

Abstract
CD22 is an inhibitory B cell co-receptor that recognizes sialic acid-containing glycoconjugates as ligands. Interactions with its glycan ligands are key to regulating the ability of CD22 to modulate B cell function, the most widely explored of which is antagonizing B cell receptor (BCR) signaling. Most importantly, interactions of CD22 with ligands on the same cell (cis) control the organization of CD22 on the cell surface, which minimizes co-localization with the BCR. In contrast with the modest ability of CD22 to intrinsically dampen BCR signaling, glycan ligands presented on another cell (trans) along with an antigen drawn CD22 and the BCR together within an immunological synapse, strongly inhibiting BCR signaling. New concepts are emerging for how CD22 controls B cell function, such as changes in glycosylation at different stages of B cell differentiation, specifically on GC B cells. Related to these changes, new players, such galectin-9, have been discovered that regulate cell surface nanoclusters of CD22. Roles of glycan ligands in controlling CD22 are the primary focus of this review as we highlight the ability of CD22 to modulate B cell function.
Keywords: B cells, Siglec, glycans, Sialic acid, Tolerance, CD22
Distinguishing self from non-self in humoral immunity
B cells as a double-edged sword in human health and disease
B cells are a critical component of the adaptive immune system, giving rise to plasma cells that ultimately produce protective antibodies, as well as memory B cells that enables a rapid response to future encounter with the pathogen [1]. In addition to being the original source of antibodies, B cells play key roles in secretion of cytokines and chemokines for the regulation of other immune cells [1], [2], [3] and contribute to differentiation of cognate CD4+ T cells through B-T cell interaction [4]. Dysregulation of B cell responses, which defines many autoimmune disorders, highlights the challenge for B cells in maintaining tolerance to self. Indeed, pathogenic autoantibodies are produced in following conditions like systemic lupus erythematosus (SLE) [5], Grave's disease [6], and Sjögren's syndrome [7]. Other key functions served by B cells can also become dysregulated; for example, strong evidence supports a role for B cells as antigen-presenting cells in rheumatoid arthritis [8]. Mechanisms that maintain B cell tolerance to self are of great interest in understanding the etiology of autoimmune diseases, which could guide the development of a new generation of immunomodulatory therapies. This review is not an exhaustive look at tolerogenic mechanisms, but an examination of one particular regulatory co-receptor on B cells, called CD22, which plays an important role in maintenance of peripheral B cell tolerance. Prior to exploring the role(s) for CD22 in tolerance to self, we will first describe the initial steps in response of B cells to antigen and the potential for inhibitory co-receptors to modulate these responses as a form of self-recognition.
The B cell receptor (BCR) complex
The initiation of antibody responses begins with the stimulation of B cell receptor (BCR) complex, which transmits downstream signals leading to B cell activation, proliferation, and differentiation. A critical component to this signaling event is the Src family kinases - one of which is Lyn - that phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) of CD79α/β, leading to recruitment of the kinase Syk and subsequent activation of downstream signaling pathways, such as ERK1/2, Akt, MAPK, NFAT, and NF-kB. Detailed information about the BCR signaling has been reviewed elsewhere [9], [10].
The need for inhibitory co-receptors to tune BCR signaling
The classical two-signal hypothesis postulates that multiple positive signals are required to ensure specificity of an immune response to an antigen, while also preventing self-reactivity [11]. For B cells, these two key signals come from encounter of BCR with antigen and interaction with their primed cognate CD4+ T cells. However, the following circumstances predict the need for additional mechanisms that help maintain B cell tolerance to self, particularly in the periphery: (1) escape of B and/or T cells from central tolerance, which is known to occur [12]; (2) encounter of highly multivalent self-antigens by the B cell that are capable of causing T-independent antibody responses [13]; (3) encounter of self-antigens under inflammatory conditions that can provide an alternative second signal (i.e. T-independent type 2 antigen) [14]; (4) bystander T cell help stemming from infection of cells with a pathogen [15]; and (5) a switch to self-reactivity following somatic hypermutation in the GCs [16]. Regulatory suppressor cells are one mechanism that contributes to the maintenance of self-tolerance under such conditions [17]. However, as is common in the immune system, redundant mechanisms are in place to prevent autoimmune responses.
The availability, or lack thereof, of T cell help and the actions of regulatory cells are both extrinsic factors imparted on the antigen-specific B cells. Other mechanisms limiting self-reactivity in the periphery are intrinsic factors - specifically, inhibitory BCR co-receptors that tune BCR signaling, including the well-studied inhibitory receptors [18], FcγRIIB [19], CD22 [20], and Siglec-G [21], and the less well-studied inhibitory receptors PECAM1, PIR-B, and PD-1 [22]. With the ability to antagonize BCR signaling, inhibitory co-receptors have the potential to help avoid reactivity to self in two ways [Fig. 1]. In the first mechanism, they create a threshold for BCR signaling, such that they prevent reactivity to weak antigens that could be considered self, such that the B cell remains silent (ignorant) or becomes unresponsive due to a very weak signal (anergic) [Fig. 1A]. While this mechanism is a commonly held view of BCR inhibitory co-receptors, it is interesting to note that germline encoded BCR clones arising from immunization to even foreign antigens are typically quite low affinity and only develop high affinity through the GC reaction [23]. A second potential mechanism involves a more active recognition of self by the inhibitory BCR co-receptors, which borrows from the well-established roles for inhibitory receptors on Natural Killer (NK) cells [Fig. 1B]. For NK cells, recognition of self by inhibitory receptors on other cells helps to ensure that they only mount effective responses in case of ‘missing self’ [24]. While data to support this mechanism on B cells are not abundant, experimental systems to examine this hypothesis are necessarily more complex. One defining feature distinguishing these two models is the context in which the antigen is displayed in a soluble form [Fig. 1A] or multivalently displayed in the context of a cell surface or immune complex [Fig. 1B].
Fig. 1.

Intrinsic and extrinsic models for BCR inhibitory co-receptors. (A) Intrinsic functioning of BCR inhibitory co-receptors wherein they constitutively dampen B cell activation by antagonizing the BCR signaling, thereby setting a threshold that prevents B cell from being activated by weak self antigens, such as soluble autoantigens. (B) Extrinsic functioning of BCR inhibitory co-receptors wherein their ability to antagonize BCR signaling is dependent on how the antigens are displayed. For example, co-expression of self-associated molecular recognition patterns with membrane-bound antigens on another cell have the potential to draw the inhibitory receptor into an immunological synapse and prevent B cell activation.
Glycans in self-recognition
One increasingly appreciated molecular determinant in self/non-self-discrimination are cell surface oligosaccharides, described more generally as glycans. Glycans are typically covalently linked to membrane-bound proteins and lipids and are present on all levels of life. Unlike the genetic code that is conserved throughout evolution, the pattern of glycosylation between humans and pathogenic organisms (i.e. parasites, fungi, and bacteria) has, for the most part, divergently evolved [25]. One unique feature of cell surface glycans in vertebrate cells is that they are capped by the monosaccharide sialic acid, which is a group of nine-carbon sugars from the nonulosonic acid family [26]. Sialic acids are ubiquitously and abundantly found on the surface of all human cells as the terminating sugar in glycolipids (gangliosides) and glycoproteins (complex N-glycans and mucin type O-linked glycans) [Fig. 2]. Due to the limited expression of sialic acid in many pathogens, our immune system has developed a means of differentiating these glycan variations and respond to them accordingly [27].
Fig. 2.

Common types of self-associated sialic acid-containing glycans on mammalian cells. Three of the most common forms of cell surface glycosylation: complex N-glycosylation, mucin-type O-glycosylation, and gangliosides. The glycosylation pattern on most pathogens is substantially different than mammalian-type glycosylation, therefore, cell surface glycosylation patterns are a unique signature of an organism that can used by the immune cells in self/non-self discrimination. One notable unique feature of mammalian glycosylation is the abundance of sialic acid sialic acid (purple diamond) that is abundant on all mammalian cells but absent on many pathogens.
Recently, an increasing number of studies suggest the potential involvement of sialic acid-containing glycans in immunological self-recognition events. This notion was first demonstrated with factor H (FH), a serum protein which controls the complement activation pathway [28]. FH binds to sialic acid on the surface of host cells and promotes the degradation of non-selectively bound complement C3b, a powerful opsonin that tags cells for phagocytosis [29]. Manipulation of antigens to express sialic acid-containing glycans substantially reduces their immunogenicity [30]. This review explores sialylated glycans as self-associated molecular patterns (SAMPs) [27] in their ability to regulate B cell function through serving as ligands for CD22.
CD22 as an inhibitory B cell co-receptor
Expression pattern of CD22
CD22 is a member of the sialic acid-binding immunoglobulin-type lectin (Siglec) family of immunomodulatory receptors. Fifteen Siglecs in human are differentially expressed predominantly throughout the hematopoietic lineage [31]. Exceptions for expression outside the hematopoietic lineage are also growing [32], [33], [34]. CD22 is one of four conserved Siglecs that share substantial sequence similarity with orthologs from other species, which are unlike the remaining Siglecs, part of the CD33-related subfamily, that are less well conserved [35], [36]. On B cells, expression of CD22 is first apparent at the pro-B cell stage of B cell development in the bone marrow, reaching its highest expression, approximately 65,000 copies [37], by the mature B cell stage in the periphery [38]. CD22 maintains its high expression on naïve B2 [39], B1 [40], marginal zone [39], GCs [41], memory [42], and regulatory [43] B cells in both mouse and man. Like many other critical B cell receptors, expression of CD22 is lost upon differentiation of B cells to plasma cells [44], with intermediate expression on plasmablasts [45].
The ability of CD22 to antagonize BCR signaling
Many studies have demonstrated that CD22 is capable of antagonizing BCR receptor signaling, which is evident by hyperactivation in response to BCR ligation in B cells lacking CD22 compared to their CD22-expressing counterpart [20]. In a recent internally-controlled competitive Ca2+ flux assay, this degree of hyperactivation was quantified as being 50% higher in CD22−/− compared to WT primary splenic B cells. Interestingly, this same degree of regulation was demonstrated for human and mouse CD22 [46] as well as a human B cell line in which CD22 was knocked out [47]. It is interesting to note this hyperactivation appears to be somewhat unique to dimeric crosslinking with anti-BCR antibodies (F(ab')2 anti-IgM) [Fig. 3A]; high affinity antigen (HEL) stimulation of IgMHEL B cells showed no difference between WT and CD22−/− B cells in two independent reports [Fig. 3B] [48], [49] and multivalent presentation of antigen can even show hypo-responsivess in CD22−/− B cells [Fig. 3C] [46], [50]. These differences reveal that the hyperactive phenotype of naïve CD22−/− B cells is not a universal principle and may relate to the degree of BCR engagement and crosslinking. Potentially related to this point is that several independent studies have reported that antibody production in response to T-dependent antigens are moderately blunted in CD22-deficient mice compared to WT mice [46], [51]; a role for CD22 in plasma cell formation could also account for this [42].
Fig. 3.

Hyperactive BCR signaling in CD22−/− B cells is unique to dimeric crosslinking of the BCR. (A) Stimulation of B cells with dimeric anti-IgM F(ab’)2 reveals a hyperactive BCR signaling in the absence of CD22. (B) Stimulation of IgMHEL B cells bearing a high affinity BCR clone specific for hen egg lysozyme (HEL) has revealed no intrinsic difference in response between WT and CD22−/− B cells. (C) Stimulation of B cells with highly multivalent antigen, such as liposomes displaying Fab fragments of anti-IgM, results in either no effect on BCR signaling in CD22−/− compared to WT B cells, or hypo-responsiveness.
A prerequisite for CD22 to antagonize BCR signaling is proximity of CD22 to the BCR [Fig. 4]. In the absence of significant recruitment of CD22 to the BCR complex, BCR signaling activates the five major aforementioned downstream pathways [Fig. 4A]. Recruitment of CD22 to the BCR results in four key steps [Fig. 4B]. First, CD22 is phosphorylated on its cytoplasmic tail immunoreceptor-based inhibitory motifs (ITIMs), primarily by the kinase Lyn [52]. Second, Shp-1 phosphatase is recruited to the phosphorylated ITIMs. Third, once Shp-1 is recruited to the BCR signaling complex, it antagonizes BCR signaling by removing phosphate residues of proximal signaling components, such as those initiated by Syk kinase [53], [54]. Finally, BCR signaling is inhibited. It is worth noting that Lyn is itself spatially regulated on the cell surface within the activatory microdomains [55], hence, it is conditions where CD22 enters these activation domains where it will become phosphorylated. Studies that have forcing CD22 and the BCR together, using polymers or liposomes co-displaying antigen and ligands of CD22, have clearly demonstrated how potently CD22 is phosphorylated when brought into an activatory microdomain along with the BCR [52], [56], [57], while establishing a clear requirement for Lyn and Shp-1 [58].
Fig. 4.

Proximity of CD22 to the BCR is required for its ability to antagonize BCR signaling. (A) Without CD22 in proximity to the BCR, ligation of the BCR leads to downstream recruitment and activation of the central kinase, Syk, through the ITAMs of CD79a and CD79b, leading to activation of 5 major BCR signaling pathways that signal through: Akt, NFAT, NFkB, ERK, and MAPK. (B) With recruitment of CD22 to the BCR during its activation, Lyn-mediated phosphorylation ITIMs on the cytoplasmic domain of CD22 recruits Shp-1 phosphatase that dephosphorylate Syk and other proximal BCR signaling components, leading to inhibited BCR signaling.
A brake that is constantly depressed or an emergency brake for threatening situations?
The earlier model depicted CD22 as acting constitutively to suppress B cell activation and create a threshold of BCR signaling that prevents inadvertent activation to weak signals that could be considered a form of self-recognition [59]. An alternative view is that CD22 acts more analogously to an emergency break to prevent unwanted B cell responses under the appropriate circumstances. These two views are by no means mutually exclusive and, indeed, could be synergistic. In the rest of this review, we explore recent work implicating CD22 in mediating B cell tolerance with a special emphasis placed on the glycan ligands of CD22 and their ability to guide the inhibitory function of CD22 as an inhibitory B cell co-receptor. Knowledge of the glycan ligands of CD22 will first be examined, followed by how these glycan ligands regulate CD22 on the surface of B cells.
Glycan ligands of CD22
Sialic acid ligands for CD22 are α2-6 sialiosides
Work dating over 25 years ago established α2-6 sialosides on complex N-glycans as ligands for CD22 [53], [54], [60], [61]. Despite the conserved specificity for α2-6 sialosides, CD22 from mouse and man have a finer ligand preference that relates to the type of glycan structures found in each respective species [Table 1]. In mice, B cells predominantly express a variant of sialic called N-glycolylneuraminic acid (Neu5Gc) linked to an underlying LacNAc (Neu5Gcα2-6LacNAc) on cell surface glycans. However, Neu5Gc is not produced in humans due to inactivation of the enzyme responsible for the biosynthesis of Neu5Gc, called CMP sialic acid hydroxylase (CMAH), meaning that all sialic acid on human cells is N-acetylneuraminic acid (Neu5Ac) [62]. Mouse CD22 (mCD22) has a strong preference for α2-6 sialosides containing Neu5Gc compared to the same structures but with Neu5Ac [63], which is on the order of 20-fold [41]. In contrast, human CD22 (hCD22) shows little discrimination between Neu5Gc and Neu5Ac [63] and increased affinity for α2-6 sialosides bearing a GlcNAc-6-sulfate residue [41].
Table 1.
Summary of glycan ligands for human and mouse CD22 and their relative binding affinities to each protein.
| Structure | Name | Relative binding |
Presence |
||||
|---|---|---|---|---|---|---|---|
| Mouse |
Man |
||||||
| hCD22 | mCD22 | Naise | GC | naive | GC | ||
 |
Neu5Gcα2-6Galβ1-4GlcNAc | ++ | ++ | + | – | – | – |
 |
Neu5Acα2-6Galβ1-4GlcNAc | ++ | + | – | + | + | + |
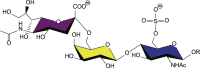 |
Neu5Acα2-6Galβ1-4(6S)GlcNAc | +++ | +++ | – | – | – | + |
Glycan ligands of CD22 on the same cell surface
Interactions between CD22 and glycan ligands on the same cell surface, denoted as cis interactions, are well established. Experimentally, cis ligand interactions are evident in two ways. The first is that removal of sialic acid on the surface of B cells - by neuraminidase digestion, mild periodate oxidation [60] or genetic ablation of St6gal1 [65] - greatly increases the ability of CD22 to engage with trans glycan ligands on another cell or particle bearing glycan ligands of CD22. The second evidence for cis ligands comes from studies with photo-crosslinkable versions of sialic acid, which can be incorporated into cell surface glycoconjugates enzymatically or metabolically [66], [67]. An important finding that came out of these crosslinking efforts, in conjugation with proteomics to identify binding partners of CD22, is that CD22 preferentially interacts with another molecule of CD22 to form homomultimers [68]. These results are in line with CD22 itself being a glycoprotein that contains 5–6 sites of complex N-glycosylation in its three most N-terminal domains [69]. It remains to be established precisely which N-glycan site preferentially acts as a ligand on a neighboring CD22 protein, but it is intriguing to speculate that it is Asn101, which is indispensable for protein folding [69]. Imaging studies have confirmed that CD22 is present in nanoclusters [37], [47] and that the size of these clusters is governed by interactions between CD22 and its glycan ligands [47]. The relationship between these CD22 nanoclusters and proximity of CD22 to the BCR will be explored below.
Glycan ligands of CD22 on another cell surface
The presence of cis interactions suggested that interactions between CD22 and glycan ligands on an opposing cell, described as a trans interaction, may only be possible upon loss of cis interactions [64], [65]. However, that was shown to not be the case, with the discovery that CD22 is drawn into the site of cell contact with other lymphocytes, which is dependent on α2-6 sialosides on the other cells [70]. Scaffolds that present synthetic high affinity CD22 ligand in a multivalent manner, have also shown to successfully engage in trans interactions with CD22 [66], [71], [72], [73]. Moreover, a photo-crosslinking study, which used a similar approach that identified cis ligands, revealed that soluble CD22 is capable of interacting with glycan ligands on the surface of a B cell [67], which is in line with staining of B cells with soluble CD22-Fc chimeric constructs [61]. Recently, the crystal structure of CD22 was determined, which in combination with single-particle electron microscopy and small angle x-ray scattering elegantly allowed Julien and co-workers to come up with a model of CD22 in which its seven extracellular domains form a rigid rod that can accommodate both cis and trans interactions [69]. Interestingly, this rigid structure has not been observed for similar cell surface proteins, such as RPTPσ [74], suggesting that this rigidity could help CD22 interact with glycan ligands in trans.
Roles of glycan ligands in controlling CD22 as a BCR inhibitory co-receptor
The ability of cis ligands to regulate nanocluster size, cellular organization, and dynamics of CD22
The first proposed model for glycan ligand-mediated regulation of the BCR by CD22 was one in which CD22 directly recognizes sialoside ligands on the BCR itself [75], [76]. This model was challenged several years later by the characterization of B cells lacking CD22 ligand, namely B cells from St6gal1−/− mice [77]. Specifically, it was discovered that St6gal1−/− B cells are hypo-responsive to BCR stimulation. Later, it was found that this hypo-responsiveness is the result of significantly increased colocalization CD22 with the BCR in St6gal1−/− B cells [78]. Consistent with the hypo-responsiveness of BCR signaling at the cellular level, St6gal1−/− mice also generate impaired antibody responses [77]. These findings were later corroborated by Nitschke and co-workers who elegantly demonstrated BCR hypo-responsiveness with a combination of neuraminidase-mediated removal of CD22 ligands from the cell surface and the development of a knock-in mice in which CD22 cannot bind sialic acid due to R130E mutation of an essential arginine [79]. The explanation for these observations is grounded in the aforementioned ability of CD22 to homomultimerize in a ligand-dependent manner [68] [Fig. 5]. In the absence of ligands, CD22 is more able to co-localize with the BCR, which could be the consequence of three effects: (1) protein–protein interactions [80]; (2) hetero-dimerization by galectins [81]; or (3) increase mobility https://www.zotero.org/google-docs/?KaqFiD [37]. In the presence of ligands, nanoclusters of CD22 effectively maintain CD22 away from the BCR [Fig. 5A]. Additional evidence for CD22 being largely maintained away from the BCR comes from studies that force CD22 with the BCR, showing how profoundly this can inhibit BCR signaling relative to the modest difference in BCR signaling comparing WT to CD22−/− B cells. A recent study using a nanomolar affinity soluble CD22 ligand also reported that this compound was capable of causing hypo-responsiveness of B cells in an St6gal1-dependent manner, which is consistent with this model [82]. Therefore, pharmacological, enzymatic, and genetic approaches to ablate CD22 ligands on the cell surface all demonstrate the same phenomenon, which is that they increase association of CD22 with the BCR, leading to hypo-responsive BCR signaling [Fig. 5B].
Fig. 5.

New players in glycan ligand-dependent organization of CD22 nanoclusters on the surface of B cells. (A) In WT B cells, CD22 nanoclusters are composed of at least CD22, CD45, and galectin-9. Glycan-dependent homomultimeric interactions between CD22 and heteromultimeric interactions with CD45, within these nanoclusters, act to keep CD22 and the BCR minimally associated on resting naive B cells. Galectin-9 appears to be involved in regulating the extent to which CD22 is kept away from the BCR since galectin-9−/− B cells show modestly hyperactive BCR signaling, while exogenously added galectin-9 dampens BCR signaling. (B) In the absence of cis glycan ligands or in knock-in B cells wherein CD22 cannot bind sialic acid - St6gal1−/− or CD22 R130E B cells, respectively - CD22 nanoclusters are smaller and more mobile, leading to increased association between CD22, potentially in galectin-9-dependent manner.
Super resolution microscopy studies have nicely confirmed a role for ligand binding in keeping CD22 away from the BCR [37]. It was also revealed that there is a ligand-dependent role for interactions between CD22 and CD45 in regulating CD22 organization and dynamics. However, this same study also reported that the cytoskeleton does not regulate CD22 and CD45 organization, which contrasts with the reported regulation of CD45 organization by the cytoskeleton in T cells [83], [84]. These results highlight that there is likely still be unanswered questions on CD22 cell surface organization. One additional layer of complexity was recently revealed by a number of studies exploring a role for galectin-9 in regulating CD22 organization [85], [86]. Galectins are another class of glycan-binding proteins that are capable of dimerizing proteins on the cell surface [87]. The most striking results linking galectins to CD22 comes from addition of exogenous galectin-9 to B cells, which demonstrated an increase in CD22 nanocluster size, increased association of CD22 with the BCR, and dampening of BCR signaling. Results comparing WT to galectin-9−/− B cells were less dramatic with activation of ERK showing hyper-responsiveness, but not CD19 or Akt [86]. More recently, it was confirmed that glycan ligands on CD22 itself regulate the ability of galectin-9 to promote association of CD22 with the BCR [47]. By taking advantage of a mutant version of CD22 lacking five N-glycan sites dispensable for protein folding expressed in a human B cell line, it was demonstrated that CD22 with deficient N-glycosylation displayed larger clusters without altering its mobility. Importantly, without the sites of N-glycosylation, exogenous galectin-9 no longer had the ability to increase association of CD22 with the BCR.
The ability of trans ligands to recruit CD22 into immunological synapse
Earlier evidence by Neuberger and co-workers first showed the involvement of CD22 trans ligands in modulating B cell responses [88]. The group observed that activation of splenic B cells upon encounter with target cells is inhibited in a CD22 trans ligand-dependent manner. Ectopic expression of St6gal1 in target cells that do not express CD22 ligands effectively dampened the stimulation of B cells in an in vitro co-culture assay, suggesting that expression of CD22 ligands on antigen-bearing cell can inhibit the activation of B cells by potentially drawing CD22 at the immunological synapse between B cell and its target cell [Fig. 6]. These findings were followed up over a decade later with the use of well-established mouse transgenic lines [89] that enabled autoreactivity to membrane-bound antigens to be investigated [50]. Incubation of hen egg-white lysozyme (HEL)-specific B cells with lymphocytes displaying membrane-bound HEL (mHEL) revealed that glycan ligands of CD22 draw CD22 into an immunological synapse. However, when CD22 ligands were destroyed either through genetic ablation of St6gal1 or mild periodate oxidation, recruitment of CD22 to the immunological synapse was abrogated. As a consequence of CD22 recruitment, BCR activation was strongly inhibited, which was observed in vitro by CD86 upregulation and in vivo through B cell proliferation and survival. These actions of CD22 not only inhibited proliferation, but also decreased the viability of B cells in vivo, although survival was not fully restored in CD22−/− B cells since an overlapping role for Siglec-G was reported, such that a full break in tolerance was only observed in CD22−/− × Siglec-G−/− B cells.
Fig. 6.

CD22 ligands displayed on the same cell as a cognate antigen for the BCR drawn CD22 into the immunological synapse. Sialic acid-containing glycan ligands of CD22 are capable of drawing CD22 in an immunological synapse formed between the B cell and a cell displaying a membrane-bound autoantigen. As a result of recruiting CD22 into the synapse with the BCR, BCR signaling is strongly inhibited.
Although these model systems demonstrate conceptual support for a role for trans ligands in preventing B cells from responding to membrane-bound autoantigens, direct evidence of this phenomenon in an intact non transgenic mouse has remained elusive. The first evidence that CD22 may be involved in maintaining self-tolerance came from autoantibodies produced in CD22-deficient mice [90]. However, this finding was later found to be strain-dependent since CD22−/− mice on a pure C57Bl/6 background do not show signs of overt autoimmunity [91]. Likewise, a mouse model carrying a CD22 mutation (R130E) that renders it incapable of binding to its glycan ligands also does not develop autoimmune-related symptoms [79]. On the other hand, CD22−/− × Siglec-G−/− mice produce aberrant levels of autoantibodies and show signs of overt autoimmunity, as documented by kidney damage, that is over and above the milder autoimmunity observed in Siglec-G−/− mice [21]. In more recent work by Nitschke and co-workers, a CD22 and Siglec-G double knock-in mouse was created with mutations at the critical arginine in both Siglecs, which allowed them to investigate the impact of abolishing sialic acid binding on B cells in immune tolerance [92]. Interestingly, these double mutant mice did not display the same autoimmunity as CD22−/− × Siglec-G−/− mice [51]. There are two possible interpretations of these findings: (1) trans ligands of CD22 (and Siglec-G) do not significantly impact B cell tolerance induction in mice or (2) CD22 R130E produces a dominant effect in suppressing B cell activation that does not enable a break in tolerance to be observed. Consistent with the latter possibility, an easier study using St6gal1 ligand-deficient B cells, which show the same hypo-reactivity as compared to CD22 R130E knock-in mice, crossed onto an autoimmune prone mouse strain prevented the development of autoimmunity [93]. More complex genetic variants of CD22 can be envisioned as a way to address this dominant effect, but the ultimate evidence for the importance of trans ligands of CD22 in maintaining peripheral B cell tolerance would be the demonstration of autoreactive antibodies arising to membrane autoantigens in mice lacking CD22 ligands on cells other than B cells.
Exploiting CD22 with synthetic trans sialosides to impact B cell fate
Current therapies for managing disorders like autoimmunity [5] largely rely on immunodepletion therapy and immunosuppressive medications that globally repress the immune system and compromise the ability to fight off infections [94]. Therefore, new therapies that specifically target autoreactive B cells are imperative. Leveraging the knowledge that particles displaying both antigen and ligands of CD22 have an extremely weak ability to activate the BCR, due to co-clustering of CD22 with the BCR [52], [95], this was taken a step further by examining the consequences of doing so with a T-dependent antigen [56]. Working with liposomal nanoparticles, which have a fluid membrane, it was shown that Siglec-engaging Tolerance-inducing Antigenic Liposomes (STALs) could induce antigen-specific tolerance of B cells in mice to protein antigens. Tolerance induction was shown to be mediated through deletion of the antigen-specific B cell from the B cell repertoire [50]. The mechanism of this depletion relates to basic B cell biology wherein resting naïve B cells, tonic BCR signaling is essential for B cell survival [Fig. 7A] [96]; STALs inhibited tonic BCR signaling, as evidenced by decreased levels of phosphorylation on Akt and several of its downstream targets, compared to resting cells [56]. The result was decreased phosphorylation of FoxO3a, which prevented it from being retained in the cytoplasm, giving rise to import of FoxO3a into the nucleus where it is capable of upregulating pro-apoptotic factors, such as Bim [Fig. 7B]. Consistent with this mechanism, B cells lacking the critical pro-apoptotic factor Bim simply remained ignorant of STALs [50].
Fig. 7.

Siglec-engaging antigenic liposomes (STALs) induce apoptosis in antigen-specific B cells through inhibiting tonic BCR signaling. (A) In resting B cells, tonic BCR signaling maintains FoxO1/3a in their phosphorylated state and maintain out of the nucleus. (B) STALs display both antigen and high affinity ligands of CD22 that co-ligate CD22 and the BCR. The consequence of enforcing ligation between CD22 and the BCR is inhibition of tonic BCR signaling, resulting in hypo-phosphorylation of Akt leads and Fox1/3a, which drives FoxO1/3a into the nucleus where it can upregulate the transcription of pro-apoptotic genes, such as BIM.
The application of STALs to induce robust suppression of antibody production, as a demonstration of their potential as a treatment strategy, was first demonstrated with FVIII, which is a blood-clotting protein and essential anti-hemophilic factor [97]. Administration of recombinant FVIII in previously STAL-tolerized FVIII-deficient mice - a mouse model for hemophilia A [98] - prevented the production of anti-FVIII antibodies, which subsequently enabled the delivery of FVIII to prevent bleeding in a tail clip assay. Two recent studies further illustrated the effectiveness of CD22 targeting-STALs in tolerizing B cells reactive towards 2S albumin (Ara h 2; the major peanut allergen) and citrulline, which are two antigens implicated in peanut allergy and rheumatoid arthritis, respectively [99], [100]. The latter of these studies suggested it is possible to induce tolerance in human antigen-specific B cells since STALs bearing a citrullinated antigen prevented the formation of anti-citrulline antibodies upon a subsequent in vitro challenge. Overall, these studies establish that liposomes targeting antigen-specific BCR and CD22 can suppress undesirable B cell-modulated humoral responses and prevent subsequent antigen sensitization. However, it remains unknown how effective STALs are at inducing long-term tolerance, especially in inflammatory conditions that could revert immune tolerance [101]. In this respect, co-administration of therapeutics aimed at dampening T cells [102] holds a lot of promise, and one study did suggest that STALs encapsulated with rapamycin may have an advantage [103].
Remodeling of CD22 glycan ligands in B cell differentiation and cell fate decision
The majority of studies looking at the functional effects of CD22 have focused on naïve B cells. However, a series of studies have documented changes in the glycan ligands of CD22 in later stages of B cell differentiation; namely, the GC [41]. The GC is a transient structure formed within the secondary lymphoid organs following immunization with a T-dependent antigen [104]. Development of GCs is a crucial event for mounting robust humoral responses against antigens, as it is the site antibody affinity maturation, with the products of the GC being long-lived memory B cells and antibody secreting cells (ASCs) [105]. It was found that in both mouse and human B cells, the high affinity cis glycan ligands for CD22 are lost in GC B cells relative to the naïve and memory B cell compartments [41]. Remarkably, these changes in CD22 glycan ligands are species-specific and cater to the specificity of CD22 from mouse and man [Table 1].
Cis ligands on resting naïve B cells ‘mask’ the ability of CD22 to interact with trans ligands, thereby creating a threshold for trans binding [64]. Conditions in which there is lower levels of expression of cis ligands are expected to unmask CD22 and increases its association with trans ligands [41]. Indeed, the consequence of loss of the higher affinity CD22 ligand on GC B cells was revealed through binding studies with fluorescent liposomes bearing a specific glycan ligand specific for CD22, where it was demonstrated that CD22 on GC B cells have an enhanced ability to bind trans ligands. Therefore, unmasking of CD22 on GC B cells is a phenomenon conserved between mouse and man, which occurs through different mechanisms. The functional consequences of unmasking of CD22 on GC B cells is still unknown, however, it is intriguing that work by Clark and colleagues demonstrated that memory B cell formation from GC B cells is impaired [42]. A defined mechanism for how unmasking of CD22 could impact memory B cell differentiation is unknown, but it was noted that subsets of GC B cells were skewed in CD22−/− mice, thus it does suggest that CD22 may play a role at this stage of B cells, albeit initial formation of GCs in CD22−/− does not appear to be significantly impaired [42], [91], [106]. Unmasking the sialic acid binding domain of CD22 should enhance the ability of CD22 to engage with trans ligands on other cells in the GC compartment, such as follicular helper T cells, follicular dendritic cells, and CXCL-12 expressing reticular cells.
Another change in glycosylation within the GC that impacts CD22 was also recently revealed by Dimitroff and co-workers, wherein they added to concept of galectin-9 mediated regulation of CD22 nanoclusters [85]. Specifically, it was shown that human GC B cells upregulate expression of a unique galactosyl transferase, called GCNT2, that initiates a type of branching within complex N-glycans known as I-branching. The consequence to galectin-9 is that I-branching destroying galectin-9 binding sites and, consistent with this, exogenous fluorescently-labeled galectin-9 staining of GC B cells is considerably lower than naïve B cells. Given the proposed role of galectin-9 in mediating CD22 nanoclusters on naïve B cells, the results suggest that loss of galectin-9 ligand on GC B cells could impact CD22 organization on GC B cells. How I-branching on glycans affects their ability to interact with CD22 remains an unanswered question.
Beyond regulation of the BCR: other roles for CD22 in control B cell function
Regulating interactions with other immune cells
Natural trans ligands of CD22 on T cells was first identified over two decades ago [61], yet studies devoted to the functional role of these interactions on T cells have been limited. Using a soluble recombinant version of CD22 (CD22Rg), Aruffo et al. [107] identified CD45RO and other CD45 isoforms as one of CD22 ligands on T cells and suggested that association of CD22 with these glycoproteins may participate in T cell function [Fig. 8]. Specifically, crosslinking of CD3 and trans CD22 ligands, using anti-CD3 and CD22Rg, resulted in inhibited intracellular Ca2+ release and phospholipase Cγ1 phosphorylation when compared with T cells ligated with anti-CD3 alone. Similarly, a later study found that blocking the interaction of CD22 and its trans glycan ligands using a monoclonal antibody diminished the capacity of activated B cells to stimulate T cell proliferation in vitro [108], suggesting that failure to bring CD22 and/or its trans ligands into an immunological synapse may alter the outcome of B-T interactions. Therefore, while it is tempting to speculate that CD22 may impact the activation of cognate T cells via trans ligand interaction, definitive experiments to directly test this hypothesis are needed.
Fig. 8.

Additional roles for CD22 in controlling B cell function. (upper portion of cell) Interactions between B and T cells, such as cognate interactions mediated through presentation of antigenic peptides on MHC-II, have been suggested to be modulated by trans interaction of CD22 with glycan ligands on T cells; the importance of these interactions remains poorly defined. (lower left portion of cell) CD22 is implicated as an adhesion receptor that binds to trans glycans on high endothelial venules (HEVs) in the gut and bone marrow to facilitate B cell homing to these locations. (lower right portion of cell) Loss of CD22 expression in B cells results in significantly hyperactive TLR signaling, suggesting that CD22 transit to the same endosomal compartment as TLRs and regulate TLR signaling; a direct role in CD22 regulating TLR signaling and the presence of CD22 in the same compartment as TLRs has yet to be established.
Beyond T cells, dendritic cells also express glycan ligands for CD22 [109]. In vitro co-culture of bone marrow-derived immature DCs (iDCs) with naïve B cells induces strong inhibition of B cell proliferation following BCR ligation in a CD22 and contact-dependent mechanism [109]. iDCs can also suppress TLR-induced B cell proliferation of B cells that through CD22-CD22 trans ligand contact [110]. Deletion of CD22 restored proliferation of stimulated B cells even in the presence of iDCs. Interestingly, abrogation of St6gal1 on iDCs, like WT iDCs, repress BCR-induced proliferation of WT B cells, suggesting that CD22 may recognize St6gal1-independent trans ligands on iDCs to initiate suppression of BCR signaling.
Regulating Toll-like receptor (TLR) signaling
Earlier studies from CD22−/− mice revealed an altered response in B cells following stimulation with a variety of TLR ligands [91], where CD22 appears to act as a negative regulator. More detailed analysis of WT versus CD22−/− B cell responses to a variety of TLR ligands have solidified these findings [82], [111]. It is intriguing that up to a 5-10-fold increase in responsiveness to TLR ligands in CD22−/− B cells is significantly more than the 50% increase in BCR stimulation. Noteworthy is that several TLRs, such as TLR3 and TLR9, are not cell surface resident, but located in cytoplasmic endosomes [112]; and uptake of CD22 through endocytic pathways [91] may present a distinct possibility, although not proven, that CD22 and TLRs co-exist at least temporarily within the same endosomal compartments [Fig. 8]. Even if CD22 does enter the same intracellular compartment as TLRs, it is not entirely obvious how CD22 would antagonize TLR signaling given that a role for Shp-1 in regulating this TLR signaling on B cells is still not well-understood [113]. Adding to the body of data for how CD22 may regulate TLRs, a recent study using high affinity soluble CD22 ligands demonstrated that treatment results in exaggerated response of B cells to TLR stimulation but, unexpectantly, these results were not St6gal1-dependent [82]. Future studies analyzing the response of knock-in mice lacking the ITIMs of CD22 [79] to TLR stimulation may prove to be informative.
Homing to specific lymphoid organs
Early on, CD22 was thought to be an adhesion receptor on B cells [114]. Supporting this concept came from studies demonstrating that CD22 facilitates physical contact with activated human endothelial cells [115] and hematopoietic stem cell-derived cells [114], [116] in vitro by recognizing its canonical glycan epitope, α2-6-linked sialic acid, on the surface of these cells. Subsequently, it was discovered that recirculating mature IgD+ B cells migrate to the bone marrow in a CD22 ligand-dependent manner. Using a soluble recombinant version of CD22 (CD22-Fc), Nitschke and co-workers identified sialylated ligands of CD22 are expressed on sinusoidal endothelial cells (ECs) in the bone marrow [117]. Administration of anti-mouse CD22 antibodies or CD22-Fc block CD22 ligand interactions, resulting in reduced migration of mature B cells in bone marrow by approximately 50 percent, recapitulating the observations from other studies using CD22-deficient mouse models [81], [118]. Furthermore, adoptive transfer of WT B cells into St6gal1 knockout mice resulted in a diminished capacity of B cells to home in the bone marrow.
Several decades later after CD22 was formally proposed as an adhesion receptor, this concept was formally revisited by Butcher and co-workers who discovered a crucial role CD22 in the recruitment and homing of B cells to the gut-associated lymphoid tissues (GALTs) [119]. GALT is comprised of Peyer's patches (PPs) and mesenteric lymph node (MLN), which are secondary lymphoid organs that are critical for immune responses to gut-derived antigens from food and microbes [120]. High endothelial venules (HEVs) from the GALT were found to express high levels of St6gal1 compared to capillary endothelial cells and HEVs of the skin-draining peripheral lymph nodes (PLN). At a finer level, it was also noted that PPs express relatively more St6gal1 than the MLN. Short-term homing of adoptively transferred WT and CD22−/− B cells into WT mice demonstrated a critical role for CD22 in trafficking to the PPs but not to the PLN, and this CD22-dependent effect was lost in St6gal1−/− mice. In line with the higher expression of St6gal1 with the PP, B cell homing to the PP was more CD22 dependent than to the MLN. These findings revealed an important role of CD22 and its trans glycan ligands in effective homing of B cells to the gut lymph nodes [Fig. 8]; however, how differential expression of St6gal1 between PPs and MLN affects intestinal immune homeostasis as well as the quality of B cell responses at these sites remains to be elucidated.
Conclusions
As a member of Siglec family, the ability of CD22 to dampen B cell activation is highly dependent on the ability CD22 to recognize α2-6 sialic acids on glycoconjugates present on surface of B cells or antigen-expressing target cells. Interactions of CD22 with cis ligand partners create highly organized CD22 nanoclusters, which sequesters CD22 away from the BCR complex in unstimulated B cells, whereby also creating a threshold for trans ligand binding. Association of CD22 with trans ligands in the context of a membrane that co-displays antigen can induce antigen-specific immunological tolerance. CD22-deficient mice or knock-in mice expressing a version of CD22 that cannot recognize its glycan ligands both do not show overt autoimmunity, which likely stems from redundant mechanisms that help enforce B cell tolerance [18], such as the other major B cell Siglec, Siglec-G.
While roles of CD22 ligands in controlling B cell activation are well-documented, it is unclear how the shared contribution of cis and trans ligands affects CD22 function at different stages of B cell development. Are CD22 nanocluster size and density exclusively controlled by cis ligands? Will chronic or transient exposure to trans ligands on other cells remodel the nanoclusters? It is speculated that these dynamic changes in CD22 nanoclusters, potentially regulated through both cis and trans ligand interactions, fine-tune the BCR activation strength resulting in tonic signaling needed for B cell survival and for the persistence of memory B cell subsets in non-lymphoid organs. Furthermore, given the changes in sialic acid-linked glycans occur during B cell differentiation at the GC, it is of great interest to understand how these changes influence nanocluster size and CD22 interactions with cis and trans ligands.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by a grant from the National Institutes of Health (NIH), AI118842 and AI128598, and tier II Canada Research Chair in Chemical Glycoimmunology to M.S.M. J.R.E is funded by the Alberta Innovates Graduate Student Scholarship. J.R.E. and J.J. are supported by scholarships from the University of Alberta.
Footnotes
Peer review under responsibility of Chang Gung University.
References
- 1.Kurosaki T., Kometani K., Ise W. Memory B cells. Nat Rev Immunol. 2015;15:149–159. doi: 10.1038/nri3802. [DOI] [PubMed] [Google Scholar]
- 2.Arkatkar T., Du S.W., Jacobs H.M., Dam E.M., Hou B., Buckner J.H. B cell–derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J Exp Med. 2017;214:3207–3217. doi: 10.1084/jem.20170580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaniel C., Pardali E., Sallusto F., Speletas M., Ruedl C., Shimizu T. Activated murine B lymphocytes and dendritic cells produce a novel CC chemokine which acts selectively on activated T cells. J Exp Med. 1998;188:451–463. doi: 10.1084/jem.188.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Pinto D., Saravia N.G., McMahon-Pratt D. CD4 T cell activation by B cells in human Leishmania (Viannia)infection. BMC Infect Dis. 2014;14:108. doi: 10.1186/1471-2334-14-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pisetsky D.S. Anti-DNA antibodies — quintessential biomarkers of SLE. Nat Rev Rheumatol. 2016;12:102–110. doi: 10.1038/nrrheum.2015.151. [DOI] [PubMed] [Google Scholar]
- 6.Menconi F., Marcocci C., Marinò M. Diagnosis and classification of Graves' disease. Autoimmun Rev. 2014;13:398–402. doi: 10.1016/j.autrev.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Shen L., Suresh L. Autoantibodies, detection methods and panels for diagnosis of Sjögren’s syndrome. Clin Immunol. 2017;182:24–29. doi: 10.1016/j.clim.2017.03.017. [DOI] [PubMed] [Google Scholar]
- 8.Derksen V.F.A.M., Huizinga T.W.J., van der Woude D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol. 2017;39:437–446. doi: 10.1007/s00281-017-0627-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monroe J.G. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat Rev Immunol. 2006;6:283. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- 10.Rickert R.C. New insights into pre-BCR and BCR signalling with relevance to B cell malignancies. Nat Rev Immunol. 2013;13:578–591. doi: 10.1038/nri3487. [DOI] [PubMed] [Google Scholar]
- 11.Bretscher P., Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 12.Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17:281–294. doi: 10.1038/nri.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamnaes J., Iversen R., du Pré M.F., Chen X., Sollid L.M. Enhanced B-cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-multimerization. PLoS One. 2015;10 doi: 10.1371/journal.pone.0134922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Enoksson S.L., Grasset E.K., Hägglöf T., Mattsson N., Kaiser Y., Gabrielsson S. The inflammatory cytokine IL-18 induces self-reactive innate antibody responses regulated by natural killer T cells. Proc Natl Acad Sci. 2011;108:20285–20286. doi: 10.1073/pnas.1107830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whiteside S.K., Snook J.P., Williams M.A., Weis J.J. Bystander T cells: a balancing act of friends and foes. Trends Immunol. 2018;39:1021–1035. doi: 10.1016/j.it.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brink R., Phan T.G. Self-reactive B cells in the germinal center reaction. Annu Rev Immunol. 2018;36:339–357. doi: 10.1146/annurev-immunol-051116-052510. [DOI] [PubMed] [Google Scholar]
- 17.Dominguez-Villar M., Hafler D.A. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19:665. doi: 10.1038/s41590-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adachi T., Wakabayashi C., Nakayama T., Yakura H., Tsubata T. CD72 negatively regulates signaling through the antigen receptor of B cells. J Immunol. 2000;164:1223–1229. doi: 10.4049/jimmunol.164.3.1223. [DOI] [PubMed] [Google Scholar]
- 19.Tamir I., Stolpa J.C., Helgason C.D., Nakamura K., Bruhns P., Daeron M. The RasGAP-binding protein p62dok is a mediator of inhibitory FcgammaRIIB signals in B cells. Immunity. 2000;12:347–358. doi: 10.1016/s1074-7613(00)80187-9. [DOI] [PubMed] [Google Scholar]
- 20.O'Keefe T.L., Williams G.T., Davies S.L., Neuberger M.S. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274:798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann A., Kerr S., Jellusova J., Zhang J., Weisel F., Wellmann U. Siglec-G is a B1 cell-inhibitory receptor that controls expansion and calcium signaling of the B1 cell population. Nat Immunol. 2007;8:695–704. doi: 10.1038/ni1480. [DOI] [PubMed] [Google Scholar]
- 22.Tsubata T. Ligand recognition determines the role of inhibitory B cell Co-receptors in the regulation of B cell homeostasis and autoimmunity. Front Immunol. 2018;9:2276. doi: 10.3389/fimmu.2018.02276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlomchik M.J., Luo W., Weisel F. Linking signaling and selection in the germinal center. Immunol Rev. 2019;288:49–63. doi: 10.1111/imr.12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anfossi N., André P., Guia S., Falk C.S., Roetynck S., Stewart C.A. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 25.Dell A., Galadari A., Sastre F., Hitchen P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int J Microbiol. 2010 doi: 10.1155/2010/148178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varki A., Schnaar R.L., Schauer R. Sialic acids and other nonulosonic acids. In: Varki A., Cummings R.D., Esko J.D., Stanley P., Hart G.W., Aebi M., editors. Essent. Glycobiol. 3rd ed. Cold Spring Harbor Laboratory Press; (NY): 2015. (Cold Spring Harbor). [PubMed] [Google Scholar]
- 27.Varki A. Letter to the Glyco-Forum: since there are PAMPs and DAMPs, there must be SAMPs? Glycan “self-associated molecular patterns” dampen innate immunity, but pathogens can mimic them. Glycobiology. 2011;21:1121–1124. doi: 10.1093/glycob/cwr087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira V.P., Pangburn M.K. Factor H–mediated cell surface protection from complement is critical for the survival of PNH erythrocytes. Blood. 2007;110:2190–2192. doi: 10.1182/blood-2007-04-083170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janssen B.J.C., Christodoulidou A., McCarthy A., Lambris J.D., Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444:213. doi: 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 30.Perdicchio M., Ilarregui J.M., Verstege M.I., Cornelissen L.A.M., Schetters S.T.T., Engels S. Sialic acid-modified antigens impose tolerance via inhibition of T-cell proliferation and de novo induction of regulatory T cells. Proc Natl Acad Sci. 2016;113:3329–3334. doi: 10.1073/pnas.1507706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crocker P.R., Paulson J.C., Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 32.Itoyama Y., Sterhberger N.H., Webster H.D., Quarles R.H., Cohen S.R., Richardson E.P. Immunocytochemical observations on the distribution of myelin-associated glycoprotein and myelin basic protein in multiple sclerosis lesions. Ann Neurol. 1980;7:167–177. doi: 10.1002/ana.410070212. [DOI] [PubMed] [Google Scholar]
- 33.Dharmadhikari G., Stolz K., Hauke M., Morgan N.G., Varki A., de Koning E. Siglec-7 restores β-cell function and survival and reduces inflammation in pancreatic islets from patients with diabetes. Sci Rep. 2017;7:45319. doi: 10.1038/srep45319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ali S.R., Fong J.J., Carlin A.F., Busch T.D., Linden R., Angata T. Siglec-5 and Siglec-14 are polymorphic paired receptors that modulate neutrophil and amnion signaling responses to group B Streptococcus. J Exp Med. 2014;211:1231–1242. doi: 10.1084/jem.20131853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angata T., Margulies E.H., Green E.D., Varki A. Large-scale sequencing of the CD33-related Siglec gene cluster in five mammalian species reveals rapid evolution by multiple mechanisms. Proc Natl Acad Sci. 2004;101:13251–13256. doi: 10.1073/pnas.0404833101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bornhöfft K.F., Goldammer T., Rebl A., Galuska S.P. Siglecs: a journey through the evolution of sialic acid-binding immunoglobulin-type lectins. Dev Comp Immunol. 2018;86:219–231. doi: 10.1016/j.dci.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 37.Gasparrini F., Feest C., Bruckbauer A., Mattila P.K., Müller J., Nitschke L. Nanoscale organization and dynamics of the siglec CD22 cooperate with the cytoskeleton in restraining BCR signalling. EMBO J. 2016;35:258–280. doi: 10.15252/embj.201593027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stoddart A., Ray R.J., Paige C.J. Analysis of murine CD22 during B cell development: CD22 is expressed on B cell progenitors prior to IgM. Int Immunol. 1997;9:1571–1579. doi: 10.1093/intimm/9.10.1571. [DOI] [PubMed] [Google Scholar]
- 39.Lajaunias F., Nitschke L., Moll T., Martinez-Soria E., Semac I., Chicheportiche Y. Differentially regulated expression and function of CD22 in activated B-1 and B-2 lymphocytes. J Immunol. 2002;168:6078–6083. doi: 10.4049/jimmunol.168.12.6078. [DOI] [PubMed] [Google Scholar]
- 40.Erickson L.D., Tygrett L.T., Bhatia S.K., Grabstein K.H., Waldschmidt T.J. Differential expression of CD22 (Lyb8) on murine B cells. Int Immunol. 1996;8:1121–1129. doi: 10.1093/intimm/8.7.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Macauley M.S., Kawasaki N., Peng W., Wang S.-H., He Y., Arlian B.M. Unmasking of CD22 Co-receptor on germinal center B-cells occurs by alternative mechanisms in mouse and man. J Biol Chem. 2015;290:30066–30077. doi: 10.1074/jbc.M115.691337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappell C.P., Draves K.E., Clark E.A. CD22 is required for formation of memory B cell precursors within germinal centers. PLoS One. 2017;12 doi: 10.1371/journal.pone.0174661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwata Y., Matsushita T., Horikawa M., DiLillo D.J., Yanaba K., Venturi G.M. Characterization of a rare IL-10–competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;117:530–541. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perfetti V., Vignarelli M.C., Bellotti V., Glennie M.J., Zorzoli I., Ubbiali P. Membrane CD22 defines circulating myeloma-related cells as mature or later B cells. Lab Investig J Tech Methods Pathol. 1997;77:333–344. [PubMed] [Google Scholar]
- 45.Jourdan M., Caraux A., Vos J.D., Fiol G., Larroque M., Cognot C. An in vitro model of differentiation of memory B cells into plasmablasts and plasma cells including detailed phenotypic and molecular characterization. Blood. 2009;114:5173–5181. doi: 10.1182/blood-2009-07-235960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bednar K.J., Shanina E., Ballet R., Connors E.P., Duan S., Juan J. Human CD22 inhibits murine B cell receptor activation in a human CD22 transgenic mouse model. J Immunol. 2017;199:3116–3128. doi: 10.4049/jimmunol.1700898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wasim L., Buhari F.H.M., Yoganathan M., Sicard T., Ereño-Orbea J., Julien J.P. N-linked glycosylation regulates CD22 organization and function. Front Immunol. 2019;10:699. doi: 10.3389/fimmu.2019.00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horikawa K., Martin S.W., Pogue S.L., Silver K., Peng K., Takatsu K. Enhancement and suppression of signaling by the conserved tail of IgG memory-type B cell antigen receptors. J Exp Med. 2007;204:759–769. doi: 10.1084/jem.20061923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferry H., Crockford T.L., Cockford T.L., Silver K., Rust N., Goodnow C.C. Analysis of Lyn/CD22 double-deficient B cells in vivo demonstrates Lyn- and CD22-independent pathways affecting BCR regulation and B cell survival. Eur J Immunol. 2005;35:3655–3663. doi: 10.1002/eji.200535247. [DOI] [PubMed] [Google Scholar]
- 50.Macauley M.S., Paulson J.C. Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells. J Immunol. 2014;193:4312–4321. doi: 10.4049/jimmunol.1401723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jellusova J., Wellmann U., Amann K., Winkler T.H., Nitschke L. CD22 × siglec-G double-deficient mice have massively increased B1 cell numbers and develop systemic autoimmunity. J Immunol. 2010;184:3618–3627. doi: 10.4049/jimmunol.0902711. [DOI] [PubMed] [Google Scholar]
- 52.Duong B.H., Tian H., Ota T., Completo G., Han S., Vela J.L. Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo. J Exp Med. 2010;207:173–187. doi: 10.1084/jem.20091873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sgroi D., Varki A., Braesch-Andersen S., Stamenkovic I. CD22, a B cell-specific immunoglobulin superfamily member, is a sialic acid-binding lectin. J Biol Chem. 1993;268:7011–7018. [PubMed] [Google Scholar]
- 54.Powell L.D., Sgroi D., Sjoberg E.R., Stamenkovic I., Varki A. Natural ligands of the B cell adhesion molecule CD22 beta carry N-linked oligosaccharides with alpha-2,6-linked sialic acids that are required for recognition. J Biol Chem. 1993;268:7019–7027. [PubMed] [Google Scholar]
- 55.Wilson B.S., Pfeiffer J.R., Oliver J.M. Observing fcεri signaling from the inside of the mast cell membrane. J Cell Biol. 2000;149:1131–1142. doi: 10.1083/jcb.149.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macauley M.S., Pfrengle F., Rademacher C., Nycholat C.M., Gale A.J., von Drygalski A. Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis. J Clin Investig. 2013;123:3074–3083. doi: 10.1172/JCI69187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Courtney A.H., Puffer E.B., Pontrello J.K., Yang Z.-Q., Kiessling L.L. Sialylated multivalent antigens engage CD22 in trans and inhibit B cell activation. Proc Natl Acad Sci U S A. 2009;106:2500–2505. doi: 10.1073/pnas.0807207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamir I., Dal Porto J.M., Cambier J.C. Cytoplasmic protein tyrosine phosphatases SHP-1 and SHP-2: regulators of B cell signal transduction. Curr Opin Immunol. 2000;12:307–315. doi: 10.1016/s0952-7915(00)00092-3. [DOI] [PubMed] [Google Scholar]
- 59.Healy J.I., Goodnow C.C. Positive versus negative signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1998;16:645–670. doi: 10.1146/annurev.immunol.16.1.645. [DOI] [PubMed] [Google Scholar]
- 60.Stamenkovic I., Sgroi D., Aruffo A. CD22 binds to alpha-2,6-sialyltransferase-dependent epitopes on COS cells. Cell. 1992;68:1003–1004. doi: 10.1016/0092-8674(92)90071-j. [DOI] [PubMed] [Google Scholar]
- 61.Stamenkovic I., Sgroi D., Aruffo A., Sy M.S., Anderson T. The B lymphocyte adhesion molecule CD22 interacts with leukocyte common antigen CD45RO on T cells and α2–6 sialyltransferase, CD75, on B cells. Cell. 1991;66:1133–1144. doi: 10.1016/0092-8674(91)90036-x. [DOI] [PubMed] [Google Scholar]
- 62.Chou H.H., Takematsu H., Diaz S., Iber J., Nickerson E., Wright K.L. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci U S A. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brinkman-Van der Linden E.C., Sjoberg E.R., Juneja L.R., Crocker P.R., Varki N., Varki A. Loss of N-glycolylneuraminic acid in human evolution. Implications for sialic acid recognition by siglecs. J Biol Chem. 2000;275:8633–8640. doi: 10.1074/jbc.275.12.8633. [DOI] [PubMed] [Google Scholar]
- 64.Razi N., Varki A. Masking and unmasking of the sialic acid-binding lectin activity of CD22 (Siglec-2) on B lymphocytes. Proc Natl Acad Sci. 1998;95:7469–7474. doi: 10.1073/pnas.95.13.7469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Collins B.E., Blixt O., Bovin N.V., Danzer C.-P., Chui D., Marth J.D. Constitutively unmasked CD22 on B cells of ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glycobiology. 2002;12:563–571. doi: 10.1093/glycob/cwf067. [DOI] [PubMed] [Google Scholar]
- 66.Feng L., Hong S., Rong J., You Q., Dai P., Huang R. Bifunctional unnatural sialic acids for dual metabolic labeling of cell-surface sialylated glycans. J Am Chem Soc. 2013;135:9244–9247. doi: 10.1021/ja402326z. [DOI] [PubMed] [Google Scholar]
- 67.Ramya T.N.C., Weerapana E., Liao L., Zeng Y., Tateno H., Liao L. In situ trans ligands of CD22 identified by glycan-protein photocross-linking-enabled proteomics. Mol Cell Proteom MCP. 2010;9:1339–1351. doi: 10.1074/mcp.M900461-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Han S., Collins B.E., Bengtson P., Paulson J.C. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat Chem Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 69.Ereño-Orbea J., Sicard T., Cui H., Mazhab-Jafari M.T., Benlekbir S., Guarné A. Molecular basis of human CD22 function and therapeutic targeting. Nat Commun. 2017;8:764. doi: 10.1038/s41467-017-00836-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins B.E., Blixt O., DeSieno A.R., Bovin N., Marth J.D., Paulson J.C. Masking of CD22 by cis ligands does not prevent redistribution of CD22 to sites of cell contact. Proc Natl Acad Sci U S A. 2004;101:6104–6109. doi: 10.1073/pnas.0400851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peng W., Paulson J.C. CD22 ligands on a natural N-glycan scaffold efficiently deliver toxins to B-lymphoma cells. J Am Chem Soc. 2017;139:12450–12458. doi: 10.1021/jacs.7b03208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Collins B.E., Blixt O., Han S., Duong B., Li H., Nathan J.K. High-affinity ligand probes of CD22 overcome the threshold set by cis ligands to allow for binding, endocytosis, and killing of B cells. J Immunol Baltim Md 1950. 2006;177:2994–3003. doi: 10.4049/jimmunol.177.5.2994. [DOI] [PubMed] [Google Scholar]
- 73.Chen W.C., Completo G.C., Sigal D.S., Crocker P.R., Saven A., Paulson J.C. In vivo targeting of B-cell lymphoma with glycan ligands of CD22. Blood. 2010;115:4778–4786. doi: 10.1182/blood-2009-12-257386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coles C.H., Mitakidis N., Zhang P., Elegheert J., Lu W., Stoker A.W. Structural basis for extracellular cis and trans RPTPσ signal competition in synaptogenesis. Nat Commun. 2014;5:5209. doi: 10.1038/ncomms6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peaker C.J., Neuberger M.S. Association of CD22 with the B cell antigen receptor. Eur J Immunol. 1993;23:1358–1363. doi: 10.1002/eji.1830230626. [DOI] [PubMed] [Google Scholar]
- 76.Leprince C., Draves K.E., Geahlen R.L., Ledbetter J.A., Clark E.A. CD22 associates with the human surface IgM-B-cell antigen receptor complex. Proc Natl Acad Sci. 1993;90:3236–3240. doi: 10.1073/pnas.90.8.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hennet T., Chui D., Paulson J.C., Marth J.D. Immune regulation by the ST6Gal sialyltransferase. Proc Natl Acad Sci U S A. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collins B.E., Smith B.A., Bengtson P., Paulson J.C. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat Immunol. 2006;7:199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- 79.Müller J., Obermeier I., Wöhner M., Brandl C., Mrotzek S., Angermüller S. CD22 ligand-binding and signaling domains reciprocally regulate B-cell Ca2+ signaling. Proc Natl Acad Sci. 2013;110:12402–12407. doi: 10.1073/pnas.1304888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang M., Varki A. Cell surface sialic acids do not affect primary CD22 interactions with CD45 and surface IgM nor the rate of constitutive CD22 endocytosis. Glycobiology. 2004;14:939–949. doi: 10.1093/glycob/cwh126. [DOI] [PubMed] [Google Scholar]
- 81.Giovannone N., Smith L.K., Treanor B., Dimitroff C.J. Galectin-glycan interactions as regulators of B cell immunity. Front Immunol. 2018;9:2839. doi: 10.3389/fimmu.2018.02839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsubara N., Imamura A., Yonemizu T., Akatsu C., Yang H., Ueki A. CD22-Binding synthetic sialosides regulate B lymphocyte proliferation through CD22 ligand-dependent and independent pathways, and enhance antibody production in mice. Front Immunol. 2018;9:820. doi: 10.3389/fimmu.2018.00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cairo C.W., Das R., Albohy A., Baca Q.J., Pradhan D., Morrow J.S. Dynamic regulation of CD45 lateral mobility by the spectrin-ankyrin cytoskeleton of T cells. J Biol Chem. 2010;285:11392–11401. doi: 10.1074/jbc.M109.075648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pradhan D., Morrow J.S. The spectrin-ankyrin skeleton controls CD45 surface display and interleukin-2 production. Immunity. 2002;17:303–315. doi: 10.1016/s1074-7613(02)00396-5. [DOI] [PubMed] [Google Scholar]
- 85.Giovannone N., Liang J., Antonopoulos A., Sweeney J.G., King S.L., Pochebit S.M. Galectin-9 suppresses B cell receptor signaling and is regulated by I-branching of N-glycans. Nat Commun. 2018;9:3287. doi: 10.1038/s41467-018-05770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cao A., Alluqmani N., Buhari F.H.M., Wasim L., Smith L.K., Quaile A.T. Galectin-9 binds IgM-BCR to regulate B cell signaling. Nat Commun. 2018;9:3288. doi: 10.1038/s41467-018-05771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rabinovich G.A., Toscano M.A. Turning “sweet” on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–352. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- 88.Lanoue A., Batista F.D., Stewart M., Neuberger M.S. Interaction of CD22 with alpha2,6-linked sialoglycoconjugates: innate recognition of self to dampen B cell autoreactivity? Eur J Immunol. 2002;32:348–355. doi: 10.1002/1521-4141(200202)32:2<348::AID-IMMU348>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 89.Hartley S.B., Crosbie J., Brink R., Kantor A.B., Basten A., Goodnow C.C. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature. 1991;353:765–769. doi: 10.1038/353765a0. [DOI] [PubMed] [Google Scholar]
- 90.O'Keefe T.L., Williams G.T., Batista F.D., Neuberger M.S. Deficiency in CD22, a B cell–specific inhibitory receptor, is sufficient to predispose to development of high affinity autoantibodies. J Exp Med. 1999;189:1307–1313. doi: 10.1084/jem.189.8.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nitschke L., Carsetti R., Ocker B., Köhler G., Lamers M.C. CD22 is a negative regulator of B-cell receptor signalling. Curr Biol. 1997;7:133–143. doi: 10.1016/s0960-9822(06)00057-1. [DOI] [PubMed] [Google Scholar]
- 92.Özgör L., Meyer S.J., Korn M., Terörde K., Nitschke L. Sialic acid ligand binding of CD22 and siglec-G determines distinct B cell functions but is dispensable for B cell tolerance induction. J Immunol Baltim Md 1950. 2018;201:2107–2116. doi: 10.4049/jimmunol.1800296. [DOI] [PubMed] [Google Scholar]
- 93.Grewal P.K., Boton M., Ramirez K., Collins B.E., Saito A., Green R.S. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.George M.P., Masur H., Norris K.A., Palmer S.M., Clancy C.J., McDyer J.F. Infections in the immunosuppressed host. Ann Am Thorac Soc. 2014;11:S211–S220. doi: 10.1513/AnnalsATS.201401-038PL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Courtney A.H., Bennett N.R., Zwick D.B., Hudon J., Kiessling L.L. Synthetic antigens reveal dynamics of BCR endocytosis during inhibitory signaling. ACS Chem Biol. 2014;9:202–210. doi: 10.1021/cb400532y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Srinivasan L., Sasaki Y., Calado D.P., Zhang B., Paik J.H., DePinho R.A. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139:573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Orlova N.A., Kovnir S.V., Vorobiev, Gabibov A.G., Vorobiev A.I. Blood clotting factor VIII: from evolution to therapy. Acta Naturae. 2013;5:19–39. [PMC free article] [PubMed] [Google Scholar]
- 98.Qadura M., Waters B., Burnett E., Chegeni R., Hough C., Othman M. Immunoglobulin isotypes and functional anti-FVIII antibodies in response to FVIII treatment in Balb/c and C57BL/6 haemophilia A mice. Haemophilia. 2011;17:288–295. doi: 10.1111/j.1365-2516.2010.02397.x. [DOI] [PubMed] [Google Scholar]
- 99.Orgel K.A., Duan S., Wright B.L., Maleki S.J., Wolf J.C., Vickery B.P. Exploiting CD22 on antigen-specific B cells to prevent allergy to the major peanut allergen Ara h 2. J Allergy Clin Immunol. 2017;139:366–369. doi: 10.1016/j.jaci.2016.06.053. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bednar K.J., Nycholat C.M., Rao T.S., Paulson J.C., Fung-Leung W.-P., Macauley M.S. Exploiting CD22 to selectively tolerize autoantibody producing B-cells in rheumatoid arthritis. ACS Chem Biol. 2019;14:644–654. doi: 10.1021/acschembio.8b01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Theofilopoulos A.N., Kono D.H., Baccala R. The multiple pathways to autoimmunity. Nat Immunol. 2017;18:716–724. doi: 10.1038/ni.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maldonado R.A., LaMothe R.A., Ferrari J.D., Zhang A.-H., Rossi R.J., Kolte P.N. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci. 2015;112:E156–E165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pang L., Macauley M.S., Arlian B.M., Nycholat C.M., Paulson J.C. Encapsulating an immunosuppressant enhances tolerance induction by siglec-engaging tolerogenic liposomes. Chembiochem. 2017;18:1226–1233. doi: 10.1002/cbic.201600702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Victora G.D., Nussenzweig M.C., Germinal Centers Annu Rev Immunol. 2012;30:429–457. doi: 10.1146/annurev-immunol-020711-075032. [DOI] [PubMed] [Google Scholar]
- 105.Shlomchik M.J., Weisel F. Germinal center selection and the development of memory B and plasma cells. Immunol Rev. 2012;247:52–63. doi: 10.1111/j.1600-065X.2012.01124.x. [DOI] [PubMed] [Google Scholar]
- 106.Poe J.C., Smith S.H., Haas K.M., Yanaba K., Tsubata T., Matsushita T. Amplified B lymphocyte CD40 signaling drives regulatory B10 cell expansion in mice. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aruffo A., Kanner S.B., Sgroi D., Ledbetter J.A., Stamenkovic I. CD22-mediated stimulation of T cells regulates T-cell receptor/CD3-induced signaling. Proc Natl Acad Sci. 1992;89:10242–10246. doi: 10.1073/pnas.89.21.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tuscano J., Engel P., Tedder T.F., Kehrl J.H. Engagement of the adhesion receptor CD22 triggers a potent stimulatory signal for B cells and blocking CD22/CD22L interactions impairs T-cell proliferation. Blood. 1996;87:4723–4730. [PubMed] [Google Scholar]
- 109.Santos L., Draves K.E., Boton M., Grewal P.K., Marth J.D., Clark E.A. Dendritic cell-dependent inhibition of B cell proliferation requires CD22. J Immunol. 2008;180:4561–4569. doi: 10.4049/jimmunol.180.7.4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sindhava V.J., Tuna H., Gachuki B.W., DiLillo D.J., Avdiushko M.G., Onami T.M. Bone marrow dendritic cell-mediated regulation of TLR and B cell receptor signaling in B cells. J Immunol. 2012;189:3355–3367. doi: 10.4049/jimmunol.1101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kawasaki N., Rademacher C., Paulson J.C. CD22 regulates adaptive and innate immune responses of B cells. J Innate Immun. 2011;3:411–419. doi: 10.1159/000322375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lee B.L., Barton G.M. Trafficking of endosomal Toll-like receptors. Trends Cell Biol. 2014;24:360–369. doi: 10.1016/j.tcb.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schweighoffer E., Nys J., Vanes L., Smithers N., Tybulewicz V.L.J. TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK. J Exp Med. 2017;214:1269–1280. doi: 10.1084/jem.20161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stamenkovic I., Seed B. The B-cell antigen CD22 mediates monocyte and erythrocyte adhesion. Nature. 1990;345:74–77. doi: 10.1038/345074a0. [DOI] [PubMed] [Google Scholar]
- 115.Hanasaki K., Varki A., Powell L.D. CD22-mediated cell adhesion to cytokine-activated human endothelial cells positive and negative regulation by α2-6-sialylation of cellular glycoproteins. J Biol Chem. 1995;270:7533–7542. doi: 10.1074/jbc.270.13.7533. [DOI] [PubMed] [Google Scholar]
- 116.Engel P., Nojima Y., Rothstein D., Zhou L.J., Wilson G.L., Kehrl J.H. The same epitope on CD22 of B lymphocytes mediates the adhesion of erythrocytes, T and B lymphocytes, neutrophils, and monocytes. J Immunol. 1993;150:4719–4732. [PubMed] [Google Scholar]
- 117.Nitschke L., Floyd H., Ferguson D.J.P., Crocker P.R. Identification of CD22 ligands on bone marrow sinusoidal endothelium implicated in CD22-dependent homing of recirculating B cells. J Exp Med. 1999;189:1513–1518. doi: 10.1084/jem.189.9.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sato S., Miller A.S., Inaoki M., Bock C.B., Jansen P.J., Tang M.L. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: altered signaling in CD22-deficient mice. Immunity. 1996;5:551–562. doi: 10.1016/s1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 119.Wen T., Mingler M.K., Blanchard C., Wahl B., Pabst O., Rothenberg M.E. The pan-B cell marker CD22 is expressed on gastrointestinal eosinophils and negatively regulates tissue eosinophilia. J Immunol. 2012;188:1075–1082. doi: 10.4049/jimmunol.1102222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Eberl G., Lochner M. The development of intestinal lymphoid tissues at the interface of self and microbiota. Mucosal Immunol. 2009;2:478–485. doi: 10.1038/mi.2009.114. [DOI] [PubMed] [Google Scholar]