

Abstract
Immune checkpoint blockade using anti-PD-1/-PD-L1 or anti-CTLA-4 monoclonal antibodies (mAbs) has revolutionized cancer treatment. However, many types of cancer do not respond and for those that do, only a minority of patients achieve durable remissions. Therefore, oncoimmunologists are working to develop adoptive cell therapies for non-hematopoietic tumors by harnessing immune effector cells such as αβ T cells and γδ T cells. In contrast to conventional αβ T cells that recognize peptides in the context of MHC class I or II molecules, γδ T cells expressing Vγ2Vδ2 T cell receptors (also termed Vγ9Vδ2) are stimulated by isoprenoid metabolites (phosphoantigens) such as isopentenyl diphosphate in a butyrophilin 3A1-dependent manner. Vγ2Vδ2 T cells kill almost all types of tumor cells that have been treated with bisphosphonates. In this study, we synthesized a series of fluorine-containing bisphosphonates based on current drugs and find that they stimulate Vγ2Vγ2 T cell killing of tumor cells. A fluorine-containing prodrug analog of zoledronate where phosphonate moities were masked with pivaloyloxymethyl groups markedly enhanced Vγ2Vδ2 T cell-mediated cytotoxicity, and also promoted the expansion of peripheral blood Vγ2Vδ2 T cells. These results demonstrate that a prodrug of a fluorine-containing zoledronate analog can sensitize tumor cells for killing as well as expand Vγ2Vδ2 T cells for adoptive cell therapy.
Keywords: cancer immunotherapy, Vγ2Vδ2 γδ T cells, fluorine, nitrogen-containing bisphosphonate, non-radioactive cellular cytotoxicity assay
Graphical abstract
Cancer immunotherapy has become a clinically validated intervention. Although much attention is focused on mAb-based immunotherapies, the efficacy of immune checkpoint therapy is limited. To develop more effective treatments, we synthesized fluorine-containing N-BPs and a prodrug 11 for use with Vγ2Vδ2 T cell-based immunotherapies.
Introduction
Standard cancer treatments comprise surgery, radiotherapy, chemotherapy, hormone therapy, and targeted therapy. Recently, immunotherapy has become a clinically validated intervention for many cancer patients with the advent of PD-1 and CTLA-4 immune checkpoint therapies.[1] CTLA-4 was identified as a negative regulator of T cells,[2–3] and it was subsequently shown that blocking CTLA-4 signaling using a monoclonal antibody (mAb) could cure established tumors in mice.[4] PD-1 was identified as another negative regulator expressed by T cells[5–6] that interacts with its ligands, PD-L1 and PD-L2. We derived an anti-murine PD-L1 mAb and demonstrated that mAb treatment augmented T cell mediated anti-tumor activity in vitro as well as in vivo in preclinical mouse models.[7] We also showed that anti-human PD-L1 mAbs augmented the anti-tumor activity of human Vγ2Vδ2 T cells in vitro.[8] PD-1 checkpoint blockade has been shown to be effective in the treatment of patients with a number of different cancers including melanoma, non-small cell lung cancer, kidney cancer, urotherlial carcinoma, head and neck carcinoma, hepatocellular carcinoma, and Hodgkin lymphoma.[9–10] However, alternatives to immune checkpoint blockade are needed because the overall response rates for immune checkpoint monotherapy range from ~20–65% with a minority of durable remissions. Moreover, a significant number of patients develop treatment-related autoimmune diseases such as pneumonitis, inflammatory arthritis, thyroid disease, inflammatory bowel disease, and hepatitis.
One of the most promising cancer immunotherapies is the adoptive transfer of immune effector cells that specifically recognize malignant tumors naturally or through expression of chimeric antigen receptors. Both T cells and NK cells can kill human tumor cells.[11] T cells are divided into αβ and γδ T cells. Whereas conventional αβ T cells recognize peptides in the context of MHC class I, Ib, or II molecules, human γδ T cells do not recognize peptides and are not restricted by MHC molecules. The most abundant human γδ T cell subset in the peripheral blood expresses Vγ2Vδ2 T cell receptors (TCR) (also termed Vγ9Vδ2). Vγ2Vδ2 T cells are stimulated by isoprenoid metabolites such as the (E)-4-hydroxy-3-methylbut-2-enyl-1-diphosphate (HMBPP) metabolite in the 2-C-methyl-D-erythritol-4 phosphate isoprenoid pathway used by bacteria and apicomplexan parasites or isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) metabolites in the mevalonate isoprenoid pathway of mammalian cells.[12–16] Stimulation requires the immunoglobulin superfamily protein, butyrophilin 3A1 (BTN3A1), that is expressed on all tumor cells, and the Vγ2Vδ2 TCR expressed by γδ T cells.[17–19]
When tumor cells are treated with nitrogen-containing bisphosphonates (N-BPs), N-BPs are internalized via fluid-phase endocytosis where they inhibit cytoplasmic farnesyl diphosphate synthase (FDPS) resulting in the accumulation of upstream IPP and DMAPP metabolites (Supporting Information, Fig. S1).[20–24] IPP and DMAPP then bind to the B30.2 intracellular domain of BTN3A1. Although the precise mechanism remains unknown, isoprenoid-metabolite binding to the B30.2 domain of BTN3A1 can be sensed at the cell surface by Vγ2Vδ2 T cells through their TCRs (Supporting Information, Fig. S2).[25] Because N-BPs were designed as therapeutics for bone-related diseases such as Paget’s disease and osteoporosis, they mimic inorganic pyrophosphate and have a high affinity for hydroxyapatite in bone.[26] The backbone structure of N-BPs is P-C(OH)-P, with the hydroxyl group (the “bone hook”) increasing N-BPs bone binding.[27] However, because bone binding is not required for stimulation of Vγ2Vδ2 T cells, it may be possible to further optimize N-BPs for their use in cancer immunotherapy.
Fluorine atoms are present in a number of drugs because they can improve bioactivity by altering physicochemical and/or pharmacokinetic properties.[28] In the present study, we sought to determine the effect of substituting a fluorine atom for the hydroxyl group in N-BP on their activity for cancer immunotherapy. We therefore synthesized a series of N-BP analogs to bisphosphonate drugs where a fluorine atom was substituted for the hydroxyl group. The activity of the analogs was compared to that of the parent compounds by treating tumor cells and measuring their killing by Vγ2Vδ2 T cells. To increase activity, a pivaloyloxymethyl derivative of one of the fluorine-containing N-BPs was synthesized and the effect of this modification on cell-mediated cytotoxicity and expansion of Vγ2Vδ2 T cells was determined.
Results and Discussion
Geminal bisphosphonates have a P-C-P backbone and are analogs of inorganic pyrophosphoric acid. The second and third generation geminal bisphosphonates are termed nitrogen-containing bisphosphonates (N-BPs) and have a hydroxyl group at the R1 position and a nitrogen-containing alkyl or aryl side chain at the R2 position (Fig. 1A).[29] The second generation N-BPs pamidronate (1), alendronate (2), and ibandronate (3) contain a nitrogen-containing alkyl group whereas the third generation N-BPs risedronate (4) and zoledronate (5) have a nitrogen-containing heterocyclic group at the R2 position (Fig. 1B).[30]
Figure 1.
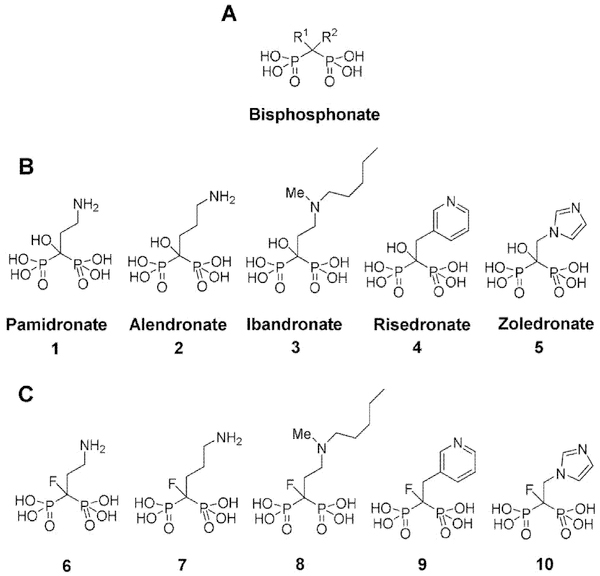
Structures of bisphosphonates. (A) General structure of geminal bisphosphonates. (B) Structures of second and third generation nitrogen-containing bisphosphonates (N-BPs). (C) Structures of fluorine-containing bisphosphonate analogs. Fluorine-containing bisphosphonates have a fluorine atom in place of the hydroxyl group at the R1 position.
We first replaced the hydroxyl group by a fluorine atom at the R1 position of second and third generation N-BPs (1-5). The details of the synthesis of fluorine-containing N-BPs (6-8) (Fig. 1C) are described in the Supporting Information.
In brief, the amino group of tetraethyl-3-aminopropylidene-1,1-bisphosphonate was protected with a Boc group and fluorine was introduced into the R1 position. Boc and ethyl groups were then removed to yield a pamidronate analog (6). For the synthesis of an alendronate analog (7), tetraethyl-4-aminobutylidene-1,1-bisphosphonate was used as a starting material and essentially the same synthetic approach was employed. To prepare an ibandronate analog (8), tetraisopropyl monofluoromethylenediphosphonate was first synthesized [31] and reacted with 2-(2-iodoethoxy)tetrahydro-2H-pyran. After the protective group was removed under acidic conditions, the nascent hydroxyl group was exposed to methanesulfonyl chloride and the resulting compound was reacted with N-methylpentylamine. Finally, isopropyl groups were removed under acidic conditions. A risedronate analog (9) was synthesized as described.[32] The synthetic scheme for a fluorine-analog of zoledronate (10) (Fig. 1C) is shown in Scheme 1 and the procedure is detailed in the chemistry part of the Experimental Section.
Scheme 1.
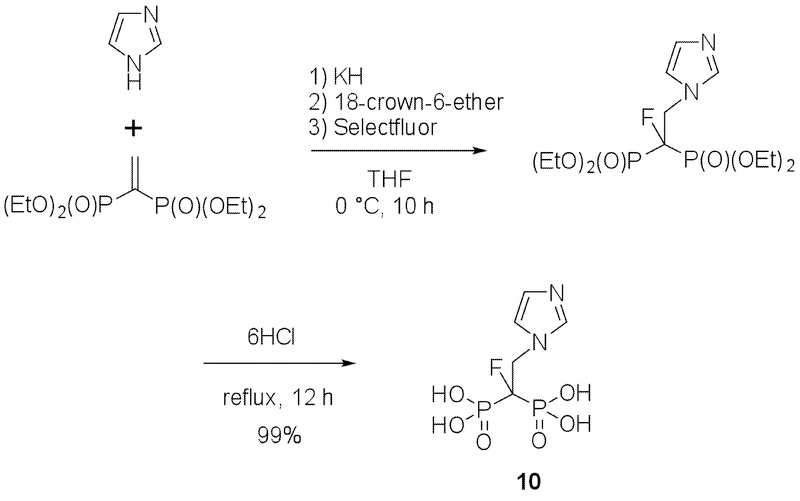
Procedure for the synthesis of 1-fluoro-2-(imidazol-1-yl)ethylidene-1,1-bisphosphonic acid (10).
The activity of the fluorine-containing N-BP analogs were compared to their parent compounds by measuring Vγ2Vδ2 T cell-mediated cytotoxicity against 786–0 human renal carcinoma cells pretreated with the compounds using a non-radioactive cellular cytotoxicity assay (Supporting Information, Fig. S3).[33] As shown in Fig. 2, Vγ2Vδ2 T cells from a healthy adult donor failed to kill untreated 786–0 cells. When the tumor cells were pretreated with compound 1 (pamidronate), however, Vγ2Vδ2 T cells exhibited potent lytic activity against the N-BP-sensitized tumor cells in a dose-dependent manner. The replacement of the hydroxyl group by a fluorine atom at the R1 position significantly reduced the lytic activity of Vγ2Vδ2 T cells against 786–0 cells (compare compound 1 with compound 6). Similar decreases in activity were observed for the other second generation N-BPs (compare compound 2 (alendronate) with 7 and compounds 3 (ibandronate) with 8). Thus, the substitution of a fluorine atom for the hydroxyl group at the R1 position of second generation N-BPs reduced their ability to stimulate killing by Vγ2Vδ2 T cells.
Figure 2.
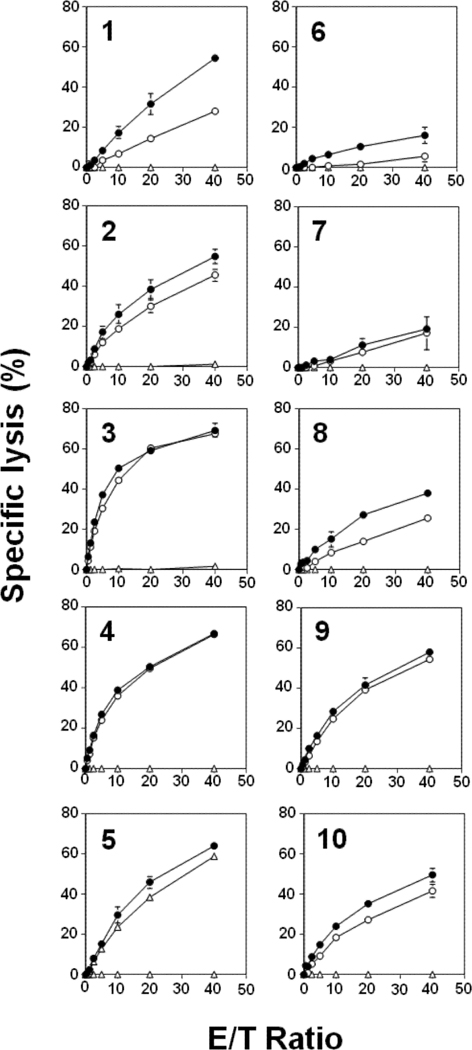
Comparison of Vγ2Vδ2 T cell-mediated cytotoxicity induced by conventional N-BPs (left panels) or their fluorine-containing analogs (right panels). 786–0 renal carcinoma cells were either not treated (Δ) or pretreated with second generation N-BPs (1, 2, or 3) at 1000 μM (○) or 3000 μM (●) or their fluorine-containing analogs (6, 7 or 8) at 1000 μM (○) or 3000 μM (●) or third generation N-BPs (4 or 5) at 300 μM (○) or 1000 μM (●) or their fluorine-containing analogs (9 or 10) at 1000 μM (○) or 3000 μM (●). 786–0 renal carcinoma cells were pretreated with the N-BPs at 37oC for 2 h. The chelate-forming BM-HT prodrug reagent was then added to 25 μM and the cells incubated at 37oC for an additional 15 min. After being washed with RPMI1640 medium, the cells were cultured with Vγ2Vδ2 T cells for 40 min. Supernatants from the cultures were then harvested and mixed with Eu3+ solution. Specific lysis was determined by measuring time-resolved fluorescence using a PHERAStar multiplate reader. Data show mean ± SD and are representative of three independent experiments.
The immunomodulatory activity of third generation N-BPs is more potent than that of second generation N-BPs. When 786–0 cells were pretreated with third generation N-BPs (compounds 4 and 5), Vγ2Vδ2 T cells exhibited potent lytic activity against the N-BP-sensitized tumor cells. Substitution of a fluorine atom for the hydroxyl group at the R1 position slightly diminished the cytolytic activity of Vγ2Vδ2 T cells (compare 4 with 9 and 5 with 10), maintaining levels of specific lysis well above 40%. The difference in the effect of fluorine substitution between second and third generation N-BPs is clearly evident when the levels of specific lysis are compared under identical conditions (effector‒to‒target ratio of 20:1 and treatment with 1,000 μM N-BP). Whereas second generation N-BPs showed large decreases in specific lysis (compounds 1/6 (14.7% versus 1.7%), 2/7 (30.1% versus 7.7%), and 3/8 (58.8% versus 14.1%)), third generation N-BPs showed only moderate decreases in specific lysis (compounds 4/9 (50.6% versus 41.9%), and 5/10 (46.0% versus 35.2%)) (Supporting Information, Table S1). The results clearly demonstrate that the incorporation of a fluorine atom into N-BPs does not improve their in vitro immunomodulatory activity.
Previously, we and others reported that some prodrugs of N-BPs have greatly increased immunomodulatory activity for Vγ2Vδ2 T cells as compared to their acid forms.[24, 34] Oldfield and co-workers earlier reported the synthesis of tetrakispyvaloyloxylmethyl 2-(pyridine-2-ylamino)ethyliene-1,1-bisphosphonate and found that the pyvaloyloxylmethyl (POM) ester compound was one of the most active N-BP‒derivatives that they tested.[34] Based on this finding, we synthesized a series of N-BP POM esters and their corresponding acid forms and compared their biologic acitivities. The immunomodulatory activity of the N-BP POM esters ranged from >20-fold to 1,100-fold more potent than that of their acid forms. Because zoledronate is the most active compound among approved N-BPs drugs, we attempted to synthesize a prodrug of zoledronate (5). However, a POM-derivative of zoledronate was unstable and decomposed immediately after synthesis.
We therefore attempted to synthesize a prodrug (11) of the fluorine-containing zoledronate analog (10). The substitution of a fluorine atom for the hydroxyl group at the R1 position of zoledronate stabilized its POM derivative and allowed its isolation. The synthesis is detailed in the chemistry part of the Experimental Section (Scheme 2). When compound 11-pretreated 786–0 renal carcinoma cells were incubated with Vγ2Vδ2 T cells, the N-BP-sensitized tumor cells were killed in a dose-dependent as well as effector-to-target ratio-dependent manner (Fig. 3). 40% of the 786–0 cells were specifically lysed by Vγ2Vδ2 T cells after pretreatment with compound 10 at 1,000 μM and an effector-to-target ratio of 40:1. Essentially the same immunomodulatory effect was achieved with 1 μM compound 11, demonstrating that its activity is ~1,000-fold higher than compound 10. Given that the zoledronate (5)-based prodrug is unstable, the incorporation of a fluorine atom at the R1 position is pivotal for the synthesis of a POM derivative.
Scheme 2.
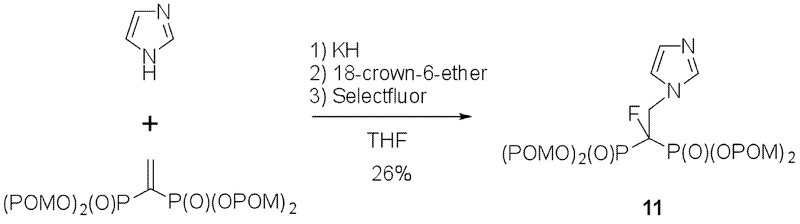
Procedure for the synthesis of tetrakispivaloxylmethyl-1-fluoro-2-(1H-imidazol-1-yl)ethylidene-1,1-bisphosphonate (11).
Figure 3.
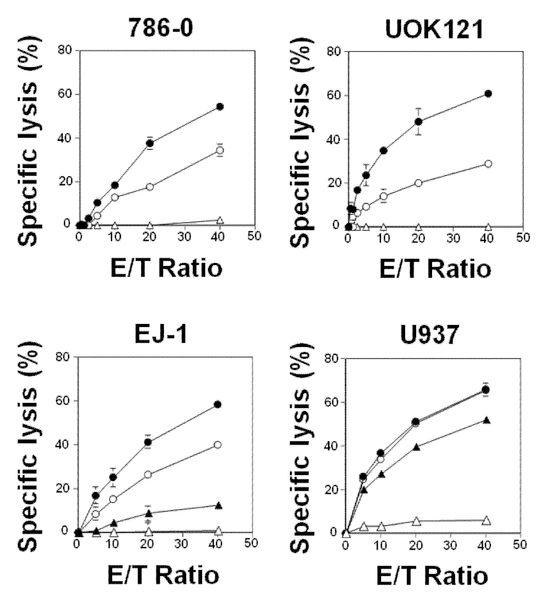
Vγ2Vδ2 T cell-mediated cytotoxicity induced by tetrakispivaloxylmethyl-1-fluoro-2-(1H-imidazol-1-yl)ethylidene-1,1-bisphosphonate (11). 786–0 and UOK121 renal carcinoma cells were pretreated with 0 μM (Δ), 1 μM (○) or 5 μM (●) of 11 at 37oC for 2 h. Similarly, EJ-1 bladder carcinoma cells and U937 histiocytic leukemia cells were pretreated with 0 μM (Δ), 1.25 μM (▲), 2.5 μM (○) or 5 μM (●) of 11. The cells were further treated with 25 μM of BM-HT at 37oC for 15 min and specific lysis by Vγ2Vδ2 T cells determined as in Fig. 2. Data show mean ± SD and are representative of three independent experiments.
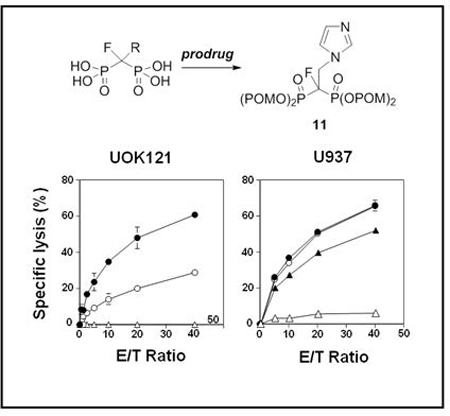
To determine if compound 11 was active with other tumor cell types, UOK121 human renal carcinoma cells, EJ-1 human bladder carcinoma cells, and U937 human pro-monocytic histiocytic lymphoma cells were treated with compound 11 and their killing by Vγ2Vδ2 T cells determined. Vγ2Vδ2 T cells efficiently killed each of the cell lines in a dose-dependent as well as effector-to-target ratio-dependent manner. The specific lysis of the cell lines by Vγ2Vδ2 T cells ranged from 20.7% to 48.2% at a compound 11 concentration of 5 μM and an effector‒to‒target ratio of 20:1 (Supporting Information, Table S2). Note that, as expected, compound 11-induced cell kiling is specific for human cells given that compound 11-treated murine tumor cells were not killed by Vγ2Vδ2 T cells whereas human tumor cell lines and virus-transformed cell lines were all killed to varying extents (Supporting Information, Table S3). These results show that fluorine-containing N-BP prodrugs such as compound 11 combined with the adoptive transfer of Vγ2Vδ2 T cells could be useful for the treatment of a wide variety of tumors.
We next examined whether compound 11 could induce the expansion of Vγ2Vδ2 T cells from a cancer patient. PBMC from a lung cancer patient were incubated with compound 11 in the presence of IL-2. Small clusters of cells were noted on day 4/5 leading to large cell clumps on day 7 (Fig. 4A) indicating that compound 11 was internalized in monocytes and dendritic cells and stimulated Vγ2Vδ2 T cell proliferation. The proportion of Vγ2Vδ2 T cells increased from 4.1% on day 0 to 98.6% on day 11 (Fig. 4B). The total number of Vγ2Vδ2 T cells increased from 2.03 × 105 cells on day 0 to 1.47 × 108 cells on day 11 (724-fold expansion). To determine if the expanded Vγ2Vδ2 T cells from the lung cancer patient were functional, expanded Vγ2Vδ2 T cells were incubated with compound 11-pretreated PC-9 lung carcinoma cells and cytotoxicity measured. Vγ2Vδ2 T cells expanded by compound 11, lysed pretreated PC-9 lung carcinoma cells in a concentration-dependent as well as effector-to-target ratio-dependent manner (Fig. 4C). Similar results were obtained using Vγ2Vδ2 T cells derived from PBMCs from a healthy adult donor where the allogeneic lung cancer cell line, H520, was efficiently killed by Vγ2Vδ2 T cells suggesting that adoptive cancer immunotherapy using allogenic Vγ2Vδ2 T cells is possible (Supporting Information, Fig. S5). Thus, compound 11 efficiently expanded Vγ2Vδ2 T cells and the expanded cells specifically killed compound 11-sensitized lung cancer cells.
Figure 4.

Immunomodulatory effects of tetrakispivaloxylmethyl-1-fluoro-2-(1H-imidazol-1-yl)ethylidene-1,1-bisphosphonate (11) on Vγ2Vδ2 T cells derived from a lung cancer patient. (A) Expansion by 11 of Vγ2Vδ2 T cells from PBMCs derived from a patient with lung cancer. PBMCs were stimulated with 11 and cell clustering was assessed on days 4, 5, 6, and 7. (B) Flow cytometric analysis of Vγ2Vδ2 T cells before (day 0) and after (day 11) expansion with 11. Cells on days 0 and 11 were stained with FITC-conjugated anti-Vδ2 mAb and PE-conjugated anti-CD3 mAb and analyzed for expression of Vδ2 and CD3 through a FACS Verse flow cytometer. (C) Specific lysis of PC-9 lung carcinoma cells by compound 11-expanded Vγ2Vδ2 T cells. (Left panel) Spontaneous and maximal time-resolved fluorescence of PC-9 cells. PC-9 cells were treated with 25 μM of BM-HT at 37oC for 15 min. After washing, the cells were incubated in the absence (spontaneous) or presence (maximum) of 0.125% detergent in DMSO for 40 min. Supernatants from the cultures were then mixed with Eu3+ solution and the spontaneous and maximum release determined by measuring time-resolved fluorescence using a PHERAStar multiplate spectrophotometer. The spontaneous release rate was 18% (Right panel) Specific lysis of PC-9 cells by Vγ2Vδ2 T cells. PC-9 cells were pretreated with 0 μM (♦), 1.25 μM (■), 2.5 μM (▲), or 5 μM (●) of 11 at 37oC for 2 h. The cells were further treated with 25 μM of BM-HT at 37oC for 15 min. The specific lysis of pretreated-PC-9 cells by Vγ2Vδ2 T cells expanded by compound 11 was determined as in Fig. 2. Data show mean ± SD and are representative of three independent experiments.
Phosphorus‒containing therapeutics are receiving increasing attention.[35] Because the cell permeability of BPs is relatively low, prodrugs of BPs have been designed and synthesized.[36] Esterification of BPs increased their direct cytotoxic activity compared with the corresponding BP acid.[37] This demonstrates the importance of the cell membrane permeability of BPs in determining their biological activity. Moreover, a bisphosphonamidate BP prodrug exhibits selectivity in tumor entry.[38] Similar approaches have been used to develop new immunomodulatory prenyl pyrophosphates with increased cell entry and stability.[39–41] Taken together, the development of BP prodrugs could open a new avenue for cancer therapy through more efficient blocking of isoprenoid biosynthesis leading to decreased tumor cell growth and the triggering of tumor immunity by Vγ2Vδ2 T cells.
Conclusions
A series of fluorine-containing N-BPs induced anti-tumor activity in Vγ2Vδ2 T cells upon treatment of tumor cells. Although the substitution of a fluorine atom for the hydroxyl group at the R1 position of N-BPs reduced their immunomodulartory activity, the introduction of a fluorine atom was essential for the synthesis and stabilization of compound 11, a prodrug of the zoledronate analog 10. The immunomoduolatory activity of the pivaloyloxymethylated prodrug 11 was ~1,000-fold more potent than its corresponding acid form 10. By using this synthetic strategy, it may be possible to synthesize N-BP prodrugs optimized for cancer immunotherapy.
Experimental Section
Chemistry
All NMR spectra were obtained through an AL400 NMR spectrometer (JEOL Ltd., Akishima, Tokyo, Japan) and a 500PS NMR spectrometer (Varian Medical Systems Inc., Palo Alto, CA) and processed using an ACD/SpecManager Enterprise platform (ACD/Labs, Tronto, Ontario, Canada). 1H, 13C and 19F NMR spectra were recorded as chemical shifts (δ) in parts per million (ppm) relative to the solvent peak using tetramethylsilane, (2, 2, 3, 3-d4) trimethylsilyl-3-propanoic acid, sodium salt (Thermo Fisher Scientific, Waltham, MA) for 1H and 13C and trichlorofluoromethane (Merck, Darmstadt, Germany) for 19F as internal standards. Chemical shifts (δ) were quoted in parts per million (ppm) and coupling constants (J) were measured in hertz (Hz). The following abbreviations were used to describe multiplicities; s: singlet, d: doublet, t: triplet, q: quartet, quint.: quintet, sext.: sextet, sept.: septet, br: broad, and m: multiplet. High resolution mass spectra (HRMS, m/z) were obtained using a JMS-700N spectrometer (JEOL Ltd.) for electron ionization or a JMS-T100TD spectrometer (JEOL Ltd.) for electrospray ionization (ESI+). All reactions were performed in apparatuses with magnetic stirring under an inert atmosphere. Flash column chromatography was performed over Silica Gel C60 (50–200 μm) (Fuji Silysia Chemical Ltd., Kasugai, Aichi, Japan) using an eluent system as described in each individual experiment. Thin-layer chromatography was performed using TLC Silica Gel 60 F254 aluminum sheets (Merck).
Tetraethyl-1-fluoro-[2-(imidazol-1-yl)ethylidene]-1,1-bisphosphonate:
Tetraethyl-1-fluoro-[2-(imidazol-1-yl)ethylidene]-1,1-bisphosphonate was synthesized according to a literature procedure. In brief, to a suspension of KH (137 mg, 30%, 1.0 mmol) in THF (7.0 mL) was added imidazole (68 mg, 1.0 mmol) at 0oC under argon atmosphere. After stirring for 1 h at 0oC and then for 30 min at room temperature, a solution of 1,1-bis(diethylphosphono)ethylene (300 mg, 1.0 mmol) in THF (2.0 mL) was added to the cooled mixture at 0oC. After stirring for 30 min, a solution of 18-crown-6-ether (54.2 mg, 0.2 mmol) in THF (2.0 mL) was added. The reaction mixture was stirred for 30 min, to which was added Selectfluor (530 mg, 1.5 mmol). The mixture was stirred for 10 h, and the reaction was quenched by the addition of aqueous sat. NH4Cl (10 mL). The aqueous layer was extracted with AcOEt (2 × 10 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo. The residue was purified by silica-gel column chromatography (eluent; Acetone:n-hexane = 1/1 to Acetone /MeOH = 10/1) to give the title compound (58 mg, 55% yield).
1H NMR (500 MHz, CDCl3) δ 1.33 (dt, J = 7.1, 11.7 Hz, 12H), 4.17‒4.27 (m, 8H), 4.67 (ddd, J = 9.6, 18.9, 25.9 Hz, 2H), 7.01 (s, 1H), 7.03 (s, 1H), 7.56 (s, 1H). 1H NMR spectrum corresponds to that of reference.[15]
Synthesis of 1-fluoro-2-(imidazol-1-yl)ethylidene-1,1-bisphosphonic acid (10):
A solution of tetraethyl-1-fluoro-[2-(imidazol-1-yl)ethylidene]-1,1-bisphosphonate (58 mg, 0.1 5 mmol) in 6N HCl (1.0 mL) was stirred under reflux for 12 hrs, and then the reaction mixture was concentrated in vacuo to remove volatiles. The residue was recrystallized from H2O/MeOH to afford the title compound as a white solid (42 mg, 99% yield).
1H NMR (500 MHz, D2O) δ 4.72−4.81 (m, 2H), 7.28 (s, 1H), 7.37 (s, 1H), 8.62 (s, 1H); 13C NMR (120 MHz, D2O) δ 51.2 (br. d, J = 18.5 Hz), 118.7, 123.6, 135.9; 19F NMR (471 MHz, D2O) δ −189.8 (tt, J = 25.7, 67.9 Hz, 1F); HRMS (ESI) m/z Calcd. for C5H8FN2O6P2 [M]- 272.9842, found 272.9807.
Synthesis of tetrakispivaloxylmethyl-1-fluoro-2-(1H-imidazol-1-yl) ethylidene-1,1-bisphosphonate (11):
The title compound was synthesized according to a slightly modified literature procedure.[15] To a suspension of KH (21 mg, 30%, 0.16 mmol) in THF (2.0 mL) was added imidazole (11 mg, 0.16 mmol) at 0 ˚C under argon atmosphere. After stirring for 1 h at 0oC and then for 30 min at room temperature, a solution of tetrakispivaloyloxymethyl vinylidene-1,1-bisphosphonate (100 mg, 0.16 mmol) in THF (1.5 mL) was added to the cooled mixture at 0oC. After stirring for 30 min, a solution of 18-crown-6-ether (8.2 mg, 0.03 mmol) in THF (1.5 mL) was added. The reaction mixture was stirred for 30 min, and then Selectfluor (82 mg, 0.23 mmol) was added. The mixture was stirred for 17 hrs, and the reaction was quenched with by the addition of aqueous sat. NH4Cl (5.0 mL). The aqueous layer was extracted with AcOEt (2 × 10 mL). The combined organic layers were dried over MgSO4, concentrated in vacuo. The residue was purified by silica-gel column chromatography (eluent; Acetone:n-hexane = 1/1 to Acetone /MeOH = 10/1) to give the title compound (30 mg, 26% yield).
1H NMR (500 MHz, CDCl3) δ 1.23 (s, 36H), 4.67 (ddd, J = 9.1, 10.3, 25.9 Hz, 2H), 5.60−5.71 (m, 8H), 6.96 (s, 1H), 7.02 (s, 1H), 7.51 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 26.8, 26.8, 38.7, 28.7, 48.1 (m), 82.8 (dt, J = 3.2, 70.2 Hz), 120.7 (d, J = 1.6 Hz), 129.2, 138.5 (d, J = 1.2 Hz), 176.5, 176.6; 19F NMR (470 MHz, CDCl3) δ −191.6 (tt, J = 26.0, 71.8 Hz); HRMS (ESI) m/z Calcd for C29H49FN2NaO14P2 [M]+ 753.2541, found 753.2502.
Biological assays
Derivation of Vγ2Vδ2 T cells:
Peripheral blood samples were obtained from a healthy adult volunteer and a lung cancer patient after approval of the institutional review board of Nagasaki University Hospital with written informed consent. The peripheral blood samples were heparinized and diluted with an equal volume of phosphate-buffered saline (PBS (−), Nissui Pharmaceutical Co., Ltd. Taito-ku, Tokyo, Japan) and peripheral blood mononuclear cells (PBMCs) were purified by density gradient centrifugation at 600 × g for 30 min at ambient temperature. PBMCs were washed two times with PBS (−) and resuspended in Yssel’s medium containing human AB serum (Cosmo Bio Co., Ltd., Koto-ku, Tokyo, Japan) at a cell concentration of 1.6 × 107 cells/mL. The cell suspension (1.5 mL each) was placed into wells of a 24-well plate, to which was added 1.5 μL each of 1 mM tetrakis-pivaloyloxymethyl 2-(thiazole-2ylamino)ethylidene-1,1-bisphosphonate (PTA in dimethylsulfoxide, Techno Suzuta Co., Ltd., Heiwa-machi, Nagasaki, Japan) or 7.5 μL each of 1 mM compound 11, tetrakispivaloxylmethyl-1-fluoro-2-(1H-imidazol-1-yl)ethylidene-1,1-bisphosphonate. After the plate was incubated at 37oC with 5% CO2 overnight, 15 μL of 10,000 U/ml interleukin-2 (IL-2, Shionogi Pharmaceutical Co., Ltd., Chuo-ku, Osaka, Japan) were added to each well. After the plate was incubated at 37oC with 5% CO2 for 24 h, the medium was replaced with new Yssel’s medium containing 100 U/mL of IL-2. IL-2 was added to each well every day until day 10. On day 11, the cell suspension was transferred into 50 mL conical tubes, which were centrifuged at 600 × g at 4oC for 5 min. After the culture supernatant was removed, the cell pellets were dispersed by tapping, and cryopreservation medium added (Cell Banker I, Nippon Zenyaku Kogyo. Co., Ltd., Koriyama, Fukushima, Japan) to give a cell concentration of 1 × 107 cells/mL. The cell suspension (1 mL each) was dispensed into cryopreservation vials and placed at −80oC overnight and then stored in liquid nitrogen.
Tumor cell lines:
The human renal carcinoma cell line 786–0 was purchased from American Type Culture Collection (Manassas, VA). The human renal cell carcinoma cell line UOK121 was a kind gift from Dr. Hirohito Kobayashi (Tokyo Women’s Medical University, Shinjuku, Tokyo, Japan). The human bladder carcinoma cell line EJ-1 and the human histiocytic lymphoma cell line U937 were purchased from Health Science Research Resources Bank (Sennan, Osaka, Japan). The human lung carcinoma cell line PC-9 was obtained from RIKEN BioResource Center (Tsukuba, Ibaraki, Japan).
Non-radioactive cellular cytotoxicity assay:
Tumor cells were pretreated with 0 – 3,000 μM of conventional or fluorine-containing N-BPs at 37oC with 5% CO2 for 2 h and treated with 25 μM of bis(butyryloxymethyl) 4’-hydroxymethyl-2,2’:6’,2”-terpyridine-6,6”-dicarboxylate (BM-HT, a non-radioactive chelate-forming proligand, Techno Suzuta Co., Ltd.) at 37oC with 5% CO2 for 15 mim. The cells were washed three times with RPMI1640 medium (Merck & Co., Inc., Kenilworth, NJ) supplemented with 10% fetal serum albumin (FCS) (Merck & Co., Inc.), 10−5 M 2-mercaptoethanol (Wako Pure Chemical Industries, Ltd., Chuo-ku, Osaka, Japan), 100 U/mL of penicillin (Meiji Seika Pharma Co., Ltd., Chuo-ku, Tokyo, Japan), 100 μg/mL of streptomycin (Meiji Seika Pharma Co., Ltd.) and resuspended in the RPMI1640 medium at a cell concentration of 5 × 105 cells/mL. To wells of a round-bottom 96-well plate (Corning Inc., Corning, NY) 100 μL of the tumor cell suspension were added. Cryopreserved Vγ2Vδ2 T cells were thawed and the labeled tumor cells were mixed with 100 μL of the thawed Vγ2Vδ2 T cells at effector to target ratios of 0, 0.625, 1.25, 2.5, 5, 10, 20, and 40. The plate was then incubated at 37oC with 5% CO2 for 40 min. For determination of the maximum release rate, the labeled cells were lysed with 0.125% digitonin (Merck & Co., Inc.) in 19% dimethylsulfoxide in H2O. After the cell suspensions were mixed well by pipetting, the plate was centrifuged at 300 × g for 2 min at room temperature and 25 μL of the supernatants were transferred into a new round-bottom 96-well plate. To each well, 250 μL of europium solution was added (Techno Suzuta Co., Ltd.), 200 μL of which were transferred into wells of a 96-well optical plate (ThermoFisher Scientific Inc., Waltham, MA). Time-resolved fluorescence was then determined using an ARVO (PerkinElmer Inc., Waltham, MA) or a PHERAstar (BMG Labtech Ltd., Allmendgruen, Ortenberg, Germany) multiplate reader. All experiments were performed in triplicate. The maximum release was calculated as [maximum release (counts) – background (counts)] and the spontaneous release as [spontaneous release (counts) – background (counts)]. Then, the spontaneous release rate (%) was calculated as [(100 x spontaneous release)/maximum release].
Microscopic analysis:
During the expansion of Vγ2Vδ2 T cells with compound 11, cell images were captured under a BZ-X700 fluorescence microscope (Keyence Corp., Higashi Yodogawa-Ku, Osaka, Japan).
Flow cytometric analysis:
Freshly isolated PBMCs and N-BP-expanded cells were plated out at 2 × 105 cells/50 μL in a round-bottom 96-well plate (Corning Inc.). The cells were incubated with 3 μL of fluorescein isothiocyanate (FITC)-conjugated TCR Vδ2 mAb (BD Biosciences, San Diego, CA) and 3 μL of phycoerythrin (PE)-conjugated anti-cluster of differentiation 3 (CD3) mAb on ice for 15 min. After being washed three times with 200 μL of PBS, the cells were resuspended in 600 μL of PBS and analyzed using a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). The stained cells were analyzed using FlowJo ver. 10 (FlowJo LLC, Ashland, Oregon).
Supplementary Material
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Culture, Sports, and Technology of Japan 16K08844 (MEXT) (to Y.T.), by the Japan Agency for Medical Research and Development 12723070 and DNW-17004 (to Y.T.), by the SUNBOR grant (to S.M.), by the Department of Veterans Affairs (Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development) Grant 1 I01 BX000972–01A1 (to C.T.M.), National Cancer Institute Grant P30CA086862 (Core Support) (to C.T.M.). C.T.M. is the Kelting Family Scholar in Rheumatology.
Footnotes
Supporting Information
Refer to Web version on PubMed Central for supporting information.
Supporting information and the ORCID identification number(s) for the authors of this article can be found under: https://doi.org/10.1002/cmdc.201800764.
Conflict of interest
Y.T. is a co-inventor of novel terpyridine-derivative proligands for measuring cytotoxicity: PCT/JP2015/059838. C.T.M. is a co-inventor of US Patent 8,012,466 on the development of live bacterial vaccines for activating γδ T cells and has no other financial or non-financial conflict of interest. The other authors declare no financial or non-financial conflicts of interest.
References
- [1].Couzin-Frankel J., Science 2013, 342, 1432–1433. [DOI] [PubMed] [Google Scholar]
- [2].Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA, Immunity 1994, 1, 405–413. [DOI] [PubMed] [Google Scholar]
- [3].Krummel MF, Allison JP, J. Exp. Med. 1995, 182, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Leach DR, Krummel MF, Allison JP, Science 1996, 271, 1734–1736. [DOI] [PubMed] [Google Scholar]
- [5].Nishimura H., Okazaki T., Tanaka Y., Nakatani K., Hara M., Matsumori A., Sasayama S., Mizoguchi A., Hiai H., Minato N., Honjo T., Science 2001, 291, 319–322. [DOI] [PubMed] [Google Scholar]
- [6].Okazaki T., Tanaka Y., Nishio R., Mitsuiye T., Mizoguchi A., Wang J., Ishida M., Hiai H., Matsumori A., Minato N., Honjo T., Nat. Med. 2003, 9, 1477–1483. [DOI] [PubMed] [Google Scholar]
- [7].Iwai Y., Ishida M., Tanaka Y., Okazaki T., Honjo T., Minato N., Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Iwasaki M., Tanaka Y., Kobayashi H., Murata-Hirai K., Miyabe H., Sugie T., Toi M., Minato N., Eur. J. Immunol. 2011, 41, 345–355. [DOI] [PubMed] [Google Scholar]
- [9].Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L., Drake CG, Pardoll DM, Chen L., Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H., Korman AJ, Jure-Kunkel M., Agrawal S., McDonald D., Kollia GD, Gupta A., Wigginton JM, Sznol M., Engl N.. J. Med. 2012, 366, 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P., Drake CG, Camacho LH, Kauh J., Odunsi K., Pitot HC, Hamid O., Bhatia S., Martins R., Eaton K., Chen S., Salay TM, Alaparthy S., Grosso JF, Korman AJ, Parker SM, Agrawal S., Goldberg SM, Pardoll DM, Gupta A., Wigginton JM, Engl N.. J. Med. 2012, 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Senju H., Kumagai A., Nakamura Y., Yamaguchi H., Nakatomi K., Fukami S., Shiraishi K., Harada Y., Nakamura M., Okamura H., Tanaka Y., Mukae H., Int. J. Biol. Sci. 2018, 14, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tanaka Y., Sano S., Nieves E., De Libero G., Rosa D., Modlin RL, Brenner MB, Bloom BR, Morita CT, Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 8175–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Constant P., Davodeau F., Peyrat MA, Poquet Y., Puzo G., Bonneville M., Fournié JJ, Science 1994, 264, 267–270. [DOI] [PubMed] [Google Scholar]
- [14].Tanaka Y., Morita CT, Nieves E., Brenner MB, Bloom BR, Nature 1995, 375, 155–158. [DOI] [PubMed] [Google Scholar]
- [15].Hintz M., Reichenberg A., Altincicek B., Bahr U., Gschwind RM, Kollas AK, Beck E., Wiesner J., Eberl M., Jomaa H., FEBS Lett. 2001, 509, 317–322. [DOI] [PubMed] [Google Scholar]
- [16].Amslinger S., Kis K., Hecht S., Adam P., Rohdich F., Arigoni D., Bacher A., Eisenreich W., J. Org. Chem. 2002, 67, 4590–4594. [DOI] [PubMed] [Google Scholar]
- [17].Bukowski JF, Morita CT, Tanaka Y., Bloom BR, Brenner MB, Band H., J. Immunol. 1995, 154, 998–1006. [PubMed] [Google Scholar]
- [18].Harly C., Guillaume Y., Nedellec S., Peigné CM, Mönkkönen H., Mönkkönen J., Li J., Kuball J., Adams EJ, Netzer S., Déchanet-Merville J., Léger A., Herrmann T., Breathnach R., Olive D., Bonneville M., Scotet E., Blood 2012, 120, 2269–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang H., Henry O., Distefano MD, Wang YC, Räikkönen J., Mönkkönen J., Tanaka Y., Morita CT, J. Immunol. 2013, 191, 1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].van Beek E., Pieterman E., Cohen L., Löwik C., Papapoulos S., Biochem. Biophys. Res. Commun. 1999, 264, 108–111. [DOI] [PubMed] [Google Scholar]
- [21].Bergstrom JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G., Arch. Biochem. Biophys. 2000, 373, 231–241. [DOI] [PubMed] [Google Scholar]
- [22].Thompson K., Rogers MJ, Coxon FP, Crockett JC, Mol. Pharmacol. 2006, 69, 1624–1632. [DOI] [PubMed] [Google Scholar]
- [23].Idrees AS, Sugie T., Inoue C., Murata-Hirai K., Okamura H., Morita CT, Minato N., Toi M., Tanaka Y., Cancer Sci. 2013, 104, 536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tanaka Y., Iwasaki M., Murata-Hirai K., Matsumoto K., Hayashi K., Okamura H., Sugie T., Minato N., Morita CT, Toi M., Sci. Rep. 2017, 7, 5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kato Y., Tanaka Y., Miyagawa F., Yamashita S., Minato N., J. Immunol. 2001, 167, 5092–5098. [DOI] [PubMed] [Google Scholar]
- [26].Fleisch H., Breast Cancer Res. 2002, 4, 30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shinoda H., Adamek G., Felix R., Fleisch H., Schenk R., Hagan P., Calcif. Tissue Int. 1983, 35, 87–99. [DOI] [PubMed] [Google Scholar]
- [28].Shah P., Westwell AD, J. Enzyme Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]
- [29].Fleisch H., Endocr. Rev. 1998, 19, 80–100. [DOI] [PubMed] [Google Scholar]
- [30].Ebetino FH, Hogan AM, Sun S., Tsoumpra MK, Duan X., Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U., Lundy MW, Boyde A., Kashemirov BA, McKenna CE, Russell RG, Bone 2011, 49, 20–33. [DOI] [PubMed] [Google Scholar]
- [31].Davisson VJ, Davis DRDR, Dixit VM, Poulter CD, J. Org. Chem. 1987, 52, 1794–1801. [Google Scholar]
- [32].Marma MS, Xia Z., Stewart C., Coxon F., Dunford JE, Baron R., Kashemirov BA, Ebetino FH, Triffitt JT, Russell RG, McKenna CE, J. Med. Chem. 2007, 50, 5967–5975. [DOI] [PubMed] [Google Scholar]
- [33].Sakai Y., Mizuta S., Kumagai A., Tagod MSO, Senju H., Nakamura T., Morita CT, Tanaka Y., ChemMedChem 2017, 12, 2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang Y., Leon A., Song Y., Studer D., Haase C., Koscielski LA, Oldfield E., J. Med. Chem. 2006, 49, 5804–5814. [DOI] [PubMed] [Google Scholar]
- [35].Rodriguez JB, Gallo-Rodriguez C., ChemMedChem 2018, 10.1002/cmdc.201800693. [DOI] [Google Scholar]
- [36].Turhanen PA, Weisell J., Vepsäläinen JJ, Beilstein J. Org. Chem. 2012, 8, 2019–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Monteil M., Migianu-Griffoni E., Sainte-Catherine O., Di Benedetto M., Lecouvey M., Eur. J. Med. Chem. 2014, 77, 56–64. [DOI] [PubMed] [Google Scholar]
- [38].Webster MR, Kamat C., Connis N., Zhao M., Weeraratna AT, Rudek MA, Hann CL, Meyers C. L. Freel, Mol. Cancer Ther. 2014, 13, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hsiao CH, Lin X., Barney RJ, Shippy RR, Li J., Vinogradova O., Wiemer DF, Wiemer AJ, Chem. Biol. 2014, 21, 945–954. [DOI] [PubMed] [Google Scholar]
- [40].Foust BJ, Poe MM, Lentini NA, Hsiao CC, Wiemer AJ, Wiemer DF, ACS Med. Chem. Lett. 2017, 8, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Davey MS, Malde R., Mykura RC, Baker AT, Taher TE, Le Duff CS, Willcox BE, Mehellou Y., J. Med. Chem. 2018, 61, 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.