

Abstract
Background
Cachexia is a paraneoplastic syndrome related with poor prognosis. The tumour micro‐environment contributes to systemic inflammation and increased oxidative stress as well as to fibrosis. The aim of the present study was to characterise the inflammatory circulating factors and tumour micro‐environment profile, as potentially contributing to tumour fibrosis in cachectic cancer patients.
Methods
74 patients (weight stable cancer n = 31; cachectic cancer n = 43) diagnosed with colorectal cancer were recruited, and tumour biopsies were collected during surgery. Multiplex assay was performed to study inflammatory cytokines and growth factors. Immunohistochemistry analysis was carried out to study extracellular matrix components.
Results
Higher protein expression of inflammatory cytokines and growth factors such as epidermal growth factor, granulocyte–macrophage colony‐stimulating factor, interferon‐α, and interleukin (IL)‐8 was observed in the tumour and serum of cachectic cancer patients in comparison with weight‐stable counterparts. Also, IL‐8 was positively correlated with weight loss in cachectic patients (P = 0.04; r = 0.627). Immunohistochemistry staining showed intense collagen deposition (P = 0.0006) and increased presence of α‐smooth muscle actin (P < 0.0001) in tumours of cachectic cancer patients, characterizing fibrosis. In addition, higher transforming growth factor (TGF)‐β1, TGF‐β2, and TGF‐β3 expression (P = 0.003, P = 0.05, and P = 0.047, respectively) was found in the tumour of cachectic patients, parallel to p38 mitogen‐activated protein kinase alteration. Hypoxia‐inducible factor‐1α mRNA content was significantly increased in the tumour of cachectic patients, when compared with weight‐stable group (P = 0.005).
Conclusions
Our results demonstrate TGF‐β pathway activation in the tumour in cachexia, through the (non‐canonical) mitogen‐activated protein kinase pathway. The results show that during cachexia, intratumoural inflammatory response contributes to the onset of fibrosis. Tumour remodelling, probably by TGF‐β‐induced transdifferentiation of fibroblasts to myofibroblasts, induces unbalanced inflammatory cytokine profile, angiogenesis, and elevation of extracellular matrix components (EMC). We speculate that these changes may affect tumour aggressiveness and present consequences in peripheral organs.
Keywords: Tumour micro‐environment, Cachexia, Epithelial–mesenchymal components, Fibrosis
Introduction
Cachexia is a multifactorial syndrome that contributes to poor prognosis and is characterised by multiple physical and metabolic alterations, including muscle and adipose tissue wasting, anorexia, negative energy balance, and inflammatory response.1, 2 Cancer‐associated cachexia (CAC) may be found in 30–80% of cancer patients.3 Clinically, CAC consists of chronic systemic inflammation and catabolism, leading to reduction of quality of life, diminished treatment tolerance, tumour metabolic reprograming, and, consequently, decreased survival.4, 5, 6
Chronic inflammation has been reported in the majority of patients with CAC7 and causes disruption of morphofunctional aspects of many tissues, such as the brain,8, 9 the adipose tissue,10, 11, 12, 13 and the muscle,14, 15 while also affecting tumour biology.5, 16 The secretory profile of the tumour may be associated with cachexia–muscle wasting, as proposed by de Matos‐Neto et al..5 On the other hand, several studies show that the presence of cachexia is not directly or exclusively related to greater tumour burden and/or with metastasis5, 17; nonetheless, it has been recently shown that metastatic cancer is associated with a higher risk for concomitant cachexia.18, 19
In the last years, data from clinical studies have implicated pro‐inflammatory cytokines in the pathogenesis of CAC. In previous studies, we demonstrated that the tumour micro‐environment and the adipose tissue contribute to systemic pro‐inflammatory profile and to the alterations of immunomodulatory function in cachectic patients.5, 10, 12, 20 Zhang et al. (2017) suggested that tumour‐derived exosomes may be responsible for the induction of cachexia.21 A plethora of studies show that inflammatory mediators, including tumour necrosis factor‐α (TNF‐α),22 interferon‐γ (IFN‐γ),23 interleukin (IL)‐1 and IL‐6, myostatin,24 and transforming growth factor β (TGF‐β)25 are involved in the etiology of the loss of skeletal muscle and adipose tissue mass. It is known that hypoxia contributes to the up‐regulation of inflammatory response, by increasing the production of reactive oxygen species (ROS), which in turn cause DNA damage, leading to increased tumour aggressiveness.26 Given the clinical significance of tumour micro‐environment rearrangement, the expression of extracellular matrix components (EMCs), including collagen, α‐smooth muscle actin (α‐SMA), and vimentin, as well as of other soluble growth factors, such as TGF‐β, epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), cytokines (IL‐8, IL‐1β, TNF‐α, and IL‐15), and also hypoxia as mediated by hypoxia‐inducible factor‐1α (HIF‐1α),27 could all play an important role in CAC.
A recent study showed that up‐regulation of HIF‐1α and TGF‐β pathways contributes to cell survival and chemoresistance in colorectal cancer.28 In line with these observations, we previously demonstrated that the adipose tissue, while playing an important role in systemic inflammation by secreting cytokines and chemokines, undergoes extensive fibrosis in CAC29, 30 and that one such effect is partly mediated by TGF‐β, which causes the rearrangement of extracellular matrix components and leads to severe tissue architecture disruption in cancer patients with cachexia.10, 29 TGF‐β signalling promotes fibroblast to myofibroblast transdifferentiation by inducing the expression of α‐SMA, a specific myofibroblast marker. In addition, TGF‐β contributes to EMT,31, 32 regulation of gut microbiota,33 and angiogenesis by a reciprocal regulatory loop with secreted protein acidic and rich in cysteine (SPARC),34, 35 which is also involved in neo‐angiogenesis.36 Interestingly, high levels of TGF‐β are associated with the proportion of tumour‐immune infiltrating cells, as mediated by p38 mitogen‐activated protein kinase (MAPK) signalling, what represents a novel putative mechanism for immune tolerance.37 However, it remains unclear whether the tumour‐associated inflammatory micro‐environment contributes to fibrosis in cachectic patients.
The aim of the present study was to assess changes in tumour micro‐environment inflammatory profile in cachectic patients and its association with fibrosis, through TGF‐β signalling. We report induction of fibrosis by TGF‐β activation in the tumour of cachectic patients through the (non‐canonical) MAPK pathway.
Materials and methods
Patient recruitment, ethical aspects, and inclusion/exclusion criteria
Enrolment of Brazilian colorectal cancer patients occurred between 2013 and 2017 at the Surgical Clinic of the University Hospital or the Santa Casa de Misericordia Hospital, after signature of the informed consent. This study was approved by the Ethics Committee on Research Involving Human Subjects of the University of the São Paulo Biomedical Sciences Institute (CEP 1151/13 CAAE n 5493116.6.0000467) and by the University Hospital (CEP 1390/14 CAAE n 54930116.6.3001.0076) and is in accordance with the Declaration of Helsinki. The exclusion criteria adopted included liver or kidney failure, acquired immunodeficiency syndrome, chronic inflammatory processes not related to cachexia, chemotherapy treatment (at the time or in the past 5 years), chronic anti‐inflammatory therapy, and body mass index (BMI) > 29.9 kg/m2. The flow chart of patient enrolment is summarised in Figure 1.
Figure 1.
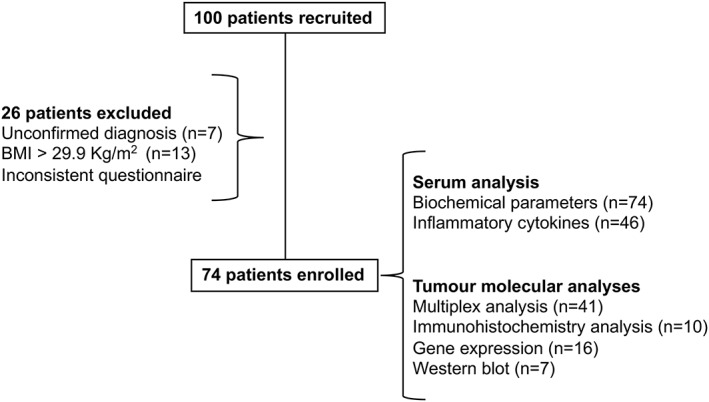
Flow chart of patient enrolment.
Cachexia classification and questionnaire assessments
The presence of cachexia was clinically assessed by employing the criteria proposed by Evans et al..6 The previous body mass was reported by the patients 6 months prior to diagnosis, and current anthropometric data were collected before the surgery. Also, quality of life and anorexia were evaluated by validated questionnaires, that is, European Organization for Research and Treatment of Cancer (EORTC QLQ‐C30)38 and Functional Assessment of Anorexia/ Cachexia Therapy Anorexia/Cachexia Subscale/ European Society for Clinical Nutrition and Metabolism (FAACT‐ESPEN), respectively.39 The patients were divided into two groups: weight‐stable cancer (WSC) and cancer cachexia (CC).
Tumour biopsies
Tumour biopsies were obtained during the surgical procedure for tumour resection. Samples were immediately snap frozen in liquid nitrogen and stored at −80°C, until analysis.
Blood collection and serum analysis
Approximately 20 mL of blood were collected in pre‐surgical fast at the hospital and centrifuged at 1000 g (RCF) for 15 min at 4°C to obtain serum and plasma. Aliquots of both were stored at −80°C for posterior analysis. All biochemical analyses were performed in the automatic LABMAX 240® equipment (Labtest, Lagoa Santa, Brazil) employing commercial kits for albumin (Cat#19), ultra‐sensitive C‐reactive protein (Cat#3331), high‐density lipoprotein cholesterol (Cat#98), triglycerides (Cat#87), low‐density lipoprotein (Cat#111), total cholesterol (Cat#76), and glucose (Cat#133). Haemoglobin concentration was obtained from the hospital records, before the surgery.
Gene expression
Total RNA was extracted with TRIzol® reagent (Invitrogen, Carlsbad, CA), following the manufacturer's recommendations. The concentration of total RNA was assessed with the Biotek® SynergyH1 spectrophotometer (Fisher Scientific, Biotek, Winooski, USA). The cDNA was obtained in a thermocycler (Veriti® Thermal Cycler; Applied Biosystems, Foster City, CA) using a high‐capacity cDNA reverse transcription kits (Invitrogen No. 4375575). Transcript levels of genes were evaluated by real‐time PCR with SYBR Green (Invitrogen) in Quant Studio™ 12K Flex real‐time PCR (Applied Biosystems Carlsbad, CA, USA). Primers employed for the amplification are described in Table 1. Gene expression was normalised against the housekeeping gene β2‐microglobulin (B2m). Relative gene expression was calculated by the comparative 2‐ΔΔCT method, described by Livak and Schmittgen.40
Table 1.
Human primer sets used for qRT–PCR in tumours of patients
| Extracellular matrix components | ||
|---|---|---|
| Gene | Forward | Reverse |
| B2m | GAATTGCTATGTGTCTGGGT | TCTTCAAACCTCCATGATGCT |
| COL1A | AGAGGTTTCCCTGGCGA | ACCAGCATCACCCTTAGCA |
| COL3A | CTCAGGGTGTCAAGGGT | CAGGGTTTCCATCTCTTCCA |
| MMP2 | TGAGACCGCCATGTCCA | TCGCACACCACATCTTTCC |
| MMP9 | ACTACTGTGCCTTTGAGTCC | GCCAGTACTTCCCATCCT |
| TGF‐β3 | AACAATTCCTGGCGATACCT | GTAGTGAACCCGTTGATGTC |
| Vimentin | AAAGGAACCAATGAGTCCCT | GCAGGTCTTGGTATTCACGA |
| HIF‐1α | CTCATCAGTTGCACTCC | ATCCAAATCACCAGCATCCA |
B2m, β2‐microglobulin; COL1A, type I collagen; COL3A, type III collagen; HIF‐1α, hypoxia‐inducible factor‐1α; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; qRT–PCR, Real‐Time Quantitative Reverse Transcription PCR; TGF‐β3, transforming growth factor‐β3.
Histological analysis and immunohistochemistry
The slides stored at the Pathology Service (SAP‐HU) were reviewed by the pathologist in regard to tumour staging, adopting the conventional tumour, node, and metastasis (TNM) staging system (seventh edition, AJCC American Joint Committee Against Cancer, 2010).41 For the histological analyses, all tissue samples were formalin fixed and paraffin embedded. The 5 μm sections were mounted onto slides (Starfrost® Knittel Glass) and stained with picrosirius red; images were obtained in a photomicroscope equipped with a Nikon DP72 CCD digital camera (Olympus BX51; Olympus Optical Co., Tokyo, Japan). For the immunohistochemical analysis, the slides were hydrated, and antigen retrieval was carried out in 10 mM of citrate buffer (pH 6.0) or Tris–EDTA buffer (pH 9.0) at 95°C for 20 min. Briefly, after blocking, sections were incubated overnight (4°C) with primary antibodies: anti‐collagen type III rabbit polyclonal antibody (1:100, Cat#19369; Rockland Immunochemicals, Pottstown, PA, EUA), α‐SMA (1:100, Ab5694; Abcam, Cambridge, MA, USA), and CD34 (Cat#3569; Cell Signalling Technology Inc. Danvers, Massachusetts, EUA), in a humidified chamber. All reactions were carried out employing Histostain‐Plus, IHC kit, and HRP broad spectrum® (Life Technologies, Brazil), following the manufacturer's instructions. The samples were developed using DAB (diaminobenzidine) solution, and tissue sections were counterstained with Mayer's haematoxylin (n = 05 for all study groups). The images were captured with Image‐Pro Plus software v.6.0 (10 images per slide/20×) and handled for better brightness and contrast in Adobe Photoshop CS6 software v.13.0 × 64 and finally quantified using the ImageJ software. Positive and negative controls were performed (Supporting Information, Figure S1).
Protein expression assessment by Luminex technology
Approximately 100 mg of the tumour samples were homogenised in 500 μL of ice‐cold extraction protein buffer (10 mM Tris base, 0.01 mM EDTA, 0.1 mM Sodium chloride, and 1% Triton X‐100), to which a protease inhibitor cocktail was added (one tablet per 50 mL extraction buffer) (Roche Diagnostics, Brazil). The homogenate was then centrifuged at 18 000 g for 40 min at 4°C, and the supernatant was stored in aliquots at −80°C. The Human Cytokine Magnetic Bead Panel Milliplex® MAP (HCYTMAG‐60 K‐PX29; Merck Millipore, Massachusetts, EUA) was employed to detect the following cytokines and growth factors: EGF, granulocyte colony‐stimulating factor (G‐CSF), granulocyte–macrophage colony‐stimulating factor (GM‐CSF), VEGF, IL‐12p40, IL‐12p40, IL‐12p70, and IL‐8. MAPK/mitogen‐activated protein kinase was analysed using a 10‐plex Signalling Magnetic Bead Panel (Cat#48‐660MAG; Millipore, Massachusetts, EUA) measuring signal transducer and activator of transcription 1 (STAT‐1) (Tyr701), c‐Jun N‐terminal kinase (JNK) (Thr183/Tyr185), mitogen‐activated protein kinase 1 (Ser217/221), mitogen‐activated and stress‐activated protein kinase 1 (MSK1; Ser212), ATF2 (Thr71), p53 (Ser15), and p38 MAPK (Thr180/Tyr182). The TGF‐β plex magnetic bead panel Milliplex MAP (TGFBMAG‐64K‐03; Merck Millipore, Massachusetts, EUA) was adopted to detect the protein content of the three TGF‐β isoforms, and the Myokines Magnetic Bead Panel Milliplex (HMYOMAG‐56K) was employed to detect SPARC, oncostatin M, and fibroblast growth factor 21. The analysis was performed following the manufacturer's recommendations. The Luminex 200™ instrument with an xMAP® technology system and xPONENT® acquisition software was employed to capture component detection. Milliplex Analyst 5.1 software was adopted integrating data acquisition and analysis.
Western blotting
The isolated total protein samples, as previously described, were used to detect SMAD2 (Cat#3122; Cell Signalling), phosphoSMAD2 (Cat#3108; Cell Signalling), SMAD3 (Cat#9523; Cell Signalling), and cell signalling and phosphoSMAD3 (Cat#9520; Cell Signalling) by Western blotting. After extraction, 80 μg of proteins was separated by 4–12% sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Protran®,GE Healthcare Biosciences). After blocking with 5% non‐fat milk in Tris‐buffered saline and Tween 20, the membranes were incubated with primary antibody at 4°C overnight (dilution 1:1000). Antibody for β‐actin (ab8227) was employed as the control. The washed membranes were incubated with rabbit anti‐goat IgG secondary antibody (dilution 1:2000) for 2 h at room temperature. The protein signals were obtained using an enhanced chemiluminescence detection system. The images obtained were quantified using the ImageJ software.
Statistical analysis
Data are expressed as mean ± standard deviation or median [first quartile; third quartile]. Initially, preliminary analysis was carried out to ensure that the assumptions of normality or homoscedasticity were not violated (D'Agostino–Pearson omnibus test, Shapiro–Wilk test, and Kolmogorov–Smirnov test). Student's t‐test or Mann–Whitney U test was applied for parametric or non‐parametric data (PRISM software 6.0; MAC, GraphPad Software, San Diego, CA, USA, www.graphpad.com). Correlation analysis was performed by Pearson's correlation coefficient analysis. χ 2 test was used for qualitative analysis. The significance level was set at P < 0.05.
Results
Characteristic of patients
Table 2 illustrates the characteristics of the patients. A total of 100 patients were recruited to this study, and twenty‐six patients were excluded. Exclusion occurred because of unconfirmed diagnosis after pathology screening (n = 7). Obese patients with BMI greater than 29.9 kg/m2 (n = 13) were excluded, as they were considered to be under chronic systemic inflammatory conditions (a confounding factor to cachexia‐derived inflammation evaluation) and also patients for whom we retrieved inconsistent data from the questionnaires (n = 6). A total of 74 patients were recruited and participated in the study (WSC: n = 31 and CC: n = 43; 59 colon and 15 rectal cancer patients). The groups showed homogenous distribution for age (P = 0.337) and height (P = 0.941). Previous body mass as informed by the patients (6 months prior to the enrolment) showed no difference between the groups (P = 0.392), while current body mass indicated significant weight loss (P = 0.011) difference in CC and WSC, in accordance with the criteria proposed by Evans et al. .6 BMI (kg/m2) of CC was also significantly lower than that of WSC (P = 0.001). C‐reactive protein plasma content was higher in CC (P = 0.039), indicating the presence of systemic inflammation. Additionally, serum haemoglobin levels of the cachectic group were significantly lower, when compared with WSC (P < 0.0001). Serum albumin concentration was decreased in CC (P = 0.069). Finally, lipid profile analysis showed lower serum levels of triglycerides (P = 0.031) and high‐density lipoprotein cholesterol (P = 0.012) in CC in relation to WSC. There was no significant difference in total cholesterol and glucose plasma concentration (P = 0.165 and P = 0.633, respectively). The presence of cachexia was paralleled by diminished quality of life, as measured by the QLQ‐C30 questionnaire (P = 0.038), and associated with anorexia (P = 0.002). Additionally, χ 2 tests show absence of correlation between T stage (P = 0.835), N stage (P = 0.217), M stage (P = 0.929), and TNM stage (P = 0.548) for the tumours of both groups. Finally, there was no significant difference in tumour dimensions between groups (P = 0.229).
Table 2.
General characteristic of patients
| Patient characteristics | WSC | CC | P |
|---|---|---|---|
| Gender (male/female) | 13/18 | 20/23 | — |
| Ethnicity | White, n = 17 | White, n = 32 | |
| Black, n = 1 | Black, n = 1 | ||
| Mixed, n = 2 | Mixed, n = 6 | ||
| Asian, n = 3 | Asian, n = 2 | ||
| Non‐declared, n = 8 | Non‐declared, n = 3 | ||
| Age (years) | 61.1 ± 2.09, n = 30 | 64 ± 2.04, n = 42 | 0.337 |
| Height (m) | 1.64 ± 0.016, n = 30 | 1.64 ± 0.014, n = 42 | 0.941 |
| Previous body mass (kg) | 68.75 ± 1.871, n = 30 | 71.33 ± 2.157, n = 42 | 0.392 |
| Current body mass (kg) | 68.17 ± 1.92, n = 30 | 60.78 ± 1.98,* n = 42 | 0.011 |
| ΔBody mass (kg) | 0 [0.0; 0.0] | 9.65 [6.45; 12.25]* | <0.0001 |
| ΔBody mass (%) | 0 [0.0.0; 0.0] | 14.29 [8.48; 17.67]* | <0.0001 |
| BMI (kg/m2) | 25.11 ± 0.503, n = 30 | 22.35 ± 0.572*, n = 42 | 0.001 |
| Biochemical parameters | |||
| C‐reactive protein (mg/dL) | 4.6[2.27; 11.25], n = 29 | 10.77[4.31; 12.64]*, n = 42 | 0. 039 |
| Haemoglobin (g/dL) | 13.73 ± 0.472, n = 27 | 11.35 ± 0.332*, n = 37 | <0.0001 |
| Albumin (g/dL) | 3.824 ± 0.183, n = 29 | 3.382 ± 0.153, n = 42 | 0. 069 |
| Total cholesterol (mg/dL) | 179.2 ± 10.5, n = 28 | 160.3 ± 8.472, n = 43 | 0. 165 |
| HDL cholesterol (mg/dL) | 39.45 ± 2.66, n = 29 | 31.81 ± 1.672*, n = 42 | 0. 012 |
| Triglycerides (mg/dL) | 126[98.5; 164.5], n = 29 | 88[70; 153]*, n = 43 | 0. 031 |
| Glucose (mg/dL) | 104[88.5; 134.5], n = 29 | 102[85; 130], n = 43 | 0. 633 |
| Questionnaire assessments | |||
| Quality of life (QLQC‐30) | 66.67 [66.67; 100], n = 27 | 66.67 [52.08; 83.33]*, n = 36 | 0. 038 |
| Anorexia score (FAACT‐ESPEN) | 36.43 ± 0.651, n = 28 | 32.2 ± 1.00*, n = 41 | 0. 002 |
| Tumour pathology | |||
| Initial tumour stage (I–II) | 11 (35.48%) | 17 (42.50%) | 0.548 |
| Final tumour stage (III–IV) | 20 (64.52%) | 23 (57.50%) | |
| T classification | |||
| T1–T2 | 14 (45.16%) | 20 (47.62%) | 0.835 |
| T3–T4 | 17 (54.84%) | 22 (52.38%) | |
| N classification | |||
| N0 | 21 (67.74%) | 23 (53.49%) | 0.217 |
| N1–N2 | 10 (32.26%) | 20 (46.51%) | |
| M classification | |||
| M0 | 29 (93.55%) | 40 (93.02%) | 0.929 |
| M1 | 2 (6.45%) | 3 (6.98%) | |
| Tumour size (cm) | 3.5 [2.5; 6], n = 30 | 4.6 [2.85; 7.13], n = 43 | 0.229 |
BMI, body mass index; CC, cancer cachexia; HDL, high‐density lipoprotein; WSC, weight‐stable cancer.
Data expressed as mean ± standard deviation or as median [first quartile; third quartile]. χ 2 test was employed for tumour staging. Sample number (n).
Significant difference between the groups was tested using unpaired t‐test and Mann–Whitney U test (P < 0.05).
Inflammatory cytokines and growth factors may contribute to fibrosis
Circulating IL‐8 and IFN‐α content was significantly higher in CC as compared with WSC (Figure 2A; P = 0.002 and P = 0.048, respectively). The circulating IL‐12p40 and IL‐12p70 levels did not differ among groups (Figure 2A; P = 0.221 and P = 0.281, respectively). In addition, no statistically significant alteration was detected in terms of serum growth factor content of EGF and VEGF between groups (Figure 2B; P = 0.740 and P = 0.418, respectively). The G‐CSF and GM‐CSF serum concentration was higher in CC relative to WSC, as illustrated in Figure 2B (P = 0.020 and P = 0.021).
Figure 2.
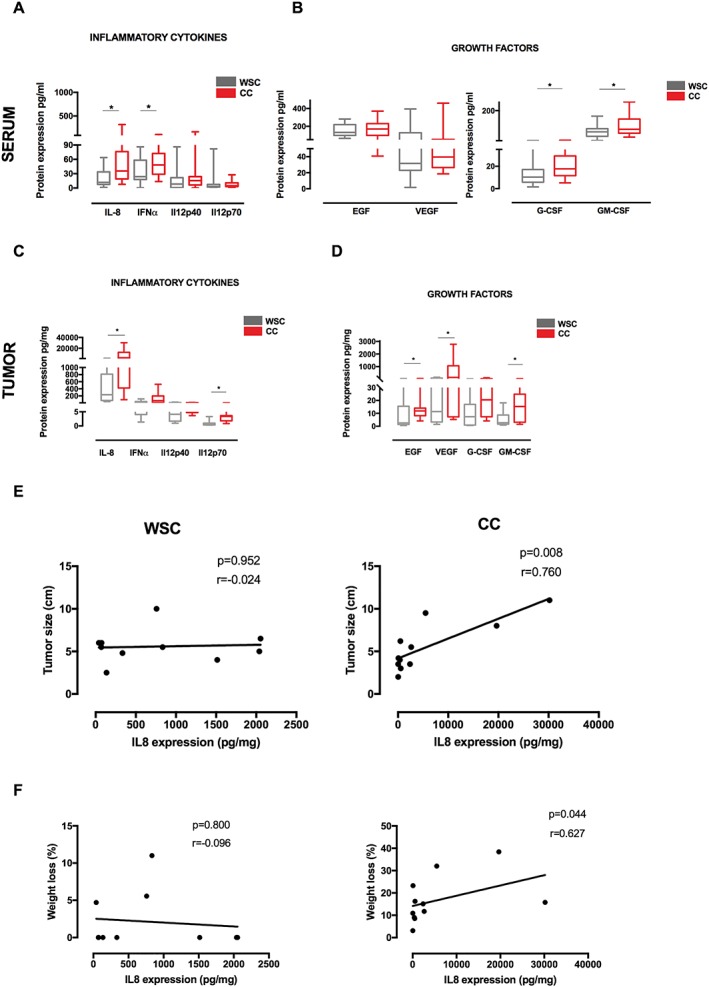
Inflammatory cytokines and growth factors in cachectic patients. Data were expressed as mean ± standard deviation or as median [first quartile; third quartile]. *Significant difference between the groups was tested using Mann–Whitney U test (P < 0.05). Pearson correlation analysis was employed between groups. (A) Protein expression of circulating inflammatory cytokines in serum of patients. (B) Protein expression of growth factors in serum of patients. Protein concentration in pg/mL of total protein. (C) Protein expression of inflammatory cytokines in tumour biopsies of patients. (D) Protein expression of growth factors in tumour biopsies of patients. Protein concentration in pg/mg of total protein. (E) Correlation between tumour size and interleukin (IL)‐8 expression in tumour. (F) Correlation between percentage of weight loss and IL‐8 expression in tumour. EGF, epidermal growth factor; G‐CSF, granulocyte colony‐stimulating factor; GM‐CSF, granulocyte–macrophage colony‐stimulating factor; IFN‐α, interferon‐α; VEGF, vascular endothelial growth factor. Sample number in serum: weight‐stable cancer (WSC) (n = 18–23) and cancer cachexia (CC) (n = 18–23). Sample number in tumour analysis: WSC (n = 8–10) and CC (n = 9–11).
Nevertheless, higher expression of IL‐8 and IL‐12p70 was found in the tumours of CC compared with WSC (Figure 2C; P = 0.036 and P = 0.001, respectively), while IFN‐α concentration showed a tendency to be higher in the tumour of CC relative to WSC (Figure 2C; P = 0.062). Analysis of IL‐12p40 protein content in the tumour presented no differences between groups, as shown in Figure 2C (P = 0.258). Moreover, expression of EGF, VEGF, and GM‐CSF was increased in the tumours of the cachectic group relative to WSC (Figure 2D; P = 0.04, P = 0.04, and P = 0.05). No statistically significant differences were observed between groups in terms of G‐CSF content (Figure 2D; P = 0.093).
Considering that weight loss is the most conspicuous clinical parameter in cachexia, we evaluated its correlation with the tumour cytokine expression (Figure 2E and 2F). We found that only IL‐8 was positively correlated with weight loss in cachectic patients (P = 0.044; r = 0.627), and this cytokine was also associated with the tumour size (P = 0.006; r = 0.760). The literature demonstrated that enhanced expression of IL‐8 is associated with cachexia progression in pancreatic cancer42 and with induction of fibrosis in several types of tumours.36, 43
Fibrosis is induced in the tumour micro‐environment of cachectic patients
To further investigate the relationship between inflammation and fibrosis, we examined well‐defined markers of fibrosis, including collagen, vimentin, matrix metalloproteinases 2 and 9, and α‐SMA in the tumour of cachectic patients and of WSC counterparts. Gene expression of COL1A and COL3A was significantly increased in CC, relative to WSC (Figure 3A; P = 0.03 and P = 0.05, respectively). Vimentin (a type III intermediate filament related to cellular motility) expression was increased in the tumour of CC (Figure 3A; P = 0.02), and this feature was shown to be correlated with the numbers of activated fibroblasts in cachexia.10 Recently, the metastatic potential of cancer cell has been correlated with cachexia development.19 To indirectly investigate whether invasion and migration potential of tumour cells was different in cachectic patients, we also analysed gene expression of matrix metalloproteinases 2 and 9 but failed to find distinction between the groups (Figure 3A; P = 0.959 and P = 0.328, for CC and WSC, respectively). TGF‐β signalling pathway can induce the expression of ECM components.44 We hereby report the gene expression of TGF‐β3 to be increased in the tumour of CC (Figure 3A; P = 0.039), as compared with WSC. Consistent with our previous findings on adipose tissue fibrosis,10, 29 collagen deposition was also higher in the tumour of CC, compared with WSC, as assessed by immunohistochemistry staining of type III collagen (Figure 3B and 3C; P = 0.0006) and by picrosirius staining (Figure 3B, representative data). Immunohistochemistry for α‐smooth muscle actin (P < 0.0001) showed this protein to present higher expression in the tumours of cachectic patients, as illustrated by Figure 3B and 3C. In the present study, the gene expression of proteins of the EMC was unrelated with TNM stage (Supporting Information, Figure S2). These results suggest that alterations in myofibroblasts are likely to be involved in fibrosis within tumour micro‐environment of cachectic patients under influence of the persistent inflammatory status.
Figure 3.
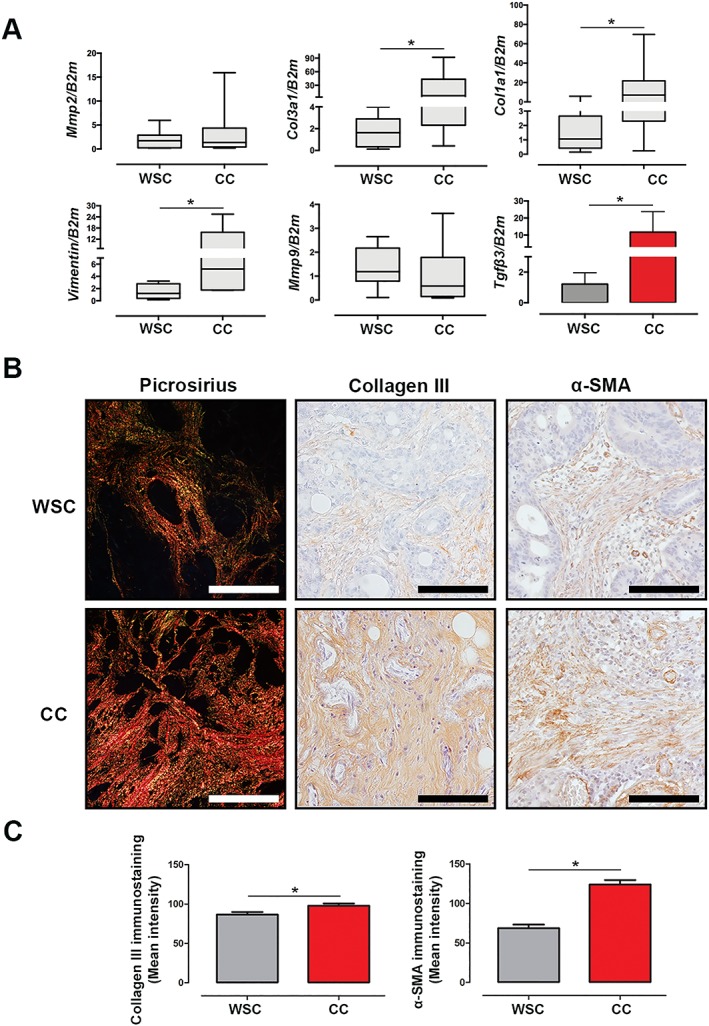
Fibrosis is induced in the tumour of cachectic patients. Data were expressed as mean ± standard deviation or as median [first quartile; third quartile]. *Significant difference between the groups was tested using unpaired t‐test and Mann–Whitney U test (P < 0.05). (A) Gene expression of components to extracellular matrix. B2m, β2‐microglobulin; COL1A, type I collagen; COL3A, type III collagen; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; TGF‐β3: transforming growth factor‐β3. Weight‐stable cancer (WSC) (n = 7–8) and cancer cachexia (CC) (n = 7–8). (B, C) Representative picrosirius red staining (n = 1 per group) and immunohistochemistry for collagen III (WSC: n = 5; CC: n = 5) and α‐smooth muscle actin (α‐SMA) (WSC: n = 5; CC: n = 5) in the tumour sample. Scale bar: 112.0 μm.
The interaction between mitogen‐activated protein kinase and transforming growth factor‐β signalling in fibrosis in cachexia
In order to establish the contribution of TGF‐β activation for tumour remodelling in CC, we evaluated the protein expression of the three isoforms to TGF‐β in the tumour biopsies. TGF‐β pathway was up‐regulated in cachexia. Protein expression analysis revealed that TGF‐β1, TGF‐β2, and TGF‐β3 expression was increased in the tumour of CC in relation to WSC, as shown in Figure 4A (P = 0.003, P = 0.050, and P = 0.047, respectively). Also, there was no difference between the protein expression of TGF‐β isoforms and tumour stage (Supporting Information, Figure S3). Moreover, TGF‐β signalling can be activated both by canonical SMAD‐dependent pathway and by non‐canonical signalling pathways, such as ERK1/2, JNK1/2, and p38 MAPKs.25 Proteins of the canonical TGF‐β pathway (SMAD2 and SMAD3 activation) showed no differences between the groups (Figure 4B; P = 0.628 and P = 0.400, respectively). In addition to provoking activation of SMAD, TGF‐β also modulates the activity of MAPK pathway, an important signalling pathway that plays a central role in the activation of muscle catabolism in animal models of CC.25 The protein expression of factors related to MAPK pathway was altered in the tumour of cachectic patients in relation to WSC: p38, JNK, and MEK1 were increased (P = 0.025, P = 0.050, and P = 0.024, respectively), while MSK1 protein expression did not show alterations between the groups (Figure 4C). Protein concentration of p53, an important tumour suppressor, showed a tendency to be significantly lower in the tumour of CC (Figure 4C; P = 0.057). These data demonstrate that TGF‐β signalling occurs through MAPK pathway activation in the tumour of cachectic patients and that this could be associated with tumour cell proliferation, angiogenesis, and protection against apoptosis and may also interfere with resistance to chemotherapy, radiotherapy, and other targeted therapies.
Figure 4.
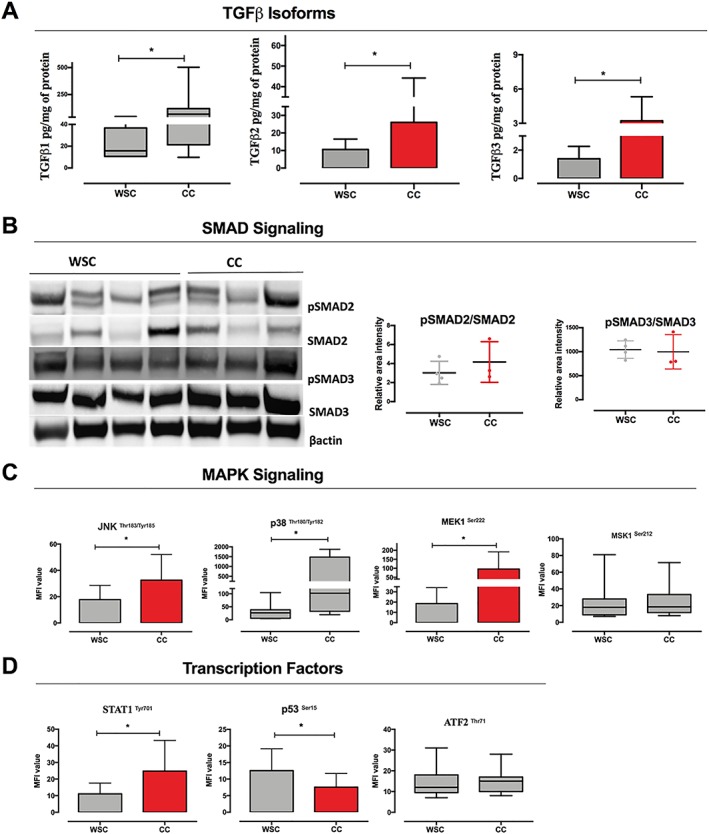
Interaction between mitogen‐activated protein kinase (MAPK) and transforming growth factor (TGF)‐β signalling in cachexia. Data were expressed as mean ± standard deviation or as median [first quartile; third quartile]. *Significant difference between the groups was tested using unpaired t‐test and Mann–Whitney U test (P < 0.05). Protein concentration in pg/mL of total protein or medium fluorescent intensity (MFI). (A) Protein expression of TGF‐β isoforms [weight‐stable cancer (WSC): n = 7–10; cancer cachexia (CC): n = 7–10]. (B) Western blot analysis of phosphoSMAD2 (pSMAD2)/SMAD2 and phosphoSMAD3 (pSMAD3)/SMAD3 and band intensity (densitometry), and data are mean ± standard deviation (WSC: n = 4; CC: n = 3). (C) Relative levels of MAPK signalling components (WSC: n = 7–10; CC: n = 8–10; MFI value). (D) Relative levels of transcription factors related to MAPK signalling (WSC: n = 7–10; CC: n = 8–10; MFI value).
Hypoxia may regulate the vascular permeability mediated by transforming growth factor‐β/mitogen‐activated protein kinase pathway
To understand the role of TGF‐β in regulating high VEGF expression in the tumour of cachectic patients, we further investigated intratumoural angiogenesis. The micro‐environment of the tumours obtained from cachectic patients showed higher presence of CD34‐labelled cells in comparison with WSC, as shown in Figure 5A (P = 0.003). HIF‐1α pathway has been shown to be a master regulator of angiogenesis by up‐regulating angiogenic factors, such as VEGF. We identify higher gene expression of HIF‐1α in tumour of cachectic patients as compared with WSC patients (P = 0.050). Besides, SPARC, an important protein involved in tumour cell survival, endothelial barrier function, and associated with hypoxia, was found to be more abundant in the tumour of cachectic patients (Figure 5B; P = 0.002). Moreover, oncostatin M, a potent tumour promoter, was increased in CC, in relation to WSC (P = 0.017). We failed to demonstrate differences in fibroblast growth factor 21 protein expression between the groups (Figure 5B; P = 0.591). These data suggest that hypoxia in cachexia could modulate TGF‐β signalling. The angiogenesis resulting from increased expression of HIF‐1α and VEGF plays an important role in tumour aggressiveness in cachexia. Tang et al. (2018) described that the HIF‐1α/TGF‐β activity is involved in resistance to therapy in colorectal cancer and proposed the analysis of this gene signature for predicting patient's outcome, as an alternative clinical approach.28 We propose that further analysis of HIF‐1α/TGF‐β pathways might be helpful in cachexia treatment.
Figure 5.
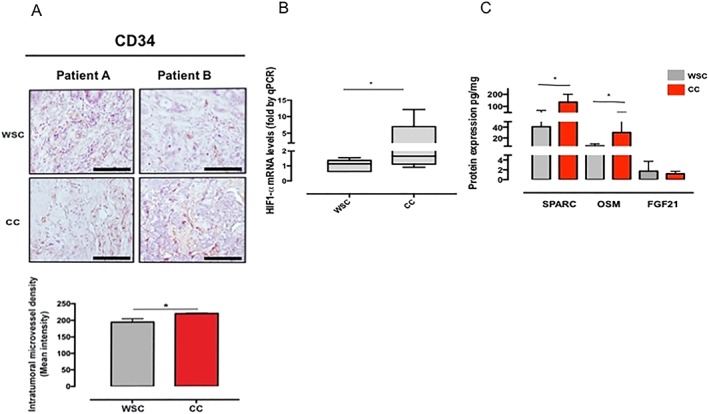
Angiogenesis mediated by hypoxia in the tumour of cachectic patients. Data were expressed as mean ± standard deviation or as median [first quartile; third quartile]. *Significant difference between the groups was tested using unpaired t‐test and Mann–Whitney U test (P < 0.05). Protein concentration expressed to medium fluorescent intensity (MFI). (A) Immunohistochemical staining for intratumoural microvessel density [CD34+; weight‐stable cancer (WSC): n = 4 and cancer cachexia (CC): n = 4]. (B) Gene expression of hypoxia‐inducible factor‐1α (HIF1‐α) (WSC: n = 7 and CC: n = 7). (C) Protein expression of myokines in tumour micro‐environment of cachectic patients (n = 7–8). FGF21, fibroblast growth factor 21; OSM, oncostatin M; qRT‐PCR, Real‐Time Quantitative Reverse Transcription PCR; SPARC, secreted protein acidic and rich in cysteine. Scale bar: 110.0 μm.
Discussion
Recent studies based on autopsy findings showed that cachexia represents the ultimate cause of death of approximately 20% of cancer patients.17, 45 Our own efforts demonstrated that cachexia is marked by aberrant fibrosis process in human adipose tissue, further supporting previous findings regarding unbalanced inflammatory response in several tissues, including adipose tissue, liver, brain, and tumour.5, 10, 29, 46 We also found that the tumours obtained from cachectic and WSC patients show differences in classical cytokine expression profile, such as TNF‐α, IL‐6, and IL1β, and postulated such differences to interfere with the development and progression of cachexia. The same study provided evidence for different macrophage phenotype percentages in the tumours of CC and WSC patients.5 Our present findings point out to higher expression (as compared with WSC) of inflammatory factors in the serum and tumour of cachectic patients, for example, IL‐8, IFN‐α, GM‐CSF, EGF, VEGF, and G‐CSF, all of which have been reported to contribute to important molecular processes including angiogenesis, inflammation, tumour malignancy, and immune evasion47, 48, 49 IL‐8 has been described to be important marker in cachexia development.42
High expression of IL‐8 and VEGF is associated with induction of angiogenesis50 and progression of prostate cancer,48 ovarian cancer,49 and melanoma,51 and thus, these factors have been recently proposed as novel players in inflammation and tumour fibrosis.47 In addition, Chen et al.52 described that the GM‐CSF acts directly in epithelial–mesenchymal transitions (EMT) by inducing MAPK activation in a murine colon cancer cell line. Unfortunately, the process of epithelial–mesenchymal transition and fibrosis in tumour micro‐environment of cachectic patients remains unclear, as well as the contribution of this process for the development of the cachexia. To further investigate the relationship between inflammation up‐regulation and fibrosis, we examined well‐defined markers of fibrosis, such as vimentin, α‐SMA, and collagen. We report increased content of mesenchymal cell markers in the tumour of cachectic patients. High expression of α‐SMA is generally considered a feature of myofibroblasts rather than tumour‐associated fibroblasts or cancer‐associated fibroblasts.53 In addition, these morphological and functional alterations lead to aberrant ECM production and to remodelling, characterised by fibrosis.53, 54 The presently obtained results suggest alterations in the tumour‐associated fibroblasts in cachectic patients, thus allowing the proposition that EMT changes may markedly contribute to the development of resistance to treatment in the patients (considering that high expression of epithelial–mesenchymal components in cancer is related to tumour progression and metastasis).55 Although we also examined the relationship of such modifications with tumour metastatic potential (Supporting Information, Figures S2 and S3), we failed to observe any differences. Moreover, bearing in mind that epithelial–mesenchymal components plays a role in cell survival and chemoresistance in cancer, we hypothesise that this finding could be associated with therapy resistance, because of augmented fibrosis in the tumours (as caused by persistent micro‐environment inflammation) of cachectic patients.56, 57
Furthermore, an increasing body of evidence shows that TGF‐β signalling is involved in EMT regulation.10, 58, 59 Recent studies suggest TGF‐β signalling may lead to muscle atrophy by a mechanism which is ROS dependent25 and plays a central role in fibrosis.60 TGF‐β signals can activate SMAD‐dependent canonical pathway,61 as well as induce the JNK/p38 MAPK signalling pathway (non‐canonical).62 However, the signalling pathway underlying TGF‐β effect upon ECM expression in the tumour of cachectic patients has not been previously identified. We also found that all isoforms of TGF‐β were increased (as compared with WSC) in the tumour samples obtained from the cachectic patients. These results confirm the interaction between TGF‐β and EMT in the tumour of CC. Increased TGF‐β expression was found to be unrelated to higher phosphorylation of SMAD, and hence, we chose to examine the non‐canonical MAPK signalling pathway. Higher expression of p38, JNK, and MEK1 was found in the samples of CC, as well as increased levels of transcription factors, such as of STAT‐1, which are involved in regulating the aggressiveness of the tumour63 and may be important for tumour resistance and immune evasion in cachectic patients. The role of STAT‐1 in cancer is unclear. Previous studies showed that STAT‐1 could present an oncogenic and tumour‐suppressing role in cancer.64 Our results show an increased expression of STAT‐1 in the tumours of cachectic patients. Recently, we demonstrated higher expression of STAT‐1 in the adipose tissue of cachectic patients, which was correlated with increased inflammation, as compared with WSC.20 STAT‐1 has been shown to present an anti‐tumourigenic and anti‐proliferative role in both human and murine studies,65 while other studies show that the increase of STAT‐1 is related to oesophageal cancer invasion associated with p53 mutation, tumour growth, and immune evasion.63, 66 Tauriello et al. (2018) showed that increased content of TGF‐β in the tumour micro‐environment could represent a primary mechanism for immune evasion.67 We also report lower levels of tumour suppressor p53 in CC, when compared with WSC. It is known that p53 dysfunction supports tumour‐immune evasion and inflammatory response.36 Our data indicate that high levels of TGF‐β in the tumour are associated with the synthesis and secretion of extracellular matrix components through non‐canonical MAPK signalling pathway and lead to EMT process in the tumour of cachectic patients, contributing to tumour malignancy, aggressiveness phenotype, unbalanced inflammatory response, and poor prognosis. KRAS mutations are related to drivers linked to aggressiveness in cancer and, consequently, correlate with poor prognosis. A recent study showed that MAPK signalling is constitutively active in KRAS‐mutated colorectal cancer,68 and other studies have shown that tumours with mutated KRAS were associated with an increased risk of weight loss.18, 69 Unfortunately, we did not examine tumour genetic profile; hence, we cannot discard an interference of this parameter in our results.
Hypoxia plays an important part in malignancy by modulating angiogenesis. Several studies point out to the fact that p38 activation may also directly induce hypoxia‐associated activation of HIF‐1α in cancer cells, leading to angiogenesis, as well as inducing VEGF expression.70, 71 As we found increased levels of VEGF in the tumour of cachectic patients, we next proceeded to identify other markers that could be involved in angiogenesis. Immunohistochemistry analysis shown an increase of CD34+ (microvascular density) in the patients. In the recent years, many studies have demonstrated that CD34+ blood vessels play a prominent part in tumour development and influence malignant severity.72, 73, 74 Tichet et al.75 reported that tumour cell‐derived SPARC instigates the disruption of the endothelial barrier by vascular cell adhesion molecule 1 and activation of p38 MAPK‐mediated signalling pathways. The present results show increase of SPARC protein levels in the tumour of cachectic patients. Interestingly, Koukourakis et al.76 showed that high levels of SPARC in the tumour are correlated with hypoxia and worsened prognosis. Higher levels of oncostantin M in CC, as compared with WSC, a member belonging to the IL‐6 superfamily, were found in the tumour of CC. These data are in accordance with a role of oncostatin M in stimulating angiogenesis and tumour malignancy.77 Finally, we found high mRNA levels of HIF‐1α in tumour of cachectic patients, suggesting that activation of p38 under hypoxia might be involved in angiogenesis during cachexia development. To the best of our knowledge, this is the first study to demonstrate that inflammation up‐regulation contributes to fibrosis, by inducing increased TGF‐β signalling in the tumour of cachectic patients as compared with the WSC counterparts. These changes in the tumour micro‐environment might be correlated with changes in peripheral organs and tissues, as previously reported that there is a significant correlation between tumour and cytokine content in the adipose tissue.5
Several limitations of this study should be considered. The number of patients was not the same between the groups (31 for WSC patients and 43 for cachectic patients), as we sought to match group patients in terms of tumour type and gender (thereby limiting the inclusion of potential participants). Similarly, not all determinations were carried out with the same sample number, as the material was scarce and thus divided among experiments. Another shortcoming was not having measured vascular permeability (nor to directly demonstrate SPARC involvement) and ROS production. Further analysis of tumour genotype needs to be performed.
Conclusions
The results obtained in this study indicate that up‐regulation of inflammation contributes to fibrosis in the tumour in cachexia. Tumour tissue remodelling, mainly by the transdifferentiation of fibroblasts to myofibroblasts (not directly assessed) induced by TGF‐β, leads to unbalanced inflammatory cytokine profile, angiogenesis, and EMC remodelling, in which hypoxia seems to play a major role. Enhanced TGF‐β signalling through p38 MAPK pathways induces fibrosis within the tumour micro‐environment of cachectic patients, as compared with tumours of weight‐stable patients. This process is summarized in Figure 6. The present results further add to the understanding of the mechanisms involved in the disruption of endothelial barrier during cachexia. Furthermore, we propose that TGF‐β signalling in tumour tissue may be viewed as a novel mechanism underlying tumour resistance that could potentially reduce the efficacy of anticancer treatment in cachectic cancer patients.
Figure 6.
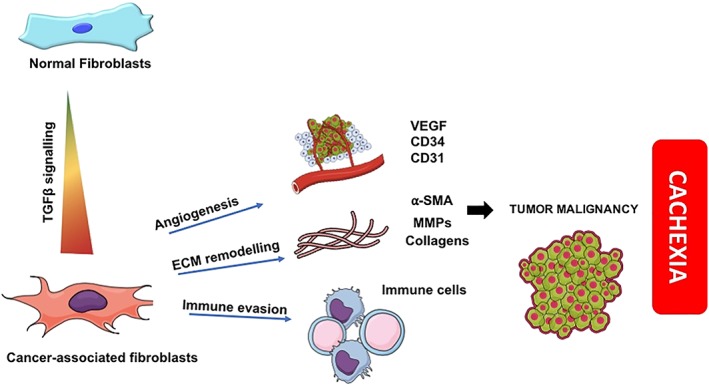
Summary of transforming growth factor‐β (TGF‐β) signalling in the tumour micro‐environment in cachexia. MMPs, matrix metalloproteinases; VEGF, vascular endothelial growth factor; α‐smooth muscle actin.
Conflict of interest
None declared.
Funding
This work was supported by the São Paulo Research Foundation (FAPESP; 2012/50079‐0, 2016/04000‐3 and 2016/08726‐1), Coordination for the Improvement of Higher Education Personnel (CAPES), and National Council for Scientific and Technological Development (CNPQ).
Author contributions
J.D.C.C.L. and M.S. designed the experiments. J.D.C.C.L. conducted all the experiments. E.M.N., E.S., and R.F. help in classification of the patients and biochemical parameters. Histological experiments were performed together with M.R.P.T.M. and T.M.T.Z. Multiplex cytokine assay was employed with E.S. and G.D.C., whereas N.I.P. and M.J.A. help in the TGF‐β analysis. E.S. help with Western blot analysis. A.S.F.‐S., F.T., J.P.O., and P.A. contributed to the patient recruitment and medical support. F.J.C., E.S.F, A.L and M.S. supervised the study. M.S. directed the study. All authors contributed to sample and blood collection and data analysis. All authors reviewed the manuscript.
Supporting information
Figure S1. Controls for antibody staining reactions. Collagen III on healthy colon mucosa (positive control) and colon cancer (negative control); CD34 on healthy colon mucosa and on colon tumour (negative control); alpha‐SMA on smooth‐muscle tissue (positive control) and colon tumour (negative control).
Figure S2. Expression of EMC components and tumour tage. Gene expression of components to extracellular matrix and tumour stage. Data expressed as mean ± SD. * Significant difference between the groups was tested using Unpaired T test. MMP2: matrix metalloproteinases 2; MMP9: matrix metalloproteinases 9; COL1A: Type I collagen; COL3A: Type III collagen. I‐II: Initial tumour stage and III‐IV: Advanced tumour stage
Figure S3. Expression of TGFβ and tumour stage. Protein expression of TGFβ isoforms and tumour stage. Data expressed as mean ± SD. * Significant difference between the groups was tested using Unpaired T test. Type III collagen. I‐II: Initial tumour stage and III‐IV: Advanced tumour stage.
Acknowledgements
The authors thank the members of the Cancer Metabolism Group for their helpful discussions and Professor Patricia Gama (Biology of Digestive Epithelia Group) for her support. We thank Emilia Ribeiro, Fernanda Barrence and Ivanir Pires for their technical assistance, as well as the clinicians, surgeons and pathologists for their support. Finally, we thank the patients and their families. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle. 78
Lima J. D. C. C., Simoes E., De Castro G., Morais M. R. P. T., de Matos‐Neto E. M., Alves M. J., Pinto N. I., Figueredo R. G., Zorn T. M. T., Felipe‐Silva A. S., Tokeshi F., Otoch J. P., Alcantara P., Cabral F. J., Ferro E. S., Laviano A., and Seelaender M. (2019) Tumour‐derived transforming growth factor‐β signalling contributes to fibrosis in patients with cancer cachexia, Journal of Cachexia, Sarcopenia and Muscle, 10: 1045–1059. 10.1002/jcsm.12441.
References
- 1. Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogene 2016;5:e200–e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mondello P, Lacquaniti A, Mondello S, Bolignano D, Pitini V, Aloisi C, et al. Emerging markers of cachexia predict survival in cancer patients. BMC Cancer 2014;14:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Donohoe CL, Ryan AM, Reynolds JV. Cancer cachexia: mechanisms and clinical implications. Gastroenterol Res Pract 2011;2011:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vanderveen BN, Fix DK, Carson JA. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev 2017;2017:24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De Matos‐Neto EM, Lima JDCC, de Pereira WO, Figuerêdo RG, Riccardi DMDR, Radloff K, et al. Systemic inflammation in cachexia—is tumor cytokine expression profile the culprit? Front Immunol 2015;6:629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 7. Seelaender M, Laviano A, Busquets S, Puschel GP, Margaria T, Batista ML. Inflammation in cachexia. Mediators Inflamm 2015;2015:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molfino A, Formiconi A, Fanelli FR, Muscaritoli M. Ghrelin: from discovery to cancer cachexia therapy. Curr Opin Clin Nutr Metab Care 2014;17:471–476. [DOI] [PubMed] [Google Scholar]
- 9. Laviano A, Seelaender M, Rianda S, Silverio R, Fanelli FR. Neuroinflammation: a contributing factor to the pathogenesis of cancer cachexia. Crit Rev Oncog 2012;17:247–252. [DOI] [PubMed] [Google Scholar]
- 10. Alves MJ, Figuerêdo RG, Azevedo FF, Cavallaro DA, Neto NIP, Lima JDC, et al. Adipose tissue fibrosis in human cancer cachexia: the role of TGFβ pathway. BMC Cancer 2017;17:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vaitkus JA, Celi FS. The role of adipose tissue in cancer‐associated cachexia. Exp Biol Med 2017;242:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Batista ML, Henriques FS, Neves RX, Olivan MR, Matos‐Neto EM, Alcântara PSM, et al. Cachexia‐associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J Cachexia Sarcopenia Muscle 2016;7:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsoli M, Swarbrick MM, Robertson GR. Lipolytic and thermogenic depletion of adipose tissue in cancer cachexia. Semin Cell Dev Biol 2016;54:68–81. [DOI] [PubMed] [Google Scholar]
- 14. Hardee JP, Montalvo RN, Carson JA. Linking cancer cachexia‐induced anabolic resistance to skeletal muscle oxidative metabolism. Oxid Med Cell Longev 2017;2017:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee DE, Brown JL, Rosa‐Caldwell ME, Blackwell TA, Perry RA, Brown LA, et al. Cancer cachexia‐induced muscle atrophy: evidence for alterations in microRNAs important for muscle size. Physiol Genomics 2017;49:253–260. [DOI] [PubMed] [Google Scholar]
- 16. Dasgupta A, Shukla SK, Gunda V, Chaika NV, Singh PK. Abstract LB‐267: metabolic alterations in tumors cause cachexia in pancreatic cancer. Cancer Res 2017; Jul;77. LB‐267 LP‐LB‐267. [Google Scholar]
- 17. Petruzzelli M, Wagner EF. Mechanisms of metabolic dysfunction in cancer‐associated cachexia. Genes Dev 2016;30:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shiono M, Huang K, Downey RJ, Consul N, Villanueva N, Beck K, et al. An analysis of the relationship between metastases and cachexia in lung cancer patients. Cancer Med 2016;5:2641–2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med 2018;24:770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Neto NIP, Murari ASP, Oyama LM, Otoch JP, Alcântara PS, Tokeshi F, et al. Peritumoural adipose tissue pro‐inflammatory cytokines are associated with tumoural growth factors in cancer cachexia patients. J Cachexia Sarcopenia Muscle 2018;9:1101–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nature Communications 2017;8(1). 10.1038/s41467-017-00726-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Patel HJ, Patel BM. TNF‐α and cancer cachexia: molecular insights and clinical implications. Life Sci 2017;170:56–63. [DOI] [PubMed] [Google Scholar]
- 23. Ma JF, Sanchez BJ, Hall DT, Tremblay AK, Di Marco S, Gallouzi I. STAT3 promotes IFNγ/TNFα‐induced muscle wasting in an NF‐κB‐dependent and IL‐6‐independent manner. EMBO Mol Med 2017;9:622–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Loumaye A, De Barsy M, Nachit M, Lause P, Frateur L, Van Maanen A, et al. Role of activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015;100:2030–2038. [DOI] [PubMed] [Google Scholar]
- 25. Ábrigo J, Campos F, Simon F, Riedel C, Cabrera D, Vilos C, et al. TGF‐β requires the activation of canonical and non‐canonical signalling pathways to induce skeletal muscle atrophy. Biol Chem 2018;399:253–264. [DOI] [PubMed] [Google Scholar]
- 26. Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, et al. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid Med Cell Longev 2016;2016:1–9, http://www.hindawi.com/journals/omcl/2016/1616781/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Anestakis D, Petanidis S, Kalyvas S, Nday CM, Tsave O, Kioseoglou E, et al. Mechanisms and αpplications of ιnterleukins in cancer immunotherapy. Int J Mol Sci 2015;16:1691–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang Y‐A, Chen Y, Bao Y, Mahara S, Yatim SM, Oguz G, et al. Hypoxic tumor microenvironment activates GLI2 via HIF‐1α and TGF‐β2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci 2018;115:E5990–E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batista ML, Peres SB, McDonald ME, Alcantara PSM, Olivan M, Otoch JP, et al. Adipose tissue inflammation and cancer cachexia: possible role of nuclear transcription factors. Cytokine 2012;57:9–16. [DOI] [PubMed] [Google Scholar]
- 30. Camargo RG, dos Reis Riccardi DM, Ribeiro HQT, Carnevali LC, de Matos‐Neto EM, Enjiu L, et al. Nf‐κBp65 and expression of its pro‐inflammatory target genes are upregulated in the subcutaneous adipose tissue of cachectic cancer patients. Nutrients 2015;7:4465–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hinz B. The extracellular matrix and transforming growth factor‐β1: tale of a strained relationship. Matrix Biol 2015;47:54–65. [DOI] [PubMed] [Google Scholar]
- 32. Papageorgis P. TGFβ signaling in tumor initiation, epithelial‐to‐mesenchymal transition, and metastasis. J Oncol 2015;2015:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bauché D, Marie JC. Transforming growth factor β: a master regulator of the gut microbiota and immune cell interactions. Clin Trans Immunol 2017;6:e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamio K, Azuma A, Usuki J, Matsuda K, Inomata M, Nishijima N, et al. XPLN is modulated by HDAC inhibitors and negatively regulates SPARC expression by targeting mTORC2 in human lung fibroblasts. Pulm Pharmacol Ther 2017;44:61–69. [DOI] [PubMed] [Google Scholar]
- 35. Ahir BK, Elias NM, Lakka SS. SPARC overexpression alters microRNA expression profiles involved in tumor progression. Genes Cancer 2017;8:453–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Z, Wang S, Lin Y, Miao Y, Zeng Y, Nie Y, et al. Epithelial–mesenchymal transition in cancer: role of the IL‐8/IL‐8R axis. Oncol Lett 2017;13:4577–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu M, Chen X, Lou J, Zhang S, Zhang X, Huang L, et al. TGF‐β1 contributes to CD8+ Treg induction through p38 MAPK signaling in ovarian cancer microenvironment. Oncotarget 2016;7:44534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, et al. The European Organization for Research and Treatment of Cancer QLQ‐C30: a quality‐of‐life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 1993;85:365–376. [DOI] [PubMed] [Google Scholar]
- 39. Arezzo di Trifiletti A, Misino P, Giannantoni P, Giannantoni B, Cascino A, Fazi L, et al. Comparison of the performance of four different tools in diagnosing disease‐associated anorexia and their relationship with nutritional, functional and clinical outcome measures in hospitalized patients. Clin Nutr 2013;32:527–532. [DOI] [PubMed] [Google Scholar]
- 40. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐ΔΔCT method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 41. Ehrmann‐Josko A, Sieminska J, Gornicka B, Ziarkiewicz‐Wroblewska B, Ziolkowski B, Muszynski J. Impaired glucose metabolism in colorectal cancer. Scand J Gastroenterol 2006;41:1079–1086. [DOI] [PubMed] [Google Scholar]
- 42. Hou Y‐C, Wang C‐J, Chao Y‐J, Chen H‐Y, Wang H‐C, Tung H‐L, et al. Elevated serum interleukin‐8 level correlates with cancer‐related cachexia and sarcopenia: an indicator for pancreatic cancer outcomes. J Clin Med 2018;7:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palena C, Hamilton DH, Fernando RI. Influence of IL‐8 on the epithelial–mesenchymal transition and the tumor microenvironment. Future Oncol 2013;8:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Papageorgis P, Stylianopoulos T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (review). Int J Oncol 2015;46:933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sesterhenn A, Szalay A, Zimmermann A, Werner J, Barth P, Wiegand S. Stellenwert der Autopsie bei Patienten mit Kopf‐Halstumoren. Laryngorhinootologie 2012. Jun;91:375–380. [DOI] [PubMed] [Google Scholar]
- 46. Lira FS, Yamashita AS, Rosa JC, Koyama CH, Caperuto EC, Batista ML, et al. Exercise training decreases adipose tissue inflammation in cachectic rats. Horm Metab Res 2012;44:91–98. [DOI] [PubMed] [Google Scholar]
- 47. Long X, Ye Y, Zhang L, Liu P, Yu W, Wei F, et al. IL‐8, a novel messenger to cross‐link inflammation and tumor EMT via autocrine and paracrine pathways (review). Int J Oncol 2016;48:5–12. [DOI] [PubMed] [Google Scholar]
- 48. Neveu B, Moreel X, Deschênes‐Rompré M‐P, Bergeron A, LaRue H, Ayari C, et al. IL‐8 secretion in primary cultures of prostate cells is associated with prostate cancer aggressiveness. Res reports Urol 2014;6:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lane D, Matte I, Rancourt C, Piché A. Prognostic significance of IL‐6 and IL‐8 ascites levels in ovarian cancer patients. BMC Cancer 2011;11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shi J, Wei PK. Interleukin‐8: a potent promoter of angiogenesis in gastric cancer. Oncol Lett 2016;11:1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jenkins MH, Brinckerhoff CE, Mullins DW. CXCR3 Signaling in BRAFWT melanoma increases IL‐8 expression and tumorigenicity. PLoS ONE 2015;10:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen Y, Zhao Z, Chen Y, Lv Z, Ding X, Wang R, et al. An epithelial‐to‐mesenchymal transition‐inducing potential of granulocyte macrophage colony‐stimulating factor in colon cancer. Sci Rep [Internet 2017;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yazdani S, Bansal R, Prakash J. Drug targeting to myofibroblasts: implications for fibrosis and cancer. Adv Drug Deliv Rev 2017;121:101–116. [DOI] [PubMed] [Google Scholar]
- 54. Manousopoulou A, Hayden A, Mellone M, Garay‐Baquero DJ, White CH, Noble F, et al. Quantitative proteomic profiling of primary cancer‐associated fibroblasts in oesophageal adenocarcinoma. Br J Cancer 2018;118:1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gleave ME, Hollier BG, Sadowski MC, Nelson CC, Ward M, Gunter JH, et al. A molecular portrait of epithelial–mesenchymal plasticity in prostate cancer associated with clinical outcome. Oncogenesis 2018. [Google Scholar]
- 56. Jung I, Fetyko A, Gurzu S, Silveanu C, Kovacs Z, Butiurca V. Systematic review of the old and new concepts in the epithelial–mesenchymal transition of colorectal cancer. World J Gastroenterol 2016;22:6764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sato M, Shames DS, Hasegawa Y. Emerging evidence of epithelial‐to‐mesenchymal transition in lung carcinogenesis. Respirology 2012;17:1048–1059. [DOI] [PubMed] [Google Scholar]
- 58. Petanidis S, Kioseoglou E, Domvri K, Zarogoulidis P, Carthy JM, Anestakis D, et al. In vitro and ex vivo vanadium antitumor activity in (TGF‐β)‐induced EMT. Synergistic activity with carboplatin and correlation with tumor metastasis in cancer patients. Int J Biochem Cell Biol 2016;74:121–134. [DOI] [PubMed] [Google Scholar]
- 59. Oruqaj G, Karnati S, Vijayan V, Kotarkonda LK, Boateng E, Zhang W, et al. Compromised peroxisomes in idiopathic pulmonary fibrosis, a vicious cycle inducing a higher fibrotic response via TGF‐β signaling. Proc Natl Acad Sci 2015;112:E2048–E2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ghafoory S, Varshney R, Robison T, Kouzbari K, Woolington S, Murphy B, et al. Platelet TGF‐β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv 2018;2:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ahmed S, Bradshaw A‐D, Gera S, Dewan M, Xu R. The TGF‐β/Smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med 2017;6:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cheruku HR, Mohamedali A, Cantor DI, Tan SH, Nice EC, Baker MS. Transforming growth factor‐β, MAPK and Wnt signaling interactions in colorectal cancer. EuPA Open Proteom 2015;8:104–115. [Google Scholar]
- 63. Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid‐derived suppressor cells. J Biol Chem 2013;288:11676–11688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meissl K, Macho‐Maschler S, Müller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017;89:12–20. [DOI] [PubMed] [Google Scholar]
- 65. Dimco G, Knight RA, Latchman DS, Stephanou A. STAT1 interacts directly with cyclin D1/Cdk4 and mediates cell cycle arrest. Cell Cycle 2010;9:4638–4649. [DOI] [PubMed] [Google Scholar]
- 66. Wong GS, Lee J, Park Y, Klein‐szanto AJ, Waldron TJ, Cukierman E, et al. Periostin cooperates with mutant p53 to mediate invasion through the induction of STAT1 signaling in the esophageal tumor microenvironment. Oncogenesis 2013;2:e59–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tauriello DVF, Palomo‐Ponce S, Stork D, Berenguer‐Llergo A, Badia‐Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 68. Haling JR, Friedman LS, Hewitt JFM, Ultsch M, Anderson DJ, Ludlam MJC, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS‐ versus BRAF‐driven cancers. Nature 2013;501:232–236. [DOI] [PubMed] [Google Scholar]
- 69. Miller A, McLeod L, Alhayyani S, Szczepny A, Watkins DN, Chen W, et al. Blockade of the IL‐6 trans‐signalling/STAT3 axis suppresses cachexia in Kras‐induced lung adenocarcinoma. Oncogene 2017;36:3059–3066. [DOI] [PubMed] [Google Scholar]
- 70. Koul HK, Pal M, Koul S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013;4:342–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carbajo‐Pescador S, Ordoñez R, Benet M, Jover R, García‐Palomo A, Mauriz JL, et al. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti‐angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer 2013;109:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Balani S, Nguyen LV, Eaves CJ. Modeling the process of human tumorigenesis. Nat Commun 2017;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kong X, Guan J, Ma W, Li Y, Xing B, Yang Y, et al. CD34 over‐expression is associated with gliomas' higher WHO grade. Medicine 2016;95:e2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Majchrzak K, Kaspera W, Szymaś J, Bobek‐Billewicz B, Hebda A, Majchrzak H. Markers of angiogenesis (CD31, CD34, rCBV) and their prognostic value in low‐grade gliomas. Neurol Neurochir Pol 2013;47:325–331. [DOI] [PubMed] [Google Scholar]
- 75. Tichet M, Prodhomme V, Fenouille N, Ambrosetti D, Mallavialle A, Cerezo M, et al. Tumour‐derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signalling to promote metastasis. Nat Commun 2015;6:6993. [DOI] [PubMed] [Google Scholar]
- 76. Koukourakis MI, Giatromanolaki A, Brekken RA, Sivridis E, Gatter KC, Harris AL, et al. Enhanced expression of SPARC/osteonectin in the tumor‐associated stroma of non‐small cell lung cancer is correlated with markers of hypoxia/acidity and with poor prognosis of patients. Cancer Res 2003;63:5376–5380. [PubMed] [Google Scholar]
- 77. Lauber S, Wong S, Cutz JC, Tanaka M, Barra N, Lhoták S, et al. Novel function of oncostatin M as a potent tumour‐promoting agent in lung. Int J Cancer 2015;136:831–843. [DOI] [PubMed] [Google Scholar]
- 78. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Controls for antibody staining reactions. Collagen III on healthy colon mucosa (positive control) and colon cancer (negative control); CD34 on healthy colon mucosa and on colon tumour (negative control); alpha‐SMA on smooth‐muscle tissue (positive control) and colon tumour (negative control).
Figure S2. Expression of EMC components and tumour tage. Gene expression of components to extracellular matrix and tumour stage. Data expressed as mean ± SD. * Significant difference between the groups was tested using Unpaired T test. MMP2: matrix metalloproteinases 2; MMP9: matrix metalloproteinases 9; COL1A: Type I collagen; COL3A: Type III collagen. I‐II: Initial tumour stage and III‐IV: Advanced tumour stage
Figure S3. Expression of TGFβ and tumour stage. Protein expression of TGFβ isoforms and tumour stage. Data expressed as mean ± SD. * Significant difference between the groups was tested using Unpaired T test. Type III collagen. I‐II: Initial tumour stage and III‐IV: Advanced tumour stage.