLR4 signals could not be an optimal choice for the treatment of inflammatory heart disease. In this study, we found that blocking TLR2 but not TLR4 significantly suppressed the HSP70‐triggered activation and nuclear translocation of NF‐κBp65 in cardiomyocytes, suggesting that the extracellular HSP70‐initiated inflammatory response is mainly mediated by TLR2 signaling. The reasons for these inconsistent results could be that TLR2‐mediated inflammation signals are almost only dependent on the MyD88‐p38‐NF‐κB pathway, whereas TLR4 has an alternative TRIF‐IRF3 pathway available,33 which is not affected by the blockade of the extracellular HSP70 activity. Taken together, our findings suggest that MyD88‐dependent TLR2 signaling pathway plays a more vital role in mediating the proinflammatory effect of extracellular HSP70.
Conclusions
As one of the representative DAMP molecules, HSP70 is a multifunctional, ubiquitous protein located both inside and outside cells. It plays pivotal roles in various physiological and pathological processes, including cell proliferation, differentiation, epithelial to mesenchymal transition, apoptosis, and death, as well as immunity and metabolism.34, 35 However, the regulatory effect of HSP70 on myocardial inflammation during the development of HF is not well characterized. Our present study highlights a novel role for extracellular HSP70 in promoting HF via activation of TLR2‐p38‐NF‐κB–mediated noninfectious inflammatory response in myocardium, which suggests a possible therapeutic approach against HF by blocking the biological functions of extracellular HSP70 or its receptor, TLR2.
Sources of Funding
This work was supported by grants from the National Key Research and Development Program of China (2017YFA0205400), the National Science and Technology Major Project of the Ministry of Science and Technology of China (2018ZX09711001‐003‐009), the National Natural Science Foundation of China (81530093 and 81773781 to Hu; 81773800 to Zhang), the Chinese Academy of Medical Sciences (CAMS), the Innovation Fund for Medical Sciences (2016‐I2M‐1‐007, 2016‐I2M‐1‐010, and 2016‐I2M‐3‐008), and the CAMS Central Public‐Interest Scientific Institution Basal Research Fund (2018PT35004 and 2017PT35001).
Disclosures
None.
(J Am Heart Assoc. 2019;8: e012338 DOI: 10.1161/JAHA.119.012338.)
References
- 1. Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812–824. [DOI] [PubMed] [Google Scholar]
- 2. Mebazaa A, Yilmaz MB, Levy P, Ponikowski P, Peacock WF, Laribi S, Ristic AD, Lambrinou E, Masip J, Riley JP, McDonagh T, Mueller C, deFilippi C, Harjola VP, Thiele H, Piepoli MF, Metra M, Maggioni A, McMurray JJ, Dickstein K, Damman K, Seferovic PM, Ruschitzka F, Leite‐Moreira AF, Bellou A, Anker SD, Filippatos G. Recommendations on pre‐hospital and early hospital management of acute heart failure: a consensus paper from the Heart Failure Association of the European Society of Cardiology, the European Society of Emergency Medicine and the Society of Academic Emergency Medicine–short version. Eur Heart J. 2015;36:1958–1966. [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y, Bauersachs J, Langer HF. Immune mechanisms in heart failure. Eur J Heart Fail. 2017;19:1379–1389. [DOI] [PubMed] [Google Scholar]
- 4. Van Linthout S, Tschope C. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep. 2017;14:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. 2015;116:1254–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng Y, Gardner SE, Clarke MC. Cell death, damage‐associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:2781–2786. [DOI] [PubMed] [Google Scholar]
- 7. Liu L, Wang Y, Cao ZY, Wang MM, Liu XM, Gao T, Hu QK, Yuan WJ, Lin L. Up‐regulated TLR4 in cardiomyocytes exacerbates heart failure after long‐term myocardial infarction. J Cell Mol Med. 2015;19:2728–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu T, Zhang DY, Zhou YH, Han QF, Wang LH, Wu L, Yao HC. Increased serum HMGB1 level may predict the fatal outcomes in patients with chronic heart failure. Int J Cardiol. 2015;184:318–320. [DOI] [PubMed] [Google Scholar]
- 9. Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y, Nakamura M. Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur J Heart Fail. 2006;8:810–815. [DOI] [PubMed] [Google Scholar]
- 10. Imbalzano E, Mandraffino G, Casciaro M, Quartuccio S, Saitta A, Gangemi S. Pathophysiological mechanism and therapeutic role of S100 proteins in cardiac failure: a systematic review. Heart Fail Rev. 2016;21:463–473. [DOI] [PubMed] [Google Scholar]
- 11. Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. [DOI] [PubMed] [Google Scholar]
- 12. Ranek MJ, Stachowski MJ, Kirk JA, Willis MS. The role of heat shock proteins and co‐chaperones in heart failure. Philos Trans R Soc Lond B Biol Sci. 2018;373: pii: 20160530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dybdahl B, Slordahl SA, Waage A, Kierulf P, Espevik T, Sundan A. Myocardial ischaemia and the inflammatory response: release of heat shock protein 70 after myocardial infarction. Heart. 2005;91:299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zou N, Ao L, Cleveland JC Jr, Yang X, Su X, Cai GY, Banerjee A, Fullerton DA, Meng X. Critical role of extracellular heat shock cognate protein 70 in the myocardial inflammatory response and cardiac dysfunction after global ischemia‐reperfusion. Am J Physiol Heart Circ Physiol. 2008;294:H2805–H2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai WF, Zhang XW, Yan HM, Ma YG, Wang XX, Yan J, Xin BM, Lv XX, Wang QQ, Wang ZY, Yang HZ, Hu ZW. Intracellular or extracellular heat shock protein 70 differentially regulates cardiac remodelling in pressure overload mice. Cardiovasc Res. 2010;88:140–149. [DOI] [PubMed] [Google Scholar]
- 16. Fujisaki G, Inokuchi C, Murashige N. Doxorubicin‐induced myocardial injury. N Engl J Med. 2004;351:1908–1909. [DOI] [PubMed] [Google Scholar]
- 17. Ma Y, Zhang X, Bao H, Mi S, Cai W, Yan H, Wang Q, Wang Z, Yan J, Fan GC, Lindsey ML, Hu Z. Toll‐like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One. 2012;7:e40763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, Kranias EG. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin‐triggered oxidative stress and cardiotoxicity. Circ Res. 2008;103:1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carvalho FS, Burgeiro A, Garcia R, Moreno AJ, Carvalho RA, Oliveira PJ. Doxorubicin‐induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med Res Rev. 2014;34:106–135. [DOI] [PubMed] [Google Scholar]
- 20. Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res. 2016;119:159–176. [DOI] [PubMed] [Google Scholar]
- 21. Austyn JM, Hankins DF, Larsen CP, Morris PJ, Rao AS, Roake JA. Isolation and characterization of dendritic cells from mouse heart and kidney. J Immunol. 1994;152:2401–2410. [PubMed] [Google Scholar]
- 22. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN. IL‐1alpha and IL‐1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–4843. [DOI] [PubMed] [Google Scholar]
- 23. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. [DOI] [PubMed] [Google Scholar]
- 24. Perretti M, Leroy X, Bland EJ, Montero‐Melendez T. Resolution pharmacology: opportunities for therapeutic innovation in inflammation. Trends Pharmacol Sci. 2015;36:737–755. [DOI] [PubMed] [Google Scholar]
- 25. Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. 2016;15:551–567. [DOI] [PubMed] [Google Scholar]
- 26. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as protagonists and targets in chronic inflammation. Nat Rev Immunol. 2017;17:248–261. [DOI] [PubMed] [Google Scholar]
- 27. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. 2016;23:915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: role of toll‐like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. [DOI] [PubMed] [Google Scholar]
- 29. Favre J, Musette P, Douin‐Echinard V, Laude K, Henry JP, Arnal JF, Thuillez C, Richard V. Toll‐like receptors 2‐deficient mice are protected against postischemic coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2007;27:1064–1071. [DOI] [PubMed] [Google Scholar]
- 30. Oyama J, Blais C Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia‐reperfusion injury in toll‐like receptor 4‐deficient mice. Circulation. 2004;109:784–789. [DOI] [PubMed] [Google Scholar]
- 31. Riad A, Bien S, Gratz M, Escher F, Westermann D, Heimesaat MM, Bereswill S, Krieg T, Felix SB, Schultheiss HP, Kroemer HK, Tschope C. Toll‐like receptor‐4 deficiency attenuates doxorubicin‐induced cardiomyopathy in mice. Eur J Heart Fail. 2008;10:233–243. [DOI] [PubMed] [Google Scholar]
- 32. Liu YY, Cai WF, Yang HZ, Cui B, Chen ZR, Liu HZ, Yan J, Jin W, Yan HM, Xin BM, Yuan B, Hua F, Hu ZW. Bacillus Calmette‐Guerin and TLR4 agonist prevent cardiovascular hypertrophy and fibrosis by regulating immune microenvironment. J Immunol. 2008;180:7349–7357. [DOI] [PubMed] [Google Scholar]
- 33. Kawai T, Akira S. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat Immunol. 2010;11:373–384. [DOI] [PubMed] [Google Scholar]
- 34. van Eden W, van der Zee R, Prakken B. Heat‐shock proteins induce T‐cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–330. [DOI] [PubMed] [Google Scholar]
- 35. Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]