

Abstract
Thymidylate synthase (TS) catalyzes the production of the nucleotide dTMP from deoxyuridine monophosphate (dUMP), making the enzyme necessary for DNA replication and consequently a target for cancer therapeutics. TSs are homodimers with active sites separated by ∼30 Å. Reports of half-the-sites activity in TSs from multiple species demonstrate the presence of allosteric communication between the active sites of this enzyme. A simple explanation for the negative allosteric regulation occurring in half-the-sites activity would be that the two substrates bind with negative cooperativity. However, previous work on Escherichia coli TS revealed that dUMP substrate binds without cooperativity. To gain further insight into TS allosteric function, binding cooperativity in human TS is examined here. Isothermal titration calorimetry and two-dimensional lineshape analysis of NMR titration spectra are used to characterize the thermodynamics of dUMP binding, with a focus on quantification of cooperativity between the two substrate binding events. We find that human TS binds dUMP with ∼9-fold entropically driven positive cooperativity (ρITC = 9 ± 1, ρNMR = 7 ± 1), in contrast to the apparent strong negative cooperativity reported previously. Our work further demonstrates the necessity of globally fitting isotherms collected under various conditions, as well as accurate determination of binding competent protein concentration, for calorimetric characterization of homotropic cooperative binding. Notably, an initial curvature of the isotherm is found to be indicative of positively cooperative binding. Two-dimensional lineshape analysis NMR is also found to be an informative tool for quantifying binding cooperativity, particularly in cases in which bound intermediates yield unique resonances.
Significance
Understanding how allosteric substrate binding correlates with allosteric function in enzymes is needed to identify the operative elements underlying allostery. Thymidylate synthases (TSs) are thought to be half-the-sites active enzymes, a property consistent with negative cooperativity in substrate binding. In this work, however, human TS is shown to bind deoxyuridine monophosphate with ∼9-fold entropically driven positive cooperativity. This quantification required globally fitting isothermal titration calorimetry isotherms collected at various c-values and temperatures and is further verified by agreement with two-dimensional lineshape analysis of NMR titration spectra, highlighting the need for rigorous analysis in cases of homotropic cooperative binding. The finding of positive cooperativity in substrate binding by human TS establishes this enzyme as a model system for investigation of allosteric mechanisms.
Introduction
Allostery in biomolecules is enabled by the ability of the molecule to undergo a long-range structural or dynamic response to a perturbation such as ligand binding or chemical modification. In the strictest interpretation of allostery, this change in the molecule results in the altered affinity of a subsequent binding event, making this phenomenon a mechanism for both regulating and, in some cases, enabling biomolecular function. However, the details of these changes are often not well-understood, and how such changes are encoded into the biomolecular sequence and structure to elicit a functionally relevant response are even less well-understood. Consequently, a major goal in current protein science is to gain a better understanding of the underlying physical mechanisms governing allosteric communication. In general, both structural and dynamic features are critical to allosteric function in proteins (1, 2, 3). Because the dynamic component has been less well studied in allosteric systems, NMR and molecular dynamics simulations have become attractive methods for studying the physical mechanisms underlying allostery (4, 5). We have recently begun work on the homodimeric enzyme thymidylate synthase (TS) from Escherichia coli (ecTS), in which activity in one protomer negatively regulates activity in the other protomer (6, 7). Although the initial NMR analysis revealed allosteric physical communication between protomers (6), a finding consistent with the latest x-ray crystallographic work (8), the two molecules of substrate were found to bind essentially without cooperativity, indicating that allosteric regulation of function must reside at later steps of the reaction mechanism (7). One of the interesting outcomes of that study is that although symmetric homodimers are attractive in their simplicity for tracking allosteric mechanisms, reliable quantification of the relative binding affinities for even just two (identical) ligands can be challenging and requires experimental care. Here, we extend our approach to characterization of allosteric binding in homodimers by 1) applying our calorimetric approach to human TS (hTS) and 2) adding two-dimensional (2D) NMR lineshape analysis using the recently developed software package, TITAN (9).
As with ecTS, the two symmetric active sites of hTS are separated by ∼30 Å. hTS differs from ecTS by additional sequence elements that increase its molecular weight from 62 to 72 kDa, and the two proteins have a sequence identity of 55% (excluding additional hTS residues). All TSs play a role in the only metabolic pathway for de novo production of the nucleotide thymidine, catalyzing the conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate with the aid of cofactor N5,N10-methylene-5,6,7,8-tetrahydrofolate, which serves as donor for the methylene group and hydride ion. TSs from several species, including human, have been shown to display half-the-sites activity (10, 11, 12), meaning that only one of the two active sites can perform catalysis at a time (13, 14). In the case of hTS, there are reports of negative substrate binding cooperativity between the two subunits; specifically, ITC experiments indicated only a single dUMP binding event to hTS (15, 16, 17). Although this appears consistent with half-the-sites activity, the crystal structures of hTS show clearly that dUMP resides in both active sites (Fig. 1). In principle, half-the-sites activity does not necessarily require negative substrate binding cooperativity because the catalytic mechanism has many steps in which half-the-sites activity might originate. We aimed to resolve these inconsistencies with an in-depth study of dUMP binding to hTS. Quantification of the two binding affinities would reveal whether cooperative binding contributes to half-the-sites reactivity in hTS. In addition, finding significant binding cooperativity would establish hTS as a useful model system for future mechanistic studies (e.g., NMR, molecular dynamics) of allosteric binding of small-molecule substrates to a homodimeric enzyme.
Figure 1.
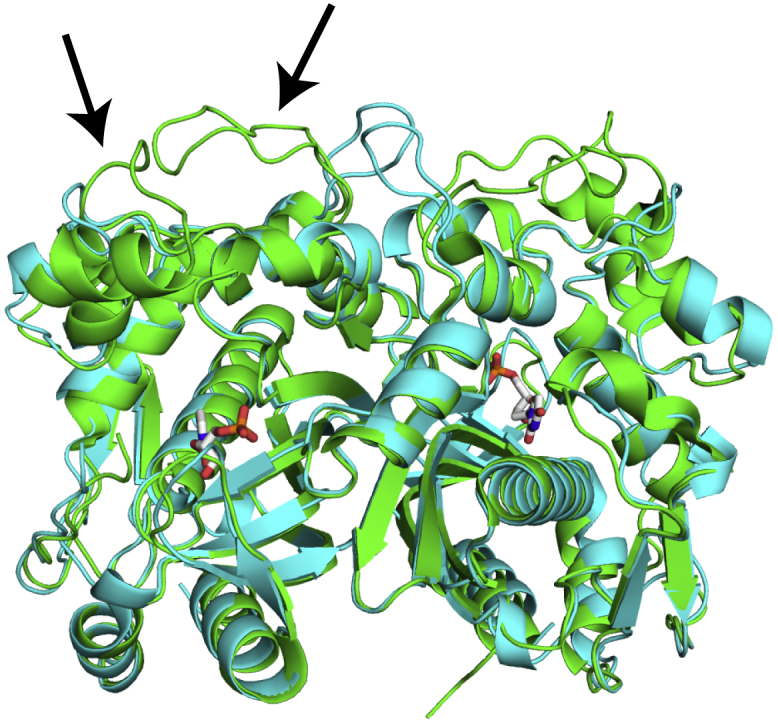
hTS and E. coli TS structures are very similar. An all-atom alignment of hTS (PDB: 5X5A, green) and E. coli TS (PDB: 1TJS, cyan) structures is shown. The only major structural differences are two loops that are extended in the human enzyme (residues 118–128, 149–156, indicated by arrows). In addition, hTS has a longer, flexible N-terminus (residues 1–25) that is not present in the crystal structure. The substrate dUMP is shown in sticks. The dUMP molecules bound to the two active sites in the enzyme are separated by ∼30 Å.
As a first step to investigating the structural and dynamic factors driving allostery in hTS, we characterize the thermodynamics of dUMP binding to hTS to quantify the extent of allosteric communication at the level of substrate binding in this enzyme using isothermal titration calorimetry (ITC) and NMR spectroscopy. We find that for homotropic ligand binding in which the cooperativity is modest and the two enthalpy changes are not strikingly different, convincing determination of the relative dUMP binding affinities requires global fitting of multiple ITC experiments. We find that in contrast to dUMP binding ecTS (7), dUMP binds hTS with ∼9-fold positive cooperativity with regard to the affinities, and that under appropriate conditions, positive cooperativity is visually apparent as a curvature in the early points of the calorimetric isotherm. In addition, we find that 2D NMR lineshape analysis offers an effective alternative to fitting cooperative ligand binding equilibria through direct observations of the populated states.
Materials and Methods
Materials
hTS was cloned in pET21a with an N-terminal 6× His tag and transformed into BL21 (DE3) rne131 cells. For preparations of unlabeled hTS for ITC, transformed BL21 cells were grown in 10 mL of LB overnight at 37°C. The 10 mL culture was used to inoculate 1 L of LB and grown to an OD600 of ∼1.0. Protein expression was then induced by addition of 1 mM isopropyl-β-D-thiogalactoside, and the culture was shaken at 20°C for ∼20 h. For use in NMR experiments, perdeuterated hTS was expressed as described previously (18), with the exception of the induction temperature (20°C), concentration of isopropyl-β-D-thiogalactoside used for induction (1 mM), and purified protein not being put through a 2H to 1H back-exchange protocol. Purification was achieved using the following workflow. His-tagged hTS was run over a nickel column (HisTrap HP 5 mL; GE Healthcare, Chicago, IL) using an ÄKTApurifier fast protein liquid chromatography system (GE Healthcare), and the His tag was subsequently cleaved by incubation with tobacco etch virus protease overnight at 4°C. Cleaved hTS was then rerun over the nickel column, and the flowthrough was collected, concentrated to 10 mL, and run over a HiLoad Superdex 200 column (GE Healthcare). Samples were then concentrated to an appropriate point using both Amicon stirred cells (MilliporeSigma, Burlington, MA) and centrifugal filters with 10 kDa cutoffs. hTS concentration was determined by 280 nm absorbance using . The concentration of dUMP (Sigma-Aldrich, St. Louis, MO) was determined using . The concentration of N5,N10-methylene-5,6,7,8-tetrahydrofolate (Merck & Cie, Schaffhausen, Switzerland) was determined using a molar ε290 of 32,000 . Protein samples for ITC, NMR titration, and mass spectrometry experiments were used within 1–2 days postpurification.
ITC
hTS samples for ITC contained 27.5 mM Na2HPO4, 1 mM tris(2-carboxyethyl)phosphine, 0.02% NaN3, and 0–165 mM NaCl at pH 7.5. Dry dUMP was dissolved in the same buffer stock to a concentration of 20 times the protein concentration. ITC experiments were performed on a MicroCal AutoITC 200 (Malvern Panalytical, Malvern, UK). Experiments yielding full isotherms were composed of 39 injections, with a small first injection of 0.2 μL followed by 38 injections of 1 μL. Experiments capturing very low mole-ratio points were composed of six injections, with a small first injection of 0.2 μL followed by five injections of 2 μL. These experiments used a syringe concentration roughly equal to the protein concentration. Heat integration was performed using the NITPIC software (19).
The data were fitted using a general two-site binding model, including a scaling parameter to adjust the protein concentration as described previously (7). The partitioning of the three different bound states in this model is described by the following binding polynomial or partition function:
| (1) |
where K1,macro and K2,macro are macroscopic association constants and L is the free ligand concentration. Note that this model does not presuppose identical independent or cooperative nature of the binding but rather utilizes separate macroscopic constants for each binding event; cooperativity, or lack thereof, between the two binding events is evaluated based on the relative fit values of these two constants, as described in the text. Nonidentical independent binding is not considered here given the homodimeric nature of the enzyme. In this fitting scheme, we fit a macroscopic affinity and enthalpy for each of the two binding events, as well as a protein concentration scaling factor. The heat of titration point k in the isotherm is constructed from these parameters in the following manner (20):
| (2) |
where V is the working volume of the sample cell, n is the number of binding events (two in this case), is the concentration of protein bound to i number of ligands after injection k, and vk is the volume of injection k. The concentration of each bound state at a given titration point is given by
| (3) |
where scaling is the fit concentration correction factor, is the initial total protein concentration, P is the binding polynomial (or partition function) given in Eq. 1, P(MLi) is the term in the partition function corresponding to bound state MLi, and v and V are the injection and working volumes as before. We define an error function, F, as a sum of residuals
| (4) |
where n is the total number of injections in titration j and σ is a constant representing the variance in the data and is based on typical residual values. In the case of the global fit at 25°C, 20 mM NaCl shown in Fig. 3, the residuals were weighted according to the moles of ligand added with each injection, , where L0(j) is the concentration of ligand in the syringe used for titration j; this serves to give differential weightings to the residuals for data sets in a global fit and was found to be necessary in cases in which the range of c-values used is large because the variance in heats across these data sets are significantly different. For a global fit, m > 1 where m is the number of titrations included in the fit. This error function was minimized by the nonlinear least-squares solver in MATLAB (The MathWorks, Natick, MA), lsqnonlin, using the Levenberg-Marquardt algorithm. For a global fit of titrations using different c-values (protein concentrations) under the same conditions, the same macroscopic affinities and enthalpies are applied to each isotherm. Different cell scaling factors within a global fit were applied only to titrations performed on protein from different preparations.
Figure 3.

hTS binds dUMP with positive cooperativity. (a) Global fit of ITC isotherms collected at various hTS concentrations in 20 mM NaCl at 25°C is shown. The low-mole-ratio points in green are designed to determine the first enthalpy, which shows a correlation with the first affinity in some of our fits (Fig. S1). Boxed points highlight the distinct initial curvature present in our high c-value data, which we find to be indicative of positive cooperativity. (b) Thermodynamic parameters from 150 Monte Carlo simulated data sets showing changes in enthalpy for the two dUMP binding events (red bars) and similarly for the corresponding two entropic contributions (blue bars) are given. A ∼9-fold increase in the second affinity relative to the first (black bars) is observed, and this is driven by the >4 kcal/mol more negative entropic contribution for the second binding event. A complete table of parameters is found in Table S1. (c) Simulation of isotherms at 400 μM protein concentration using the fitted enthalpies shows the presence of a large initial curvature only in the case of positive cooperativity. This curvature is clearly seen in 246 μM data in Fig. 3a. For each curve, the fitted first macroscopic affinity is used and the second macroscopic affinity is adjusted to give the indicated cooperativity. (d) The reason for this initial curvature is seen by looking at the change in the equilibrium populations with each injection (Δlig1 and Δlig2) during titration; Δlig1 has a large negative slope at low mole ratio. Although there is also a large positive slope in Δlig2, the slope of Δlig1 is greater in magnitude, leading to the large initial curvature seen in the Δlig1 + Δlig2 curve. Note that the isotherms are simply this Δlig1 + Δlig2 curve with Δlig1 weighted by ΔH1 and Δlig2 weighted by ΔH1 + ΔH2.
Globally fitting isotherms collected at various temperatures using the van’t Hoff equation to determine the temperature dependence of the affinities has also been described previously (21). In this fitting scheme, macroscopic affinities are fitted at a single temperature and enthalpies are fitted at all temperatures, in addition to the usual protein concentration scaling factor. Macroscopic affinities for all the other temperatures were determined by trapezoidal integration of the van’t Hoff equation as follows:
| (5) |
Determination of heats and minimization of the error function were done in the same way as described above. Note that these two fitting schemes can be combined within a single global fit such that any number of the temperatures involved in the fit can include isotherms with multiple c-values, as we have done here.
NMR assignments and titration
Apo and lig2 resonances were assigned using a set of six triple resonance experiments on both unbound and dUMP-saturated hTS samples (unpublished data) as described previously (18). Assignment of select lig1 resonances was based on proximity to the corresponding apo and lig2 resonances. Notably, the fitted binding parameters are unaffected by swapping assignment of lig1 resonances. The U-[2H-15N] hTS sample for NMR titration contained 27.5 mM Na2HPO4, 20 mM NaCl, 0.02%NaN3, and 5 mM dithiothreitol at pH 7.5, differing from ITC samples only in reducing agent. Before experiment, D2O was added to 5%. TROSY-HSQC (transverse relaxation optimized spectroscopy- heteronuclear single quantum coherence) spectra were collected on an 850 MHz Bruker Avance III spectrometer (Bruker, Billerica, MA) equipped with a TCI H-C/N-D 5 mm cryoprobe using the trosyf3gpphsi19.2 Bruker pulse program. Data sets were traditionally sampled with (140, 1699) complex points and (54 ms, 100 ms) acquisition times in (t1, t2), respectively, along with eight scans per complex t1 point and an interscan delay of 2 s at 25°C.
TITAN (9) was used to fit binding affinities and kinetics, along with proton and nitrogen chemical shifts and transverse relaxation rates of individual resonances, from the dUMP titration spectra. TITAN performs quantum mechanical simulations of NMR pulse sequences to fit resonance lineshapes from 2D spectra. These simulations are performed in the Liouville vector space (22), allowing the effects of relaxation and exchange processes on the coherence(s) of interest to be easily accounted for. By incorporating these effects on the detected signals that occur during the pulse sequence, this approach enables more accurate analyses of NMR spectra than previous methods, which fitted lineshapes using only the spectra themselves. This 2D lineshape analysis software requires exponential apodization to maintain Lorentzian lineshapes; line broadenings of 0.75 and 1 Hz were used in the direct and indirect dimensions, respectively. We created a new, to our knowledge, symmetric dimer binding model and TROSY pulse program for the trosyf3gpphsi19.2 sequence for use with TITAN, which were used here. Creation of the TROSY-HSQC pulse program required making several significant modifications to the TITAN HSQC pulse program: 1) incorporating additional pulses (in the Liouville space used in TITAN’s calculations, these are matrices that act on the vectorized density “matrix”); 2) extending the operator basis set, and thus the Liouvillian matrix, to include multiple quantum operators that are present during the TROSY sequence but not the HSQC sequence; and 3) isolating the TROSY component of the proton multiplet at the end of the sequence via transformation from a basis set of cartesian operators to one of single-transition cartesian operators (, ). To avoid fitting an additional parameter, the multiple quantum terms were given the same relaxation rate as the Sx(y) and 2IzSx(y) terms.
The symmetric binding model includes four states: apo, bound subunit of singly bound dimer (lig1,bound), unbound subunit of singly bound dimer (lig1,empty), and doubly bound (lig2). Such a model allows different chemical shifts and relaxation rates to be applied to resonances from the lig1,bound and lig1,empty states. The apo state is connected to each of the singly bound states through kon(off),1, the on/off rate of the first binding event, and each of the singly bound states is connected to the doubly bound state through kon(off),2, as is shown in the exchange matrix, Kexch (Eq. S1). The populations of each of the singly bound states are set to be one half the total population of singly bound dimer. The populations of the bound states are determined by the same partition function (Eq. 1) used in the ITC fitting presented here. Consequently, this protocol fits macroscopic affinities, which were converted to microscopic affinities as was done in the ITC analysis. We did not fit a protein concentration scaling factor, but a scaling factor of 0.75—based on the PULCON-NMR measurement—was applied. Additionally, minor modifications were made to the software to only include spectral points with signal/noise ratio greater than three in computation of residuals and points with signal/noise ratio greater than two in bootstrap resampling for error determination. MATLAB scripts are available upon request.
Results
hTS binds dUMP with positive cooperativity
As a first step toward investigating structural and dynamic allosteric mechanisms in hTS, we aimed to characterize the thermodynamics of dUMP binding. ITC is an attractive technique because it allows determination of not only the binding free energy but also its enthalpic and entropic components. The challenge of obtaining a single set of best-fitting parameter values when using multisite binding models has been described previously (23), along with several potential solutions. These solutions essentially boil down to globally fitting multiple data sets, collected either from ITC under different conditions or from different binding experiments altogether (21, 24, 25). Here, we have chosen to take the former approach, globally fitting ITC data sets collected at both different temperatures as well as various concentrations (or c-values). In our case, globally fitting only two isotherms collected at different c-values yielded multiple sets of affinity values giving comparable fits to the data, as well as a strong correlation between the first macroscopic affinity and enthalpy, highlighting the need for larger amounts of data in these fits (Fig. S1).
Several conditions were tested for quantifying dUMP binding using ITC and NMR. We found that at 35°C, highly concentrated samples of hTS are prone to aggregation, precluding a multiple c-value global fit approach to quantify the binding thermodynamics under physiologically relevant conditions. Instead, we chose to investigate substrate binding at 25°C, at which we observe no signs of aggregation at the high concentrations necessary both for globally fitting ITC isotherms at a wide range of c-values and for NMR study. Additionally, because salt decreases the sensitivity of NMR experiments, we chose to work in low-salt conditions. We have found, however, that more physiologically relevant salt conditions do not qualitatively change our result (Fig. S2; Table S1). Thus, to identify a single set of thermodynamic parameter values describing the binding of two molecules of dUMP to hTS, we globally fitted four ITC data sets collected at hTS concentrations ranging from 36 to 246 μM under conditions of 20 mM NaCl and at 25°C using the general two-site model described in Fig. 2 and Eq. 1 (Fig. 3 a). Representative thermograms are shown in Fig. S3.
Figure 2.

General two-site binding model. The binding polynomial, P, is used to determine the populations of bound species (apo, lig1, lig2) throughout titration based on the two macroscopic binding affinities for both ITC and NMR studies presented here. Note that this model fixes the stoichiometry at n = 2, which is justified by the x-ray model (Fig. 1), as well as our NMR titration spectra (Fig. 5). This model does not presume cooperative or independent binding. Rather, separate macroscopic affinities are fitted, and the presence or absence of cooperativity is assessed afterwards. In the case of independent binding, the two microscopic affinities would be identical because of the homodimeric nature of the enzyme. On the other hand, cooperativity in the binding would introduce a difference in the values of the two microscopic affinities, as discussed in the text. In addition to the two macroscopic affinities and enthalpies, our ITC fits include a parameter that scales the given protein concentration, accounting for error in the predicted extinction coefficient and/or presence of binding incompetent fraction. This parameter is not fitted for the NMR titration, but rather, a fixed scaling factor of 0.75 is used based on our PULCON-NMR result.
This is a completely general model that assumes nothing about the mechanism of the binding, only that there are two binding events (20). The model allows basic detection of the two binding affinities and their differences; with additional information, these data can be reanalyzed within the context of a more complex model that incorporates a mechanism of the binding (see Discussion). We report microscopic association constants (Kmicro), which are the reciprocal of microscopic dissociation constants (Kd,micro) and differ from the directly fitted macroscopic association constants (Kmacro) (dissociation constant Kd,macro) by factors of two (Eq. 6) because of the sequential nature of binding. It should be noted that the influence on the affinity of the number of binding sites available to a particular binding event, which is removed here to generate the microscopic constants, is purely entropic in nature. As a result, although we will distinguish between “macroscopic” and “microscopic” affinities and entropies in this text, we will not make this distinction for the enthalpies because they are the same in either case. The ratio K2,micro/K1,micro is reflected by the parameter ρ, which is a simple measure of binding cooperativity (20).
| (6) |
Additionally, ITC data were fitted using a scaling factor that corrects the ultraviolet-absorbance-determined protein concentration (see Supporting Materials and Methods). In contrast to previous studies of dUMP binding (15, 17), the global calorimetric fit at 25°C yielded a significant increase in binding affinity for the second dUMP compared to the first, ρ = 9 ± 1 , with K1,micro and K2,micro equal to 39,000 ± 3000 and 360,000 ± 40,000 M−1, respectively (Fig. 3 b).
This positive binding cooperativity is visually evident in the ITC isotherms only at high c-values as a downward curvature in the first few titration points (Fig. 3 a). Simulation of isotherms with varying relative affinities and enthalpies confirms this signature curvature (Fig. 3 c). Notably, the appearance of this curvature is dependent on the relative values of the enthalpies (Fig. S4). A detailed discussion of this feature of positively cooperative isotherms can be found in the Supporting Materials and Methods. This feature was also observed in the isotherm of a positively cooperative system, human I-BABP, reported previously (26). The ninefold positive binding cooperativity, as well as the enthalpic and entropic contributions, are shown in bar plots in Fig. 3 b.
The −TΔS values plotted (Fig. 3 b) are derived from the Kmicro values (and corresponding ΔGmicro values). Although both binding events are dominated by enthalpy, ΔH2 provides half the binding energy of ΔH1, and the entropic contribution turns from unfavorable in the first binding event (−TΔS1,micro = 2.5 kcal/mol) to favorable in the second binding event (−TΔS1,micro = −2.4 kcal/mol). This ∼5 kcal/mol swing (and countermoving enthalpy) indicates that dUMP binding cooperativity in hTS is driven by entropic effects.
Temperature dependence of binding thermodynamics suggests a coupled equilibrium
To ensure the validity of positively cooperative dUMP binding, as well as investigate the temperature dependence of the binding thermodynamics, we globally fitted seven ITC data sets collected at 0 mM NaCl and temperatures ranging from 5 to 35°C, including four experiments at 25°C with various hTS concentrations (Figs. 4 and S5; Table 1). Globally fitting ITC data collected at various temperatures was carried out utilizing the van’t Hoff equation as described previously (21). This approach involves fitting enthalpies at each temperature, but fitting affinities at only one temperature (5°C in our case)—the van’t Hoff equation is then used to compute the affinities at every other temperature (see Materials and Methods). Using this van’t Hoff fitting method with the general two-site binding model (Fig. 2), we find that dUMP binds with ∼5-fold positive cooperativity under these conditions, consistent with the single temperature fit at 20 mM NaCl. This corresponds to a cooperative free energy, Δgmicro, of about −1 kcal/mol (Fig. 4). Further, the fit shows a modest increase in the magnitude of the cooperativity with increasing temperature (Table 1). Notably, the cooperativity is maximized at the more physiologically relevant 35°C condition. Additionally, a dramatic increase in the cooperative enthalpy, Δh (= ΔΔH), was observed from 25 to 35°C relative to the other temperature changes, indicative of a significant divergence of the two binding heat capacities (ΔCp) in this temperature range. Note that the temperature dependence of ΔCp,1 and ΔCp,2 between 25 and 35°C, −18 ± 2 and , respectively (Table S2), are significantly larger than the temperature dependence of the heat capacity of the second proton dissociation of phosphate at 25°C, reported as (27), indicating that this behavior is not due to a change in protonation. This is consistent with the presence of a conformational equilibrium coupled to dUMP binding (28, 29), though this behavior could be caused by any sort of coupled equilibrium. Additional evidence consistent with an equilibrium coupled to dUMP binding is present in the thermogram collected at 5°C; these data show a slower exothermic process occurring in addition to dUMP binding (Fig. S3). Such a conformational equilibrium has been proposed in hTS by Lebioda and co-workers based on fluorescence measurements (30) and may be related to active and inactive conformations of the active site loop of this enzyme reported previously (15), given that the active site loop contains a tryptophan.
Figure 4.

Temperature dependence of dUMP binding thermodynamics. Global fit of binding isotherms at temperatures ranging from 5 to 35°C using van’t Hoff temperature dependence of the macroscopic affinities is shown (top). The low-mole-ratio points in yellow are designed to determine the first enthalpy, which shows a correlation with the first affinity in some of our fits (Fig. S1). A bar graph of cooperative thermodynamic parameters from 150 Monte Carlo simulated data sets (bottom) shows positive cooperativity in dUMP binding across this temperature range. Cooperative parameters are defined as the difference between these parameters for the two binding events (Δh = ΔH2 − ΔH1, etc.). Note that Δgmicro = −RTln(ρ). The observed Δgmicro values of ∼−1 kcal/mol correspond to ρ ∼ 5. The large jump in Δh (and TΔsmicro) from 25 to 35°C results from a significant change in the binding heat capacities in this range. This is consistent with an equilibrium coupled to the binding, as described in the text. A complete table of parameters is found in Table S2.
Table 1.
Temperature Dependence of dUMP Binding Thermodynamics in kcal/mol at 0 mM NaCl
| Temperature (°C) | ΔG1,microa | ΔG2,micro | ρ | ΔH1 | ΔH2 | −TΔS1,micro | −TΔS2,micro |
|---|---|---|---|---|---|---|---|
| 5 | −6.59 ± 0.02 | −7.37 ± 0.01 | 4.0 ± 0.2 | −3.71 ± 0.05 | −2.73 ± 0.07 | −2.88 ± 0.06 | −4.63 ± 0.07 |
| 15 | −6.66 ± 0.03 | −7.50 ± 0.01 | 4.4 ± 0.3 | −5.98 ± 0.05 | −4.30 ± 0.07 | −0.67 ± 0.07 | −3.21 ± 0.08 |
| 25 | −6.64 ± 0.03 | −7.59 ± 0.01 | 4.9 ± 0.3 | −8.22 ± 0.06 | −6.12 ± 0.09 | 1.58 ± 0.08 | −1.5 ± 0.01 |
| 35 | −6.53 ± 0.03 | −7.63 ± 0.02 | 6.1 ± 0.5 | −12.2 ± 0.2 | −6.6 ± 0.2 | 5.7 ± 0.2 | −1.1 ± 0.1 |
Minor disagreement between ΔGmicro and enthalpy and entropy values results from differences in the number of significant figures present in these values.
2D lineshape analysis of NMR titration spectra
As an alternate method for measuring dUMP binding cooperativity, NMR spectroscopy was employed because it has been shown previously that it can quantify homotropic cooperative binding (7). We determined the two binding affinities by fitting dUMP titration spectra using the 2D lineshape analysis software TITAN (9). TITAN fits binding affinities and off rates, as well as chemical shifts and transverse relaxation rates for individual resonances, by reconstructing the Lorentzian lineshapes of NMR signals in titration spectra. TITAN provides an ideal approach to fitting NMR titration data because, unlike previous one-dimensional lineshape analysis methods (31, 32), it accounts for relaxation occurring during the pulse sequence by performing quantum mechanical simulations of the entire 2D pulse sequence, with explicit terms describing chemical exchange processes (see Materials and Methods). Although TITAN comes with several pulse programs ready for use, the TROSY-HSQC sequence used here is not among them. To analyze the lineshapes as accurately as possible, we created a TITAN pulse program for this sequence by modifying TITAN’s premade HSQC pulse program and incorporated the general two-site binding model (see Materials and Methods).
A number of residues in hTS display “quartet” resonance patterns in the dUMP titration (Fig. 5 a), which has only recently been reported as a strategy for monitoring two-site binding in homodimers (7, 33). A “quartet” for a particular residue is composed of four resonances, one from each of the symmetric species (apo, lig2) and two distinct resonances from the asymmetric lig1 species (bound and empty). Such a resonance pattern indicates that a residue and its symmetric counterpart in the other protomer are in distinct chemical environments in the lig1 state. Additionally, it is highly informative from the perspective of determining binding affinities because it provides distinct signals for each of the bound species. Use of TITAN/2D lineshape analysis of peak quartets here extends this monitoring strategy for the characterization of ligand binding. Assignment of apo and lig2 resonances was accomplished using a set of six triple resonance experiments (18) on both unbound and saturated hTS samples. Assignment of lig1 resonances was done based on proximity to apo and lig2 resonances.
Figure 5.

Positive binding cooperativity determined by NMR titration. (a) Quartet resonance pattern for Asp48 of hTS at 1:1 ratio of substrate/dimer, as seen in our titration, is shown. The presence of distinct resonances for the singly bound state makes these NMR signals well suited for determination of macroscopic affinities for multiple binding events. (b) Fit of 170 μM (after application of 0.75 scaling factor) hTS dimer titration spectra for Asp48 using TITAN software is shown. For each titration point, three-dimensional plots (left) as well as 2D contour plots (right) of the Asp48 resonances from our data (blue) overlaid with reconstructed resonances from our fit parameters (red) are shown. The number of resonances present at each mole ratio, as well as their linewidths and intensities, are well-captured by our fit. Other spin systems used in the global fit can be seen in Fig. S6. (c) A box plot of ρ-values obtained from 150 Monte Carlo simulated ITC data sets and 100 bootstrap resampled NMR titration spectra shows excellent agreement in the relative microscopic affinities between the two approaches. A table of parameters for the NMR titration fit is found in Table S3.
Asp-48 is one residue in hTS that shows a clear quartet peak pattern during titration with dUMP and has resonances with high signal/noise ratio, making it well suited for this analysis. A dUMP titration was performed at dUMP/hTS dimer ratios of 0, 0.4, 0.7, 1, 1.5, 2, 3, and 4.5 using a TROSY-HSQC 2D acquisition. The Asp-48 quartet was observed at ratios 0.7, 1, and 1.5 (Fig. 5), indicating that apo, lig1, and lig2 are clearly distinguished, and the quartet resonances can in principle be used to quantify these states in a TITAN analysis to measure K1,macro and K2,macro, which we convert to microscopic constants (Eq. 6). A global fit of titration spectra of Asp-48 along with several other spin systems (not showing a quartet pattern) using the general two-site binding model and TROSY-HSQC TITAN pulse program shows relative microscopic affinity of the two binding events very similar to that determined by ITC, with ρ values of 7 ± 1 and 9 ± 1 from NMR and ITC, respectively (Fig. 5 c; Table S3). Additionally, reconstruction of NMR titration spectra allows fitting of binding kinetics because the NMR lineshape is influenced by the rates of ligand binding and dissociation. Off rates were determined to be koff,1 = 23 ± 3 s−1 and koff,2 = 10 ± 1 s−1. This ∼2-fold change in the off rate indicates that the majority of the ∼7-fold increase in K2,micro comes from the on rate.
Discussion
In this work, the binding of two molecules of dUMP to the human TS dimer has been characterized thermodynamically using ITC and with respect to thermodynamics and kinetics using quantitative 2D NMR lineshape analysis. Collectively, the results show that the hTS homodimer binds dUMP substrate with ∼9-fold positive cooperativity at 25°C and low salt (pH 7.5). We are not able to determine the mechanism of the cooperativity at this point because the binding model used to fit our data is general in nature. The model does show, however, that the cooperativity is driven by changes in entropy, which could derive from differential changes in flexibility of the protein in response to each of the two binding events (34, 35, 36), as well as from differential release of ordered water molecules (37). For example, this result is consistent with a mechanism in which there is a global reduction in the flexibility of the protein in response to the first binding event alone. Thus, the first binding event pays an entropic cost that the second binding event does not have to pay, leading to entropically driven positive cooperativity. This is analogous to the dynamically driven negatively cooperative binding mechanism of CAP described previously (34).
Alternatively, previous crystallographic reports of active and inactive conformations of the apo enzyme (38) and our finding of a significant temperature dependence of the binding heat capacities between 25 and 35°C are suggestive of a conformational equilibrium of the protein that is coupled to dUMP binding. These distinct conformations could possess different flexibilities or number of ordered waters, leading to the observed entropy difference driving the cooperativity. Within the context of this more mechanistically informative model, the microscopic binding parameters reported here can be further decomposed as follows (if we assume that ) (28):
| (7) |
| (8) |
| (9) |
where i = (1, 2) for the first or second binding event, Kactive is the association constant of dUMP binding to the active conformation, is the equilibrium constant of the conformational equilibrium of the unbound subunit(s) in the apo protein (first binding event, i = 1) or singly bound protein (second binding event, i = 2), ΔHactive is the enthalpy of dUMP binding to the active conformation, ΔHconf is the enthalpy of the conformational change, ΔCp,active is the heat capacity of binding to the active conformation, and ΔCp,conf is the heat capacity of the conformational change. Notably, this model provides a more detailed interpretation of the ρ parameter:
| (10) |
Within the context of this model, the parameter ρ—which we found to take a value of ∼9 in our experimental conditions—tells us about the relative values of the equilibrium constant of the conformational equilibrium coupled to each of the two binding events. Put another way, a ρ-value > 1 tells us that the mechanism of positive cooperativity is a biasing of the conformational equilibrium of the unbound subunit toward the active conformation upon the first binding event (Kconf,1 > Kconf,2).
Also of note is that Eq. 9 demonstrates how the presence of such a coupled equilibrium can introduce (or contribute to) a temperature dependence of ΔCp; this is accomplished through the temperature dependence of Kconf, ΔHconf, and/or ΔCp,conf. This equation also indicates that differences in Kconf,1 and Kconf,2 can lead to significant differences between the two binding heat capacities. Such behavior of the binding heat capacities has been reported previously in a case of cooperative binding involving a coupled equilibrium (39). Interestingly, despite observing positive cooperativity from 5 to 35°C, we only see this large difference in the binding heat capacities between 25 and 35°C. We note that the aforementioned aggregation of hTS at 35°C was observed at significantly higher concentrations than that used in the fit presented here (∼175 μM compared to 44 μM). Preliminary attempts to determine the values of the parameters listed in Eqs. 7, 8, and 10 using Kmicro-values, ΔH-values, and ρ from the 25°C 20 mM NaCl fit show that many possible sets of values satisfy these equations. Notably, we find that is consistent with a wide range of possible effects on the unbound subunit of the singly bound state. For example, one possible solution is and . This means that although the population of the inactive state in the apo subunits is ∼89%, the unbound subunit of the singly bound state has an inactive population of ∼0% (in other words, an Monod-Wyman-Changeux-like mechanism). Alternatively, and represent another viable solution. In this case, the population of the inactive state goes from ∼96% in the apo subunits to ∼67% in the unbound subunit of the singly bound state, indicating a much smaller perturbation of the inactive population than that seen in the previous solution. Thus, further experimentation is required to determine the correct binding mechanism. Although this information is necessary to develop a complete thermodynamic understanding of the binding of dUMP to hTS, the data and analysis presented here clearly show that there are thermodynamic differences between the two binding events, demonstrating that such experimentation is warranted.
The finding of positive cooperativity is unexpected given previous reports of strong negative cooperativity in dUMP binding by hTS (15, 17), as well as reports of half-the-sites activity in TSs (10, 12). The reliability of our finding is supported by the quality of samples used, as confirmed by mass spectrometry (Fig. S7), careful determination of the proper sample concentration scaling factor by two independent methods (Fig. S8), and corroboration of the ITC result by quantitative NMR analysis. From this work and our previous efforts to quantify binding cooperativity in ecTS, accurate determination of ligand binding cooperativity can be challenging, particularly when the degree of cooperativity is less than 10- to 20-fold and if the enthalpies for multiple binding events are not drastically different (7, 23). Thus, it is important to establish reliable strategies for measuring ligand binding cooperativity in cases of homotropic allosteric systems. In the case of hTS here, dUMP binding cooperativity only became clear upon careful analysis of multiple isotherms. Even with the global fit of two ITC data sets (varying c-values) at 25°C, analysis of the error surface using a parameter grid search showed two distinct minima: one corresponding to positive cooperativity and the other with negative cooperativity (Fig. S3). The set of parameter values corresponding to negative cooperativity, which qualitatively agrees with previous reports (15, 16, 17), reproduces the data less well as additional data sets are added to the fit (Fig. S5). Those studies, which only detected a single dUMP binding event, only analyzed a single ITC isotherm and thus may be less reliable than the findings reported here. The fitted scaling factor obtained from this negative cooperativity minimum of the error surface is ∼1.5, inconsistent with the value of 0.75 determined by PULCON-NMR (Fig. S8). In addition, single dUMP binding is inconsistent with the observation of two dUMP molecules in the cocrystal structure (Fig. 1, Protein Data Bank [PDB]: 5X5A). Given our findings, it seems likely that by only analyzing a single isotherm, it was not possible to obtain an error surface (regarding the affinities) with a single minimum in those works.
Positive cooperativity in dUMP binding is further supported by the 2D NMR lineshape analysis using TITAN. TITAN is a sophisticated new tool that enables quantification of both binding affinities in a homodimeric system by analyzing quartet resonance patterns. This approach was necessary to determine the positively cooperative nature of dUMP binding; a simpler analysis—based purely on resonance intensities, for example—would not lead one to this conclusion because the intensities of the singly bound resonances are comparable to that of the apo and doubly bound resonances at 1:1 mol ratio of dUMP/hTS dimer (Fig. 5 b). Our analysis using TITAN, however, reveals that the unexpectedly large intensities of the singly bound peaks result from reduced relaxation rates of these resonances relative to the apo and doubly bound signals (Table S3).
The basis for the difference in dUMP binding cooperativity between ecTS (ρ ∼1) and hTS (ρ ∼9) remains unclear. Even though the core structures of the two enzymes are extremely similar, having a sequence identity of 55%, hTS has two loop extensions (residues 118–128, 149–156) and a longer, flexible N-terminus (residues 1–25) that may contribute to the binding cooperativity. In fact, these regions have been implicated in the equilibrium between active and inactive conformations of the active site loop in hTS (15, 38). Further study is needed to determine whether this conformational equilibrium is responsible for the cooperativity in dUMP binding, i.e., to determine whether this conformational equilibrium is the coupled equilibrium observed in the temperature dependence of the binding thermodynamics as discussed above.
Elimination of the possibility of negative binding cooperativity is important in the case of hTS because this might be naively expected, given that TSs have been shown to have half-the-sites reactivity. Our finding of positive cooperativity in substrate binding shows that the mechanism of half-the-sites reactivity in this enzyme must involve some other step of the reaction cycle. Although further study is necessary to assess the functional relevance of this positive binding cooperativity, one might speculate that this cooperativity relates to the translational autoregulation of hTS by binding its own mRNA (40). It is known that substrate binding prevents mRNA binding (40), so the positive cooperativity in dUMP binding may serve to amplify translation of hTS transcripts when dUMP is abundant.
Conclusions
hTS is a homodimeric enzyme that binds two molecules of dUMP at active sites separated by ∼30 Å with ∼9-fold positive cooperativity. Notably, this positive cooperativity stems from a ∼5 kcal/mol more favorable entropy of the second binding event relative to the first at 25°C. The difference in binding entropy could be the result of differences in protein dynamics, solvent-exposed surface area, etc. associated with each of the two binding events and is the subject of further investigation. Comparison of hTS with the structurally similar ecTS, which lacks cooperativity in dUMP binding, suggests that two extended loops in hTS may play a key role in this difference in binding entropy. Quantification of this homotropic allostery required globally fitting multiple ITC isotherms collected at various c-values and temperatures, as well as careful determination of protein concentration using PULCON-NMR. A key finding was the distinct initial curvature of our isotherms at high c-values that is indicative of positive cooperativity, allowing cooperativity to be clearly visualized in the data itself. Our work illustrates a potential solution to the challenge of calorimetric characterization of multisite binding and shows the utility of 2D lineshape analysis NMR toward this end in cases in which bound intermediates yield unique resonances.
Author Contributions
Research was designed by A.L.L., P.J.S., and J.P.B.; J.P.B. performed and analyzed ITC and NMR experiments. E.W., D.G., L.W., L.H., X.C., and M.B.M. performed and analyzed mass spectrometry experiments. J.P.B. and A.L.L. wrote the manuscript.
Acknowledgments
We thank Dr. Ashutosh Tripathy of the University of North Carolina Macromolecular Interactions Facility for assistance in ITC data collection and Dr. Bradley Falk for initial work preparing hTS samples. We also thank Dr. Gregory Young of the University of North Carolina Biomolecular NMR Facility for assistance in NMR data collection.
This work was funded by National Institutes of Health (NIH) grant GM083059 to A.L.L. and NIH grant 5 T32 GM 8570-22 to J.P.B. This work was supported in part by the National Cancer Institute of the NIH under award number P30CA016086 Cancer Center Support Grant. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Editor: Doug Barrick.
Footnotes
Supporting Material can be found online at https://doi.org/10.1016/j.bpj.2019.08.015.
Supporting Material
References
- 1.Cui Q., Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 3.Motlagh H.N., Wrabl J.O., Hilser V.J. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo J., Zhou H.X. Protein allostery and conformational dynamics. Chem. Rev. 2016;116:6503–6515. doi: 10.1021/acs.chemrev.5b00590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lisi G.P., Loria J.P. Solution NMR spectroscopy for the study of enzyme allostery. Chem. Rev. 2016;116:6323–6369. doi: 10.1021/acs.chemrev.5b00541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falk B.T., Sapienza P.J., Lee A.L. Chemical shift imprint of intersubunit communication in a symmetric homodimer. Proc. Natl. Acad. Sci. USA. 2016;113:9533–9538. doi: 10.1073/pnas.1604748113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sapienza P.J., Falk B.T., Lee A.L. Bacterial thymidylate synthase binds two molecules of substrate and cofactor without cooperativity. J. Am. Chem. Soc. 2015;137:14260–14263. doi: 10.1021/jacs.5b10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finer-Moore J.S., Lee T.T., Stroud R.M. A single mutation traps a half-sites reactive enzyme in midstream, explaining asymmetry in hydride transfer. Biochemistry. 2018;57:2786–2795. doi: 10.1021/acs.biochem.8b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waudby C.A., Ramos A., Christodoulou J. Two-Dimensional NMR lineshape analysis. Sci. Rep. 2016;6:24826. doi: 10.1038/srep24826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson A.C., O’Neil R.H., Stroud R.M. The structural mechanism for half-the-sites reactivity in an enzyme, thymidylate synthase, involves a relay of changes between subunits. Biochemistry. 1999;38:13829–13836. doi: 10.1021/bi991610i. [DOI] [PubMed] [Google Scholar]
- 11.Johnson E.F., Hinz W., Anderson K.S. Mechanistic characterization of Toxoplasma gondii thymidylate synthase (TS-DHFR)-dihydrofolate reductase. Evidence for a TS intermediate and TS half-sites reactivity. J. Biol. Chem. 2002;277:43126–43136. doi: 10.1074/jbc.M206523200. [DOI] [PubMed] [Google Scholar]
- 12.Maley F., Pedersen-Lane J., Changchien L. Complete restoration of activity to inactive mutants of Escherichia coli thymidylate synthase: evidence that E. coli thymidylate synthase is a half-the-sites activity enzyme. Biochemistry. 1995;34:1469–1474. doi: 10.1021/bi00005a001. [DOI] [PubMed] [Google Scholar]
- 13.Bernhard S.A., MacQuarrie R.A. Half-site reactivity and the “induced-fit” hypothesis. J. Mol. Biol. 1973;74:73–78. doi: 10.1016/0022-2836(73)90356-2. [DOI] [PubMed] [Google Scholar]
- 14.Nichols C.E., Dhaliwal B., Stammers D.K. High-resolution structures reveal details of domain closure and “half-of-sites-reactivity” in Escherichia coli aspartate beta-semialdehyde dehydrogenase. J. Mol. Biol. 2004;341:797–806. doi: 10.1016/j.jmb.2004.05.073. [DOI] [PubMed] [Google Scholar]
- 15.Chen D., Jansson A., Nordlund P. Structural analyses of human thymidylate synthase reveal a site that may control conformational switching between active and inactive states. J. Biol. Chem. 2017;292:13449–13458. doi: 10.1074/jbc.M117.787267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dev I.K., Dallas W.S., Yates B.B. Mode of binding of folate analogs to thymidylate synthase. Evidence for two asymmetric but interactive substrate binding sites. J. Biol. Chem. 1994;269:1873–1882. [PubMed] [Google Scholar]
- 17.Lovelace L.L., Gibson L.M., Lebioda L. Cooperative inhibition of human thymidylate synthase by mixtures of active site binding and allosteric inhibitors. Biochemistry. 2007;46:2823–2830. doi: 10.1021/bi061309j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sapienza P.J., Lee A.L. Backbone and ILV methyl resonance assignments of E. coli thymidylate synthase bound to cofactor and a nucleotide analogue. Biomol. NMR Assign. 2014;8:195–199. doi: 10.1007/s12104-013-9482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller S., Vargas C., Schuck P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012;84:5066–5073. doi: 10.1021/ac3007522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freire E., Schön A., Velazquez-Campoy A. Isothermal titration calorimetry: general formalism using binding polynomials. Methods Enzymol. 2009;455:127–155. doi: 10.1016/S0076-6879(08)04205-5. [DOI] [PubMed] [Google Scholar]
- 21.Freiburger L., Auclair K., Mittermaier A. Global ITC fitting methods in studies of protein allostery. Methods. 2015;76:149–161. doi: 10.1016/j.ymeth.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bain A.D., Berno B. Liouvillians in NMR: the direct method revisited. Prog. Nucl. Magn. Reson. Spectrosc. 2011;59:223–244. doi: 10.1016/j.pnmrs.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Brautigam C.A. Fitting two- and three-site binding models to isothermal titration calorimetric data. Methods. 2015;76:124–136. doi: 10.1016/j.ymeth.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houtman J.C., Brown P.H., Schuck P. Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 2007;16:30–42. doi: 10.1110/ps.062558507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H., Schuck P. Global multi-method analysis of affinities and cooperativity in complex systems of macromolecular interactions. Anal. Chem. 2012;84:9513–9519. doi: 10.1021/ac302357w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toke O., Monsey J.D., Cistola D.P. Determinants of cooperativity and site selectivity in human ileal bile acid binding protein. Biochemistry. 2006;45:727–737. doi: 10.1021/bi051781p. [DOI] [PubMed] [Google Scholar]
- 27.Fukada H., Takahashi K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins. 1998;33:159–166. [PubMed] [Google Scholar]
- 28.Vega S., Abian O., Velazquez-Campoy A. On the link between conformational changes, ligand binding and heat capacity. Biochim. Biophys. Acta. 2016;1860:868–878. doi: 10.1016/j.bbagen.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Eftink M.R., Anusiem A.C., Biltonen R.L. Enthalpy-entropy compensation and heat capacity changes for protein-ligand interactions: general thermodynamic models and data for the binding of nucleotides to ribonuclease A. Biochemistry. 1983;22:3884–3896. doi: 10.1021/bi00285a025. [DOI] [PubMed] [Google Scholar]
- 30.Phan J., Steadman D.J., Lebioda L. Structure of human thymidylate synthase suggests advantages of chemotherapy with noncompetitive inhibitors. J. Biol. Chem. 2001;276:14170–14177. doi: 10.1074/jbc.M009493200. [DOI] [PubMed] [Google Scholar]
- 31.Arai M., Ferreon J.C., Wright P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012;134:3792–3803. doi: 10.1021/ja209936u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Günther U.L., Schaffhausen B. NMRKIN: simulating line shapes from two-dimensional spectra of proteins upon ligand binding. J. Biomol. NMR. 2002;22:201–209. doi: 10.1023/a:1014985726029. [DOI] [PubMed] [Google Scholar]
- 33.Lee A.L., Sapienza P.J. Thermodynamic and NMR assessment of ligand cooperativity and intersubunit communication in symmetric dimers: application to thymidylate synthase. Front. Mol. Biosci. 2018;5:47. doi: 10.3389/fmolb.2018.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Popovych N., Sun S., Kalodimos C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caro J.A., Harpole K.W., Wand A.J. Entropy in molecular recognition by proteins. Proc. Natl. Acad. Sci. USA. 2017;114:6563–6568. doi: 10.1073/pnas.1621154114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tzeng S.R., Kalodimos C.G. Protein activity regulation by conformational entropy. Nature. 2012;488:236–240. doi: 10.1038/nature11271. [DOI] [PubMed] [Google Scholar]
- 37.Huggins D.J. Quantifying the entropy of binding for water molecules in protein cavities by computing correlations. Biophys. J. 2015;108:928–936. doi: 10.1016/j.bpj.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lovelace L.L., Minor W., Lebioda L. Structure of human thymidylate synthase under low-salt conditions. Acta Crystallogr. D Biol. Crystallogr. 2005;61:622–627. doi: 10.1107/S0907444905005895. [DOI] [PubMed] [Google Scholar]
- 39.Freiburger L., Miletti T., Mittermaier A. Substrate-dependent switching of the allosteric binding mechanism of a dimeric enzyme. Nat. Chem. Biol. 2014;10:937–942. doi: 10.1038/nchembio.1626. [DOI] [PubMed] [Google Scholar]
- 40.Tai N., Schmitz J.C., Chu E. Translational autoregulation of thymidylate synthase and dihydrofolate reductase. Front. Biosci. 2004;9:2521–2526. doi: 10.2741/1413. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.